Article Text

Abstract
Objectives To investigate convergence of endoplasmic reticulum stress pathways and enhanced reactive oxygen species (ROS) production, due to intracellular retention of mutant tumour necrosis factor receptor 1 (TNFR1), as a disease mechanism in TNFR-associated periodic syndrome (TRAPS).
Methods Peripheral blood mononuclear cells from patients with TRAPS (n=16) and healthy controls (HC) (n=22) were studied alongside HEK293T cells expressing wild type-TNFR1 or TRAPS-associated mutations. Unfolded protein response (UPR)-associated proteins (protein kinase-like endoplasmic reticulum kinase, PERK), phosphorylated-PERK (p-PERK), phosphorylated inositol-requiring enzyme 1α (p-IRE1α) and spliced X-box binding protein 1 (sXBP1)) were measured by flow cytometry. XBP1 splicing and UPR-associated transcript expression were assessed by reverse transcription PCR/quantitative real-time PCR. ROS levels were measured using CM-H2DCFDA and MitoSOX Red in patients' monocytes or HEK293T cells by flow cytometry.
Results Mutant TNFR1-expressing HEK293T cells had increased TNFR1 expression associated with intracellular aggregation. TRAPS patients had increased sXBP1 transcripts (p<0.01) compared with HC. Raised p-PERK protein was seen, indicative of an UPR, but other UPR-associated transcripts were normal. Increased ROS levels were observed in TRAPS monocytes compared with HCs (p<0.02); these increased further upon IL-6 stimulation (p<0.01). Lipopolysaccharide-stimulated peripheral blood mononuclear cells of patients with TRAPS, but not HCs, demonstrated increased sXBP1 levels (p<0.01), which were reduced by antioxidant treatment (p<0.05).
Conclusions Patients with TRAPS have evidence of increased sXBP1 and PERK expression but without other signs of classical UPR, and also with high ROS generation that may contribute to the pro-inflammatory state associated with TRAPS. The authors propose a non-traditional XBP1 pathway with enhanced sXBP1 as a novel disease-contributing mechanism in TRAPS.
Statistics from Altmetric.com
Introduction
The autoinflammatory disease, tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS), is characterised by recurrent fever attacks and organ-localised inflammation.1 Although rare,2 autoinflammatory disorders have major implications for understanding the role of innate immunity in initiating and sustaining inflammation against self, and for optimal therapy of inflammatory disorders.3,–,5 Options for treatment of TRAPS are varied, including corticosteroids and biological agents targeting TNF,6 interleukin (IL)-1β,7 and IL-6,8 but are not always successful.9 ,10
Many hypotheses have been proposed to explain how TNFRSF1A mutations cause TRAPS,11 including defective TNF receptor 1 (TNFR1) shedding,1 ,12 TNF-induced apoptosis13 and nuclear factor (NF)-κB activation,14 aberrant activation of c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinases (MAPK),15 and mitochondrial reactive oxygen species (ROS) controlling pro-inflammatory cytokine production.16
One of the most widely accepted disease mechanisms is impaired TNFR1 trafficking.17 ,18 Intracellular protein accumulation can cause an associated unfolded protein response (UPR), whereby cells respond to stress by inducing expression of genes to restore proper endoplasmic reticulum (ER) function.19 The UPR relies on coordinated responses involving three major ER stress sensors, protein kinases inositol-requiring enzyme 1α (IRE1α) and protein kinase-like ER kinase (PERK), as well as activating transcription factor 6 (ATF6), to restore homeostasis within the ER. Consequences include reduced global protein synthesis, chaperone induction to assist with folding and protein degradation or cellular apoptosis.20 When activated through BiP dissociation, IRE1α and PERK homodimerise, becoming autophosphorylated, while ATF6 translocates to the golgi where it is cleaved into an active transcription factor. IRE1α cleaves XBP1 mRNA to generate the transcription factor sXBP1.21 This pathway is rapidly attenuated, whereas PERK activation, which shuts down translation, is sustained under ER stress conditions.22 ATF6 signalling clears and degrades accumulated misfolded proteins,23 in part by increasing transcription of XBP1 mRNA.21
An additional role for XBP1 in control of Toll-like receptor (TLR) signalling has been described.24 In response to TLR 2 and 4 stimulation, activation of IRE1α/XBP1 occurred without PERK or ATF6 activation. Furthermore, XBP1 was essential for pro-inflammatory cytokine responses in macrophages and is dependent upon nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2. Induction of ER stress enhanced TLR responses, with synergistic induction of IL-6, indicating that ER stress can enhance pro-inflammatory signalling downstream of TLR2 and 4. This is of considerable relevance to disease mechanisms in TRAPS since a recent study reported hyper-responsiveness of these patients' cells to lipopolysaccharide (LPS), the classical TLR4 agonist.15
A number of studies on TRAPS have reported low cell surface expression of TNFR1 with intracellular aggregation and ER retention.17 ,18 ,25 ,26 A patient-based study demonstrated significantly increased total TNFR1 expression without differences in UPR-associated transcripts BiP and CHOP.15 Conversely, an earlier study described co-localisation of mutant receptors with BiP in HEK293 cells.26 Therefore, it remains unclear whether a classical UPR is activated in TRAPS or whether a non-traditional UPR involving XBP1 could be operative.
