Article Text

Abstract
BACKGROUND/AIMS Impaired colonocyte metabolism of butyrate has been implicated in the aetiopathogenesis of ulcerative colitis. Colonocyte butyrate metabolism was investigated in experimental colitis in mice.
METHODS Colitis was induced in Swiss outbred white mice by oral administration of 4% dextran sulphate sodium (DSS). Colonocytes isolated from colitic and normal control mice were incubated with [14C]butyrate or glucose, and production of 14CO2, as well as of intermediate metabolites (acetoacetate, β-hydroxybutyrate and lactate), was measured. The effect of different substrate concentrations on oxidation was also examined.
RESULTS Butyrate oxidation (μmol/h per mg protein; mean (SEM)) was significantly reduced in DSS colitis, values on day 7 of DSS administration being 0.177 (0.007) compared with 0.406 (0.035) for control animals (p<0.001). Glucose oxidation (μmol/h per mg protein; mean (SEM)) on day 7 of DSS administration was significantly higher than in controls (0.06 (0.006) v 0.027 (0.004), p<0.001). Production of β-hydroxybutyrate was decreased and production of lactate increased in DSS colitis compared with controls. Increasing butyrate concentration from 10 to 80 mM enhanced oxidation in DSS colitis (0.036 (0.002) to 0.285 (0.040), p<0.001), although it continued to remain lower than in controls. Surface and crypt epithelial cells showed similar ratios of butyrate to glucose oxidation. When 1 mM DSS was added to normal colonocytes in vitro, it did not alter butyrate oxidation. The initial histological lesion of DSS administration was very patchy and involved crypt cells. Abnormal butyrate oxidation became apparent only after six days of DSS administration, at which time histological abnormalities were more widespread.
CONCLUSIONS Colonocyte metabolism of butyrate, but not of glucose, is impaired in DSS colitis, and may be important in pathophysiology. Histological abnormalities preceded measurable defects in butyrate oxidation.
- butyrate
- colonocytes
- dextran sulphate sodium
- cell metabolism
- short chain fatty acids
- ulcerative colitis
Statistics from Altmetric.com
The cause of human ulcerative colitis remains unknown. It has been proposed that epithelial abnormalities are the central defect, and that they underlie the development of mucosal inflammation and its chronicity.1 The short chain fatty acid (SCFA), butyrate, is the preferred energy source for colonocytes.2Colonocytes from patients with ulcerative colitis have a specific defect in butyrate oxidation.3 Diversion colitis, occurring in the surgically bypassed colon, histologically resembles ulcerative colitis,4 and is postulated to be due to lack of luminal SCFAs, because luminal instillation of SCFAs reverses the abnormalities.5 Both these factors suggest that colitis may be caused by impaired colonocyte oxidation of butyrate. Several animal models of experimental ulcerative colitis have been described and, of these, colitis induced in mice by oral dextran sulphate sodium (DSS) has been widely used because of many similarities to human ulcerative colitis.6 7 Shortening of crypts occurs early after exposure to oral DSS and precedes development of significant inflammation,6 suggesting that the primary defect in DSS colitis is an abnormal colonic epithelium. This study was carried out to investigate oxidative metabolism of butyrate and glucose by colonocytes in DSS colitis and to compare this with normal control mice.
Materials and methods
INDUCTION OF COLITIS
Swiss white mice (Rockefeller strain; 8 weeks old) were used for these studies. Colitis was induced by administering DSS (approximate molecular mass 40 kDa, 19% sulphation) for either one cycle (DSS I) or two cycles (DSS II). DSS (4%) was administered along with the laboratory chow. For DSS I studies, DSS was administered for seven days, at the end of which animals were killed. For DSS II studies, mice were fed two cycles of DSS as described above with seven days of normal feeding between cycles. Mice were killed at the end of the second cycle. These schedules were initially standardised using various DSS concentrations in the feed. Control mice were pair fed with normal feed (no added DSS) for seven days and then killed. To determine whether metabolic changes preceded histological changes, mice were killed and colonocytes isolated after feeding 4% DSS for two, four, five, and six days. Variables measured during standardisation included daily weight loss, faecal occult blood (Hemoccult; Smith Kline Diagnostics, San Jose, California, USA ), shortening of the colon, and histological appearance of sections of proximal and distal colon. The histological evaluation was performed independently by two pathologists on paraffin wax embedded sections stained with haematoxylin and eosin.
