

Abstract
The central nervous system (CNS) is endowed with several immune-related mechanisms that contribute to its protection and maintenance in homeostasis and under pathology. Here, we discovered an additional mechanism that controls inflammatory responses within the CNS milieu under injurious conditions, involving CD200 ligand (CD200L) expressed by newly formed endothelial cells. We observed that CD200L is constitutively expressed in the mouse healthy CNS by endothelial cells of the blood–cerebrospinal fluid barrier and of the spinal cord meninges, but not by the endothelium of the blood–spinal cord barrier. Following spinal cord injury (SCI), newly formed endothelial cells, located only at the epicenter of the lesion site, expressed CD200L. Moreover, in the absence of CD200L expression by CNS-resident cells, functional recovery of mice following SCI was impaired. High throughput single-cell flow cytometry image analysis following SCI revealed CD200L-dependent direct interaction between endothelial and local CD200R+ myeloid cells, including activated microglia and infiltrating monocyte-derived macrophages (mo-MΦ). Absence of CD200L signaling, both in vitro and in vivo, resulted in a higher inflammatory response of the encountering macrophages, manifested by elevation in mRNA expression of Tnfα and Il1β, increased intracellular TNFα immunoreactivity, and reduced expression levels of macrophage factors that are associated with resolution of inflammation, Dectin-1, CD206 (mannose receptor), and IL-4R. Collectively, our results highlight the importance of CD200-mediated immune dialogue between endothelial cells and the local resident microglia and infiltrating mo-MΦ within the lesion area, as a mechanism that contributes to regulation of inflammation following acute CNS injury.
SIGNIFICANCE STATEMENT This manuscript focuses on a novel mechanism of inflammation-regulation following spinal cord injury (SCI), orchestrated by CD200-ligand (CD200L) expressed by newly formed endothelial cells within the lesion site. Our study reveals that, in homeostasis, CD200L is expressed by endothelial cells of the mouse blood–cerebrospinal fluid barrier and of the blood–leptomeningeal barrier, but not by endothelial cells of the blood–spinal cord barrier. Following SCI, newly formed endothelial cells located within the epicenter of the lesion site were found to express CD200L at time points that were shown to be critical for repair. Our results reveal a direct interaction between CD200L+ endothelial cells and CD200R+ microglia and macrophages, resulting in attenuated inflammation, biasing macrophage phenotype toward inflammation-resolving cells, and promotion of functional recovery following SCI.
Introduction
The fate of the central nervous system (CNS), in homeostasis and following injury, is influenced by circulating and resident immune cells (Moalem et al., 1999; Ziv et al., 2006; Rolls et al., 2007; Shechter et al., 2009; London et al., 2011; Ron-Harel et al., 2011; Kunis et al., 2013; Shechter et al., 2013b; Baruch et al., 2014; Raposo et al., 2014; Benhar et al., 2016). The activation state of the CNS resident immune cells, the microglia, is critically dependent on specialized regulatory mechanisms within the CNS microenvironment. Among those unique mechanisms are the constitutive expression of the anti-inflammatory molecule TGFβ1 (Cohen et al., 2014), MAFB expression by microglia (Matcovitch-Natan et al., 2016), and neuron-microglia interactions, such as that of CX3CL1/CX3CR1 and the CD200-ligand (CD200L)/CD200-receptor (CD200R) (Biber et al., 2007; Ransohoff and Perry, 2009; Kierdorf and Prinz, 2013; Linnartz and Neumann, 2013; Limatola and Ransohoff, 2014; Lampron et al., 2015).
CD200L (OX2) is a membrane glycoprotein widely expressed by a variety of cells, including thymocytes, circulating B cells, some peripheral T cells, follicular dendritic cells, endothelium, placental trophoblasts, smooth muscle cells, lung epithelium, neurons, astrocytes, and oligodendrocytes (Barclay et al., 2002; Snelgrove et al., 2008; Koning et al., 2009). The CD200R, which contains cytoplasmic tyrosine residue domains that regulate the inflammatory response (Gorczynski et al., 2004; Zhang et al., 2004; Mihrshahi et al., 2009; Mihrshahi and Brown, 2010), is expressed by mouse macrophages, dendritic cells, neutrophils, basophils, mast cells, and by human and mouse T- and B-lymphocytes (Rijkers et al., 2008) and NK cells (Wright et al., 2003). The CD200R expressed by myeloid cells can modulate their function, via the interaction with CD200L, by attenuating inflammatory responses (Hoek et al., 2000; Snelgrove et al., 2008; Rygiel et al., 2012; Vaine and Soberman, 2014).
In the CNS, the interaction between neuronal CD200L and microglial CD200R is important for regulating microglial function (Hoek et al., 2000; Broderick et al., 2002; Zhang et al., 2011). Impaired CD200L-CD200R signaling leads to tissue damage derived from microglia-induced inflammation, and is associated with increased susceptibility to autoimmune and neurodegenerative diseases (Hoek et al., 2000; Broderick et al., 2002; Frank et al., 2006; Koning et al., 2007; Walker et al., 2009; Zhang et al., 2011).
Repair processes within the CNS following spinal cord injury (SCI) differ from those taking place in peripheral wound healing. Peripheral wound healing involves an efficient proinflammatory to anti-inflammatory phenotype switch, or timely regulated recruitment of anti-inflammatory cells, such as monocyte-derived macrophages (mo-MΦ), leading to resolution of the inflammatory response, needed for an efficient spontaneous repair process in term of regeneration and tissue restoration (Martin, 1997; Singer and Clark, 1999; Auffray et al., 2007; Shechter et al., 2013a). In contrast, following acute CNS insult, the primary spontaneous inflammation, mediated by the microglia, often fails to be adequately resolved, leading to secondary degeneration of the surrounding tissue and resulting in additional neuronal tissue loss. Under such conditions, infiltrating mo-MΦ have been shown to exert beneficial function by promoting repair (Rapalino et al., 1998; Shechter et al., 2009, 2013a; London et al., 2011, 2013a; Cohen et al., 2014).
Here, we identified a novel mechanism of inflammation-resolution, orchestrated by newly formed endothelial cells via their expression of CD200L following acute CNS injury in mice. CD200L was found to be differentially expressed by endothelial cells at the various CNS borders during homeostasis. Thus, while the endothelial cells of the mouse blood–CSF barrier and of the meninges constitutively express CD200L, the blood–spinal cord barrier endothelium hardly expresses this ligand. Following SCI, CD200L expression was upregulated by only newly formed endothelial cells, located within the epicenter of the lesion site. CD200L+ CNS resident cells were found to be important in recovery from SCI. Moreover, we detected direct interaction between endothelial cells and CD200R+ myeloid cells (mo-MΦ or microglia) at the epicenter of the lesion site, which involved CD200L signaling. This interaction was shown here to play an important role in regulation of inflammation and promotion of inflammation-resolving processes.
Materials and Methods
Animals.