In addition to UPR-induced NF-κB activation19 and pro-inflammatory cytokine secretion,27 increased ROS production may also be a key UPR component,28 ,29 and a role for mitochondrial ROS has been reported in TRAPS, mediating prolonged MAPK activation and increased IL-6 secretion.16 ROS are heavily implicated in TNF signalling, promoting cell death,30 ,31 NF-κB activation32 and JNK activation.31 Therefore ROS generation may serve as a readout for TRAPS-associated hyper-inflammatory states.
We explored the hypothesis that mutations in extracellular domains of TNFR1 interfere with normal receptor folding, causing intracellular accumulation, which may activate the XBP1 pathway independently of the classical UPR. This response may enhance cytokine release and ROS generation, and confer hyper-responsiveness to LPS.15 Convergence of these pathways may contribute to the pro-inflammatory phenotype of TRAPS.
Material and methods
Patient cohort
A total of 16 patients with TRAPS and 22 controls were studied. Patients recruited locally in Leeds (n=5), and from Merlin Park Regional Hospital, Galway (Ireland; n=3), in addition to the healthy controls (HC) (n=22); all gave written informed consent under the Leeds (East) Research Ethics Committee approved protocol. Additional patients' samples were obtained from the University of Siena (Italy; n=3) and the Royal Free Hospital (London, UK; n=5), with appropriate written informed consent. Patients' characteristics, including TRAPS-associated mutations, are summarised in table 1. Peripheral blood mononuclear cells (PBMC) were collected by sucrose density gradient centrifugation (Lymphoprep, Axis-Shield UK, Dundee, Scotland).
Demographics of patients with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) and healthy controls (HCs)
HEK293T cells
Wild type (WT)-TNFRSF1A or TRAPS-associated mutant TNFRSF1A cDNA (C33Y, T37I, P46L, T50K, T50M, C88R and c.472+1G>A), were cloned into pcDNA6/V5-His B (Invitrogen, Paisley, UK) and transfected into HEK293T cells using Fugene HD (Roche, Hertfordshire, UK), selecting stable clones by blasticidin-HCL resistance (Invitrogen).
Cell stimulation
For ROS stimulation, PBMCs were incubated for 1 h with 50 ng/ml each of TNF and IL-6 (both PeproTech, London, UK) or ultrapure LPS (E.coli K12, Invivogen, Nottingham, UK). For UPR stimulation, PBMCs were incubated in 2 mM dithiothreitol (Invitrogen) or1 μM thapsigargin (Sigma-Aldrich). For XBP1 splicing assays, PBMCs were cultured for 6 h with 10 ng/ml ultrapure LPS and 5 μM diphenyleneiodonium chloride antioxidant (DPI; Caymen Chemical, Tallin, Estonia).
XBP1 splicing PCR
Total RNA extracted from PBMCs using TRIzol (Invitrogen) was DNase1 treated (Ambion, Applied Biosystems, Warrington, UK), and reverse transcribed into complementary DNA (cDNA) using Superscript II (Invitrogen). Reverse transcription PCR (RT-PCR) primers; forward 5′-CTGAAGAGGAGGCGGAAGC -3′ and reverse 5′-AATACCGCCAGAATCCATGG-3′, as previously published, at final concentration of 1 μM.33 ,34 Cycling parameters: 1×5 min 94°C, 35×30 s 94°C, 30 s 55°C and 1 min 72°C, followed by 1×10 min 72°C. PCR products were analysed by 3% (w/v) agarose gel electrophoresis.
