Article Text

Abstract
Background: Wilson’s disease is a rare inborn disease related to copper storage, leading to liver cirrhosis and neuropsychological deterioration. Clinical data on larger cohorts are limited owing to low disease frequency.
Objective and methods: We performed a retrospective analysis of 163 patients with Wilson’s disease, examined at the University of Heidelberg, Heidelberg, Germany, to determine clinical presentation, diagnostic course and long-term outcome.
Results: Diagnostic criteria for non-caeruloplasmin-bound serum copper, serum caeruloplasmin, 24-h urinary copper excretion, liver copper content, presence of Kayser–Fleischer rings and histological signs of chronic liver damage were reached in 86.6%, 88.2%, 87.1%, 92.7%, 66.3% and 73% of patients, respectively. By analysis of the coding region of ATP7B (except exons 2, 3 and 21), disease-causing mutations were detected in 57% and 29% of patients with Wilson’s disease on both chromosomes and on one chromosome, respectively. No mutations were detected in 15% of patients with Wilson’s disease. No significant differences were found in clinical parameters or initial presentation between patients grouped according to their mutations. The patients with neurological symptoms were significantly older at the onset of symptoms than patients with hepatitic symptoms (20.2 v 15.5 years of age, p<0.05), and the neurological symptoms were associated with a significantly longer time from onset to diagnosis than hepatic symptoms (44.4 v 14.4 months, p<0.05). After initiating treatment, 76.1% of the patients had a stable or improved course of the disease. Disease progression under treatment was more likely for neuropsychiatric than for hepatic symptoms. Side effects of treatment occurred in 74.4% of patients.
Conclusions: Patients with Wilson’s disease having predominantly neuropsychiatric symptoms manifest symptoms later, have a longer time delay from onset of symptoms until definitive diagnosis and have a poorer outcome than patients with hepatic symptoms.
- MRI, magnetic resonance imaging
Statistics from Altmetric.com
Wilson’s disease is a rare autosomal recessive disorder of copper metabolism, with a prevalence of about 1 in 30 000 people. It is characterised by a decreased biliary copper excretion and a defective incorporation of copper into caeruloplasmin, leading to copper accumulation in the liver, brain and kidneys.1 In 1993, the Wilson’s disease gene ATP7B was cloned. This gene codes for a membrane-bound, P-type copper-transporting ATPase expressed primarily in the liver.2,3 Wilson’s disease may exhibit a variety of clinical symptoms, the most common being liver disease and neuropsychiatric disturbances.4–6 Wilson’s disease typically begins with a presymptomatic period, during which copper accumulation in the liver causes subclinical hepatitis, and progresses to liver cirrhosis and development of neuropsychiatric symptoms. The type of hepatic and neurological symptoms can be highly variable. Wilson’s disease may also present as fulminant hepatic failure with an associated Coombs-negative haemolytic anaemia and acute renal failure.
The diagnosis of Wilson’s disease is based on the results of several clinical and biochemical tests (table 1).7–9 Each of the diagnostic tests has its limitations, and only the combination of clinical, biochemical and genetic tests provides a powerful and reliable tool for the diagnosis of Wilson’s disease.
Diagnostic variables in patients with Wilson’s disease
The available treatments are chelating agents (d-penicillamine and trientine) and zinc salts. In general, the approach for treatment is dependent on whether the patient is asymptomatic or has symptoms, and on the predominant manifestation of the symptoms (neurological or hepatic). Owing to the possible side effects of d-penicillamine, trientine and zinc are being increasingly used as preferred or second-line treatment. Trientine, particularly, has proved to be effective in the treatment of decompensated neurological or hepatic disease.8,10 For asymptomatic or presymptomatic patients, zinc salts or maintenance dosages of chelating agents may be used.11
As data are rare on larger cohorts with Wilson’s disease, we analysed 163 patients with Wilson’s disease for their initial clinical presentation and diagnostic findings, including laboratory results, liver histology and mutational analysis. The course of neurological and hepatic symptoms while under treatment and the development of side effects were analysed by long-term follow-up.
