Article Text

Abstract
Background Leukodystrophies are genetic white matter disorders affecting the formation or maintenance of myelin. Among the recently discovered genetic defects associated with leukodystrophies, several genes converge on a common mechanism involving protein transcription/translation and ER stress response.
Methods The genetic basis of a novel congenital leukodystrophy, associated with early onset spastic paraparesis, acquired microcephaly and optic atrophy was studied in six patients from three unrelated Ashkenazi-Jewish families. To this end we used homozygosity mapping, exome analysis, western blot (Hikeshi, HSF1-pS326 and b-actin) in patient fibroblasts, indirect immunofluorescence (HSP70 and HSF1) in patient fibroblasts undergoing heat shock stress, nuclear injection of plasmids expressing Hikeshi or EGFP in patient fibroblasts, in situ hybridization and Immunoblot analysis of Hikeshi in newborn and adult mouse brain.
Results All the patients were homozygous for a missense mutation, p.Val54Leu, in C11ORF73 encoding HSP70 nuclear transporter protein, Hikeshi. The mutation segregated with the disease in the families and was carried by 1:200 Ashkenazi-Jewish individuals. The mutation was associated with undetectable level of Hikeshi in the patients' fibroblasts and with lack of nuclear HSP70 during heat shock stress, a phenomenon which was reversed upon the introduction of normal human Hikeshi to the patients cells. Hikeshi was found to be expressed in central white matter of mouse brain.
Conclusions These data underscore the importance of Hikeshi for HSP70 relocation into the nucleus. It is likely that in the absence of Hikeshi, HSP70 cannot attenuate the multiple heat shock induced nuclear phenotypes, leaving the cells unprotected during heat shock stress. We speculate that the sudden death of three of the six patients following a short febrile illness and the life-threatening myo-pericarditis in the fourth are the result of excess extra-nuclear HSP70 level which initiates cytokine release or provide target for natural killer cells. Alternatively, nuclear HSP70 might play an active role in stressed cells protection.
- Neurology
Statistics from Altmetric.com
Introduction
Myelination, the tightly regulated process of lipid and protein deposition in a multilamellar membranous sheath by glial cells around axons, is of major importance during the development and maturation of the central nervous system. Leukodystrophies are genetic white matter disorders affecting the formation or maintenance of myelin with variable peripheral nervous system involvement.1 Clinical suspicion of leukodystrophy is often raised by the presence of combined abnormalities in muscle-tone and developmental delay or regression. The definitive diagnosis rests on MRI features, such as distribution and type of white matter abnormality (eg, hypomyelination), age of onset and accompanying features such as peripheral nervous system involvement, dental or skin abnormalities and deviations of head-size growth. Schiffman and Van der Knaap have suggested to classify white matter disease primarily into either hypomyelinating or those with prominent T2-hyperintensity of the white matter.2
The molecular basis of the leukodystrophies is unknown in about half of the patients despite the increasing rate of diagnosis enabled by next generation sequencing. Among the recently discovered genetic defects associated with leukodystrophies, several genes converge on a common mechanism involving protein transcription/translation (POLR3A, POLR3B and DARS), especially within the mitochondria (DARS2 and EARS2).3–6 During translation, the nascent polypeptide chain is exposed in a partially folded state for a prolonged period of time because the narrow ribosomal exit channel allows only α helix formation and prevents tertiary interactions.7 ,8 A further delay of the translation process, imposed by the above-mentioned mutant proteins, further exposes this inherent vulnerability, culminating in the accumulation of unfolded/misfolded proteins which trigger the ER unfolded protein stress response (UPR). In yet another leukodystrophy, Pelizaeus-Merzbacher disease, the accumulation of the mutant protein itself in the ER induces UPR,9 resulting in the apoptosis of oligodendrocytes.10 ,11 The clinical relevance of these cellular processes is demonstrated by the recurrent encephalopathic episodes associated with shallow breathing and lethal apneic spells observed in patients with leukodystrophy due to mitochondrial HSP60 chaperonopathy during acute febrile illnesses12 suggesting an abnormal response to heat shock stress.
Here, we present six patients from three unrelated Ashkenazi-Jewish (AJ) families with a distinct combination of congenital leukodystrophy, early onset spastic paraparesis, acquired microcephaly, optic atrophy and risk of early death. Whole exome sequencing led to the identification of a missense mutation in the HSP70 nuclear transporter gene C11ORF73 and subsequent experiments confirmed the pathogenicity of the mutation, thereby linking leukodystrophy with abnormal heat-shock stress response.
