Article Text

Abstract
Background Non-alcoholic fatty liver disease (NAFLD) is characterised by accumulation of excessive triglycerides in the liver. Obesity is usually associated with NAFLD through an unknown mechanism.
Objective To investigate the roles of Yin Yang 1 (YY1) in the progression of obesity-associated hepatosteatosis.
Methods Expression levels of hepatic YY1 were identified by microarray analysis in high-fat-diet (HFD)-induced obese mice. Liver triglyceride metabolism was analysed in mice with YY1 overexpression and suppression.
Results YY1 expression was markedly upregulated in HFD-induced obese mice and NAFLD patients. Overexpression of YY1 in healthy mice promoted hepatosteatosis under high-fat dietary conditions, whereas liver-specific ablation of YY1 using adenoviral shRNA ameliorated triglyceride accumulation in obese mice. At the molecular level, YY1 suppressed farnesoid X receptor (FXR) expression through binding to the YY1 responsive element at intron 1 of the FXR gene.
Conclusions These findings indicate that YY1 plays a crucial role in obesity-associated hepatosteatosis, through repression of FXR expression.
- Nonalcoholic Steatohepatitis
- Signal Transduction
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Obesity is tightly associated with an increased risk of non-alcoholic fatty liver disease (NAFLD).
-
FXR (farnesoid X receptor) plays a critical role in liver triglyceride metabolism.
-
FXR inhibits the progression of liver steatosis through upregulation of small heterodimer partner, which downregulates the transcription of SREBP-1c, a key transcription factor involved in the lipogenesis.
What are the new findings?
-
Hepatic Ying Yang 1 (YY1) expression levels were upregulated in obese mice.
-
YY1 overexpression leads to hepatosteatosis in C57BL/6 mice, as well as upregulation of lipogenic genes.
-
YY1 suppresses FXR transcription through binding to the YY1 responsive element at intron 1 of the FXR gene.
-
Liver-specific ablation of YY ameliorates liver triglyceride accumulation in obese mice.
How might it impact on clinical practice in the foreseeable future?
-
In light of the roles of YY1 in hepatosteatosis, therapeutic interventions targeting the YY1–FXR axis may be a promising strategy for treating NAFLD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) has emerged as one of the most common public health problems over the past few decades.1 ,2 NAFLD, characterised by aberrant hepatocellular accumulation of lipids, is tightly associated with multiple comorbid conditions, including insulin resistance and type 2 diabetes.3 ,4 Moreover, it can trigger a progressive cascade of lipid disorders, ranging from hepatosteatosis to non-alcoholic steatohepatitis, liver fibrosis, cirrhosis and eventually hepatocellular carcinoma.5
The amount of triglycerides accumulated in hepatocytes represents a complex interaction between hepatic fatty acid uptake, de novo lipogenesis (DNL), fatty acid oxidation and export within very low-density lipoprotein. Hepatosteatosis develops when the rate of triglyceride input is greater than that of the output. Obesity, a well-described epidemic in recent decades, has been considered the most prevalent cause for NAFLD.6 The prevalence of hepatosteatosis and steatohepatitis is much higher in obese than in non-obese populations.7 ,8 Despite a close relationship between obesity and NAFLD in population studies, the molecular determinants of hepatosteatosis in the obese remain largely unknown. It has been discovered that the rate of hepatic fatty acid uptake is higher in obese than in lean subjects.9 ,10 In addition, obesity-induced NAFLD in rodents and humans is associated with increased hepatic expression of several genes involved in the DNL pathway.11 ,12 Therefore, investigating the molecular mechanisms of obesity-induced NAFLD would help identify unique targets for therapeutic intervention of hepatosteatosis.
In this study, we discovered that Yin Yang 1 (YY1) was upregulated in the liver in obese mice and NAFLD patients. YY1 is a ubiquitous and multifunctional zinc-finger transcription factor of the polycomb group protein family, which can act as a transcriptional repressor, activator or initiator element binding protein.13 Previous studies have identified a myriad of potential YY1 target genes, which are important for cell proliferation and differentiation.13 It was shown that YY1 null mouse died during embryonic development around implantation.14 Moreover, the findings of YY1 overexpression in multiple cancer cells suggested that YY1 might play an important role in the regulation of tumour cell proliferation and apoptosis.15 Besides, YY1 could promote triglyceride accumulation in adipocytes through repression of Chop10 transcription, suggesting its potential role in obesity.16 YY1 was also identified to repress genes associated with the insulin/insulin-like growth factor (IGF) signalling pathway, such as IGF1-2, IRS1-2 and Akt1-3 in skeletal muscles.17 In addition, a recent study indicated that YY1 could be a candidate gene associated with body weight, glucose, cholesterol or free fatty acid levels.18 However, the exact cellular functions of YY1 in hepatic metabolic disorders remains largely unexplored.