Adult 8- to 10-week-old male C57BL/6J and CD200L−/− (Hoek et al., 2000) mice, a kind gift of Prof. Neil Barclay (Sir William Dunn School of Pathology, University of Oxford, Oxford, UK), were used. C57BL/6-Tg (ACTbEGFP)1Osb/J mice (Okabe et al., 1997) provided a source of β-actin-GFP bone marrow (BM). Animals were supplied by the Animal Breeding Center of the Weizmann Institute of Science. All animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee.
SCI.
The spinal cords of deeply anesthetized mice were exposed by laminectomy at T12, and contusive (200 kdynes) centralized injury was performed using the Infinite Horizon spinal cord impactor (Precision Systems), causing bilateral degeneration without complete penetration of the spinal cord. The animals were maintained on twice-daily bladder expression. Animals that were contused in a nonsymmetric manner (two or more grades in 0–9 score difference) were excluded from the experiments that tested functional recovery. All animals were included in the histological and flow cytometry analyses.
BM radiation chimeras.
CD200L−/− > wild-type (WT) and WT > CD200L−/− BM chimeras were prepared by subjecting mice to lethal whole-body irradiation (950 rad) while shielding the brain. On the day following irradiation, the mice were injected with 5 × 106 BM cells harvested from the hindlimbs (tibia and femur) and forelimbs (humerus) of WT or CD200L−/− donor mice. BM cells were obtained by flushing the bones with Dulbecco's PBS under aseptic conditions, and were then collected, washed, and centrifuged (10 min, 1250 rpm, 4°C). After irradiation, mice were maintained on drinking water fortified with cyproxin for 1 week to limit opportunistic infection. The chimeric mice were subjected to SCI 8–10 weeks after BM transplantation.
Assessment of functional recovery from spinal cord contusion.
Functional recovery was evaluated by hindlimb locomotor performance, assessed according to the open-field Basso Mouse Scale (BMS) (Basso et al., 2006), with nonlinear scores ranging from 0 (complete paralysis) to 9 (normal mobility); each score represents a distinct motor functional state. Blind observers, who were not aware of the identity of the mice or the treatments that they received, examined the animal motor function, and the final scores were calculated as the average of both legs. In cases in which the position of the leg did not correspond exactly to the distinct motor functional scale values of the BMS, half scores were given. Locomotor activity in an open field was monitored twice a week by placing the mouse for 4 min at the center of a circular enclosure (diameter 90 cm, wall height 7 cm) made of molded plastic, with a smooth, nonslippery floor. Before each evaluation, the mice were carefully examined for peritoneal infection, wounds in the hindlimbs, and tail and foot autophagia.
Immunohistochemistry.
Mice were killed by an overdose of anesthesia and were perfused with PBS via the left ventricle. For whole-mount staining of the choroid plexus and meninges, isolated tissues were fixed with 4% PFA for 1 or 24 h, respectively, and subsequently transferred to PBS containing 0.05% sodium azide. Before staining, the dissected tissues were washed with PBS, blocked with a mixture of 20% horse serum, 0.3% Triton X-100 (Sigma-Aldrich), and stained with the following primary antibodies: goat anti-CD200L (1:40; R&D Systems) and rat anti-CD31 (1:40; BD Biosciences PharMingen) in the presence of 2% horse serum and 0.3% Triton X-100. Secondary antibodies included Cy2-conjugated donkey anti-rat (1:150; Jackson ImmunoResearch Laboratories) and Cy3-conjugated donkey anti-goat (1:200; Jackson ImmunoResearch Laboratories). Each step was followed by three washes in PBS. The tissues were mounted onto slides, using Immu-Mount (Thermo), and sealed with coverslips.
For staining of sectioned spinal cords and brains, two different tissue preparation protocols (paraffin-embedded and floating sections) were applied. The tissues were blocked with M.O.M. immunodetection kit reagent (Vector Laboratories) containing 0.1% Triton X-100 (Sigma-Aldrich), and stained with the following primary antibodies: rabbit anti-Glial fibrillary acidic protein (GFAP) (1:400; Dako), rabbit anti-Ionized calcium binding adapter molecule-1 (Iba-1) (1:300; Wako), goat anti-CD200L (1:40; R&D Systems), goat anti-CD200R1 (1:100; R&D Systems), rat anti-CD31 (1:40; BD Biosciences PharMingen), rabbit anti-βIII tubulin (1:5000; Covance), mouse anti-Neuronal Nuclei (NeuN) (1:150; Millipore Bioscience Research Reagents International), rabbit anti-Ki67 (1:100; Abcam), and chicken anti-vimentin (1:500; Millipore); Fluorescein isothiocyanate (FITC)-conjugated Bandeiraea simplicifolia isolectin B4 (1:40; Sigma-Aldrich) was added for 1 h to the secondary antibody solution. Secondary antibodies included Cy-2-conjugated donkey anti-mouse, Cy-2-conjugated donkey anti-rabbit, Cy-2-conjugated donkey anti-rat (1:150; Jackson ImmunoResearch Laboratories), Cy-3-conjugated donkey anti-goat (1:200; Jackson ImmunoResearch Laboratories), Cy-5-conjugated donkey anti-rabbit (1:150; Jackson ImmunoResearch Laboratories), and Cy-5-conjugated donkey anti-rat (1:150; Jackson ImmunoResearch Laboratories). In some staining experiments, the slides were exposed to Hoechst for nuclear staining (1:2000; Invitrogen) for 30 s.
Flow cytometry analysis and sorting.
Mice subjected to SCI were killed by an overdose of anesthesia, and their spinal cords were prepared for flow cytometric analysis by perfusion with PBS via the left ventricle. The injured segments of the spinal cords were dissected from individual mice (parenchymal segments spanning 0.3 mm from each side of the lesion site), and tissues were homogenized using a software controlled sealed homogenization system (Dispomix, Medic Tools; Miltenyi). The cells were separated using 40% Percoll (GE Healthcare) and centrifuged at 2000 rpm for 20 min at room temperature. All samples were filtered through an 80 μm nylon mesh and blocked with Fc-block CD16/32 (BD Biosciences). Next, samples were stained using the following antibodies: FITC-conjugated CD11b, peridinin chlorophyll protein (PerCP) Cy5.5-conjugated Ly6C, and Allophycocyanin (APC)-conjugated CD200R1 (eBioscience); Pacific Blue-conjugated CD45.2, APC-conjugated CD45.2, FITC-conjugated CD206, phycoerytherin (PE)-conjugated TNFα, APC-conjugated Ly6C, and PE-conjugated CD200L (Biolegend); PE-conjugated CD200L, PE-conjugated isotype control Rat IgG1, and AlexaFluor-645-conjugated Dectin-1 (AbD Serotec); PE-conjugated IL-4Rα (R&D Systems). Cells were analyzed on a FACS-LSRII cytometer (BD Biosciences) using BD FACSDIVA software (BD Biosciences) and FlowJo software (FlowJo). For intracellular TNFα detection, cells were purified from spinal cords 7 d following SCI, and incubated with DMEM (Biological Industries) supplemented with 5% FCS, 1 mm l-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin, and Golgi-stop (1:1000; BD Biosciences) for 3 h at 37°C, to enable expression of intracellular cytokines and to prevent their extracellular secretion. Cells were washed, fixed, permeabilized, and stained for surface and intracellular proteins, using Cytofix/Cytoperm kit, according to the manufacturer's instructions (BD Biosciences).