Patients
Patients A-II-1, A-II-2 and A-II-9, two males and a female, were born to distantly related AJ couple (figure 1A). The parents and their other six children were healthy. The affected children were born after uneventful pregnancies and deliveries. Initial symptoms consisted of feeding difficulty, eventually necessitating placement of feeding gastrostomy, irritability and increased muscle tone, which were noted at a few months of age. Significant developmental delay without regression was present in all of them. The oldest surviving patient, A-II-2, lived until age 15 years, and at the time of his death, had not achieved voluntary motor milestones or expressive language. Patient A-II-1 could stand with support and drink from a training cup at 5 years; at that age he understood simple commands and had an expressive vocabulary of few words. Head circumference was normal at birth in all the affected children but dropped to around −2SD by 5–8 months of age. Physical examination of the three patients was notable for spasticity, mainly of the lower limbs, with eventual contractures, optic atrophy (in patients A-II-1 and A-II-2), visual impairment and nystagmus. Patient A-II-2 had a central conduction delay on brainstem auditory evoked response. Brain MRI performed on patient A-II-2 at 8 years disclosed a leukodystrophic pattern in a periventricular distribution (figure 2B). Electromyography, nerve conduction studies and EEGs were all normal. In all three patients, there was no indication of system involvement other than the central nervous system. The patients passed away after short febrile illness at 1 (A-II-9), 5.5 (A-II-1) and 15 years (A-II-2); heart failure was documented in patient A-II-9, but no autopsy was performed.

Pedigree and genotype of the p.V54L mutation (A–C). Affected represented by filled symbols. (D) Evolutionary conservation of the Val54 residue in the Hikeshi protein.
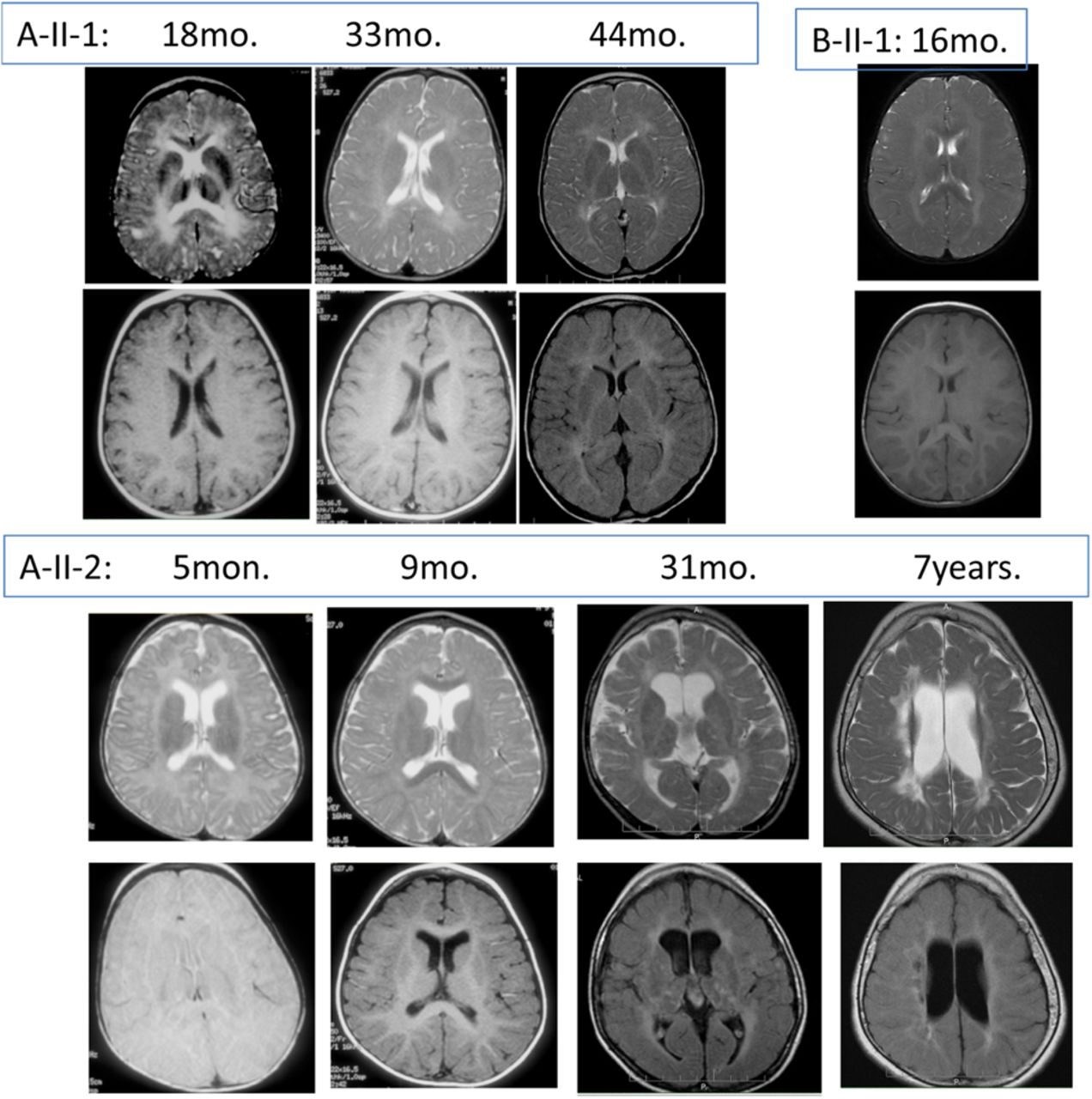
Brain MRI of the patients at ages indicated, top row: T2-weighted, bottom row: T1-weighted. All the patients had abnormal periventricular white matter as well as delayed myelination. Patient A-II-2 at age 7 years (bottom, right) had cystic changes of periventricular white matter and loss of white matter.