COLONOCYTE ISOLATION
Colonocytes were isolated by EDTA chelation.8 9Briefly, the colon was excised, washed with oxygenated Ca2+-free Krebs-Henseleit Ringer, ligated at one end, distended with Ca2+-free Krebs-Henseleit Ringer containing 0.1 M EDTA and the other end sealed. The distended colon was incubated in oxygenated Ca2+-free Krebs-Henseleit Ringer in a shaking water bath at 37°C for 20 minutes. At the end of the incubation, the colon was removed and gently palpated with fingers to loosen the cells. Cells were collected and centrifuged at 3000 rpm for five minutes. The cell pellet was washed twice, and resuspended in Ca2+-containing Krebs-Henseleit. Viability of cells was measured by trypan blue dye exclusion as well as by assay of lactate dehydrogenase (LDH) release.10 LDH levels in 1 ml supernatant were measured at each time period, and expressed as a percentage of the LDH content of the cell pellet from a 1 ml suspension immediately after cell isolation. The above value was subtracted from 100 to give percentage viability of cells. Protein in the cell pellet was estimated by the method of Lowry et al.11 To assess contamination of isolated colonocytes by infiltrating cells, cell smears were stained by haematoxylin and eosin and a differential count of cells was performed on the basis of nuclear morphology.
Differential isolation of surface and crypt cells was carried out using a modification of the above procedure. The colon was incubated with EDTA-containing solution for 17 minutes, and the first fraction of cells then collected from the lumen without manipulating the colon. This fraction contained predominantly surface cells. The colon was then incubated for a further three minutes with EDTA-containing solution, and palpated with the fingers to loosen the remaining cells, which were then collected. This fraction contained predominantly crypt cells. Purity of the fractions was assessed by alkaline phosphatase activity12 and verified by histological examination of the colon at every step of the sequential isolation procedure (see fig 3). Paraffin wax sections stained with haematoxylin and eosin were examined by light microscopy.

Light micrographs of normal mouse colon before and during sequential isolation of surface and crypt cells. (A) At the start of colonocyte isolation; (B) after isolation of surface cells; (C) after isolation of crypt cells. Haematoxylin and eosin stain; original magnification × 40.
METABOLIC STUDIES WITH COLONOCYTES IN VITRO
Metabolic studies with colonocytes were carried out in metabolic flasks as described previously.8 A 1 ml portion of the cell suspension was incubated, in a final volume of 2 ml, with either radiolabelled butyrate or glucose (10 mM concentration) as substrate, for 45 minutes at 37°C in a shaking water bath. [14C]Glucose and [14C]butyrate were obtained from the Board of Radiation and Isotope Technology, India.14CO2 released by cell metabolism was trapped in 3 M NaOH, and quantified by liquid scintillation spectrometry using an LKB Rackbeta counter. The intermediate metabolites,l-lactate (for cell incubations with glucose), acetoacetate, and β-hydroxybutyrate (for incubations with butyrate), were assayed enzymically in the cell supernatant, after neutralisation with 20%KOH.10 To test the direct effect of DSS on colonocyte substrate oxidation, metabolic studies were also carried out using colonocytes isolated from normal mice in the absence and presence of 1 mM DSS added in vitro.
KINETICS OF BUTYRATE AND GLUCOSE OXIDATION
Kinetic studies of substrate oxidation were carried out using colonocytes isolated from normal mice and mice that had been fed two cycles of DSS (DSS II mice). The procedure used was similar to the metabolic studies described above, using 10–80 mM concentrations of labelled butyrate or glucose as substrate.14CO2 production was measured, other intermediate metabolites not being measured for these studies.