METHODS
Between 2000 and 2005, 163 patients with clinical, biochemical or histological evidence of Wilson’s disease were either diagnosed or had a previously established diagnosis confirmed at the Department of Gastroenterology, University Hospital of Heidelberg, Heidelberg, Germany. The study was approved by the local ethics committee. After obtaining informed consent from participants, data were primarily collected retrospectively by record analysis and patient interviews. Follow-up visits, including the evaluation of potential side effects, were scheduled at least once a year.
Clinical analysis
Wilson’s disease was diagnosed on the basis of typical symptoms and the presence of conventional biochemical indicators as previously published (table 1).6 Patients were examined by slit lamp for the presence of Kayser–Fleischer rings. In 23% of the patients, magnetic resonance imaging (MRI)of the brain was performed and the scan was evaluated for focal high-intensity lesions and atrophy.12 Hepatic and neuropsychiatric symptoms were assessed in a semiquantitative way. Neuropsychiatric symptoms, such as tremor, dysarthria, ataxia, rigidity, dyskinesia, cognitive impairment and mood disturbances, were assessed semiquantitatively according to the method given by Oder et al,13 and ranged from 0 (completely normal) to 3 (severely impaired). Hepatic symptoms were also classified into four categories: 0, completely normal; 1, increased levels of liver enzymes, without signs of liver cirrhosis or impaired liver function; 2, compensated liver cirrhosis; 3, decompensated liver cirrhosis or acute liver failure. Patients with a neurological and hepatic score of 0 were classified as asymptomatic. Patients with a hepatic or neurological score ⩾1 were classified according to the higher score. If both scores were equal, patients were classified as a neurological case.
Mutational analysis
DNA analysis for the presence of ATP7B mutations was performed as given by Ferenci et al.,9 as part of an ongoing protocol to study genotype–phenotype correlations in patients with established diagnosis of Wilson’s disease. Mutation analysis was carried out stepwise. Firstly, a rapid, seminested polymerase chain reaction was used to detect the H1069Q mutation as described previously.14 Patients who were not homozygotic for this mutation were further analysed exon by exon by denaturating high performance liquid chromatography (WAVE mutation detection system model 4000; Transgenomics, Crewe, UK). So far, the analysis has been completed for exons 3–20. Exons were amplified with primers as described previously.15 Samples with potential mutations identified by this approach were sequenced by the ABI Prism 310 Genetic Analyser (Perkin Elmer; Norwalk, Connecticut, USA).
Statistical analysis
Data were statistically analysed with SPSS for Windows, V.10.05. Quantitative variables were compared using the unpaired t test. Data are expressed as means with standard deviation in parenthesises (SD); p<0.05 was considered to be significant.
RESULTS
In all, 163 patients (65 men and 98 women) with an established diagnosis of Wilson’s disease were analysed. The course of Wilson’s disease was followed for a mean of 16.7 years (range 1–51 years). This study therefore encompasses 2476 patient-years of Wilson’s disease.