An unrelated fourth patient, B-II-1 (figure 1B), was a 3-year-old female, the first child to non-consanguineous AJ parents. She was born after an uneventful pregnancy with a birth weight of 2600 g. Head circumference was reported to be normal at birth and dropped to 2nd percentile (−2SD) at 6 months. Throughout infancy, she had feeding difficulties, vomiting and failure to thrive. At the age of 1 year, lower limb spasticity was noted. Neurological evaluation revealed increased muscle tone, hyper-reflexia and clonus, more prominent in the lower limbs. Ophthalmological examination was normal with no sign of optic atrophy or nystagmus. Developmental delay, more prominent in the motor aspect, was noted with no evidence of regression. The child was able to sit unsupported and pronounce a few words at 2 years. Brain MRI at 16 months revealed diffuse abnormal white matter signal in periventricular and subcortical distribution (figure 2A). Extensive metabolic workup, including determination of enzymatic activities of the respiratory chain complexes in muscle mitochondria and determination of cerebospinal fluid (CSF) neurotransmitter levels, was negative. Chromosomal microarray analysis in blood leucocytes was normal as well.
At the age of 2 years, she was hospitalised in the paediatric intensive care unit due to a febrile illness, tachycardia and tachypnea. At that time, the level of creatine phosphokinase and liver transaminases were abnormally elevated. Chest X-ray disclosed increasing heart size, and cardiac echocardiogram revealed a pericardial effusion and left ventricle hypertrophy with a decreased contractility of the left ventricle, compatible with acute perimyocarditis. She was treated with diuretics, afterload reduction and ionotropic drugs with a favourable response. Cardiac echocardiogram normalised 6 weeks after the acute illness.
On her last examination at the age of 3 years, she had microcephaly and progressive spasticity of the lower limbs. She was still unable to walk even when supported; her cognitive development was mildly delayed, and she could compose two-word sentences.
Patient B-II-2 (figure 1B) was diagnosed molecularly at early infancy before any symptoms have appeared. Her neurological examination was normal at birth and at 2 months. However, on repeated examination at 5 months of age, increased muscle tone with lower limb spasticity was evident.
The sixth patient, a female, (C-II-3 in figure 1C), currently 20 months old, presented in early infancy with feeding difficulties, nystagmus, increased startle reflex and delayed development without regression. At 18 months, she could sit unsupported, did not have a pincer grasp or any expressive language. Head circumference fell in percentiles from median at birth to −2 SD at 18 months. Examination at that age revealed spasticity of the lower limbs, nystagmus and ataxia as well as increased startle reflex at 18 months. Brain MRI disclosed a diffuse, abnormally increased T2 signal throughout the white matter.
Taken together, these six patients shared early feeding difficulties, global developmental delay, nystagmus, postnatal progressive microcephaly, truncal hypotonia and lower limb spasticity.
Methods
Whole exome analysis
Exonic sequences were enriched in the DNA samples of patient A-II-2 using SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, California, USA). Sequences were determined by HiSeq2000 (Illumina, San Diego, California, USA) and 100-bp were read paired-end. Reads alignment and variant calling were performed with the DNAnexus software (Palo Alto, California, USA) using the default parameters with the human genome assembly hg19 (GRCh37) as a reference. We removed variants which were called less than X8, were off-target, heterozygous, synonymous, minimal allele frequency (MAF)>0.1% at dbSNP138 and MAF>1% in the Hadassah in-house dbSNP. Parental consent was given for DNA studies. The study was performed with the approval of the ethical committees of Hadassah Medical Center and the Israeli Ministry of Health.
Cell culture
A skin biopsy was obtained from patient A-II-2 under local anaesthesia. Fibroblasts were cultured in DMEM/10% fetal bovine serum (FBS) at 37°C in a humidified incubator with 5% CO2.
Immunofluorescence microscopy
Detection of endogenous HSP70 and heat shock transcription factor 1 (HSF1) were performed as described previously.13 For heat treatment, cells grown on poly-Lys-coated coverslips were incubated at 43°C for 1 h with pre-warmed DMEM/10% FBS/20 mM HEPES (pH 7.3) as described previously.13
Western blotting
After washing with ice-cold phosphate buffered saline (PBS), fibroblast were lysed using ice-cold RIPA (1% NP-40, 0.5% Na-deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris-Cl, pH7.4, 150 mM NaCl, 1 mM EDTA)/1 µg/mL each of aprotinin, leupeptin and pepstatin on ice for 15 min. The extracted proteins were separated on 12% polyacrylamide gels, transferred to polyvinylidene fluoride (PVDF) membranes and immunoblotted with primary antibodies and HRP-conjugated secondary antibodies (Bio-Rad) using the enhanced chemiluminescence technique. Images were recorded using an LAS-3000 Imager (Fuji Film).