STATISTICAL ANALYSIS
Numerical data are expressed as mean (SEM). Student'st test was used for assessing significance of differences between groups. A p value less than 0.05 was considered statistically significant.
Results
CLINICAL AND HISTOPATHOLOGICAL CHARACTERISTICS OF DSS COLITIS
A substantial weight loss (about 10% decrease) in mice receiving DSS was evident on day 7. Weight loss was greater in DSS II mice. Loose stools, which were Hemoccult positive, were first noted around day 3 of DSS administration, and became more common thereafter until day 7, when all mice showed Hemoccult positive stool. Overt bleeding in the stool was noted in 30% around day 5. Grossly, the colon showed shortening and areas of bleeding. Colon length was shortened to a similar extent (about half that of control mice) in both DSS I and DSS II mice.
Histologically, DSS treated mice showed characteristic lesions similar to those described in the literature.16 The earliest changes, noted around day 3, involved disruption of the muscularis mucosa, with changes developing at the base of 5% of the crypts. Mild inflammatory infiltration was noted on day 4. Crypt loss and damage started at the bases of the crypts, where crypt cells were replaced by fibroblasts and collagen connective tissue. The remaining crypts showed increased mitotic activity. An inflammatory infiltrate, consisting of lymphocytes, neutrophil polymorphs, and plasma cells, was noted in the mucosa and submucosa. Changes were evenly distributed in both proximal and distal colon. By day 5, there was crypt abscess formation along with increased infiltration. Surface epithelium was preserved. Mucosal erosion was more extensive on day 6. DSS II mice showed increased incidence of regenerative epithelium, crypt branching, and hyperplasia of crypt epithelium with increased mitotic activity, mimicking an adenomatous dysplastic change.
METABOLISM OF BUTYRATE AND GLUCOSE BY COLONOCYTES
Characteristics of isolated colonocytes
Colonocytes isolated from both control and colitic tissue always showed a viability greater than 80%. There was no significant fall in viability after 45 minutes incubation. Viability of control cells by trypan blue dye exclusion after 45 minutes incubation of isolated cells in the presence of 0, 10, 20, 40, and 80 mM butyrate was 95.1 (0.84), 96.25 (0.61), 93.94 (0.53), 89.84 (1.6), and 88.96 (0.63)% respectively. As assessed by this method, cell viability decreased significantly when butyrate concentration was increased above 20 mmol/l. Measurement of LDH release showed cell viability after 45 minutes incubation of 83.2 (0.09), 82.36 (0.38), 82.1 (0.75), and 81.79 (0.25)% in the presence of 0, 10, 40, and 80 mM butyrate respectively. For cells isolated from colitic tissue, the trypan blue dye exclusion method demonstrated a viability of 83.60 (0.41)%. The epithelial cell suspension obtained from colitic mice showed that >75% of the cells were colonocytes, while <25% (21.6 (2.14%)) comprised inflammatory cells of non-epithelial origin.
Butyrate and glucose oxidation
Table 1 shows the results for butyrate and glucose oxidation to CO2. Butyrate oxidation was significantly decreased in colonocytes from DSS I and DSS II mice compared with control pair fed mice. A decrease in butyrate oxidation by 56.40% in DSS I (after seven days of DSS feeding) and by 83.99% in DSS II was observed in comparison with controls. Glucose oxidation was significantly increased in DSS treated mice compared with controls. The ratio of butyrate to glucose oxidation was 15:1 in controls, 4.6:1 in DSS I, and 1:1 in DSS II mice. As shown in table 1, a significant reduction in butyrate oxidation was first observed on day 6 of DSS feeding. On the other hand, significantly increased glucose oxidation was first noted on day 7 of DSS feeding.