For sorting experiments, GFP+ BM-MΦ (CD45+CD11b+) were sorted from in vitro cocultures using SORP-FACS (BD Biosciences) into 100 μl of FACS buffer (PBS supplemented with 1% FCS, 0.5% NaO3, and 0.04 mm EDTA).
High-throughput single-cell flow cytometry image analysis (ImageStream).
Mice subjected to spinal cord injury were killed after 7 d by an overdose of anesthesia and perfused with PBS via the left ventricle. A parenchymal segment spanning 0.3 mm from each side of the lesion sites was dissected, and tissues were homogenized using a software controlled sealed homogenization system (Dispomix, Medic Tools; Miltenyi). Cells were treated with a mixture of Collagenase IV (1 mg/ml; Worthington) and DNase (30 U/ml; Sigma-Aldrich) and diluted with PBS for 45 min on a shaker at 37°C. The cells were washed with cold PBS and treated with a mixture of Collagenase IV (1 mg/ml; Worthington), Dispase (1 mg/ml; Roche), and DNase (10 U/ml; Sigma-Aldrich), for 1 h on a shaker at 37°C. Following washing with cold PBS, cells were centrifuged with 30% Percoll (GE Healthcare) at 2000 rpm, 20 min, at 20°C. Cells were washed and incubated with RPMI medium (Biological Industries), supplemented with 10% FCS, 1 mm l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin for 20 min at 37°C, to enable conjugate formation. Cells were washed gently, centrifuged, and fixed using Cytofix/Cytoperm (BD Biosciences) for 20 min at room temperature. Following two washes with BD-perm wash buffer (BD Biosciences), cells were FC blocked, and stained for the specific surface markers: FITC-conjugated CD11b, Allophycocyanin (APC)-conjugated CD200R1, Pacific Blue-conjugated CD45.2, PE-conjugated CD200L, and PE-cy7-conjugated CD31. Each sample, which was derived from a pool of 5 mice, was examined by imaging flow cytometry using the ImageStreamX (Amnis-part of EMD Millipore). At least 105 cells were collected from each sample. Images were analyzed using IDEAS 6.0 software (Amnis). Cells were gated for the conjugated population using the Gradient RMS feature (George et al., 2006). CD45+CD11b+ cells were gated according to their pixel intensities and then further gated for CD31+ cells. To identify cell conjugates of CD45+CD11b+ myeloid cells with CD11b−CD31+ endothelial cells, the DeltaCentroid XY feature was used, which calculates the geometric distance between the centers of two staining foci. The gate was verified by visual inspection to include only cell conjugates that had the appropriate distance between the CD31-CD45 and CD11b-CD45 staining. Because of the large heterogeneity in size and morphology of the cells and the conjugates, the identified conjugates were further visually inspected by two examiners in a blinded manner.
BM macrophage culture.
GFP+ BM progenitors were harvested from C57BL/6-Tg (ACTbEGFP)1Osb/J mice and cultured for 7 d on Petri dishes (0.5 × 106 cells/ml) in RPMI-1640 supplemented with 10% FCS, 1 mm l-glutamine, 1 mm sodium pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, and 50 ng/ml monocyte-colony-stimulating factor (M-CSF; Peprotech). On day 4, the cells were treated with fresh M-CSF (50 ng/ml). On day 7, cells were mechanically detached with cold PBS and replated on 24-well tissue culture plates (0.2 × 106 cells/ml; Corning) for 1 d before treatments (described below).
In vitro cultures.
The b.END3 cell line (a kind gift of Dr. Dan Frenkel, Tel-Aviv University, Tel-Aviv, Israel) was grown in 25 cm2 cell-culture flasks (Greiner bio-one) in culture medium [DMEM (high glucose) medium (Biological Industries) supplemented with 10% FCS, 1 mm l-glutamine, 1 mm sodium pyruvate, 100 U/ml penicillin, 100 mg/ml streptomycin, and 1% MEM nonessential amino acids (Biological Industries)]. The GFP+ BM-MΦ were cultured in 24-well dishes with or without b.END3 cells, in BM-MΦ media with 10 ng/ml M-CSF, with or without 50 μg/ml CD200L inhibitor (LFNTFGSQKVSGT, based on the CDR3 g′ peptide; GL Biochem) (Chen et al., 2005; Gorczynski et al., 2008). After 24 h, the cells were treated with 100 ng/ml lipopolysaccharide (LPS) (Escherichia coli 055:BS, Sigma-Aldrich) for 4 h (CD200L inhibitor was added again). The GFP+ BM-MΦ were sorted, based on their GFP expression using SORP-FACS (BD Bioscience), RNA was extracted, and qRT-PCR was performed as described below.
qRT-PCR.
Total RNA of in vitro GFP+ BM-MΦ was extracted with the miRNeasy kit according to the manufacturer's instructions (QIAGEN). RNA was reverse-transcribed using the high-capacity cDNA reverse transcription kit (Applied Biosystems), amplified using SYBR green I Master Mix (Roche) and detected by StepOnePlus (Applied Biosystems), in duplicates. Results were normalized to the expression of the housekeeping gene, peptidylprolyl isomerase-A (Ppia), and then expressed as fold change relative to the control sample. The following primers were used: Ppia forward, 5′-AGCATACAGGTCCTGGCATCTTGT-3, and reverse, 5′-CAAAGACCACATGCTTGCCATCCA-3′; Tnfα forward, 5′-CCCTCACACTCAGATCATCTTCT-3′, and reverse, 5′-GCTACGACGTGGGCTACAG-3′; Il-β forward, 5′-ACCTGTCCTGTGTAATGAAAGAC-3′, and reverse, 5′-TGGGTATTGCTTGGGATCCA-3′; Il-6 forward, 5′-TGCAAGAGACTTCCATCCAGTTG-3′, and reverse, 5′-TAAGCCTCCGACTTGTCAAGTGGT-3′.
Statistical analysis.
Data were analyzed using Student's t test to compare between two groups. One-way or two-way ANOVA tests were used to compare several groups; the Bonferroni post hoc tests (p = 0.05) were used for follow-up pairwise comparison of groups. The specific tests used to analyze each set of experiments are indicated in the figure legends. The results are presented as mean ± SEM.
Results
CD200L is differentially expressed by endothelial cells within different CNS barriers
To understand the role of CD200L in repair processes following acute insult in the CNS, its spatial distribution at different compartments within the CNS territory was first examined in homeostasis. We found that, in the brain parenchyma, neuronal cell bodies located at the hippocampus, cerebellum, and cortex were immunoreactive for CD200L, as was shown by other groups (Hoek et al., 2000; Wright et al., 2001; Koning et al., 2009) (Fig. 1A). In contrast, in the spinal cord parenchyma, CD200L expression by neurons was negligible (Fig. 1B), in line with previous studies (Chitnis et al., 2007; Koning et al., 2009). However, neurons at the dorsal root ganglia (DRG) were found to express CD200L (Fig. 1B).