Clinical presentation and diagnostic course
In all, 137 (84.1%) patients were diagnosed as symptomatic index cases, 26 (15.9%) were diagnosed by family screening, 96 (58.9%) presented with predominantly hepatic symptoms and 55 (33.7%) patients presented with predominantly neurological symptoms. All the 8 (4.9%) patients who presented with a fulminant hepatic failure were women. In all, 14 of the 26 patients diagnosed by family screening already had disease-associated symptoms at the time of diagnosis, whereas 12 patients were asymptomatic at the time of diagnosis. The mean (SD) age at the appearance of first symptoms was 17.4 (10.0) years. Patients with mainly hepatic symptoms showed an overall earlier onset of symptoms (15.5 (9.6) years) than patients with predominantly neurological symptoms (20.2 (10.8) years; p = 0.031 (fig 1A). Diagnosis was established at a mean age of 20.4 (SD 10.6, range 4–56) years, after a mean time delay of 25.3 months from the occurrence of symptoms until diagnosis. In all, 52.8% of patients were diagnosed before the age of 18 years. The time lag from the appearance of first symptoms or clinical signs to diagnosis showed a great variation, from 0 to 360 months. Neurological presentation was associated with a significantly longer time from onset of symptoms to diagnosis than hepatic presentation (44.4 v 14.4 months, p = 0.004), with a mean (SD) age at diagnosis of 23.9 (10.6) versus 17.4 (9.6) years, p = 0.001) (fig 1A). Of all patients with symptoms, 60.3% could be diagnosed within 1 year and 68.2% within 2 years. In 22.5% of all patients, diagnosis had not been established 3 years after the appearance of initial symptoms (fig 1B). Family screening was effective in diagnosing Wilson’s disease early, as the mean age of patients diagnosed by family screening was lower than that of patients diagnosed as a result of symptomatic disease (15.5 v 20.4 years, p = 0.021).
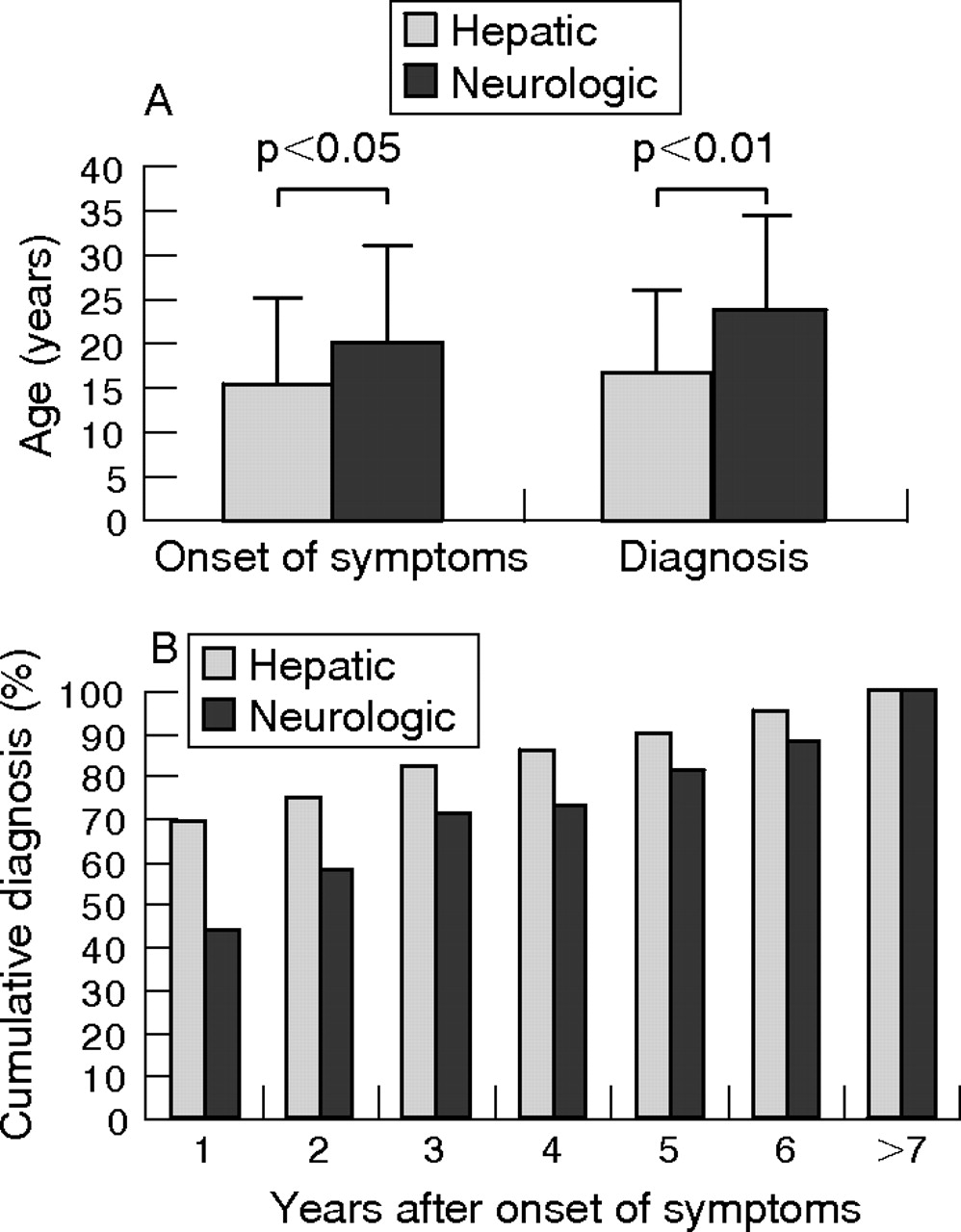
(A) Age (years) of patients with Wilson’s disease at the onset of symptoms and at diagnosis. (B) Cumulative diagnosis of Wilson’s disease dependent on the years after onset of symptoms.
Diagnosis
