Article Text

Abstract
Background Retinitis pigmentosa in combination with hearing loss can be a feature of different Mendelian disorders. We describe a novel syndrome caused by biallelic mutations in the ‘exosome component 2’ (EXOSC2) gene.
Methods Clinical ascertainment of three similar affected patients followed by whole exome sequencing.
Results Three individuals from two unrelated German families presented with a novel Mendelian disorder encompassing childhood myopia, early onset retinitis pigmentosa, progressive sensorineural hearing loss, hypothyroidism, short stature, brachydactyly, recognisable facial gestalt, premature ageing and mild intellectual disability. Whole exome sequencing revealed homozygous or compound heterozygous missense variants in the EXOSC2 gene in all three patients. EXOSC2 encodes the ‘ribosomal RNA-processing protein 4’ (RRP4)—one of the core components of the RNA exosome. The RNA exosome is a multiprotein complex that plays key roles in RNA processing and degradation. Intriguingly, the EXOSC2-associated phenotype shows only minimal overlap with the previously reported diseases associated with mutations in the RNA exosome core component genes EXOSC3 and EXOSC8.
Conclusion We report a novel condition that is probably caused by altered RNA exosome function and expands the spectrum of clinical consequences of impaired RNA metabolism.
- Clinical genetics
- EXOSC2
- RNA processing
- exosome component 2 protein
- novel Mendelian disease
Statistics from Altmetric.com
Introduction
A broad application of whole exome sequencing (WES) has revolutionised the field of rare genetic disease research by facilitating the discovery of the causative genes.1 However, highly heterogenic disorders, such as intellectual disability (ID), remain challenging with the diagnostic yield rarely exceeding 30%.2 Early studies of WES-based discovery were focused on homogenous groups of patients with a well-defined phenotype,3 demonstrating the power of WES in gene discovery for rare monogenic disorders even if WES is applied in small but well-characterised cohorts.4
In this study, we describe a novel Mendelian disease and associate it with recessive mutations in the EXOSC2 gene.
Material and methods
Human subjects
Within two unrelated German families we ascertained three patients with an overlapping phenotype that is not consistent with any known Mendelian disorders. The patients underwent a standard evaluation in a medical genetics facility; all individuals participating in this study have signed a written informed consent and agreed to publication of the results. The patients were ascertained in two adjacent genetic clinics in Dresden, Germany. Striking facial minor anomalies in patient 1 strongly resembling her affected aunt (patient 2, II.3 at figure 1A) forced the family to seek genetic counselling. The phenotypic similarities with patient 3 were recognised by NDD as patient 3 was presented during a regular discussion of unsolved cases.


Pedigrees, segregation, localisation and modelling of the EXOSC2 mutations are shown. (A) family 1: III.1—patient 1, II.3—patient 2; (B) family 2: II.1—patient 3. Solid square (male) and solid circles (female) indicate the affected individuals. A single-letter code for amino acids and International Union of Pure and Applied Chemistry (IUPAC) ambiguity code for nucleotides are used to indicate the mutations in the EXOSC2 gene. (C) Domain organisation of the EXOSC2 protein (RRP4) with the localisation of the discovered mutations is shown. ExAC represents the frequency of the mutation on the Exome Aggregation Consortium; CADD shows a PHRED-like scaled C-score; a score of ≥20 indicated the 1% most deleterious substitutions; SNAP—pathogenicity prediction by SNAP2 on a scale from −100 (neutral) to +100 (predicted functional effect). Schematic representation of the secondary structure: α helix (green), β turns dark grey and turns orange. Predicted protein domains (Pfam database) are shown in blue (N-terminal domain) and red boxes (KH). (D) Structural modelling of the mutation p.Gly30Val: three-dimensional representation of the native (left) and mutant (right) EXOSC2 protein (yellow) in complex with the EXOSC4 (white) is shown. Key amino acids are represented as sticks coloured by atoms. The mutation p.Gly30Val (cyan) would shorten the interatomic distance between this EXOSC2 residue and the neighbouring amino acids of EXOSC4 (Arg153, Asp154 and Phe155, dark grey). Interatomic distances are represented in coloured dashes: wild-type 5.2–4.5Å (left) and mutant 3.5–2.2Å/3.1–2.7Å (right). The side chain of the mutant Val30 amino acid requires more molecular interspace than the wild-type Gly30 and may conflict with the molecular surface of the Asp154 of EXOSC4. (E) Structural modelling of the mutation p.Gly198Asp: molecular structure of a section of the EXOSC2 KH domain is shown. The wild-type residue Gly198 (pink, left) is located at the end of a β-strand which is composed from AA 193 to 198 (green). The change from Gly to Asp (pink, right) would shorten the wild-type β-strand from six to four amino acids (left panel: wild-type AA 193 to 198, right panel: mutant AA 193 to 196). Thereby the whole β-hairpin structure would be affected by this change; the intermediate turn (dark grey) would be extended from three to four amino acids (left panel: wild-type AA 199 to 201, right panel: mutant AA 197 to 200) and the antiparallel β-strand would also extend from five to six amino acids (left panel: wild-type AA 202 to 206, right panel: mutant AA 201 to 206). 1000G, 1000 Genome project; CADD, Combined Annotation Dependent Depletion; KH, K homology domain type 1; PP2, pathogenicity prediction by PolyPhen2.