Antibodies
Primary antibodies used: mouse 1H5–1 monoclonal antibodies (specific to Hsc70/Hsp70),14 mouse anti-β-actin (Sigma, A5441), rat anti-HSF1 (Santa Cruz Biotechnology, 10H8), mouse anti-GSTπ (oligodendrocyte marker) (BD, 610718), rabbit anti-HSF1-pS326 (abcam, EP1713Y), and rabbit anti-C11orf73 (Hikeshi, l7Rn6) (Proteintech, 14808-1-AP) antibodies, rabbit anti-green fluorescent protein (GFP) antibodies (MBL, 598). Secondary antibodies used: Alexa Fluor 488-labelled goat anti-rabbit or anti-rat antibodies (Molecular Probes, A11037, A11007), Alexa Fluor 594-labelled goat anti-mouse antibodies (Molecular Probes, A11032).
Intra-nuclear microinjection
Patient fibroblasts were grown on poly-Lys-coated coverslips 24 h prior to microinjection. Plasmids (pcDNA/FLAG/Hikeshi and pEGFP-C1) expressing human Hikeshi and enhanced green fluorescent protein (EGFP) (as an injection marker), respectively, were co-injected through a glass capillary into the nucleus of fibroblasts. After injection, the cells were incubated for 24 h at 37°C in a humidified incubator with 5% CO2, and then subjected to heat treatment and immunofluorescence microscopy studies.13
Hikeshi expression in mouse brain
In situ hybridisation was performed according to the method of Hisatsune et al. (2013).15 Paraffin-embedded (5 μm thick) sagittal sections of P0 and P60 mice brain were treated with proteinase K (10 μg/mL, Wako) for 10 min at room temperature. The sections were probed with antisense or sense l7Rn6 and Myelin Basic Protein (MBP) probes overnight at 68°C. Antisense or sense l7Rn6 and MBP digoxigenin-labelled RNA probes were transcribed with T7 or SP6 RNA polymerase from SpeI or NcoI-linearised pGEM (Promega)-l7Rn6 or -MBP. Bright-field images were obtained using a fluorescence microscope BZ-9000 (KEYENCE). Image analysis was performed using the BZ-9000, Common Use Equipment, in the Support Unit for Bio-Material Analysis, RIKEN BSI, Research Resources Center (RRC). For immunoblot analysis, extracts of brain regions were prepared from adult mouse brain (ZYAGEN).
Results
Identification of the homozygous p.V54L mutation in the C11ORF73 gene
The exome analyses of the DNA of patient A-II-2 yielded 187.94 million confidently mapped reads with a mean coverage of X137. Following alignment to the reference genome, 245 662 variants were noted and after filtering 10 variants remained (see online supplementary table), however, only Chr11:86017416 G>C, NM_016401:c.G160C, p.Val54Leu (V54L) in the C11ORF73 gene segregated with the disease in family A (figure 1A). C11ORF73 consists of 5 exons encoding the 197 amino acid Hikeshi protein and the Val54 residue is conserved throughout evolution (figure 1D). Genotyping polymorphic single nucleotide polymorphism (SNPs) around the mutation site yielded the identification of ∼4.5 Mb homozygous region (chr11:82359847-86912169), shared by the patients but not by their unaffected sibs. We genotyped 1012 healthy individuals of AJ origin and identified 5 carriers suggesting a carrier rate of about 1/200.
We therefore searched all exome analyses available at our as well as at GeneDx laboratories for additional patients with C11ORF73 mutations. This search yielded patient B-II-1 and patient C-II-3 who were similarly homozygous for the p.V54L mutation in the C11ORF73 gene. The same homozygous mutation was detected at birth in patient B-II-2.
V54L mutation site in Hikeshi protein
We recently showed the crystal structure of human Hikeshi.16 Hikeshi forms an asymmetric homodimer, which is important for interaction with Hsp70. Further, the N-terminal domain of Hikeshi contains a unique extended loop (E-loop) and hydrophobic pockets, which are important for the recognition of phenylalanine-glycine nucleoporins (FG-Nup). Phe97 in the E-loop is buried in the hydrophobic pocket, and its association is likely to regulate interaction of Hikeshi with FG-Nup, resulting in the regulation of Hikeshi for nuclear pore complexes (NPC) passage activity. Val54 resides within its own hydrophobic pocket, and its residue faces inside the molecule (figure 3). The substitution of Val54 by Leu could affect an intra-molecular interaction between the E-loop and the hydrophobic pocket, which in turn might alter the dynamic movement of Hikeshi.