Diagnosis of Wilson’s disease was based on the diagnostic tests (table 1). Kayser–Fleischer rings were detected in 66.3% of patients with Wilson’s disease, and were more frequently seen in patients with neurological symptoms rather than those with hepatic symptoms (85.5% v. 52.1%, p<0.001). Reduced serum caeruloplasmin levels (<200 mg/l) were seen in 88.2% of all patients at diagnosis. Serum caeruloplasmin level was <50 mg/l in 29.5% of the patients. Non-caeruloplasmin-bound serum copper levels were raised in 86.6% of all patients. A liver biopsy specimen was necessary for the determination of liver copper content and was obtained in 55 patients. In all, 92.7% of the patients examined had a liver copper content ⩾250 μg/g dry weight. The range of liver copper content was 95–3776 μg/g dry weight, with a mean (SD) value of 898.6 (631.5) µg/g dry weight. In 87.1% of all patients, levels of 24-h urinary copper excretion were raised. The range of urinary copper excretion was 0.15–33.6 μmol/day, with a mean (SD) value of 4.9 (5.4) µmol/day. Mean values of serum caeruloplasmin, serum copper, liver copper content and urinary copper excretion were characteristic of Wilson’s disease, however, they did not differ significantly (p>0.05) between patients with hepatic symptoms, neurological symptoms and asymptomatic patients. Mutation analysis of the coding region of ATP7B (except exons 2, 3 and 21) was performed in 150 of 163 patients with the disease. Disease-causing mutations were detected in 85 (57%) and 43 (29%) patients on both chromosomes and on one chromosome, respectively. No mutations were detected in 22 (15%) patients (table 2). We found no significant differences in the frequencies of pathological laboratory test values between patients disease-causing ATP7B mutations on both chromosomes and those with at least one unknown mutation.
Distribution of ATP7B mutations according to clinical presentation
Liver biopsy for histological examination was performed in 78 patients and the specimen was evaluated for the presence of steatosis, fibrosis and cirrhosis. At the time of diagnosis, signs of chronic liver damage were present in 73% of patients, with fibrosis occurring in 36% and cirrhosis in 37% of patients. Steatosis was found in 54% of liver biopsy specimens. Liver biopsy specimens of three patients showed an essentially normal histological examination. MRI of the brain was performed in 37 patients. In all, 3 of the 19 patients showing focal high-intensity lesions on MRI, and 4 of the 8 patients with focal atrophy, were neurologically asymptomatic. Three of the 10 patients in whom MRI showed no pathological indicators had mild to moderate neurological symptoms.