Conventional and molecular karyotyping
After excluding numeric and gross structural chromosomal aberrations by conventional karyotyping (550 band-resolution), we performed microarray analysis for all three patients by hybridising labelled genomic DNA from venous blood to Agilent 2×400 K genomic arrays, following standard and manufacturer’s recommendations (Agilent Technologies, Santa Clara, California, USA).
Whole exome sequencing
The exomes of five individuals (three affected patients; two unaffected parents of patient 1) were enriched from total genomic DNA, using either the ‘Agilent Human All Exon 50 Mb’ kit or the ‘Illumina Nextera Rapid Capture Expanded Exome’ kit. Subsequently, paired-end sequencing (2×100 nt) was performed on the Illumina HiSeq-2000 sequencer. For each sample, over 90% of the exome was covered at least 25-fold.
Data analysis
We analysed the data, assuming an autosomal recessive inheritance of the disease. The ‘Biomedical Genomics Workbench’ from Qiagen was used for alignment (mapping to UCSC hg19), variant identification (SNPs and indels), variant annotation and filtering.
Only rare non-synonymous coding variants (ExAC frequency <0.001) or variants predicted to have a splice effect were taken into consideration. The ‘Combined Annotation Dependent Depletion’ tool was used for scoring the deleteriousness of variants.5
Homozygosity mapping
We used ALLEGRO (V.1.2c)6 to identify overlapping runs of homozygosity (ROH; autosomal regions with two identical chromosomal copies). ROH can be detected by analysing the genotypes of informative single nucleodite variant (SNV) markers. In order to locate ROH, high quality (DP>15; QUAL>40; 0.38>AF<0.62; QD>5; MQ>55; FS>60; MQRankSum>−3.2; ReadPosRankSum>−8; HaplotypeScore<13) and informative (MAF <0.85) SNV calls were taken from the GATK UnifiedGenotyper programme and used as ALLEGRO (V.1.2c) input. ALLEGRO was run with a pseudo-consanguine pedigree of first-degree cousins relationship and a full penetrant autosomal recessive model with a disease allele frequency of 0.0001 (a model which is also known as homozygosity mapping). All regions with positive LOD scores (the maximal LOD score for the given model is ∼1.2) were extracted.
Kinship estimation was done using the kinship tool of the Varbank pipeline (https://varbank.ccg.uni-koeln.de) analysing the sharing proportion of the autosomal rare SNV calls (MAF <0.001 based on frequencies in the 1000 Genomes database). The sharing proportion between each two individuals was calculated in both directions and combined into a mean value. Due to the rare nature of the chosen SNVs, the percentage of shared rare variations, here called percSRV, is a direct reflection of kinship. Depending of the sequencing depth, total target size and error rate, the number of filter-passed SNVs derived by exome sequencing is sufficient to estimate kinship up to five generations back.
Variant verification and analysis of variant frequency
Verification and familial segregation analysis of candidate variants was accomplished by conventional Sanger sequencing. Allele-specific PCR was used to determine the frequency of our candidate variants within a cohort of 500 anonymous German control individuals.
Protein alignment and structural modelling
A multiple alignment of EXOSC2 amino acid sequences was done according to HomoloGene (NCBI) in order to assess the amino acid conservation of the detected variants in 21 species with homologous proteins.
Both mutations were modelled with the mutagenesis wizard of PyMOL molecular graphics software (http://www.pymol.org) by placing the most common side chain rotamer for the mutated residues. Predicted protein domains were extracted from the Pfam Database (http://pfam.xfam.org/).