Materials and methods
Animal studies and human liver tissues
Male C57BL/6 lean and db/db mice aged 8–12 weeks were purchased from the Shanghai Laboratory Animal Company (SLAC, Shanghai, China). Farnesoid X receptor (FXR) heterozygous mice were obtained from Jackson Laboratories Bar (Harbor, Maine, USA). Mice were housed at 21±1°C with a humidity of 55±10% and a 12-hour light–dark cycle. High-fat-diet (HFD)-induced obese mice were maintained with free access to a high-fat chow (D12492, Research Diets, New Brunswick, New Jersey, USA) and drinking water. The HFD contains 60 Kcal% fat, 20 Kcal% carbohydrate and 20 Kcal% protein. The normal diet (ND) contains 10 Kcal% fat, 70 Kcal% carbohydrate and 20 Kcal% protein. The animal protocol was reviewed and approved by the Animal Care Committee of Shanghai Jiao Tong University School of Medicine. The human liver tissues were commercially obtained from Alena Bio. Company (Xi'an, China) as described previously.19 The protocol was reviewed and approved by the Ethics Committee of Shanghai Jiao Tong University School of Medicine.
Adenovirus preparation
Adenovirus expressing murine YY1 or FXR (Ad-YY1, Ad-FXR) was constructed by Invitrogen (Shanghai, China) with a full-length YY1 or FXR complementary DNA (cDNA) coding sequence. Overexpression of hepatic YY1 or FXR was achieved by means of tail-vein injection of Ad-YY1 or FXR (4 or 2×109 plaque-forming units) in normal C57BL/6 mice. To silence YY1 expression in db/db mice, adenoviruses expressing YY1 shRNA were generated using pAD_BLOCK_IT_DEST vectors (Invitrogen, Grand Island, New York, USA). All viruses were purified by the caesium chloride method and dialysed in phosphate-buffered saline (PBS) containing 10% glycerol prior to animal injection.
Hepatic and cellular triglyceride measurement
Mice were sacrificed at 09:00 after an overnight fasting (16 h). Liver tissues (weighed ∼100 mg) were harvested and homogenised in chloroform/methanol (2 : 1 v/v) using a polytron tissue grinder. The extracts were dried under nitrogen flow and resuspended in isopropanol. For the in vitro model of cellular steatosis, HepG2 or mouse primary hepatocyte cells were exposed to palmitate at a concentration of 0.5 mM. Triglyceride concentrations were measured using commercial kits (Sigma, St. Louis, MO, USA; Biovision, Milpitas, California, USA) according to the manufacturer's instructions.
Glucose and insulin tolerance tests
Glucose tolerance tests were performed by intraperitoneal injection of D-glucose (Sigma, USA) at a dose of 2.0 mg/g body weight after a 16-hour fast. For insulin tolerance tests, mice were injected with regular human insulin (Eli Lily, Indianapolis, Indiana, USA) at a dose of 0.75 U/kg body weight after a 6-hour fast. Blood glucose was determined using a portable blood glucose meter (Lifescan, Johnson & Johnson, New Brunswick, New Jersey, USA).
Microarray analysis and quantitative real-time PCR
Total RNA was isolated from hepatic tissues or cell lysates using the standard TRIzol method according to the manufacturer's instructions (Invitrogen, Shanghai). Affymetrix array hybridisation and scanning were performed using Mouse Genome 430 2.0 chips by Gene Tech Company Limited (Shanghai, China). Total RNA samples obtained from six mice per group (ND and HFD) and pooled by each of the two were used for microarray analysis. In order to quantify the transcripts of the genes of interest, quantitative real-time PCR was performed using a SYBR Green Premix Ex Taq (Takara, Japan) on Light Cycler 480 (Roche, Switzerland). The primer sequences used are available upon request.