Genotype–phenotype correlation
For genotype–phenotype correlations, patients were grouped according to the results of the ATP7B mutation analysis (table 2). Statistical analysis between these subgroups showed no significant differences with regard to the initial presentation (p>0.5). Patients were also grouped according to their ATP7B H1069Q genotype, for presence of a mutation in exon 8, and for the presence of an ATP7B mutation in general. Analysis among patients with (a) ⩾1 H1069Q mutation compared with no H1069Q mutation, (b) ⩾1 mutation in exon 8 compared with patients with no mutation in exon 8, and (c) mutations on both chromosomes compared with no detectable mutation showed no statistically significant differences with regard to clinical presentation.
Analysis of the diagnostic parameters, serum caeruloplasmin, non-caeruloplasmin-bound serum copper, 24-h urinary copper excretion, liver copper and presence of Kayser–Fleischer rings showed no significant differences between patients with Wilson’s disease having mutations on both chromosomes and those with no detectable ATP7B mutation. We also found no significant difference in the groups with respect to initial clinical presentation.
Treatment
Patients were treated with d-penicillamine (900–1800 mg/day), trientine (900–2100 mg/day) or zinc salts (150–250 mg/day). In all, 138 patients were treated initially with d-penicillamine, 9 with trientine, 13 with zinc salts and 3 had to undergo high-urgency liver transplantation due to acute liver failure without previous treatment (fig 2A). Treatments at end of the study (160 patients alive) were as follows: 63 patients treated with d-penicillamine, 44 with trientine, 45 with zinc salts and 8 patients with successful liver transplantations (fig 2B).

Treatment of patients with Wilson’s disease (A) at diagnosis and (B) at the end of the study.
Of the 163 patients, 85 (52.2%) patients remained with their initial treatment and required no change in treatment. Of these 85 patients, 63 (74.1%) patients were treated continuously with d-penicillamine (mean treatment duration 16.2 years), 7 (8.2%) with trientine (2.3 years), 12 (14.1%) with zinc (4.2 years) and 3 patients were successfully liver transplanted. Changes in treatment regimen were necessary in 47.9% of patients. The treatment was changed once in 65 patients and twice or more in 18 patients.
In all, 138 of the 163 patients analysed were treated initially with d-penicillamine; 97 (70.3%) of these patients developed side effects. Of the patients treated with d-penicillamine, 31.3% had severe side effects such as neurological worsening, pancytopenia, nephrotoxicity, polyneuropathy, optic neuritis and polymyositis. In 39 patients with side effects, treatment was changed to trientine, in 34 patients to zinc and 24 patients were kept on d-penicillamine. Patients who were switched to an alternative treatment with trientine or zinc commonly showed improvement of the side effects developed while under treatment with d-penicillamine. However, in some patients, d-penicillamine-related side effects such as neurological worsening or polyneuropathy did not improve after changing the treatment. Side effects were recorded in 19 of 59 (32.2%) patients and in 25 of 67 (37.3%) patients treated at any time with trientine or zinc salts (zinc acetate and zinc sulphate), respectively. Neurological worsening was recorded while under all three treatments. For d-penicillamine, neurological worsening commonly occurred early after the initiation of treatment, whereas neurological worsening while under treatment with trientine and zinc occurred at any time. Relevant side effects recorded while under treatment with d-penicillamine, such as nephrotoxicity, polyneuropathy, pancytopenia, polymyositis and optic neuritis, were not seen in patients treated with trientine or zinc. Table 3 summarises the recorded side effects.
Side effects recorded while under treatment with d-penicillamine, trientine and zinc salts
In all, 136, 59 and 67 patients received, at any time of their course of Wilson’s disease, treatment with d-penicillamine, trientine and zinc salts, respectively.