Results
Clinical assessment
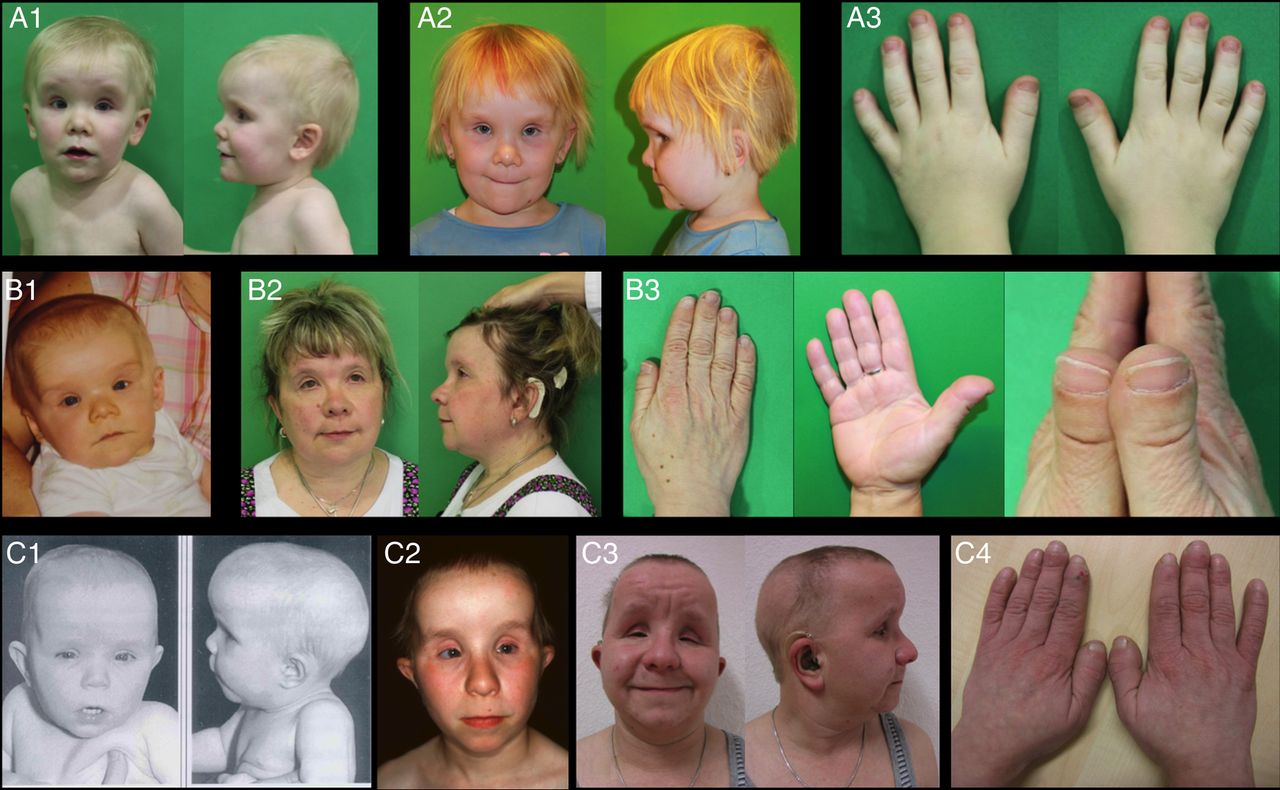
Patient 1 (family 1, figure 2A, figure 1A III.1 and supplementary table 1) was a 6-year-old girl who was referred to the clinical genetics facility due to developmental delay, short stature, brachydactyly with broad thumbs (see online supplementary figure S2A), myopia, hearing loss, sparse hair and minor facial anomalies. She did not closely resemble her parents, but rather strongly resembled her paternal aunt (figure 2A, B, figure 1A II.3). She showed relatively sparse and fine hair, high prominent forehead, deep set eyes, short upslanted palpebral fissures, short nose with concave nasal ridge, anteverted nares, wide nasal base with broad nasal tip and broad columella, long philtrum, thin upper lip and low set and posteriorly rotated ears. She also had broad terminal phalanges in all fingers, particularly in both thumbs. Brain MRI showed mildly enlarged extra-axial spaces and borderline cerebellar hypoplasia (see online supplementary figure S1A–C). The parents as well as other family members were healthy and unrelated.