Western blotting
Hepatic tissues and cells were lysed in radioimmunoprecipitation buffer containing 50 mM Tris-HCl, 150 mM NaCl, 5 mM MgCl2, 2 mM EDTA, 1 mM NaF, 1% NP40 and 0.1% sodium dodecyl sulfate. Western blotting was performed using antibodies against FXR (Santa Cruz, USA), YY1 (Santa Cruz, California, USA), phospho-insulin receptor β (Cell Signaling, USA), phospho-insulin receptor substrate 1 (Cell Signaling), phosphor-AKT (Cell Signaling), AKT (Cell Signaling, Danvers, Massachusetts, USA) and GAPDH (Cell Signaling). Protein expression levels were quantitated with the use of ImageJ.
Histological examination
For H&E staining, liver tissues were fixed overnight in 4% formalin, embedded in paraffin and sectioned at 5 μm. Immunohistochemical staining was performed according to a standard protocol. In brief, the sections were deparaffinised, rehydrated and treated with 3% hydrogen peroxide in methanol for 30 min to quench endogenous peroxidase activity. The pretreated sections were then blocked in PBS containing 2.5% horse serum for 1 h and then incubated with primary antibodies in a humidified chamber at 4°C overnight. Images were acquired using an Olympus BX51 microscope.
Luciferase assays
YY1 oligo-siRNA was obtained from Dharmacon (Thermo Scientific, Brookfield, Wisconsin, USA). FXR intron 1 was amplified from the mouse genomic DNA and inserted into pGL4.15 vector (Promega, Madison, Wisconsin, USA). For the luciferase reporter assays, HepG2 cells were seeded in 24-well plates and transfected with YY1 plasmids and reporter vectors, using Lipofectamine 2000 (Invitrogen). Cells were harvested 30 h after transfection. Luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega).
Chromatin immunoprecipitation assays
A chromatin immunoprecipitation (ChIP) assay kit was used (Upstate, Billerica, Massachusetts, USA). In brief, HepG2 cells or homogenised mouse liver tissues (nuclear lysates) were fixed with formaldehyde. DNA was sheared to fragments at 200–1000 bp using sonications. Chromatin was incubated and precipitated with antibodies against YY1 (Santa Cruz), histone deacetylase 1 (HDAC1), HDAC2, HDAC3 (Abcam, Cambridge, Massachusetts, USA) or IgG (Santa Cruz).
Statistical analysis
Values were shown as mean±SEM. Statistical differences were determined by a Student t test. Statistical significance is displayed as *p<0.05, **p<0.01 or ***p<0.001.
Results
Upregulation of hepatic YY1 in mice with obesity
Obesity is closely associated with fatty liver. C57BL/6 mice that were fed a HFD containing 60 Kcal% of fat for 12 weeks displayed severe hepatosteatosis (see online supplementary figure 1A). The clustering analysis of Affymetrix arrays revealed that a number of genes involved in DNL were markedly upregulated in the livers of HFD-fed mice (figure 1A), including ATP citrate lyase (Acly), fatty acid synthesis (Fasn) and stearoyl-coenzyme A desaturase 1 (Scd-1). The activation of the hepatic lipogenesis programme was accompanied by an increase in SREBP1-c expression, a master hepatic transcriptional regulator. Intriguingly, we found that YY1, which is abundantly expressed in hepatocytes (see online supplementary figure 1B), was also upregulated. We further confirmed that hepatic YY1 mRNA and protein levels were increased by real-time PCR and western blotting (figure 1B). Interestingly, YY1 expression was not changed in either white adipose tissues or skeletal muscles in obese mice, compared with lean mice (see online supplementary figure 1C,D).

Yin Yang 1 (YY1) expression in the liver. (A) Cluster analysis of hepatic gene expression in C57BL/6 mice aged 8 weeks fed with a normal diet (ND) or high-fat diet (HFD) for 12 weeks. Cluster in red indicates upregulation and in green indicates downregulation. (B,C) Hepatic YY1 expression by real-time PCR and western blotting analysis in HFD-fed mice (B) and in db/db mice aged 12 weeks (C) (n=5–8).
To determine whether YY1 upregulation represents a common feature of obesity-related fatty liver, we examined hepatic YY1 expression in leptin receptor-deficient (db/db) mice and patients with NAFLDs. YY1 was markedly increased in the livers of db/db mice (figure 1C) and NAFLD patients (see online supplementary figure 1E,F). Thus, abnormal expression of YY1 represents a typical feature of hepatosteatosis in obese animals and humans.