Clinical course under treatment
To evaluate long-term outcome while under treatment, we compared the presence of hepatic and neurological symptoms at diagnosis and course of symptoms while under treatment. After initiation of treatment, 76.1% of patients had a stable or improved course of disease. From 12 initially asymptomatic patients, all but one patient (who developed mild hepatic symptoms) remained asymptomatic. During long-term follow-up, only 3 of 163 patients died, two as a result of multiple-organ failure after liver transplantation and one as a result of the development of a cholangiocellular carcinoma while under zinc treatment. These results reconfirm our previous survival analysis on a cohort of 51 patients with the disease, showing a similar survival of patients with Wilson’s disease when compared with a control population matched for age and sex.4
Course of neurological symptoms
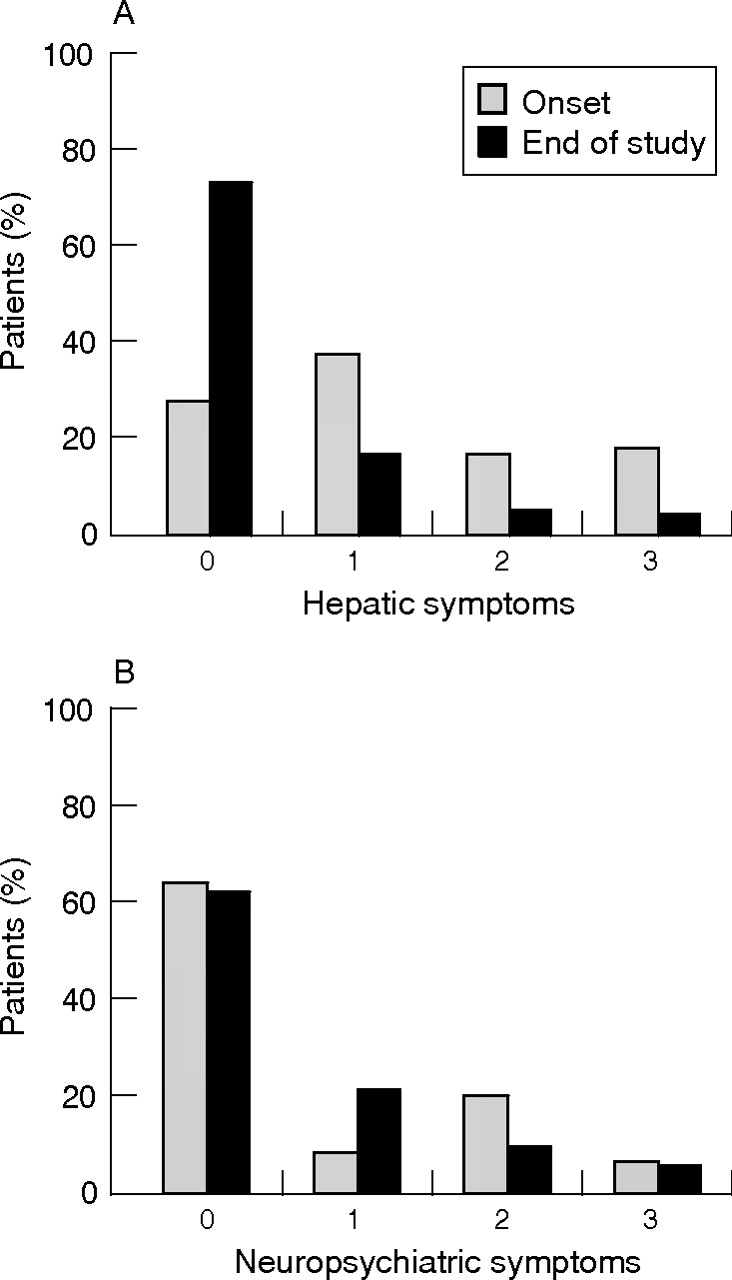
In all, 20 of 105 (19.1%) patients without initial neurological symptoms developed neurological symptoms while under treatment, whereas 85 (80.9%) patients remained neurologically asymptomatic. Of the 58 patients with initial neurological symptoms, 31 (53.5%) patients improved neurologically, 13 (22.4%) showed a stable neurological condition and 14 (24.1%) experienced worsening of the symptoms. Neurological worsening was witnessed in treatment with all three drugs. Of the 34 patients with progressive neurological symptoms, only five experienced a parallel deterioration of hepatic function, seven maintained a stable hepatic condition and 22 had an improvement in hepatic condition. Of the 12 patients whose liver condition worsened, five experienced a parallel deterioration in neurological symptoms, whereas four showed a stable and three patients even showed an improving neurological condition. Statistical analysis showed no significant correlation between the course of hepatic and neurological symptoms (p>0.05; fig 3A).