View of Val54 residue (yellow) in the hydrophobic pocket of the N-terminal domain (NTD). Structure of the NTD of Hikeshi is shown by ribbon representation. The E-loop on the surface of the NTD is coloured orange. Val54 and key hydrophobic residues are shown as sticks and coloured yellow and blue, respectively.
The effect of p.V54L mutation on the Hikeshi protein
In order to characterise the effect of the p.V54L on the Hikeshi protein, we first investigated the protein level in patient A-II-2 cells. As shown in figure 4, Hikeshi-V54L proteins were hardly detected in cell extracts from the patient's fibroblasts with or without heat stress.

Expression levels of the Hikeshi-V54L mutant protein in patient A-II-2 fibroblasts. Extracts of the patient's and control cells with or without heat-shock stress were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to western blotting. HSF1-pS326 and β-actin were used as the heat shock response and loading control, respectively.
Nuclear accumulation of Hsp70 in patient's cells occurs inefficiently under heat stress conditions
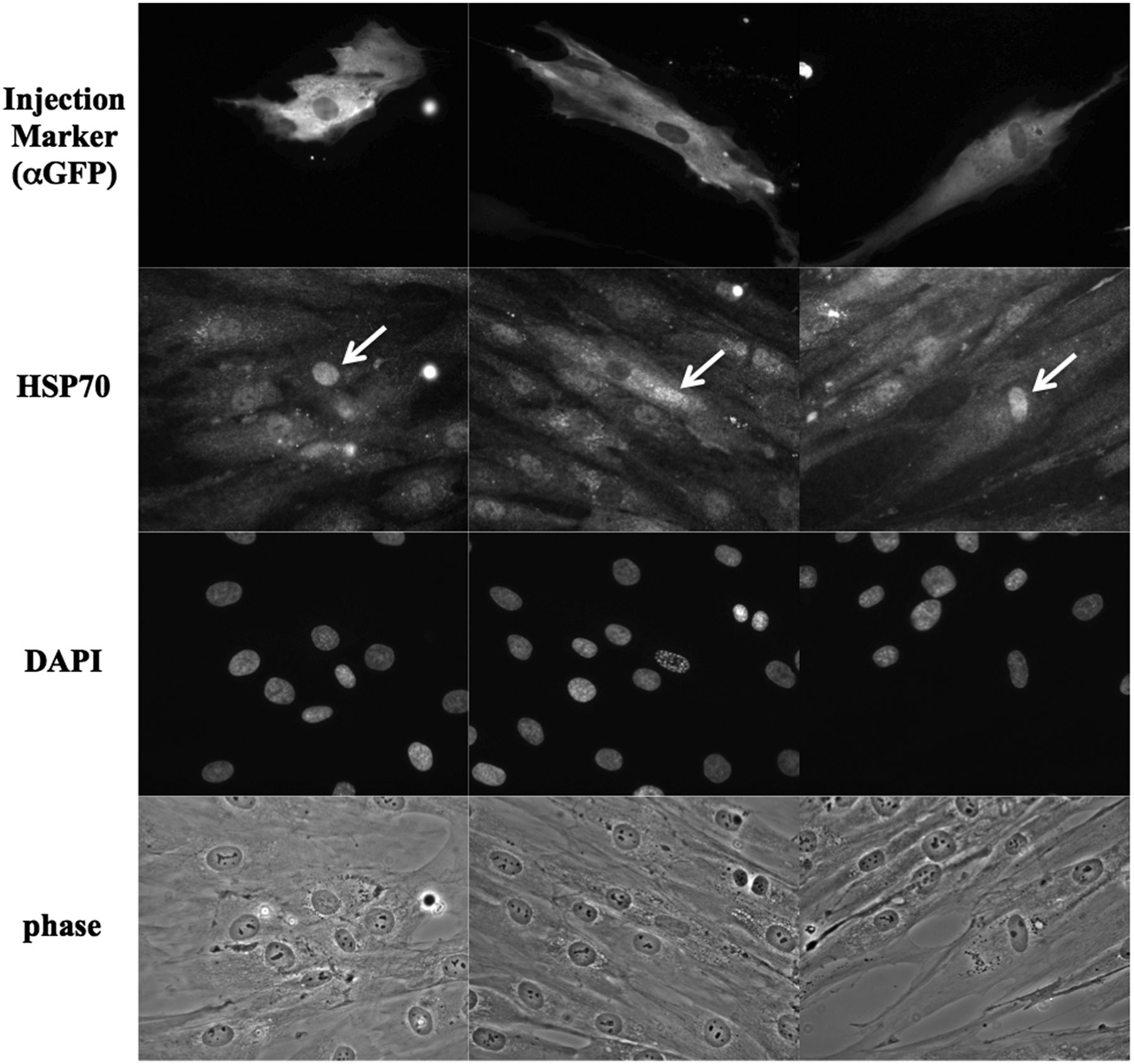
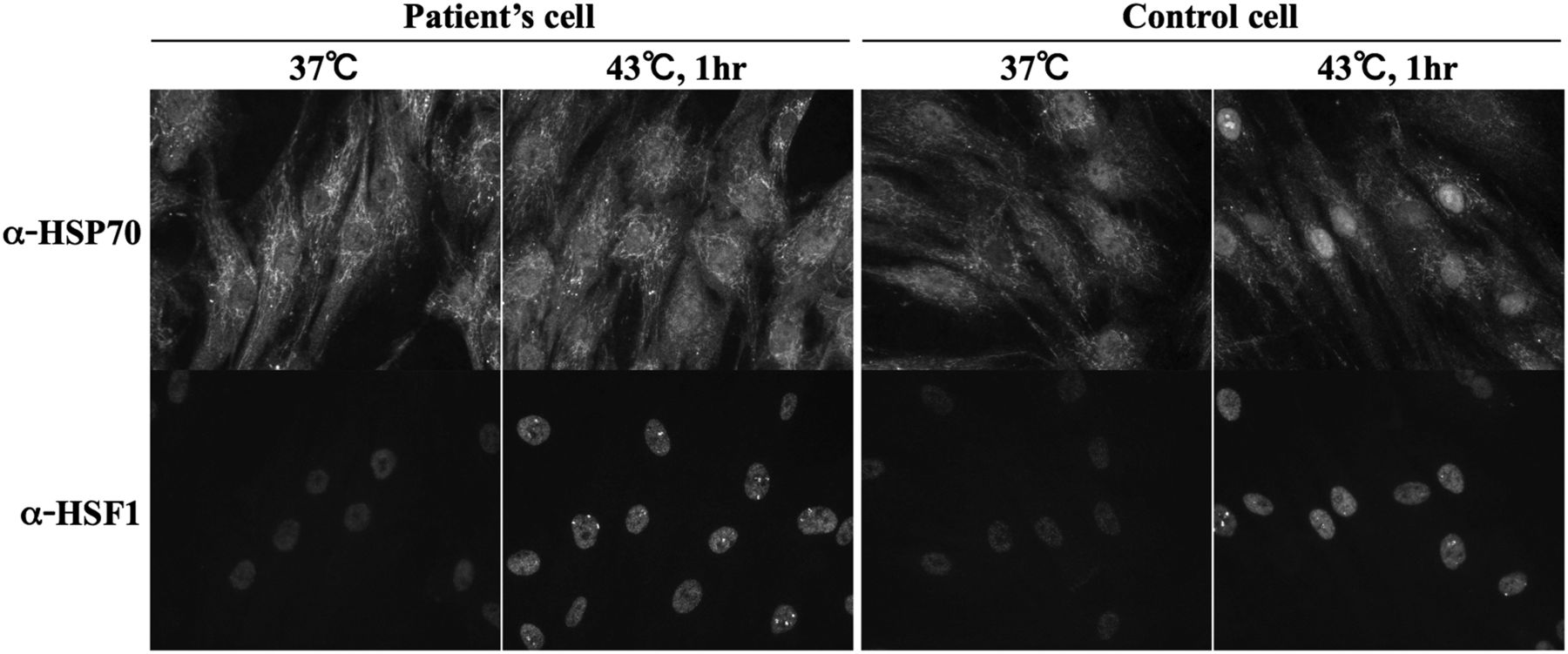
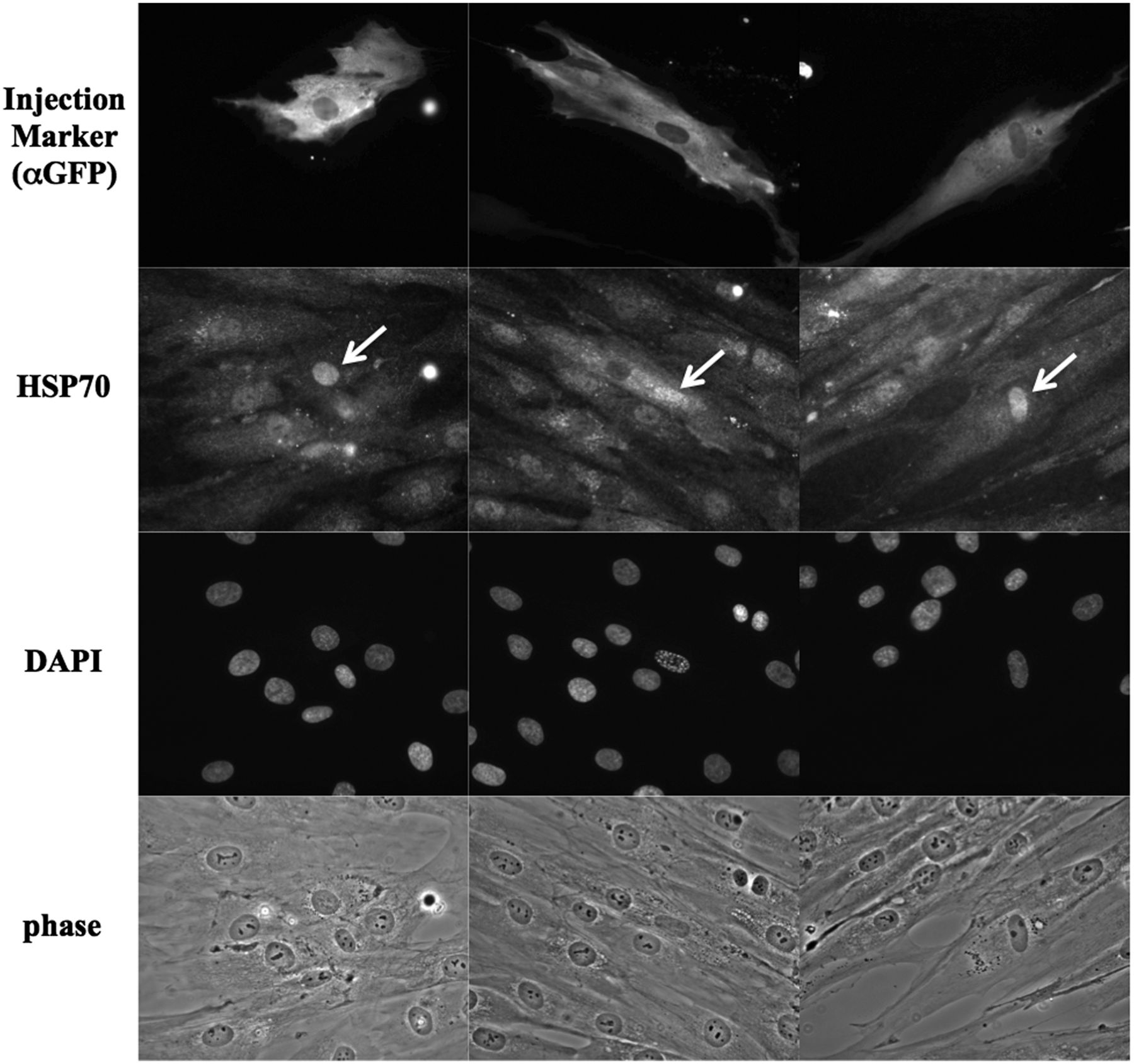