YY1 overexpression promotes hepatosteatosis in C57BL/6 mice
The elevated expression of YY1 in the livers in obesity prompts us to investigate whether YY1, as a causal factor, leads to hepatosteatosis. We generated adenoviral YY1 and delivered it via tail-vein injection to C57BL/6 mice. As shown in figure 2A, YY1 was dramatically increased in the livers, but not in other tissues such as white adipose tissues (data not shown). In mice fed with normal chow, YY1 overexpression had little effect on hepatic triglyceride contents (data not shown). However, in YY1 overexpressed mice fed with a HFD for 1 week, the liver showed a pale appearance and prominent hepatosteatosis by H&E staining (figure 2B). Moreover, liver weights and triglyceride contents were significantly elevated (figure 2C,D). In parallel, overexpression of hepatic YY1 resulted in an increase in serum triglyceride and free fatty acid levels (figure 2E). However, body weight, food intake and body fat remained unaffected in YY1 overexpressed mice (see online supplementary figure 2A–C). Besides, serum alanine aminotransferase levels were not changed in the adenoviruses-infected group (Ad-Green Fluorescent Protein (GFP) and Ad-YY1) as compared with non-adenoviral infected mice (PBS), suggesting that adenovirus delivery caused no obvious signs of liver damage (see online supplementary figure 2D).

Overexpression of Yin Yang 1 (YY1) promotes hepatosteatosis. (A) Overexpression of hepatic YY1 in C57BL/6 mice aged 10 weeks infected with adenoviral YY1 by western blotting. (B) Representative livers at macroscopic (left) and histological (H&E, right) examination in mice overexpressing GFP or YY1. At day 3 after virus injection, mice were fed with high-fat diet for another 7 days. Magnification: 400×; scale bar: 50 μM. (C–E) Liver weight (C), hepatic triglyceride (TG) contents (D) and serum TG and free fatty acid (FFA) levels (E) in mice infected with Ad-GFP and Ad-YY1. After a 16-hour fast, mice were sacrificed at day 10. Serum and liver tissues were collected for further analysis (A–E) (n=7–9). (F) Glucose tolerance test and insulin tolerance test and the corresponding area under curve (AUC) were determined at day 6 and day 8, respectively.
Due to a close association between fatty liver and systematic insulin resistance, we observed that C57BL/6 mice with hepatic YY1 overexpression demonstrated a moderate reduction in insulin sensitivity by glucose and insulin tolerance tests (figure 2F). In addition, phosphorylation of the insulin receptor β (p-IRβ), insulin receptor substrate 1 (p-IRS1) and Ser473 AKT (p-AKT), key molecules in the insulin-signalling pathway, was also partially impaired (see online supplementary figure 2E). Moreover, mice with YY1 overexpression displayed hyperinsulinaemia (see online supplementary figure 2F). Collectively, these data underline the notion that YY1 overexpression results in fatty liver development and hypertriglyceridaemia under conditions of chronic energy surplus.
YY1 downregulates FXR expression
Next, we sought to investigate the molecular basis for the observed phenotypic changes in mice with YY1 overexpression. Except for peroxisome proliferator-activated receptor γ (PPARγ), quantitative real-time PCR showed that SREBP1-c, Fasn and Scd-1 were increased in YY1 overexpressed liver tissues (figure 3A), suggesting an enhancement of lipogenesis. SREBP1-c is tightly regulated by several nuclear receptors,20 including liver X receptor (LXR), which upregulates SREBP1-c,21 and FXR, which downregulates SREBP1-c via induction of the orphan nuclear receptor, short heterodimer partner (SHP).22 As shown in figure 3B, hepatic FXR and SHP expression was significantly reduced in YY1 overexpressed mice, while LXR expression was not changed. The reduction of hepatic FXR expression was further confirmed by western blotting analysis (figure 3C). Besides, we observed an increased bile acid pool as well as upregulation of bile acid synthesis genes (Cyp7A1, Cyp8B1) in mice with YY1 overexpression (see online supplementary figure 3A,B), which is consistent with the notion that FXR also plays a critical role in the negative feedback control of bile acid synthesis.23 Therefore, these results indicate that YY1 enhances hepatic triglyceride accumulation, probably through repression of FXR expression.