Production of CO2 by colonocytes from control mice and mice treated with dextran sulphate sodium (DSS) incubated with either 10 mM butyrate or glucose
Intermediate metabolite production
As shown in table 2, β-hydroxybutyrate production was significantly reduced in DSS II colitis (p<0.001) compared with that in controls. There was a reduction in DSS I, which was not statistically significant. Acetoacetate production by colonocytes was slightly reduced in DSS I and DSS II colitis (statistically not significant). Lactate production was increased by fourfold and threefold in DSS I and in DSS II mice respectively compared with control cells.
Production of ketone bodies and lactate by colonocytes from control mice and mice treated with dextran sulphate sodium (DSS) incubated with either 10mM butyrate or glucose
Effect of substrate concentration on oxidation
Figure 1 shows the effect of substrate concentration on butyrate oxidation in colonocytes from control and DSS II mice. As butyrate concentration was increased from 10 to 80 mM, colonocytes from both DSS treated and control mice showed a linear increase in butyrate oxidation. Control colonocytes showed a 15-fold increase in butyrate metabolism while DSS colonocytes showed 7.9-fold increase in oxidation. The absolute values were always significantly lower in DSS colitis compared with controls.

Effect of substrate concentration on butyrate and glucose oxidation (measured as CO2 production) by colonocytes. Shown here are the absolute number of substrate molecules oxidised. DSS, colonocytes from mice subjected to two cycles of feeding with dextran sulphate sodium; controls, colonocytes obtained from pair fed mice maintained on normal feed. Values are mean (SEM) from four experiments. ***Significantly different from respective controls (Student's t test, p<0.001).
Figure 1 also shows the effect of glucose concentration in the medium on glucose oxidation by control and DSS colonocytes. In both, an increase in concentration from 10 to 80 mM led to increased glucose oxidation, amounting to a sixfold increase in controls and a 6.8-fold increase in DSS colonocytes. Glucose oxidation levels were not statistically significantly different in DSS treated mice compared with controls.
Effect of DSS on colonocyte metabolism of butyrate and glucose in vitro