Spatial distribution of CD200L in the CNS compartments in homeostasis. A, Immunostaining for βIII-tubulin, NeuN, and CD200L of neurons located in the hippocampus, cerebellum, and cortex of brains derived from noninjured WT mice. Scale bar, 500 μm. Data are representative of two different experiments; n = 2 mice in each group, on each experiment. B, Immunostaining for βIII-tubulin and NeuN of neurons expressing CD200L in the DRGs, but not in the spinal cord parenchyma of noninjured WT mice. Scale bar, 100 μm. C, Whole-mount immunostaining for CD31 and CD200L in the choroid plexus derived from noninjured WT mice. D, Immunostaining for CD200L and vimentin+ ependymal cells in longitudinal sections of the central canal in the spinal cord of noninjured WT mice. Inset, Immunostaining of coronal section of the central canal. E, F, Whole-mount immunostaining for CD31 and CD200L in the dura (E) and pia (F) mater of meninges derived from noninjured WT mice. G, Immunostaining for CD31 and CD200L in spinal cord parenchyma of noninjured WT mice. H, Whole-mount immunostaining for CD31 and CD200L in the choroid plexus derived from noninjured CD200L−/− mice. C–H, Scale bar, 50 μm. Data are representative of 2–5 independent experiments; n = 3–5 mice in each group.
Because of the vital role of infiltrating cells in repair processes following acute CNS insult, and the reported involvement of CD200 signaling in the regulation of myeloid cell activity (Barclay et al., 2002; Broderick et al., 2002), we also assessed CD200L expression at the CNS barriers and entry routes, which represent an interface between the CNS and periphery (Shechter et al., 2013a). In healthy WT mice, we detected CD200L expression by CD31+ endothelial cells of the choroid plexus (Fig. 1C), an interface that was shown to serve as a gateway for immune-regulatory cells to the CNS territory for immune surveillance and repair (Kivisäkk et al., 2002; Kunis et al., 2015), and in close proximity to vimentin+ cells of the central canal (Fig. 1D). In addition, we searched for CD200L expression in the blood–leptomeningeal barrier. Whole-mount immunohistochemistry of the spinal cord meningeal layers, including the dura (Fig. 1E) and pia (Fig. 1F) mater, revealed expression of CD200L by CD31+ endothelial cells within the meninges. In contrast, the tightly joined endothelial cells of the blood–spinal cord barrier (Fig. 1G) of WT mice did not express CD200L in homeostasis. The lack of staining in specimens derived from CD200L−/− mice was used as a control for the specificity of CD200L antibodies (Fig. 1H).
CD200L is involved in the functional recovery following SCI and is upregulated by newly formed endothelial cells within the lesion site
We next examined whether CD200L expression by endothelial cells of the blood–spinal cord barrier in the area of the lesion site was altered following SCI. We used GFAP immunoreactivity to delineate the margins of the injury site (Shechter et al., 2009). CD200L immunoreactivity was found in close proximity to the GFAP+ reactive astrocytes 3 d after injury, when the lesion site area was not yet fully demarcated by the reactive astrocytes. From day 7 onward, CD200L-expressing cells were found inside the epicenter of the lesion site, and this expression was reduced by day 14 following SCI (Fig. 2A). Further analysis of the lesion site area 7 d following SCI revealed that cells that expressed CD200L coexpressed CD31, thereby suggesting that the injury-induced expression of CD200L could be associated with endothelial cells within the epicenter of the lesion site (Fig. 2B). No staining could be detected in the injury site of CD200L−/− animals (Fig. 2C). Notably, expression of CD200L by the blood–spinal cord barrier endothelial cells 7 d following SCI was restricted to the lesion site area and could not be detected in CD31+ endothelium located distally to the lesion site (Fig. 2D). In addition to the endothelial cells, at 7 d following the injury, we detected a small number of neurons (βIII tubulin+ and NeuN+) that were positive for CD200L at the margins of the lesion site, but not distally (Fig. 2E). No changes were found with respect to DRG neuronal expression of CD200L following SCI (data not shown).
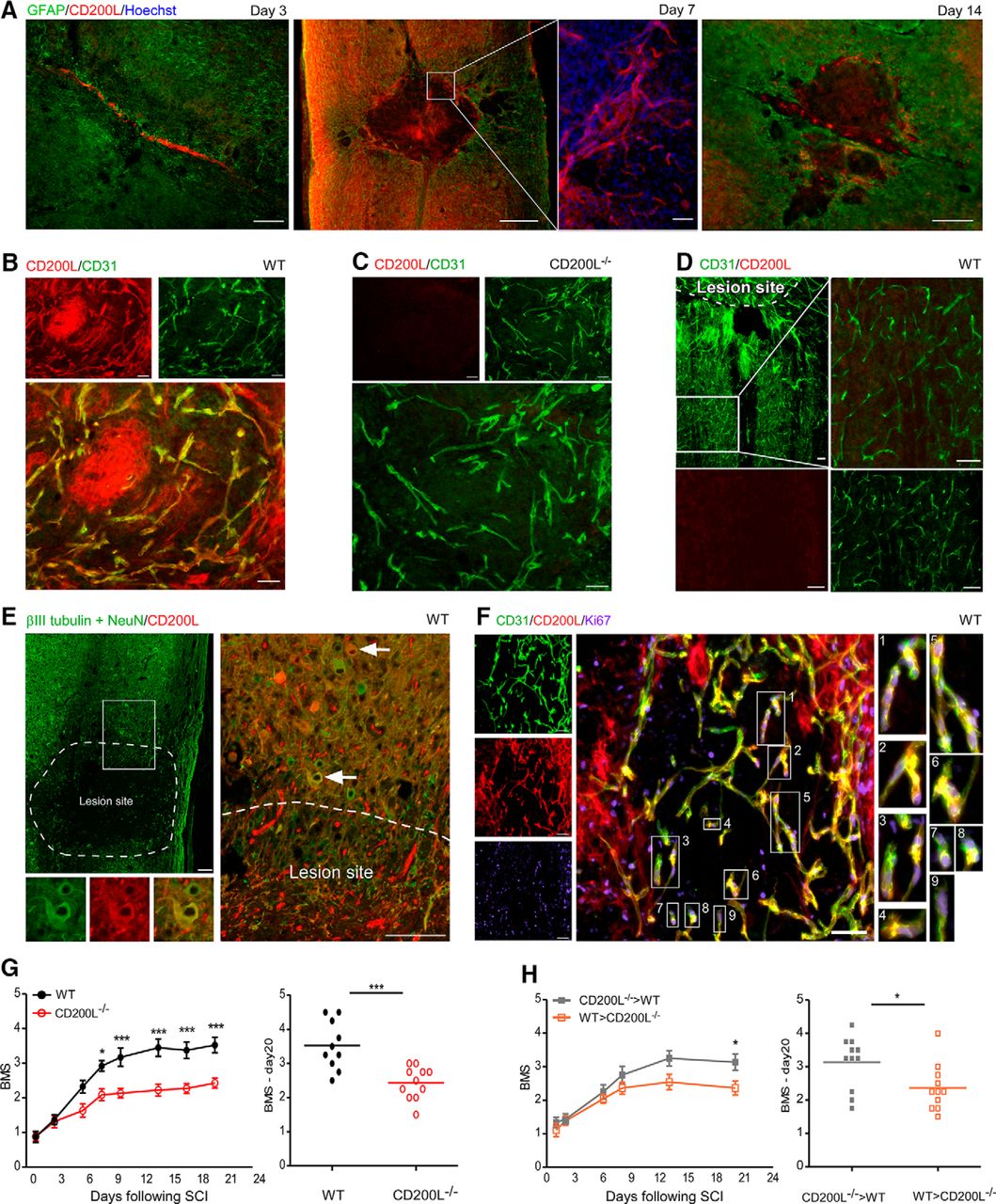
CD200L is involved in the functional recovery following SCI and is upregulated by newly formed endothelial cells within the lesion site. A, Immunostaining for GFAP, to detect reactive astrocytes that surround the lesion site, together with CD200L on days 3, 7, and 14 following SCI. Scale bar, 500 μm. Day 7: Enlarged image of CD200L+ Hoechst+ cells. Scale bar, 100 μm. B, C, Immunostaining for CD200L and CD31 at the epicenter of the lesion site 7 d following SCI in WT (B) and CD200L−/− (C) mice. D, Immunostaining for CD200L and CD31 distal to the lesion site 7 d following SCI in WT mice. E, Immunostaining for βIII-tubulin, NeuN, and CD200L showing neurons that are located adjacent to the lesion site 7 d following SCI. Scale bar, 100 μm. F, Immunostaining for CD200L+CD31+ endothelium positive for Ki67 at the epicenter of the lesion site 7 d following SCI in WT mice. B–D, F, Scale bar, 50 μm. A–F, Data are representative of 2–5 independent experiments; n = 3–5 mice in each group. G, Hindlimb motor function assessed according to the BMS for WT (filled black circles) and CD200L−/− (hollowed red circles) mice. Left, Motor score as a function of time. FInteraction(1,152) = 10.2; ***p < 0.001 (repeated-measures two-way ANOVA). Right, Scores of individual mice (d20). ***p < 0.001 (Student's t test). H, Hindlimb motor function, assessed according to the BMS for [WT > CD200L−/−] (hollowed orange squares) and [CD200L−/− > WT] (filled gray squares) BM chimeras. Left, Motor score as a function of time. FInteraction(1,60) = 4.94; ***p < 0.001 (repeated-measures two-way ANOVA). Right, Scores of individual mice (d20). *p = 0.0252 (Student's t test). G, H, Data are representative of two different experiments; n = 10 – 11 mice in each group. *p < 0.05. ***p < 0.001. Data are mean ± SEM.