Yin Yang 1 (YY1) downregulates farnesoid X receptor (FXR) expression in vivo and in vitro. (A) Real-time PCR analysis of lipogenic gene expression in mice transfected with Ad-GFP or Ad-YY1. (B,C) Hepatic FXR expression was determined by real-time PCR and western blotting. (D) HepG2 cells were transfected with Ad-GFP or Ad-YY1. Cell lysates were immunoblotted for YY1 and FXR. (E) Influence of YY1 overexpression on intracellular triglyceride in HepG2 cells. Cells were treated with palmitate (0.5 mM) after 24 h of virus transfection and harvested after 24 h incubation with palmitate. (F) FXR was upregulated as YY1 was silenced by YY1 siRNA in HepG2 cells. At 24 or 48 h post-transfection, cell lysates were harvested for RNA or protein analysis, respectively. LXR, liver X receptor; SHP, short heterodimer partner.
To further confirm this regulation in an independent setting, HepG2 cells were transfected with Ad-GFP or Ad-YY1. As shown in figure 3D, YY1 overexpression led to a dramatic decrease of FXR, as well as cellular triglyceride accumulation in the presence of palmitate (figure 3E). Moreover, HepG2 cells were transfected with siRNA oligos targeting YY1. Knockdown of hepatic YY1 led to upregulation of FXR mRNA and protein expression (figure 3F). In addition, palmitate induced substantial lipid accumulation in HepG2 cells, while YY1 knockdown prevented cellular triglyceride accumulation significantly (see online supplementary figure 3C). Together, our data implicate that FXR could be a transcriptional target of YY1.
YY1 negatively regulates FXR through an intronic binding site
To seek the molecular basis for this regulation, we identified a potential YY1 binding site in the first intron of FXR gene using an online transcription factor scanning system (http://www.cbil.upenn.edu/cgi-bin/tess/tess) (figure 4A). It is of interest that this binding site is conserved in many species (figure 4A). Indeed, mutation of this site completely abolished the inhibitory effect of YY1 on FXR transcriptional activity in a transient transfection assay (figure 4B). A ChIP assay also showed that YY1 is bound to the intron 1 region of the FXR gene in HepG2 cells (figure 4C). Furthermore, YY1 bound to this site on FXR was more abundant in the liver extracts from obese than lean mice, which indicates that FXR represents a direct YY1 target gene in vivo (figure 4D). In this regard, we also found that hepatic FXR was reduced to some extent in obese mice (see online supplementary figure 4A,B). To examine whether FXR downregulation could contribute to hepatosteatosis in obesity, we placed FXR heterozygous mice on a HFD for 4 weeks. Compared with wild-type mice, hepatic FXR expression was downregulated about 45%–55% in FXR heterozygous mice (data not shown). Of note, these mice demonstrated hepatosteatosis, hyperlipidaemia and insulin resistance, with a dysregulation of SREBP1-c expression as compared with wild-type littermates (see online supplementary figure 5A–E).

Yin Yang 1 (YY1) bound to an intronic binding site of farnesoid X receptor (FXR) gene. (A) Intron 1 of FXR contains a potential conserved binding site for YY1. H, human; M, mouse; R: Rat. (B) Luciferase assays. HepG2 cells were co-transfected with YY1 expression plasmids (0, 200, 400 ng) and luciferase reporter plasmids (200 ng) containing wild type (WT-Luc) or mutant (Mut-Luc) binding site of FXR intron 1. (C,D) Chromatin immunoprecipitation assays showing representative YY1 binding to FXR intron 1 in HepG2 cells (C) or hepatic tissue lysates from lean, db/db and high-fat-diet-fed mice (D) and quantified by real-time PCR (lower panel).
Negative regulation of gene transcription by YY1 has been partly attributed to its interaction with histone deacetylase (HDAC) proteins.24 Consistent with this, the HDAC class I and class II inhibitor trichostatin A but not the class III (SirT1) inhibitor EX527 abolished YY1-mediated repression of FXR transcription (see online supplementary figure 6A,B), suggesting that YY1 recruits nuclear HDAC to confer its transcriptional repression roles. In addition, simultaneous transfection of YY1 and HDAC1, but not HDAC2 or HDAC3, further inhibited FXR transcription (see online supplementary figure 6C and data not shown). Consistently, YY1 overexpression recruited much more HDAC1, but not HDAC2 or HDAC3, to the YY1 binding site in the livers (see online supplementary figure 6D). Thus, these studies suggested that YY1 repressed FXR transcription via its recruitment of HDAC1.