Primers for quantitative real-time PCR (qPCR); unspliced XBP1 (uXBP1) forward 5′-TCCGCAGCACTCAGACTACG-3′, sXBP1 forward 5′-CTGAGTCCGCAGCAGGTG-3′ and reverse primer for u/sXBP1 5′-AGTTGTCCAGAATGCCCAACA-3′), final concentration 50 fmol/μl. Forward uXBP1 primer spans the splice site to avoid amplifying sXBP1 and primer pairs span different exons. Relative gene expression (2(–ΔCT)) was analysed using SDS 2.3 (Applied Biosystems), normalising to hypoxanthine phosphoribosyltransferase 1 (HPRT):
Forward 5′-GGAAAGAATGTCTTGATTGTGGAAG-3′ and reverse 5′-GGATTATACTGCCTGACCAAGGAA-3′, final concentration 500 fmol/μl. All reactions were carried out in duplicate using SYBR green on the ABI 7900 (Applied Biosystems), using standard cycling parameters. Standard SYBR green PCR conditions were used, with an annealing temperature of 59°C and 40 cycles. Optimisation steps were carried out to ensure specificity of primer pairs to amplify individual u/sXBP1 cDNA products.
UPR flow cytometry analysis
PBMCs were fixed in 5% (w/v) methanol-free paraformaldehyde (Thermo Fisher Scientific, Rockford, USA) and permeabilised using BD Phosflow permeabilisation Buffer III (BD Biosciences, Oxford, UK). Primary antibodies: PERK and phosphorylated PERK (p-PERK) (sc-13073 and sc-32577, respectively; Santa Cruz Biotechnology, Heidelberg, Germany), sXBP1 (Clone 143F, 647502, BioLegend UK, Cambridge, UK) and p-IRE1 (AB48187, Abcam, Cambridge, UK). Isotype controls: mouse IgG2a negative control MCA929 (AbD Serotec) and rabbit Ig fraction X0903 (Dako). Secondary antibodies: goat antimouse Alexa Fluor-488 and goat antirabbit Alexa Fluor-488 (A11029 and A11008 respectively; Invitrogen). The LSRII flow cytometer (BD Biosciences) was used to analyse fluorescence, gating around monocyte/lymphocyte populations by scatter profiles. Median fluorescent intensity (MFI) data analysed with FACSDiva V.5.0.2 (BD Biosciences) and histogram overlays (normalised to cell number) generated with FlowJo V.8.87.
ROS detection
PBMC/HEK293T cells were incubated with 10 µM CM-H2DCFDA (total ROS) or 5 μM MitoSox Red (mitochondrial ROS; both molecular probes, Invitrogen) for 30 min at 37°C, before measuring fluorescein isothiocyanate (CM-H2DCFDA) or phycoerythrin (MitoSox Red) fluorescence. Data was analysed as previously described.
UPR gene expression analysis
A custom TaqMan low-density array (TLDA) was used to analyse expression of UPR genes relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Each TLDA port contained 200 ng cDNA and TaqMan gene expression master mix (Applied Biosystems) with samples run in duplicate. Exon spanning, ‘3’ most' TaqMan assays were selected for the array wherever possible; these were DDIT3/CHOP Hs99999172_m1, ERN1/IRE1α Hs00176385_m1, HSPA5 Hs99999174_m1, PPP1R15A Hs00169585_m1, SYVN1 Hs00381211_m1, PDIA4 Hs01115905_m1 and GAPDH Hs99999905_m1 (for functions and full gene names see online supplementary text). Data was analysed using RQ manager 1.2 (Applied Biosystems), applying an automatic cycle threshold (Ct). Relative gene expression was analysed with the 2(–ΔCT) method, normalising to GAPDH.
Statistical analysis
All statistical analyses were conducted using GraphPad Prism 5 software. A Shapiro-Wilk normality test was performed prior to Mann–Whitney, paired/unpaired t test, Wilcoxon matched-pairs signed rank and Kruskal-Wallis tests with Dunn's multiple comparison test when required. Significance was defined as p<0.05.
Results
Mutant TNFR1 aggregates within HEK293T cells
Studies in transfected HEK293T cells replicated previous findings of both increased expression and intracellular aggregation of mutant TNFR1 (online supplementary figure S1).26 Cell surface expression of mutant TNFR1, similar or greater than that in the WT-TNFR1 cells, was seen in T37I, T50K, T50M and c.472+1G>A cells (online supplementary figure S2). The C33Y, P46L and C88R mutants displayed lower surface expression of TNFR1 than WT, more equivalent to untransfected HEK293T cells (online supplementary figure S3). P46L cells displayed a small shift in fluorescence compared with untransfected cells, but this was low in comparison with WT cells.