To verify whether the injury-induced elevation of CD200L was associated with the presence of newly formed endothelial cells within the lesion site, we used antibody to Ki67, which is expressed by proliferating cells (Gerdes et al., 1983). We found that CD200L+ endothelial cells, located inside the lesion site, coexpressed Ki67 (Fig. 2F). Notably, endothelial cells located within the injury site of CD200L−/− mice also expressed the Ki67 marker (data not shown). These results suggest that acute SCI activates a mechanism whereby newly formed endothelial cells within the lesion site express CD200L.
To determine whether CD200L could be functionally involved in the repair process following SCI, motor functional recovery of WT and CD200L−/− mice was evaluated, based on a scale measuring hindlimb motor ability in an open field (BMS) (Basso et al., 2006), in which a score of 0 indicates complete paralysis and a score of 9 represents full mobility. Mice lacking CD200L exhibited impaired recovery relative to the spontaneous recovery exhibited by WT mice (Fig. 2G). Because CD200L can be expressed following injury not only by CNS-resident cells but also by BM-derived cells, which infiltrate following the insult, we used chimeric mice to confirm that CNS CD200L+ resident cells contributed to the functional repair process. Specifically, BM cells of CD200L−/− mice were replaced at adulthood with WT BM cells (WT > CD200L−/−). As control, we used WT mice that their BM cells were replaced at adulthood with CD200L−/− BM cells (CD200L−/− > WT). We found that the absence of CD200L in CNS-resident cells (WT > CD200L−/−) resulted in impaired recovery following SCI (Fig. 2H). These results substantiate the role of CD200L+ CNS-resident cells in the recovery following SCI, and primarily, the CD200L+ endothelial cells within the lesion site.
CD200R is expressed by different myeloid cell subpopulations within the lesion site
The important role of CD200L expressed within the CNS in the functional recovery process following SCI, and the temporally regulated expression of CD200L inside the lesion site, encouraged us to search for cell populations that might express CD200R, and whose interaction with CD200L+ endothelial cells could explain the effect on recovery. To this end, spinal cords of WT mice were excised at days 3, 7, and 14 following SCI and analyzed by immunohistochemistry for CD200R expression (Fig. 3A,B). At days 7 and 14 following SCI, when the lesion site area was already defined by GFAP+ reactive astrocytes, CD200R+ cells were found to be located at the epicenter and at the margins of the lesion site (Fig. 3A). We used the activation marker IB4 to identify activated myeloid cells (microglia and infiltrating mo-MΦ) within the lesion site (Galin et al., 1963; Hoek et al., 2000; Rijkers et al., 2008). As early as 3 d after the injury, IB4+ activated cells within the spinal cord parenchyma were found to highly express CD200R; moreover, at later time points after the injury (days 7 and 14), all the IB4+ activated myeloid cells expressed CD200R and were located at the epicenter of the lesion site (Fig. 3B). We further analyzed CD200R expression by different subpopulations of myeloid cells (microglia and mo-MΦ) within the lesion site by flow cytometry. To this end, we excised a 6 mm segment of the spinal cord parenchyma at the area of the lesion sites on different days after the injury, and dissociated it to a single-cell suspension. In line with the immunohistochemical results, both CD11blowCD45.2low cells, which mainly represent resident microglia, and CD11bhighCD45.2high cells, which mainly represent infiltrating mo-MΦ and activated microglia, were found to be CD200R immune-reactive (Fig. 3C). The CD45.2−CD11b− population did not express CD200R+, and was used as a negative-control for this staining (Fig. 3D,F). Notably, at all tested time points following SCI, CD11bhighCD45.2high cells (mainly infiltrating mo-MΦ and activated microglia) showed higher mean fluorescent intensity (MFI) levels of CD200R compared with the CD11blowCD45.2low population (mainly nonactivated microglia) (Fig. 3C). More detailed characterization of CD200R expression among different subpopulations of infiltrating mo-MΦ revealed that both M1-like proinflammatory cells (CD11bhighCD45.2highCX3CR1lowLy6C+) and M2-like anti-inflammatory cells (CD11bhighCD45.2highCX3CR1highLy6C−) (Arnold et al., 2007; Nahrendorf et al., 2007; Shechter et al., 2013b) expressed CD200R following SCI (Fig. 3E–G), although CD2000R was expressed by a higher percentage of the M1-like population compared with the M2-like population (Fig. 3G). These results indicate that myeloid populations, including activated microglia and infiltrating mo-MΦ, could directly interact with the CD200L+ endothelial cells at the epicenter of the lesion site.