Endogenous Hsp70 proteins are well known to accumulate rapidly in the nucleus in response to heat stress. Since Hikeshi binds to ATP-form of Hsp70 and carries its Hsp70 into the nucleus, we assessed nuclear localisation of Hsp70 in the fibroblasts of a patient bearing homozygous p.V54L mutation of Hikeshi protein, under heat stress conditions. In control cells, in response to heat stress, nuclear stress granules (nSGs) of HSF1 formed within the nucleus by the activation of HSF1, and Hsp70 highly accumulated within the nucleus. In contrast, nuclear accumulation of Hsp70 was obscure in the patient A-II-2 cells even after heat treatment (figure 5) although the activation of HSF1 and formation of nSGs occurred normally, suggesting that heat shock response in the patient's cells is normal. Of note, nuclear accumulation of HSP70 in patient cells upon heat treatment was somewhat heterogenous: some cells accumulated Hsp70 in the nucleus weakly, whereas in other cells Hsp70 nuclear accumulation was entirely abolished. The experiment was repeated multiple times and in all cases, nuclear accumulation of Hsp70 in patient cells upon heat treatment was much weaker than that of control cells. To further substantiate the importance of Hikeshi for this cellular phenotype, we injected Hikeshi cDNA along with the marker EGFP cDNA into the nucleus of the patient fibroblasts and incubated for 24 h to allow protein expressions from the injected cDNAs; then, cells were treated with heat shock. In cells expressing EGFP, endogenous HSP70 accumulated strongly within the nucleus (figure 6), underscoring the role of Hikeshi in HSP70 relocation into the nucleus.

Nuclear accumulation of Hsp70 in patient A-II-2 fibroblasts occurs inefficiently in response to heat shock stress. Localisation of endogenous HSP70 and HSF1 was detected by indirect immunofluorescence.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Exogenously expressed Hikeshi proteins facilitate nuclear accumulation of HSP70 in the patient fibroblasts under heat stress condition. Twenty-four hours after nuclear coinjection of plasmids expressing Hikeshi or EGFP (as an injection marker), patient fibroblasts were subjected to heat shock treatment and stained with anti-Hsc70/Hsp70 and anti-GFP antibodies. In cells expressing EGFP, endogenous HSP70 were accumulated into the nucleus strongly. Arrows indicate microinjected cells.