Figure 2 shows butyrate and glucose oxidation by normal mouse colonocytes in the presence and absence of DSS. Butyrate oxidation was not significantly altered by addition of DSS. Glucose oxidation was marginally increased in the presence of DSS. The ratio of butyrate to glucose oxidation was not altered either in the presence or absence of DSS.
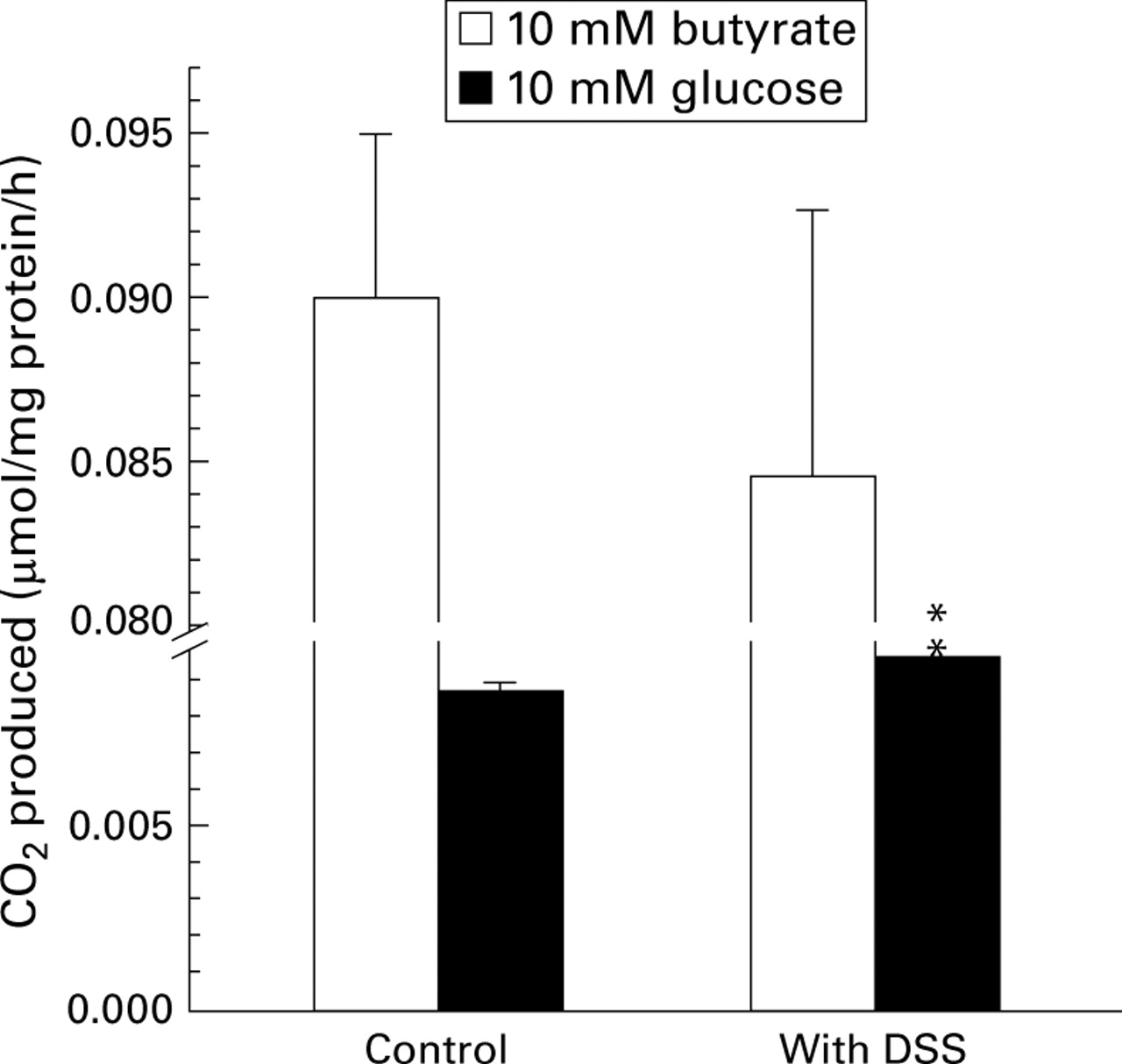
Effect of 1 mM dextran sulphate sodium (DSS), added in vitro, on CO2 production by colonocytes using either 10 mM butyrate or 10 mM glucose as substrate. Values are mean (SEM) from four experiments. **p<0.01 compared with controls (paired t test).
METABOLISM OF BUTYRATE AND GLUCOSE BY SURFACE AND CRYPT COLONOCYTES
Colonocytes sequentially isolated into predominantly surface and crypt cell fractions showed 2.06-fold enrichment of alkaline phosphatase activity in the surface cell fraction compared with the crypt cell fraction (1.63 (0.21) v 0.79 (0.11) units/mg protein, p<0.05). Light microscopy confirmed isolation of surface cells in the first step and removal of entire epithelium in the subsequent step (fig 3). Figure 4 shows butyrate and glucose oxidation by these two cell fractions. Absolute values for butyrate and glucose oxidation were significantly higher in surface cells than in crypt cells (p<0.01). However, the ratio of butyrate to glucose oxidation was similar in the two fractions, being 12:1 and 10:1 for surface and crypt cells respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Butyrate and glucose oxidation (measured as CO2 production) by surface and crypt colonocytes isolated from normal mice. Values represent the mean (SEM) from four experiments. **p<0.01 compared with surface cells (paired t test).
Discussion
SCFAs, including acetate, propionate, and butyrate, produced by bacterial fermentation in the colon, influence colonic epithelial cell function in diverse ways. They are the principal source of metabolic energy for colonocytes and are necessary for maintenance of normal mucosal function.2 13 14 SCFAs, principally butyrate, provide the colonocyte with about 70% of its energy. In healthy colonocytes, the order of utilisation of various substrates available to the colonic mucosa is butyrate>glucose>ketone bodies>glutamine.2 In the event of a specific metabolic impairment, as is hypothesised to occur in ulcerative colitis,3 colonocytes will avail themselves of alternative substrates to make up for the energy deficit.