{kind=link}
{kind=link}
{kind=link}
Severity of symptoms before and after long-term treatment (end of study) in patients with Wilson’s disease. Symptoms are assessed semiquantitatively and score ranges from 0 to 3 (A) Hepatic symptoms; (B) Neuropsychiatric symptoms.
Course of hepatic symptoms
Only 2 of 41 (4.9%) patients who were initially free of major hepatic symptoms developed hepatic symptoms while under long-term treatment. Of the 119 patients with initial hepatic symptoms, 94 (79.0%) patients improved their liver function under treatment, 10 (8.4%) maintained a stable hepatic condition and 15 (12.6%) showed decreasing liver function (fig 3B). Of the 17 patients with progressive liver disease, eight were treated with liver transplantation (three at diagnosis and five because of decreasing liver function due to non-compliance with treatment). Progressive liver disease in those patients compliant with their maintenance treatment was seen with only trientine and zinc (n = 7). These patients were categorised as “treatment failures”. Stable and improving liver function was seen in maintenance treatment with all three drugs, with no considerable differences among them.
DISCUSSION
Our study compiles data on 163 patients with Wilson’s disease, and is therefore one of the largest clinical studies on Wilson’s disease. Long-term observation is reflected in a mean follow-up of 16.7 years. The analysed study cohort comprises more patients with hepatic than with neuropsychiatric symptoms (58.9 v 33.7%). This might be owing to the enrolment of patients at the Department of Gastroenterology. A predominance of women (60.1%) in our study cohort is notable. A gender difference with a predominance of women has been documented previously in an Austrian cohort with Wilson’s disease and in the occurrence of fulminant hepatic failure in patients with Wilson’s disease.14,16,17
One aspect of our study was to discriminate between the age at symptom onset and at diagnosis. Patients with hepatic symptoms showed a considerably earlier onset of symptoms and a shorter diagnostic delay before definitive diagnosis than those with neuropsychiatric symptoms. Family screening was effective in diagnosing patients with Wilson’s disease at an early, and most often asymptomatic or pre-symptomatic, stage of disease. By family screening, patients were diagnosed an average of 4 years earlier, with an excellent long-term outcome.
The analysis of the diagnostic findings underscores the difficulties in diagnosing Wilson’s disease. Neither the absence of Kayser–Fleischer rings nor normal values for serum copper or caeruloplasmin can exclude Wilson’s disease.4,18,19 Hepatic copper content >250 μg/g dry weight remains the best biochemical evidence for Wilson’s disease. The major problem with hepatic parenchymal copper concentration is that in later stages of Wilson’s disease, distribution of copper in the liver is often inhomogeneous.20 Sampling error may be partly responsible for the striking variability of hepatic copper content in our study.21
The wide spectrum of symptoms and of age at onset in patients with Wilson’s disease raised the question whether these features are determined by the type of the ATP7B mutation. The ATP7B mutational analysis showed no clear genotype–phenotype correlations. However, previously published studies on genotype–phenotype correlations have shown variable results. A recent meta-analysis showed an association of the ATP7B H1069Q mutation with a late neurological presentation in Wilson’s disease,22 although the underlying studies are quite controversial.15,23–25 Most reports on mutations other than H1069Q show no clear-cut phenotype–genotype correlations.26–28 In our study, mutations on both chromosomes were identified in 57% of patients. No mutation was detectable in 15% of patients with an established diagnosis of Wilson’s disease. This may be as a result of undetected mutations in the promoter region, in other non-coding regions or in exons not analysed (exons 2, 3 and 21). However, a distinct pathophysiological mechanism of copper accumulation rather than an ATP7B defect cannot be ruled out completely in these patients. We speculate that in the group of patients who are currently clinically diagnosed with Wilson’s disease, people with other, as yet unidentified, genetic defects may be included.