Ablation of YY1 improves hepatosteatosis in obese mice
Next, we explored whether ablation of YY1 reversed hepatosteatosis in obese mice. Adenoviral shRNA against an YY1 coding region (Ad-shYY1) was adopted to silence hepatic YY1 expression in db/db mice. Ad-shYY1 or Ad-shLacZ as a control was delivered to db/db mice via tail-vein injection. Hepatic YY1 was almost completely abolished in Ad-shYY1 mice as compared with Ad-shLacZ mice (figure 5A). As expected, hepatosteatosis was greatly improved (figure 5B) and hepatic triglyceride contents were reduced (figure 5C) in Ad-shYY1-infected db/db mice. The liver weight was also reduced in db/db mice treated with Ad-shYY1 (figure 5D). Consistently, abrogation of hepatic YY1 resulted in a reduction in serum triglyceride and free fatty acid levels (figure 5E), as well as an improvement of systemic insulin resistance (figure 5F). Body weight, food intake and total body fat contents were comparable in the two groups (see online supplementary figure 7A–C). Moreover, knockdown of hepatic YY1 in db/db mice resulted in a pronounced increase of FXR expression and a reduction of SREBP1-c, Fasn and Scd-1 (see online supplementary figure 7D). We also observed an increase of FXR protein levels in db/db mice with YY1 knockdown (see online supplementary figure 7E).

Yin Yang 1 (YY1) ablation improves hepatosteatosis in obese mice. (A) Hepatic YY1 expression was markedly reduced in db/db mice aged 12 weeks infected with adenoviral YY1 shRNA by real-time PCR and western blotting (n=8). shRNA virus targeting LacZ was used as a negative control. (B) Representative hepatic histology by H&E staining. Magnification: 400×; scale bar: 50 μM. (C–E) Hepatic triglyceride (TG) contents (C), liver weight (D) and serum TG and free fatty acid (FFA) levels (E). (F) Insulin tolerance test and area under curve (AUC) at day 8 after adenoviral injection. All the above experiments were conducted in db/db mice infected with adenoviral YY1 shRNA or control (LacZ) shRNA. After a 16-hour fast, db/db mice were sacrificed at day 12. Liver and serum samples were collected for further analysis of gene expression (A–E) (n=8).