Monocytes from patients with TRAPS have increased PERK expression
Flow cytometric analyses of PERK, p-PERK and p-IRE1α were carried out in HCs (n=6) and patients with TRAPS (n=5) lymphocyte and monocyte populations (figure 1). PERK levels were significantly higher in TRAPS monocytes (p<0.01; figure 1A) with no significant differences in lymphocytes (figure 1D). The p-PERK levels were not significantly different from controls in either cell population; however, both cell types displayed a trend towards higher p-PERK levels in patients with TRAPS than the controls (figure 1B and 1E). The p-IRE1α levels in monocytes (figure 1C) and lymphocytes (figure 1F) showed a trend towards lower expression in patients with TRAPS. These findings suggest that TRAPS monocytes could potentially have low-level activation of the PERK pathway.

Analysis of unfolded protein response activation by flow cytometry in healthy controls (HCs) and tumour necrosis factor receptor associated periodic syndrome (TRAPS) peripheral blood mononuclear cells. The levels of protein kinase-like endoplasmic reticulum kinase (PERK) (A,D), phosphorylated PERK (p-PERK) (B,E) and phosphorylated inositol-requiring 1α (p-IRE1α) (C,F) were measured by flow cytometry in monocyte (A–C) and lymphocyte (D–F) populations from HCs (n=6) and patients with TRAPS (n=5). Shapiro-Wilk normality tests and Mann–Whitney tests were carried out to check for statistical significance between groups (**p<0.01). The dot plot shows the median value of each group. Histogram overlays depicting an example of data normalised for cell number from one HC and one patient with TRAPS were created using FlowJo V.8.87. FITC, fluorescein isothiocyanate; MFI, median fluorescent intensity.
Cells from patients with TRAPS display evidence of XBP1 splicing
To further investigate the presence of an active UPR, splicing of XBP1 transcript was determined in PBMCs. Figure 2A highlights examples of amplified sXBP1 cDNA from patients with TRAPS, which was not evident in HCs. HC PBMCs stimulated with thapsigargin was a positive control. All samples were processed immediately after phlebotomy to avoid undue cellular stress. Expression of uXBP1 (figure 2B) and sXBP1 (figure 2C) was quantified by qPCR. There were no significant differences between uXBP1 transcripts levels in patients with TRAPS compared with HCs, whereas, sXBP1 transcripts were significantly higher in patients with TRAPS (p<0.01).

Patients with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) have evidence of increased XBP1 splicing. (A) Splicing of XBP1 transcripts, as demonstrated by reverse transcription PCR. Total RNA was extracted from peripheral blood mononuclear cells (PBMCs) of four patients with TRAPS (one T50M patient, two c.472+1G>A patients and one C88R patient) and four healthy control (HC). As an endoplasmic reticulum stress control, one PBMC sample was incubated with 1 μM Thapsigargin for 4 h to induce endoplasmic reticulum stress (positive control). A ‘no RT’ control was carried out, but no XBP1 product was amplified (data not shown). The uXBP1 product was 150 bp, whereas, sXBP1 was 124 bp. The relative levels of XBP1 splicing were determined using quantitative PCR analysis. PBMCs were isolated from HCs (n=12) and TRAPS (n=9). Levels of (B) uXBP1 and (C) sXBP1 were quantified relative to hypoxanthine phosphoribosyltransferase (HPRT). Data are plotted as relative expression units (REU); expression of the transcript, relative to HPRT, using the 2(–ΔCT) method. The levels of sXBP1 protein expression were measured by flow cytometry in monocyte (D) and lymphocyte (E) populations from HCs (n=6) and patients with TRAPS (n=5). Histogram overlays depicting data normalised for cell number from 1 HC and 1 patient with TRAPS, were created using FlowJo V.8.87. A Shapiro-Wilk normality test and unpaired t test were carried out to identify significant differences between groups (**p<0.01). The dot plots display median fluorescent intensity (MFI) values for each patient/control along with the median value for each group. . FITC, fluorescein isothiocyanate.
The sXBP1 protein expression, measured by flow cytometry, was higher in two out of five TRAPS monocytes compared with HCs; however, as a group, this was not significant (figure 2D). Similar levels of sXBP1 protein were expressed in lymphocyte populations in both groups (figure 2E).