Hikeshi is expressed in central white matter of mouse brain
In order to understand the preferential involvement of central myelin in Hikeshi depletion, we studied the expression of the mouse Hikeshi homologue (l7Rn6) in mouse brain. The l7Rn6 RNA signal was uniform in the adult mouse brain; in the newborn mouse brain the pattern was essentially similar, but the l7Rn6 signal was higher in the corpus callosum of the newborn when compared with the adult animal, indicating a higher abundance of Hikeshi homologue in regions where oligodendrocytes are generated (see online supplementary figure S1A, B). The Hikeshi (17Rn6) protein was ubiquitously expressed in mouse brain, including in regions where the oligodendrocyte marker GSTp is expressed (see online supplementary figure S1C).
Discussion
Six patients with a neurodegenerative disease of infantile onset, affecting mainly the central white matter, are reported. Three died suddenly and one survived an acute perimyocarditis. Using whole exome analysis and Sanger sequencing validation we identified in all the patients homozygosity for a missense mutation, p.V54L, in C11ORF73, the Hikeshi protein gene. The mutation was associated with an undetectable Hikeshi protein in the patient fibroblasts, likely due to its accelerated degradation.
Recently, we reported that Hikeshi was essential for the entry of the co-chaperone Hsc70/Hsp70 (Hsp70s) to the nucleus under stress condition.13 Regulation of the nucleocytoplasmic transport of macromolecules through NPCs is crucial for various cellular functions in eukaryotic cells. Members of the importin β family, referred to as importins, transportins, exportins and karyopherins, are nucleocytoplasmic carrier molecules and are thought to mediate most of the selective nucleocytoplasmic protein transport.17–19 Hikeshi which is conserved from yeast to human is a unique nuclear import carrier since it does not belong to this well-characterised importin β family.
Stresses such as heat shock and oxidative stress induce down-regulation of all importin β family-mediated pathways, preventing their normal operation under stress conditions.14 ,20 The importance of Hikeshi is underscored exactly under such stress; in response to heat shock, Hsp70s are rapidly and transiently relocated from the cytoplasm into the nucleus and nucleolus.21 Inside the nucleus, the Hsp70 dissociates from Hikeshi and binds native or non-native client proteins and functions as a molecular chaperone, likely reversing and attenuating the multiple heat shock-induced nuclear phenotypes and therefore protecting the cells from heat shock damage.22 Consistently, the viability of Hikeshi-depleted cells during heat shock stress was significantly reduced.13
Here we show that homozygosity for the p.V54L mutation in Hikeshi is associated with lack of detectable Hikeshi protein with the resultant absence of nuclear HSP70 during heat shock stress. We speculate that the observed leukoencephalopathy is underlined either by nuclear Hsp70 deficiency or by its excessive cytoplasmic accumulation during stress. The possibility that Hikeshi has other cargo molecules besides Hsp70s was previously declined by the rescue of viability of Hikeshi-depleted by the expression of NLS-tagged Hsp70.13 The sudden death of three of the patients perhaps via acute inflammation of myocardial and pericardial tissue supports the possibility of an adverse effect of HSP70 when localised extranuclearly during stress; HSP70 has an immunogenic role when present in the extracellular compartment or is membrane-bound. These molecules, which are actively released in exosomes,23 were shown to either act as carriers for immunogenic peptides, initiate cytokine release or provide target structures for natural killer (NK) cells.24 Furthermore, recent evidence indicates that the absence of nuclear Hsp70 does not relieve the inflammatory signalling.25
As mentioned above, leukodystrophy has been proposed to result from perturbation of one of several mechanisms. It may occur as a result of impaired protein transcription/translation, where the accumulation of unfolded/misfolded proteins triggers the UPR, or by accumulation of a mutant protein in the ER, again inducing UPR with the resultant apoptosis of oligodendrocytes. We suggest that inefficient heat shock stress response is a new disease mechanism for leukoencephalopathy , probably in a similar sequence of events as that observed in patients with leukodystrophy due to mitochondrial HSP60 chaperonopathy who also die during acute febrile illnesses.
Finally, the significant carrier rate of this mutation among AJ and the identification of homozygotes in three unrelated families indicate that this variant should be included in parental screening panels used for the AJ community.
Acknowledgments
We thank the families for their contributions and participation. This work was supported in part by a trilateral project grant from The German Research Foundation (Ga354/9-1), and by Japan MEXT grant-in-aids.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure
Footnotes
SE, SK, CJ and AF-V are contributed equally.
Contributors AFV and SBS undertook patient management, collected samples and delineated the phenotype, analysed the data and wrote the paper. AW, YO, HM, AMF, AS and SB performed the experiments, analyzed the data and wrote the paper. SE, SK, CJ, NT, SB, JG, NI and OE conceived and designed the experiments, analyzed the data and wrote the paper, undertook patient management, collection of samples and delineation of the phenotype.
Competing interests SB is an employee of GeneDx. AMF and NRT are employees of Reproductive Medicine Associates of New Jersey Basking Ridge, New Jersey.
Patient consent Obtained.
Ethics approval The ethical committees of Hadassah Medical Center and the Israeli Ministry of Health.
Provenance and peer review Not commissioned; externally peer reviewed.