Myeloid cell subpopulations expressing CD200R following SCI. A, B, Immunostaining for CD200R together with (A) GFAP, to detect reactive astrocytes, and (B) with isolectin B4 (IB4), to detect the activated myeloid cells (microglia and mo-MΦ), on days 3, 7, and 14 following SCI. Scale bar, 50 μm. A, B, Data are representative of two different experiments; n = 5 mice in each group, on each experiment. C, Flow cytometry histograms and quantification (percentages of CD200R-expressing population), comparing differential CD200R expression at different time points following SCI, between CD11blowCD45.2low resident microglia (orange) and CD11bhighCD45.2high mo-MΦ (blue), compared with negative control (CD45.2−CD11b− population (filled gray); Fbetween microglia and mo-MΦ(1,19) = 126.25; p < 0.001 (repeated-measures ANOVA). n = 2–6 mice per time point. D, Flow cytometry dot-plots and histograms showing CD200R+ and CD200R− gates in the CD45.2−CD11b− population. E, Left, Flow cytometry gates of CD11bhighCD45.2high mo-MΦ (blue), CD11blowCD45.2low microglia (orange), and CD45.2−CD11b− population (gray). Right, Flow cytometry gates of the proinflammatory mo-MΦ population (CX3CR1lowLy6C+; green) and of anti-inflammatory mo-MΦ population (CX3CR1highLy6C−; red). F, Flow cytometry dot-plots showing CD200R+ and CD200R− gates in the nonexpressing population, CD45.2−CD11b−, and in proinflammatory and anti-inflammatory mo-MΦ on day 3 following SCI. G, Percentages of CD200R+ cells of the proinflammatory and of the anti-inflammatory mo-MΦ on days 1, 3, 7, and 17 following SCI. Fbetween groups (days 3 and 7)(1,4) = 21.47; **p = 0.0036 (repeated-measures ANOVA). n = 2–6 mice per time point. C–G, Data are merged from two different experiments. *p < 0.05. Data are mean ± SEM.
Newly formed endothelial cells within the lesion site directly interact with CD200R+ myeloid cells via CD200 ligand
To test the possible direct interaction between CD200R+ myeloid cells (microglia and mo-MΦ) and CD200L+ endothelial cells at the lesion site, spinal cords were excised from CD200L−/− and WT mice 7 d after injury, and immunohistochemical staining was performed. We found in injured spinal cord of WT specimens, but not in CD200L−/−, a ring-like organization of Iba1+ activated myeloid cells (microglia and mo-MΦ) inside the epicenter of the lesion site, located in close proximity to the CD200L+CD31+ endothelial cells. This spatial organization was lacking in specimens excised from CD200L−/− mice (Fig. 4A). We next used high-throughput single-cell flow cytometry image analysis for assessment of the direct interaction between spinal cord-derived endothelial and CD200R+ myeloid cells (microglia and mo-MΦ). For this purpose, the lesion site area (6 mm) was excised from spinal cords 7 d following the injury and mechanically and enzymatically processed to obtain a single-cell suspension. The isolated cells were left in medium for 20 min to allow cell–cell interactions and were subsequently fixed and analyzed using ImageStream (see Materials and Methods). We searched for CD45.2+CD11b+ myeloid cells (both microglia and mo-MΦ) that expressed CD200R and which were in direct contact with CD11b−CD31+ endothelial cells that expressed CD200L, at their contact area (Fig. 4B). Subsequently, to test the involvement of CD200L in this direct interaction, we quantified the number of conjugating cells in the presence of CD200L inhibitor peptide (CDR3 g′; LT-13) (Chen et al., 2005; Gorczynski et al., 2008). As control for the specificity of the inhibitor, we also tested the number of conjugating cells in the presence of ovalbumin (OVA) peptide, and in PBS only. The gating strategy of the flow cytometry for quantification of microglia/mo-MΦ-endothelial cell conjugates was manually determined (see Materials and Methods; Fig. 4C). In the presence of the CD200L inhibitor, the number of conjugates was significantly reduced, compared with the uninhibited control samples (PBS) (Fig. 4D). Notably, OVA peptide did not change the number of conjugates, compared with the control sample (PBS) (Fig. 4E). Examples of conjugates between myeloid cells (microglia/mo-MΦ) and a single endothelial cell (Fig. 4F, left), or with endothelial cell clusters (Fig. 4F, right) are shown by the single-cell flow cytometry images. Notably, endothelial cells and macrophages/microglia have the same circular shape; however, in some conjugates, the macrophages/microglia were characterized by a larger diameter, probably due to their activation state. These results indicate that newly formed endothelial cells could directly interact with CD200R+ microglia and mo-MΦ within the lesion site, and that this interaction is mediated by CD200L signaling.

Endothelial cells form conjugates with CD200R+ myeloid cells via CD200L following SCI. A, Immunostaining for CD31+ endothelium, CD200L, and Iba1+ activated microglia and infiltrating mo-MΦ at the lesion site 7 d following SCI in WT and CD200L−/− mice, to examine the spatial distribution of myeloid cells relative to CD31+CD200L+ endothelium. Scale bar, 100 μm. Data are representative of three different experiments; n = 3–5 mice in each group. B, Image of conjugate between CD11b+CD45.2+CD200R+ myeloid cell and CD11b−CD31+CD200L+ endothelial cell derived from the lesion site 7 d following SCI, acquired by high-throughput single-cell flow cytometry image analysis (ImageStream). Data are representative of two different experiments; n = 5 mice. C, CD45.2+CD11b+CD31+ reads were first gated, then analyzed for distances between cell centers (see Materials and Methods), and finally CD11b−CD31+ and CD45.2+CD11b+CD31− cells with definite contact were classified as conjugates (purple gate). D, E, ImageStream quantification of CD11b−CD31+ and CD45.2+CD11b+CD31− conjugates incubated ex vivo with PBS (control) compared with (D) cells incubated with CD200L inhibitor peptide (Student's t test(one tailed), *p = 0.042) or (E) cells incubated with OVA peptide. F, ImageStream images of conjugation between (left) single or (right) clusters of CD11b−CD31+ endothelial cells and CD45.2+CD11b+CD31− myeloid cells. D, E, Data are pooled from two different experiments; n = 5 mice in each group. *p < 0.05. Data are mean ± SEM.