This study shows that colonocyte metabolism in mice is similar to that in man, rat, and other species reported in the literature15 16 in utilising butyrate for oxidative metabolism in preference to glucose. The study also shows that butyrate oxidation by colonocytes is impaired in DSS colitis in mice. The impairment is specific, because glucose oxidation continues at normal (or even increased) levels in DSS colitis. The reduction in butyrate oxidation by 80% in DSS II colitis will translate to an overall decrease of about 55% of cell energy, which is very slightly compensated for by increased glucose oxidation. The colonocyte carries out many energy dependent processes vital to health, including electrolyte exchange, mucin synthesis, lipid synthesis, structural protein synthesis, and detoxification.17-20 Loss of cellular energy may impair all these processes, and may contribute to epithelial cell damage and loss in DSS colitis. Colonic epithelial starvation may lead to atrophy in the short term and to colitis in the long term.
Impairment of butyrate oxidation was substantially greater in mice exposed to two cycles of DSS (DSS II) than in those exposed to only one cycle (DSS I). Greater reduction of butyrate oxidation in DSS II mice correlated with more severe histological changes, consisting of appreciable crypt loss, dysplasia, and regenerative atypia. The time course of development of impaired butyrate metabolism in relation to histological changes is important in determining the significance of changes in metabolism. The earliest changes in histology were noted after three days of DSS administration, although such changes were limited and patchy. The earliest changes included loss of cells at some crypt bases and defects in the muscularis mucosa, which were followed almost immediately by inflammatory infiltration. Substantive impairment of metabolism was detectable only by day 6, at which time histological changes were quite pronounced. This, together with the fact that DSS incubation with colonocytes did not alter cell metabolism, raises the strong possibility that changes in epithelial cell metabolism were secondary to mucosal inflammation. However, if cell metabolism were to be affected in a non-specific manner by inflammation, both butyrate and glucose metabolism would be expected to be decreased. The compensatory increase in glucose metabolism noted in DSS colitis suggests specific impairment of butyrate metabolism. As the initial lesion of DSS colitis is patchy, it is possible that altered metabolism (even if it were a primary event) may not become measurably apparent until the later stages of the disease.
Colonocytes mature as they migrate up the crypt-surface axis. Associated with this process are a number of changes in expression of proteins and cell function.21 In any condition resulting in epithelial damage, the surface epithelium may be functionally immature. Any functional impairment of the epithelium (such as impaired butyrate oxidation) in this situation may hypothetically be secondary to immaturity of the epithelium. We therefore examined the possibility that surface (mature) and crypt (immature) cells have different capacities to metabolise butyrate. Surface cells showed a quantitatively greater capacity to oxidise both butyrate and glucose than crypt cells (about 4.5 times and 3.5 times respectively). In this study, we did not examine the utilisation of glutamine, which, although not quantitatively very important in normal colonocytes, may be enhanced in colonic explants from patients with ulcerative colitis.22 The ratio of butyrate to glucose oxidation was equally high in both surface and crypt cells (12:1 and 10:1), suggesting that both crypt and surface cells show a preference for butyrate as the substrate for energy production. The appreciably reduced butyrate to glucose utilisation ratio noted in DSS colitis suggests that diminished butyrate oxidation in DSS colitis is not related to the maturity of the colonocytes.
Ketone body production, especially β-hydroxybutyrate production, was also affected in DSS colitis compared with normal control mice. The concomitant inhibition of CO2 production and ketone body production from butyrate in DSS colitis suggests an alteration in the β-oxidation pathway rather than the Lynen (ketone body) pathway or Krebs cycle. The detrimental change in fatty acid oxidation but not glucose oxidation in colonocytes from DSS treated mice also suggests maintenance of the Krebs cycle and therefore mitochondrial functions in the cell.