{kind=link}
{kind=link}
{kind=link}
Clinical pictures of the patients. (A) Patient 1 at the age of 3 years (A1, A3) and at the age of 6 years and 4 months (A2); (B) Patient 2 at the age of 1 year (B1) and 41 years (B2, B3); (C) Patient 3 at the age of 1 year (C1), 13 years (C2) and 28 years (C3, C4). Note the minor anomalies present in all three patients: high prominent forehead, deep set eyes, short palpebral fissures, wide nasal base with broad nasal tip and broad columella, long philtrum, thin vermilion of the upper and lower lips and low set and posteriorly rotated ears. Nose was short with concave nasal ridge along with anteverted nares in all three patients, but only in the early childhood. Note the sparse hair in patients 1 and 3; additionally, patient 3 developed partial occipital alopecia (C3). Brachydactyly with broad terminal phalanges and especially broad thumbs was universally seen in all three patients (A3, B3 and C4).
Patient 2 (family 1, figures 1A II.3, 2B and supplementary table 1) is a paternal aunt of patient 1. She resembled patient 1 and presented with similar facial features, short stature (−3.5 SD), brachydactyly (see online supplementary figure S2B), progressive bilateral hearing loss (required cochlea implant), complex eye phenotype (severe myopia, retinitis pigmentosa (RP), corneal dystrophy and glaucoma), borderline ID and hypothyroidism. Facial minor anomalies included broad and prominent forehead, deep set eyes, short palpebral fissures, wide nasal base with broad nasal tip and broad columella, long philtrum, thin upper lip and low set ears. The patient showed mild brachydactyly with short and broad terminal phalanges and remarkably broad thumbs. Moreover, she looked significantly older than her biological age and required medication due to arterial hypertension before the fourth decade. Brain MRI at the age of 39 years showed mild cortical and cerebellar atrophy with unremarkable white matter (see online supplementary figure S1H,I).
Patient 3 (family 2, figure 2C, figure 1B II.1 and supplementary table 1) was first referred to genetic counselling during his first year of life because of congenital hypothyroidism and short stature not compensated by adequate thyroid hormone replacement. He also showed myopia, nystagmus, strabismus, multiple facial minor anomalies, brachydactyly with broad thumbs (see online supplementary figure S2C) and sparse hair. He was followed up as a teenager when he presented with multiple additional features such as progressive hearing loss, nyctalopia further diagnosed as RP, arterial hypertension, alopecia areata of the scalp and premature aged appearance. His facial features as well as hand anomalies strikingly resembled the gestalt of the patients 1 and 2 and included broad and prominent forehead, deep set eyes, short palpebral fissures with the reduction of the vertical distance between the upper and lower eyelids, wide nasal base with broad nasal tip and broad columella, long philtrum, thin upper lip and low set ears. Brain MRI was abnormal with diffuse dysmyelination, bilateral calcifications in the basal ganglia and thalamus and mild cortical and cerebellar atrophy (see online supplementary figure S1D–F). The history of family 2 was unremarkable with no evidence of consanguinity and no known relationship with family 1.
The pedigree of family 1, however, was unusual for a classic Mendelian inheritance (figure 1A), especially taking into account the non-consanguineous background of the German population.
Karyotyping and WES
Conventional karyotype analysis as well as high-resolution chromosomal microarray (copy number variantion (CNV) detection up to 5 kb) showed normal results in all three patients and thus ruled out gross genomic imbalances and cryptical structural aberrations as a cause of the disorder.
We proceeded with WES and analysed the resulting data, assuming a recessive inheritance mode. At first we performed a trio analysis (patient 1 plus parents) and filtered for rare variants which were (1) homozygous in the patient and heterozygous in the parents or (2) compound heterozygous in the patient with each parent being heterozygous for one of these variants. This left us with six mutated genes. Then we checked these genes in patient 2 (II.3 at figure 1A) and found that only three of them shared the variants observed in the niece. Finally, after analysing the unrelated patient 3, we were left with only one candidate gene: EXOSC2.
Familial segregation
All patients in this study share the missense variant NM_014285.5:c.89G>T (p.(Gly30Val)) in the first exon of EXOSC2 (chr9:133,569,267 G>T); patients 1 and 2 (III.1 and II.3 at figure 1A) were homozygous for this variant, whereas patient 3 was heterozygous. Additionally, patient 3 had a second heterozygous nucleotide exchange in EXOSC2, namely NM_014285.5:c.593G>A (p.(Gly198Asp)) in exon 7 (chr9:133,577,618 G>A).