Absence of CD200L signaling abrogates the ability of endothelial cells to regulate the inflammatory phenotype of macrophages
To gain an insight as to whether CD200L expressed by the endothelial cells could affect the inflammatory response of the encountering microglia and infiltrating mo-MΦ, we used both in vitro and in vivo assays. Because of the vital role of the infiltrating mo-MΦ in the repair process following SCI (Rapalino et al., 1998; Shechter et al., 2009) and because of the high expression levels of CD200R by this cell type (Fig. 3C), we used mo-MΦ population in the following experiments. Thus, BM-derived cells were isolated from β-actin-GFP mice and differentiated to macrophages in the presence of M-CSF. The obtained GFP+ BM-derived macrophages (BM-MΦ) were cultured in the presence or absence of a brain endothelial cell line (b.END3), and LPS (100 ng/ml) was added for 4 h to induce inflammatory conditions (Fig. 5A,B). At the end of the incubation period, the GFP+ BM-MΦ were sorted from the cultures using FACS, and gene expression profile of proinflammatory cytokines was analyzed by qRT-PCR (Fig. 5A). Upon exposure to LPS, GFP+ BM-MΦ derived from cocultures with b.END3 cells showed significantly lower expression levels of the proinflammatory cytokines Tnfα and Il-1β, compared with GFP+ BM-MΦ that were cultured without endothelial cells (Fig. 5C). Notably, Il-6 expression was not significantly affected by the presence of b.END3 cells. In a second set up of these experiments, we assessed the effect of the brain endothelial cells (b.END3) on the inflammatory response of GFP+ BM-MΦ that were cultured with CD200L inhibitor (Fig. 5A,D). The b.END3 cells failed to reduce the inflammatory response of GFP+ BM-MΦ that were cultured with the CD200L inhibitor (Fig. 5D,E). These results indicate that endothelial cells can directly reduce macrophage-derived inflammation in a CD200-dependent signaling pathway.

CD200L inhibitor abrogates the ability of endothelial cells to regulate the inflammatory phenotype of macrophages in vitro. A, B, GFP+ BM-MΦ were cultured with or without brain endothelial cell-line cells (b.END3) for 24 h. To mimic injury-induced inflammation in vitro, 0.1 μg/ml LPS was added to the cultures for 4 h. GFP+ BM-MΦ were collected using FACS sorting, RNA was extracted, and gene expression was analyzed by qRT-PCR. C, GFP+ BM-MΦ gene expression of proinflammatory cytokines Tnfα, Il-1β, and Il-6 before and following LPS induction, with (filled black) or without (empty black) b.END3 cells. Tnfα: F(2,14) = 119.5, ***p < 0.001 (one-way ANOVA). Il-1β: F(2,14) = 45.28, ***p < 0.001 (one-way ANOVA). Il-6: F(2,12) = 27.71, ***p < 0.001 (one-way ANOVA). Asterisks above bars without horizontal line indicate significance relative to the [mo-MΦ without LPS] sample. A, D, GFP+ BM-MΦ + CD200L inhibitor were cultured with or without b.END3 cells for 24 h. To mimic injury-induced inflammation in vitro, 0.1 μg/ml LPS was added to the cultures for 4 h. GFP+ BM-MΦ were collected using the FACS sorter, RNA was extracted, and gene expression was analyzed by qRT-PCR. E, GFP+ BM-MΦ gene expression of proinflammatory cytokines Tnfα, Il-1β, and Il-6 before and following LPS induction, with (filled red) or without (empty red) b.END3 cells. Tnfα: F(2,14) = 78.44, ***p < 0.001 (one-way ANOVA). Il-1β: F(2,14) = 23.71, ***p < 0.001 (one-way ANOVA). Il-6: F(2,15) = 7.137, **p = 0.0066 (one-way ANOVA). Asterisks above bars without horizontal line indicate significance relative to the [mo-MΦ without LPS] sample. Data shown from two different experiments were combined, by normalization based on [BM-MΦ+LPS] samples; n = 5 – 6 replicates in each group. **p < 0.01. ***p < 0.001. Data are mean ± SEM.
To confirm in vivo the functional relevance of CD200 regulatory pathway to the activity of the infiltrating mo-MΦ within the lesion site, we analyzed the phenotype of WT and CD200L−/− lesion-site derived infiltrating mo-MΦ by flow cytometry, 7 d following SCI (the peak of endothelial CD200L expression; Fig. 2) and of inflammation-resolving activities by mo-MΦ (Shechter et al., 2009). Previous documentation revealed the inability of proper discrimination between activated microglia and mo-MΦ in CD200L−/− mice, due to the increased expression levels of CD11b and CD45 molecules by microglia derived from CD200L−/− mice (Hoek et al., 2000). Therefore, we used a pregate of CD45+CD11b+ cells for the analysis of the following in vivo experiments. We examined proinflammatory activity by measuring the levels of the intracellular cytokine TNFα, using anti-TNFα and isotype-control antibodies (Fig. 6A). Among the proinflammatory CD45+CD11b+Ly6Chigh mo-MΦ that were isolated from the lesion site of CD200L−/− mice, a higher percentage of cells were positive for the intracellular cytokine-TNFα, compared with cells derived from lesion site of WT mice (Fig. 6B,C). Moreover, the proinflammatory mo-MΦ derived from CD200L−/− injured mice were characterized by a higher MFI of TNFα, compared with those isolated from WT mice (Fig. 6B,D). Next, we analyzed the expression of markers commonly associated with macrophages exhibiting anti-inflammatory resolving activities: Dectin-1, CD206 (mannose receptor) and IL-4R. CD45.2+CD11b− cells were used as control for the staining of these markers (Fig. 6E,F). Overall, lower levels of all tested markers associated with anti-inflammatory and inflammation-resolving activities were found to be expressed by mo-MΦ isolated from CD200L−/− mice relative to WT (Fig. 6G,H). These results support our contention that CD200L-expressing endothelial cells contribute to the downregulation of the inflammatory response and to the induction of inflammation resolving phenotype of CD200R+ macrophages following acute insult in the CNS.

Absence of CD200L in vivo promotes a stronger mo-MΦ-derived inflammatory response and prevents anti-inflammatory activities. A, Flow cytometry gates of CD45.2+CD11b+Ly6Chigh mo-MΦ, and their staining for TNFα and its isotype control expression levels at day 7 following SCI. B–D, Flow cytometry: (B) histogram, (C) percentage quantification, and (D) MFI quantification of the CD45.2+CD11b+Ly6Chigh mo-MΦ population expressing TNFα in WT (black) and CD200L−/− (red) mice at day 7 following SCI. Percentage TNFα: *p = 0.03 (Student's t test). MFI TNFα: *p = 0.03 (Student's t test). Each sample represents a pool of 2 animals; n = 2 – 3 replicates in each group. E, Flow cytometry gates of CD45.2+CD11b+ myeloid cells (microglia and mo-MΦ) and CD45.2+CD11b− population at day 7 following SCI. F, Flow cytometry dot-plots showing gating strategy for the tested surface markers (Dectin-1, CD206, and IL-4R) in the CD45.2+CD11b− population. G, H, Flow cytometry: (G) histograms and (H) quantification of the CD45.2+CD11b+ myeloid cell population expressing Dectin-1, CD206 (mannose receptor) and IL-4R in WT (black), and CD200L−/− (red) mice at day 7 following SCI. Dectin-1: *p = 0.05 (Student's t test). CD206: **p = 0.01 (Student's t test). IL-4R: *p = 0.04 (Student's t test). Data are representative of two different experiments; n = 2 – 3 mice per group. *p < 0.05. **p < 0.01. Data are mean ± SEM.