FXR is required for YY1-mediated hepatosteatosis
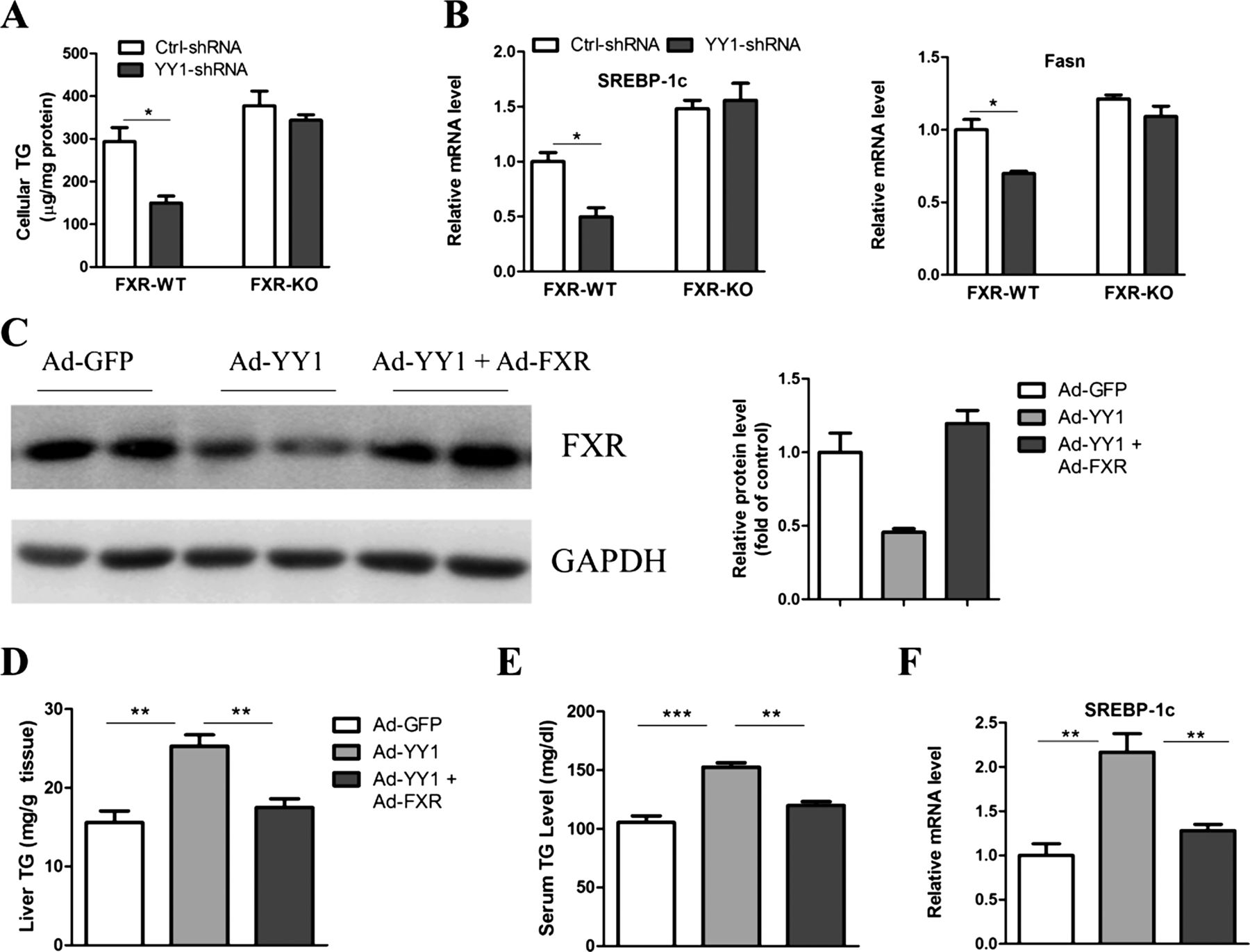
If YY1 promotion of triglyceride accumulation in the liver relies mainly on FXR downregulation, it would be anticipated that YY1 should have no further effect when FXR is depleted in the livers. To evaluate this possibility, we infected YY1 shRNA adenovirus equally in both wild-type and FXR-null hepatocytes. Our data showed that knockdown of YY1 reduced triglyceride contents in wild-type but not in FXR-null hepatocytes (figure 6A). Consistently, lipogenic genes were downregulated in wild-type but not in FXR-null cells (figure 6B). Next, we tested whether a reduction of hepatic FXR was responsible for the hepatosteatosis in YY1-overexpressed mice by restoring hepatic FXR to physiological levels using an adenoviral vector. Notably, FXR restoration completely reversed the hepatic triglyceride accumulation (figure 6C,D), increased serum triglyceride levels and hepatic SREBP-1c expression displayed in YY1-overexpressed mice (figure 6E,F). Therefore, our data indicate that the role of YY1 in the promotion of triglyceride accumulation is dependent on FXR downregulation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Farnesoid X receptor (FXR) is essential for Yin Yang 1(YY1)-mediated hepatosteatosis. (A) FXR wild-type (WT) and knockout (KO) primary hepatocytes were infected with adenovirus expressing control or YY1-specific shRNA for 16 h. Cells were then incubated with palmitate (0.5 mM) for another 24 h. The cellular triglyceride (TG) contents were measured. (B) Real-time PCR analysis of SREBP-1c and Fasn in hepatocytes as described in (A). (C–F) C57BL/6 mice aged 10 weeks were exposed to adenoviral GFP, YY1 or YY1 plus FXR. At day 3 after virus injection, mice were fed with high-fat diet. Ten days after virus infusion, mice were fasted for 16 h and sacrificed for further analysis. Hepatic FXR protein levels (C), liver and serum TG content (D,E), and SREBP-1c expression (F) were analysed (n=5–6).