Segregation analysis showed that all healthy family members of family 1 were heterozygous carriers of p.(Gly30Val) as well as the mother of patient 3. The second variant, p.(Gly198Asp), was inherited from the father (figure 1B).
Homozygosity mapping and kinship estimation
Homozygosity mapping in family 1 revealed only two ROH regions >500 kb shared by both affected individuals (III.1 and II.3): an 850 kb region on chromosome 6 (see online supplementary table S2) and a 7.7 Mb ROH block on chromosome 9 (rs1129169-rs35803302) that encompasses EXOSC2. Kinship estimation showed the expected degree of relationship between III.1 and II.3, who are first-degree relatives. The parents of patient 1 (figure 1A II.1 and II.2) revealed a percSRV score <1%, meaning that there is no consanguine relationship up to five generations back. Patient 3 was not related to family 1 (perscSRV <1% in comparison with patients 1 and 2).
Haplotype analysis
Neither patient 3 nor the parents of patient 1 (figure 1A II.1 and II.2) showed ROH blocks >500 kb. However, all three individuals shared a 6.7 Mb haplotype (rs28428946–rs35803302) that was present as a large, homozygous ROH block in patients 1 and 2. This haplotype included the c.89G>T (p.(Gly30Val)) mutation in EXOSC2.
Variant frequency in healthy controls
Allele-specific PCR detected one heterozygous carrier of the c.89G>T (p.(Gly30Val)) variant among 1000 local German control individuals, that is, the frequency of this allele is 0.0005. The second allele, c.593G>A (p.(Gly198Asp)) has not been found in any of the 1000 controls.
A retrospective analysis of WES data from over 100 unrelated patients with non-syndromic RP with unknown genetic cause did not reveal any additional mutations in EXOSC2.
Protein alignment and structural modelling
Multiple sequence alignment of the EXOSC2 N-terminal (NT) domain and K homology (KH) domain between 21 species showed that both mutated residues p.Gly30 and p.Gly198 are well conserved across nearly all species (see online supplementary figure S3).
The p.Gly30Val and p.Gly198Asp changes are located within the NT and the KH domains, respectively. The substitution of p.Gly30 by Valine would shorten the interatomic distance between this EXOSC2 residue and the neighbouring amino acids of EXOSC4 (Arg153, Asp154 and Phe155, dark grey). The side chain of the mutant Val30 amino acid requires more molecular interspace than the wild-type Gly30 and may conflict with the molecular surface of the Asp154 of EXOSC4 (figure 1D).
The wild-type residue Gly198 is located at the end of a β-strand which is composed from amino acids 193 to 198. The change from Gly to Asp would shorten the wild-type β-strand from six to four amino acids. Thereby the whole β-hairpin structure would be affected by this change (figure 1E).
Differential diagnoses
We also assessed the WES data for rare exonic or splice-site variants in genes causative of possible differential diagnoses (see online supplementary note) and found no variants with pathogenic potential.
Discussion
Novel Mendelian disorder
We describe a complex autosomal-recessive entity observed in three patients from two unrelated families. The specific combination of phenotypic features does not closely match any known Mendelian disorders. Although, some of the key clinical features occur in different combinations in other syndromes (Usher syndrome, Cockayne syndrome, full list under online supplementary note), none of these syndromes comprises all the key features. Furthermore, WES did not identify a mutation in any of the OMIM genes with an acknowledged disease association. It remains unclear if the MRI features seen in patient 3 are part of the syndrome, or a ‘double-trouble’ representing a form of the primary familial brain calcification, and are not related to the rest of the symptoms.
EXOSC2 mutations as a likely cause of the novel syndrome
The pattern of inheritance in family 1 is consistent with a recessive condition, however unusual, considering the non-consanguineous background of the family. After ruling out structural aberrations and gross genomic imbalances, we proceeded with WES and were able to identify an association of the disease with mutations in a novel gene, namely EXOSC2. EXOSC2 is encoding for RRP4—one of the three cap proteins of the RNA exosome. Mutations in other exosome components (EXOSC3 and EXOSC8) have already been shown to cause severe disability and lethality. Therefore, we considered EXOSC2 to be a convincing candidate gene in the context of ID. The absence of bi-allelic EXOSC2 mutations in the healthy family members (as far as they were available for testing) further supports the clinical relevance of EXOSC2.