Discussion
In the present study, we show that, in homeostasis, CD200L is selectively expressed by the endothelial cells of the mouse choroid plexus (comprising part of the blood–cerebrospinal fluid barrier; BCSFB) and the lepto-meninges (blood–leptomeningeal barrier), but not by the endothelial cells that form the blood–spinal cord barrier. However, following SCI, newly formed endothelial cells within the lesion site upregulate CD200L expression. We further found that these newly formed endothelial cells directly interact with CD200R+ myeloid cells within the lesion site (resident microglia and infiltrating mo-MΦ) and regulate the inflammatory phenotype of mo-MΦ via the CD200 ligand.
Endothelial expression of CD200L was shown to vary between tissues, and depending on the type of capillary. For instance, its expression was observed in both fenestrated and nonfenestrated endothelium, and may also be upregulated following LPS challenge (Ko et al., 2009). It was suggested that endothelial CD200L is involved in modulation of T cell adhesion and migration into tissues (Ko et al., 2009; Denieffe et al., 2013). In the CNS, extensive expression of CD200L on retinal neurons and retinal vascular endothelium has been reported (Dick et al., 2001; Broderick et al., 2002). However, endothelial expression of CD200L in the brain parenchyma has been controversial. Whereas some groups reported that CD200L is exclusively expressed by neuronal cell bodies, others also found its expression on the luminal surface of blood–brain barrier endothelium in the hippocampus, and on the luminal and abluminal surfaces of non-blood–brain barrier endothelium in the area postrema (Broderick et al., 2002). Here, we found the selective endothelial expression of CD200L at the borders of the CNS in homeostasis.
The neovascularization process in wound healing following an acute insult has been mainly associated with enhanced supply of oxygen and nutrients into the lesion area, and outside the CNS, also enables extravasation of leukocytes (Tonnesen et al., 2000; Dray et al., 2009; Avraham-Davidi et al., 2013). However, no role has been attributed to newly formed endothelial cells in regulation of the inflammatory responses of the local microglia or the infiltrating mo-MΦ. The limited recovery following acute insult in the CNS, relative to the efficient healing of wounds in peripheral tissues, has been often attributed to the incomplete resolution of inflammation, which might cause secondary damage and neuronal loss (Shechter et al., 2009; Shechter and Schwartz, 2013). Under severe acute and chronic inflammation, microglia fail to bring repair to completion, and recruitment of mo-MΦ was found to be beneficial (Rapalino et al., 1998; Simard et al., 2006; Shechter et al., 2009, 2011; London et al., 2011, 2013b; Gliem et al., 2012, 2016; Cohen et al., 2014; Ritzel et al., 2015). One of the factors that were found to be critical for skewing the recruited mo-MΦ toward an inflammation resolving phenotype following SCI is the glial scar matrix, chondroitin sulfate proteoglycan (Rolls et al., 2008; Shechter et al., 2011). The present study highlights an additional mechanism whereby inflammation within the privileged CNS could be regulated following acute injury. This mechanism involves a unique population of newly formed endothelial cells located within the epicenter of the lesion site, and which directly interact with CD200R+ myeloid cells (microglia and infiltrating mo-MΦ) via CD200L signaling, and thereby controls the mo-MΦ phenotype switch from proinflammatory to resolving activity. In the absence of CD200L signaling, inflammatory mo-MΦ in the injured CNS showed higher expression levels of TNFα, lower expression levels of inflammation-resolving activities, and the extent of functional recovery was reduced. Our results are consistent with published data linking CD200 signaling with protection from extensive chronic inflammation under different pathological conditions outside and inside the CNS (Hoek et al., 2000; Chitnis et al., 2007; Copland et al., 2007; Snelgrove et al., 2008; Koning et al., 2009; Rygiel et al., 2009; Walker et al., 2009; Luo et al., 2010; Zhang et al., 2011), and with “alternative macrophage activation” (Koning et al., 2010; Mukhopadhyay et al., 2010). Notably, while indirect communication between endothelial and myeloid cells was shown to occur during regular wound healing processes (Martin and Leibovich, 2005; Mahdavian Delavary et al., 2011; Avraham-Davidi et al., 2013), no evidence for CD200-dependent direct cell–cell interactions has been described previously. Interestingly, in a cancer model, it was proposed that, in cases in which the tumor benefits from inflammation, CD200L+ endothelial cells in the tumor microenvironment might interact with CD200R+ myeloid cells, reduce their inflammation, and thereby promote antitumor immune responses (Belkin et al., 2013).
In CNS homeostasis, the communication between microglia and the surrounding glial cells as well as with neighboring neurons is supported by a versatile subset of cell surface molecules on the microglial cell membrane. Unlike surface molecules, which their ligand recognition results in a proinflammatory activation, there is a small subset of molecules, including CD200, in which ligand binding is crucial for maintaining the resting ramified phenotype of microglia in the healthy CNS, and these receptors are therefore categorized as “inhibitory molecules.” Another example of such a fundamental signaling pair in neurophysiology is the interaction between CX3CR1-expressing microglia (Jung et al., 2000) and CX3CL1+ neuronal subsets (Kim et al., 2011). This route of neuron–microglia communication was shown to play different roles under inflammatory and neurodegenerative conditions (Harrison et al., 1998; Prinz and Priller, 2010).
The present study suggests that newly formed endothelial cells within the CNS parenchyma following acute injury in mice contribute to regulation of the local inflammatory response following SCI. This regulation was found to be dependent on the CD200 pathway. Of note, thus far, CD200L expression within the brain has been mainly associated with neurons that regulate activation and restore the resting state of CD200R+ microglia in homeostasis and during pathology (Hoek et al., 2000; Kierdorf and Prinz, 2013). Here we attribute a role to newly formed endothelial cells within the spinal cord parenchyma in controlling mo-MΦ inflammatory response via upregulation of CD200L in acute pathology. Such a mechanism might be unique to immune privileged tissues, such as the CNS; yet, further studies to identify similar mechanisms in tissues that are not immune privileged are also needed. Moreover, the essential role of the neovascularization process that was shown here following SCI, calls for studies regarding additional activities of endothelium during repair processes outside and within the CNS.
Footnotes
This work was supported by the European Union Seventh Framework Program HEALTH-2011 Grant 279017 to M.S. M.S. holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology. We thank Shelley Schwarzbaum for proofreading and Margalit Azoulay for handling the animals.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Prof. Michal Schwartz or Dr. Merav Cohen, Weizmann Institute of Science, 7610001 Rehovot, Israel. michal.schwartz{at}weizmann.ac.il or merav.cohen{at}weizmann.ac.il





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}