Discussion
In the present study, we identified YY1 as a novel transcription factor involved in hepatic triglyceride metabolism in obesity. This is suggested by multiple lines of evidence. First, we observed an upregulation of YY1 expression in the fatty livers in obese mice and NAFLD patients. Second, selective overexpression of hepatic YY1 resulted in massive triglyceride accumulation and moderate insulin resistance in mice fed with HFD, while hepatic silence of YY1 significantly attenuated hepatosteatosis in db/db mice. Third, our results demonstrated that FXR could be a transcriptional target to mediate the effects of YY1. Thus, we propose that YY1 could be a crucial mediator for obesity-related hepatosteatosis. However, the limitations of our study consist of a lack of YY1 knockout mice due to embryonic lethality. Besides, potential mechanisms that may underlie the upregulation of YY1 in obese liver tissue remain to be determined. Earlier studies have shown that YY1 could be upregulated by inflammatory cytokines such as TNFα in rhabdomyosarcoma.24 ,25 It is known that chronic inflammation states exist in obesity.26 ,27 Thus, we further investigated whether hepatic YY1 could be upregulated by inflammatory cytokines. We employed C57BL/6 wild-type mice receiving a lipopolysaccharide (LPS) injection as a standard model for systematic inflammation. Indeed, hepatic YY1 expression was markedly increased in LPS-treated mice as compared with control littermates (see online supplementary figure 8A). Moreover, TNFα treatment also increased YY1 expression in mouse primary hepatocytes (see online supplementary figure 8B). Therefore, we speculated that inflammatory cytokines, such as TNFα, at least in part, underline the mechanisms linking obesity and YY1 upregulation.
Recently, hepatic YY1 expression was reported to be increased in diabetic rats.28 Here, we proposed that hepatic YY1 could be a critical endogenous regulator to promote triglyceride accumulation in the liver and lead to the fatty liver. We further found that YY1 suppressed FXR expression via interaction with the YY1 binding site in the first intron of the FXR gene. Similar regulatory models were also reported previously regarding CD21 and HSD3B2 expression regulated by YY1.29 ,30
YY1 has been shown to regulate the expression or activity of several nuclear receptors such as PPARδ and androgen receptor.31 ,32 FXR, a metabolic nuclear receptor abundantly expressed in the liver, intestine and kidney, was first identified as a key regulator of cholesterol and bile acid homeostasis.33 FXR is also a major transcriptional factor involved in the regulation of glucose and lipid metabolism in the liver. Previous studies showed that mice with FXR deficiency displayed hepatic steatosis and hyperlipidaemia, as well as glucose and insulin intolerance, whereas FXR activation or overexpression could efficiently prevent hepatic triglyceride accumulation and hyperlipidaemia in many obese mice models.22 ,34 At molecular levels, FXR controls hepatic triglyceride homeostasis, at least through the repression of lipogenesis.22 In addition, some key molecules involved in lipid and glucose homeostasis, such as PGC-1α, Sirt1 and FoxO1, have been shown to modulate hepatic triglyceride metabolism through regulation of FXR.35–37 Indeed, abnormal expression or activity of FXR has been regarded as an important cause for aberrant metabolism and liver function in obesity.38 In addition, we showed that hepatic FXR was reduced in obese animal models, which was also supported by other studies in Zucker fat rats and NAFLD patients.39 ,40 Therefore, together with other studies, our data suggest that FXR could be an emerging treatment target for NAFLD.41–43
In summary, our data demonstrate that YY1 is a key metabolic regulator in the liver. Hepatic YY1 promotes hepatosteatosis and insulin resistance in obesity, mainly through repression of FXR expression. Thus, the YY1–FXR regulatory axis may be a promising therapeutic target for fatty liver diseases and related metabolic disorders.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
- Data supplement 5 - Online figure 4
- Data supplement 6 - Online figure 5
- Data supplement 7 - Online figure 6
- Data supplement 8 - Online figure 7
- Data supplement 9 - Online figure 8
Footnotes
-
YL and ZM equally contribute to this work.
-
Correction notice This article has been corrected since it was published Online First. The statement ‘YL and ZM equally contribute to this work’ has been added to this article.
-
Contributors GN and XL conceived the research ideas, supervised the project and wrote the manuscript. YL and ZM performed animal and cellular experiments, analysed the data and wrote the manuscript. ZZ, XX, XW, HZ and GS performed cellular experiments and analysed the data. XX provided technical advice on the animal and cellular studies.
-
Funding This study is supported by grants from the China Natural Science Foundation (Nos 30890043, 81030011, 81070681, and 81000320), National Key Basic Research Program of China (973 Program) (No. 2012CB524902) and Shanghai Committee for Science and Technology (No. 09XD1403400).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethics Committee of Shanghai Jiao Tong University School of Medicine.
-
Provenance and peer review Not commissioned; externally peer reviewed.