Both EXOSC2 variants identified in this study are very rare, affect highly conserved amino acids in RRP4 and were predicted to be deleterious by four different in silico prediction programmes (figure 1C).
The residue p.Gly30 is strictly conserved from yeast to human and resides within the NT domain. Protein modelling predicted the change of the interatomic distance between the mutated EXOSC2 residue and neighbouring amino acids of EXOSC4 and, therefore, a destabilised EXOSC2–EXOSC4 interaction. A similar mechanism was proposed for the recurrent mutation p.Gly31Ala in the NT domain of EXOSC3.7 Our second EXOSC2 mutation, p.(Gly198Asp), resides in the β-strand of the hnRNP-(KH domain and leads to the change of the β-hairpin structure. Therefore, this amino acid change might alter the RNA recruiting and binding abilities of RNA exosome.
RNA exosome
The RNA exosome complex is the main cellular machinery responsible for degrading RNA molecules in 3′ to 5′ direction,8 as well as for processing and surveillance of a broad spectrum of coding and non-coding RNAs.9 The human RNA exosome consists of a non-catalytic core—a two-layered barrel-like structure of nine polypeptides (Exo-9) 9 and active nuclease(s). The upper layer is composed of a cap of three proteins (encoding genes in brackets): CSL4 (EXOSC1), RRP4 (EXOSC2) and RRP40 (EXOSC3). The cap rests on a ring of six other proteins: RRP41 (EXOSC4), RRP42 (EXOSC7), RRP45 (EXOSC9), RRP46 (EXOSC5), RRP43 (EXOSC8) and MTR3 (EXOSC6). This structural arrangement creates a continuous channel through Exo-9, which is necessary for the unwinding and binding of the single-stranded RNA—steps required for further degradation.10 An additional subunit RRP44 (DIS3) associates with Exo-9 to form the catalytically active Exo-10 complex that is ubiquitously present in the cytoplasm and nucleus of eukaryotic cells.8 In the nucleus, Exo-10 recruits an additional nuclease: RRP6 (EXOSC10).10 Finally, RNA exosome function requires two cofactors—the nuclear TRAMP complex 11 and the cytoplasmic SKI (superkiller) complex.12
So far three human Mendelian diseases have been associated with mutations in subunits or cofactors of the RNA exosome: the first one is pontocerebellar hypoplasia (PCH) with spinal motor neuron disease (PCH1B, OMIM 614678), which is caused by EXOSC3 deficiency.7 Homozygous missense mutations in EXOSC8 cause another type of PCH (PCH1C, OMIM 616081). Mutations in the genes encoding the proteins of the SKI complex lead to a tricho-hepato-enteric syndrome.13 ,14 Mutations in both EXOSC3 and EXOSC8 cause severe neurodegeneration associated with a significantly reduced lifespan (EXOSC3) or even death within the first years of life (EXOSC8).
EXOSC2 versus EXOSC3 and EXOSC8
Our three patients showed no neurological decline, neither in childhood nor during adult life. Brain MRI scans, however, showed mild brain and cerebellar atrophy in all three patients and abnormal white matter in patient 3 that suggested a mild asymptomatic demyelination. Other features observed in our patients, such as progressive changes in the retinal pigment epithelium and cochlear hair cells, have not been reported in patients with PCH1B and PCH1C.15 However, we cannot rule out that patients with PCH1B/C might also develop RP or other EXOSC2-related features if they survived the second or third decades. The facial gestalt of all patients in our study was very distinct already in early infancy and thus, remains clearly a specific, EXOSC2-related feature.
Taking into account the similarity, adjacent protein localisation and ubiquitous expression of EXOSC2 and EXOSC3, we were surprised that missense mutations with similar localisation in these genes are associated with so different phenotypes. According to the data from the GTEx Portal, EXOSC2 shows the highest level of expression in the cerebellar hemispheres, but our patients had only borderline cerebellar atrophy and no clinical signs of neurodegeneration. This might indicate that loss of RRP4 (EXOSC2) function in neuronal cells can be compensated by other components of the RNA exosome, for example, RRP40 (EXOSC3), but not vice versa. Both genes show very similar ubiquitous expression profile (GTEx). Knockdown of EXOSC3 in zebrafish embryos disrupts normal hindbrain development and strongly correlates with the human phenotype.7 Unfortunately, no animal models have been created for EXOSC2 so far.