Impaired oxidation of butyrate, but not of glucose, has been noted in colonocytes isolated from patients with active and quiescent ulcerative colitis.3 20 22-24 The failure of other investigators25 26 to confirm such a finding has been ascribed to possible differences in methodology.1 The pattern of abnormal metabolism described in ulcerative colitis is similar to that noted in DSS colitis in this study. Ibuprofen, a non-steroidal anti-inflammatory drug implicated in the development of colitis, decreases butyrate oxidation but not glucose oxidation when added to normal colonocytes in vitro.27 Similarly, reducing sulphur compounds, also implicated in the development of ulcerative colitis, selectively impair butyrate oxidation in human and rat colonocytes, with a slight compensatory increase in glucose oxidation.28-30 In each of the above situations, the pattern of metabolism is similar to that in DSS colitis. Starvation of colonocytes, secondary to lack of luminal butyrate, is the postulated cause for diversion colitis.5 In this disease condition, glucose derived from the circulation is presumably available to, and metabolised normally by, colonocytes. On the other hand, luminal SCFA deficiency in rats causes atrophy but not the other changes of colitis.31 32 Hence it appears that impaired energy metabolism alone may not be sufficient to induce colitis, and that other factors, presumably luminal in origin, are necessary for all the manifestations of colitis.
Butyrate enemas are effective in improving the histological picture in both diversion colitis and ulcerative colitis.5 33 Their utility in ulcerative colitis has been particularly difficult to explain, and a mass action has been invoked.31 In the present studies, substrate concentration was noted to quantitatively increase substrate oxidation not only in normal colonocytes, but also in DSS colitis. While the increase in butyrate oxidation was about 15-fold in control colonocytes, it was only about eightfold in colonocytes from DSS treated animals, as butyrate concentration increased from 10 to 80 mM. This increase, in practical terms, would amount to substantially greater availability of cellular energy, and may explain the benefit observed with butyrate enemas in ulcerative colitis. Although butyrate at high concentrations may induce apoptosis in cell lines, these studies used only short term incubations (45 minutes) and cell viability was not demonstrably altered during this time.
DSS has been reported to have a direct cytotoxic effect on intestinal epithelial cells and intraepithelial lymphocytes.34 In the present study, a direct effect of DSS on colonocyte metabolism was excluded because DSS added in vitro to normal colonocytes did not significantly impair butyrate metabolism. Reducing sulphur compounds, principally sulphide, impair butyrate oxidation by human and rat colonocytes.28-30 This impairment may occur at the level of butyryl-CoA dehydrogenase.35 Reducing sulphur compounds may be produced by the action of intestinal bacteria on sulphated polysaccharides.30 36 DSS, a sulphated polysaccharide, may increase colonic availability of sulphate and may stimulate sulphate reducing bacteria to generate higher sulphide levels, which are eventually toxic to the colonic mucosa. Sulphate reducing bacteria are increased in the colon of patients with ulcerative colitis.37 The possibility that luminal bacteria are necessary in the pathogenesis of DSS colitis is supported by the observation that metronidazole protects against DSS colitis.38 Similarly, carageenan, a sulphated polysaccharide, induces colitis in normal but not in germ-free mice.39 On the other hand, DSS colitis has been reported to occur in germ-free mice.40 It is obvious that the pathogenesis of impaired butyrate oxidation in DSS colitis will need further explanation.
In conclusion, DSS colitis causes a defect in colonocyte butyrate oxidation, with a compensatory increase in glucose oxidation, closely resembling abnormalities in human ulcerative colitis. Owing to the patchy nature of the initial mucosal lesion, it remains unclear whether alteration of cell metabolism is a primary phenomenon or secondary to mucosal inflammation. In either case, as colonic epithelial cells have important barrier and detoxicative functions, impaired butyrate metabolism may contribute significantly to the pathogenesis of colitis.
Acknowledgments
This project was funded by a grant from the Department of Science and Technology, Government of India. S K is the recipient of a Research Associateship from the Council of Scientific and Industrial Research, Government of India.
References
Footnotes
- Abbreviations used in this paper:
- DSS
- dextran sulphate sodium
- LDH
- lactate dehydrogenase
- SCFA
- short chain fatty acid