One main aspect of our analysis was long-term follow-up with regard to the course of hepatic and neuropsychiatric symptoms while under treatement. Treatment with d-penicillamine, trientine or zinc is effective, as 76.1% of the analysed patients experienced a stable or improved course of disease. However, analysis might be biased, because symptoms might be partly unrelated to Wilson’s disease. This is especially true for neuropsychiatric disturbances such as depression or anxiety disorders that have high lifetime prevalence even in the normal population. In our study cohort, liver disease was stable or improving in most of the patients, and development of progressive hepatic symptoms while under treatment was a rare event. The development of new symptoms while under treatment or progression of pre-existing symptoms was more often recorded for neurological than for hepatic symptoms. Most surprisingly, our data showed no marked correlation of the course of neurological and hepatic symptoms while under treatment. In some patients, severe neurological symptoms developed, although their hepatic condition improved. We speculate that the pathophysiological disturbances leading to organ damage in Wilson’s disease are not completely the same for the liver and the brain. Here, differential expression or polymorphisms of other genes related to copper and oxidative stress may influence organ susceptibility. For apolipoprotein E3 homozygosity a correlation with higher age at presentation was seen in the subgroup of patients with Wilson’s disease homozygotic for the ATP7B H1069Q mutation.29 Another possible candidate modifier gene is the copper-binding prion protein, as the human prion protein polymorphism M129V influences the age of onset in Wilson’s disease. The onset of symptoms is considerably delayed in patients homozygotic for the 129M allele compared with patients with at least one V allele.30
In non-compliant patients, deterioration of the disease paralleled by increasing copper excretion was seen irrespective of the kind of treatment with which they were non-compliant. In patients adhering to their treatment, signs of treatment failure such as progressive hepatic symptoms paralleled by an increasing urinary copper excretion were reported for only trientine and zinc. These patients were quoted as treatment failures. By switching these patients to an alternative treatment, amelioration of hepatic symptoms and normalisation of urinary copper excretion could be achieved. Owing to potential treatment failures, we recommend verification of sufficient treatment in all patients at regular intervals. If signs of treatment failure occur, patients should be switched to an alternative treatment at an early stage to prevent liver transplantation.
Three patients died during the follow-up period, one owing to a cholangiocellular carcinoma. Development of other malignancies was not observed in our study cohort. The diagnosis of a cholangiocellular carcinoma in a patient with Wilson’s disease is rare, and to date has been reported only once.31 Interestingly, a recent study on ATP7B knock-out mice showed occurrence of cholangiocellular carcinomas as a common event at older ages.32
A substantial proportion of the patients developed side effects. This was especially true in d-penicillamine treatment, where severe side effects were recorded in >30% of all treated patients. In the light of our data, and especially considering that effective alternative drugs available, the role of d-penicillamine as a standard treatment for Wilson’s disease must be questioned.8,11,33,34 In our study cohort, development and worsening of neurological symptoms were reported while under treatment with all three drugs, but most frequently (13.8%) for d-penicillamine. This may be partly as a result of the longer summarised follow-up period for d-penicillamine. Nevertheless, in our opinion, d-penicillamine should not be the drug of choice for patients with neurological symptoms.
REFERENCES
Footnotes
-
↵* These authors contributed equally to this work.
-
Published Online first 18 May 2006
-
Competing interests: None.