Tissue-specific expression or cell-type-specific compensation mechanisms can be possible explanations of the remarkable differences in EXOSC2-related versus EXOSC3-related disorders. Different substrate specificity might be another reason for the different consequence of the EXOSC2 and EXOSC3 mutations. The substrate specificity has been demonstrated in the archaeal exosome with the Rrp4-exosome preferring poly(A)-RNA.16
Lack of additional patients with EXOSC2-related features
Since the facial gestalt is obvious and patients present with a unique set of anomalies, we believe that non-geneticists would also be able to recognise it. We ascertained two families in the same institution, but found no reports of similar cases in the literature. We could show a founder effect in the local German Saxonian population by the demonstration of the specific haplotype segregating with the p.Gly30Val mutation. Moreover, the variant c.89G>T (p.(Gly30Val)) had a considerably higher allele frequency in the local German population—0.0005 versus 0.00 002 shown in ExAC (both variants are absent in 1000 Genomes browser). However, the German c.89G>T frequency is still consistent with variant frequencies for rare Mendelian diseases (0.01 per 10 000).
EXOSC2 and premature ageing
The two adult patients in families 1 and 2 showed several aspects of premature ageing, such as aged skin appearance, arterial hypertension and diabetes type 2, as well as diffuse joint space narrowing around the carpal bones seen in patient 2. Over 60 years ago Peter Medawar proposed the first mutation accumulation ageing theory stating that mutated genes accumulate over time and promote ageing. This view was integrated into many subsequent theories.17 Many syndromes with a defective DNA damage response and genomic instability show multiple progeroid aspects resembling premature ageing. Defective RNA degradation with accumulation of unprocessed RNA products has not been discussed as a possible ageing mechanism so far. However, P. Brooks has suggested that the defects in transcription by both RNA polymerases II and I may be responsible for the severe growth and neurodevelopmental defects in patients with Cockayne syndrome.18 The observation of several ageing aspects in our patients with presumably impaired RNA degradation and consequent accumulation of abnormal RNA products may as well suggest the involvement of an abnormal RNA metabolism in the ageing process.
Summary and conclusion
In summary, we report an apparently novel syndrome of RP, hearing loss, hypothyroidism, premature ageing and distinctive facial gestalt associated with biallelic mutations in EXOSC2. WES helps to identify disease-causing genes even in pedigrees that at first glance do not suggest a classical Mendelian inheritance. The unique combination of the symptoms, as well as the distinct facial appearance should help identifying similarly affected patients and consequently expand our knowledge of this rare disorder.
Open access resources
UCSC Genome Browser—http://genome-euro.ucsc.edu/cgi-bin/hgGateway
GTEx Portal—http://www.gtexportal.org/home/
PolyPhen2—http://genetics.bwh.harvard.edu/pph2/
MutationTaster—http://www.mutationtaster.org/
Acknowledgments
The authors are grateful to the patients and their families for participating in this study and their permission to publish the results. Carl Gustav Carus Faculty of Medicine, TU Dresden and Medical Genetics Center Munich provided funding for the project. The analysis of exomes was performed on a high performance computer cluster (CHEOPS), located at regional data center of the University of Cologne (RRZK) and supported by the Deutsche Forschungsgemeinschaft (DFG). NDD was a recipient of the research fellowship grant provided by the Deutsche Forschungsgemeinschaft.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online table 1
- Data supplement 4 - Online table 2
Footnotes
Contributors NDD, TN, A-KK, AB and AR summarised the data and drafted the manuscript; NDD, A-KK, IN, BN, JS, IAG, SEP, WBD and AB collected and analysed specific clinical data; NDD, TN, KH, CK, AB-P, JA, ES and AR performed laboratory work and analysed the primary data; WB and BHFW provided data regarding additional retinitis pigmentosa cohorts; BK performed a haplotype analysis; AB-P and AMN made the protein modelling; HT performed homozygosity mapping and kinship estimation.
Funding This work was supported by the Deutsche Forschungsgemeinschaft, grant number DI 2170/2-2. Faculty of Medicine Carl Gustav Carus, TU Dresden and Medical Genetics Center Munich provided funding for the project.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethikkommission TU Dresden.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All available data are presented in the original article. No additional unpublished data were generated during this study.