Patients with TRAPS have similar levels of UPR-associated gene transcripts as healthy controls
Expression of selected UPR-associated transcripts was investigated in PBMCs from HCs (n=22) and patients with TRAPS (n=8). When PBMCs from four HCs were treated with dithiothreitol to induce ER stress, increased expression of transcripts was observed (figure 3A). However, no significant differences were observed between the HCs and patients with TRAPS, (figure 3B), indicating similar downstream UPR signalling activity, despite an apparent increase in sXBP1 levels (figure 2C) and potential for increased PERK activation (figures 1B and 1E).

Analysis of transcript levels associated with the activation of the unfolded protein response in peripheral blood mononuclear cells (PBMCs) from patients with tumour necrosis factor-associated periodic syndrome (TRAPS) and healthy controls. Transcript expression of DDIT3, ERN1, HSPA5, PPP1R15A, SYNV1 and PD1A4 were measured by quantitative PCR with TaqMan low density arrays. Data are plotted as relative expression units (REU); expression of the transcript, relative to GAPDH using the 2(–ΔCT) method. (A) PBMCs from healthy controls (HCs) (n=4) were stimulated with 2 mM dithiothreitol for 2 h to induce an artificial endoplasmic reticulum stress. (B) A comparison of transcript levels in PBMCs from patients with TRAPS (n=8; two clinic time points for T50M ⊠) and HCs (n=22). A Shapiro-Wilk normality test and a Mann–Whitney test were carried out to analyse significant differences between the groups. The dot plots show the median values measured.
TRAPS cells have increased ROS production which is further increased in response to IL-6
Increased total ROS levels were observed in TRAPS monocytes (n=9) compared with HCs (n=8) (p<0.02; figure 4A), as reported by Bulua et al.16 For each experiment, at least one HC was analysed alongside each patient to correct for inter-experimental variability; ROS levels in patients were compared with HCs in the same experiment to generate a fold change in ROS production. Mitochondrial ROS production was elevated in two patients with TRAPS, one c.472+1G>A mutation and one T50M mutation, when compared with an HC (online supplementary figures S4B and S4C). The c.472+1G>A patient was responding to etanercept, with normalisation of C reactive protein and total ROS levels similar to HCs (online supplementary figure S4A). Increased total ROS were also demonstrated in all mutants tested in the in vitro model, HEK293T cells with stable expression of WT or mutant TNFR1 (figure 4B, p<0.0001 across the group with a Kruskal-Wallis test; T50M and C88R p<0.05 and P46L p<0.01).

Monocytes of patients with tumour necrosis factor receptor (TNFR)-associated periodic syndrome (TRAPS) have elevated total reactive oxygen species (ROS) levels, which further increase with interleukin 6 (IL-6) stimulation. (A) Total basal ROS levels, measured with CM-H2DCFDA, in the monocyte population from healthy controls (HCs) (n=8) and patients with TRAPS (n=8; >1 clinic time point for T50M patients ⊠ and ◊). ROS levels measured in patients were compared with the levels in the HCs from each experiment to calculate the fold change. A Wilcoxon signed rank test compared the column median value with the normalised value of 1.0 (*p<0.05). (B) Analysis of total ROS levels in HEK293T cells, untransfected, or with stable expression of WT, or mutant TNFR1 (n≥6 for each cell line). Graph illustrates fold change in ROS levels from the WT median value. A Shapiro-Wilk normality test and a Kruskal-Wallis test with a Dunn's multiple comparison correction were carried out to analyse any significant differences (ANOVA p<0.0001, *p<0.05, **p<0.01). PBMCs from HCs (n=8) and patients with TRAPS (n=9) were stimulated with 50 ng/ml of (C) TNF or (D) IL-6; for up to 1 h, and total ROS production in the monocyte population was measured. A paired t test was carried out to analyse differences between treatment groups (*p<0.05, *p<0.01). The monocytes' values for HCs and TRAPS for 60 min stimulations were directly compared with an unpaired t test; the dot plots show the median value of the group.
PBMCs were stimulated with pro-inflammatory cytokines, TNF and IL-6, as well as LPS. The monocytes of HCs demonstrated significantly increased ROS in response to TNF (figure 4C; p=0.0296), whereas, the monocytes of TRAPS had significantly higher ROS after IL-6 treatment (p=0.0044; figure 4D). Direct comparison of ROS levels between HCs and patients with TRAPS at the 1 h time point did not demonstrate any significant differences with either TNF or IL-6. LPS stimulation did not have any significant effects on ROS levels either (data not shown).
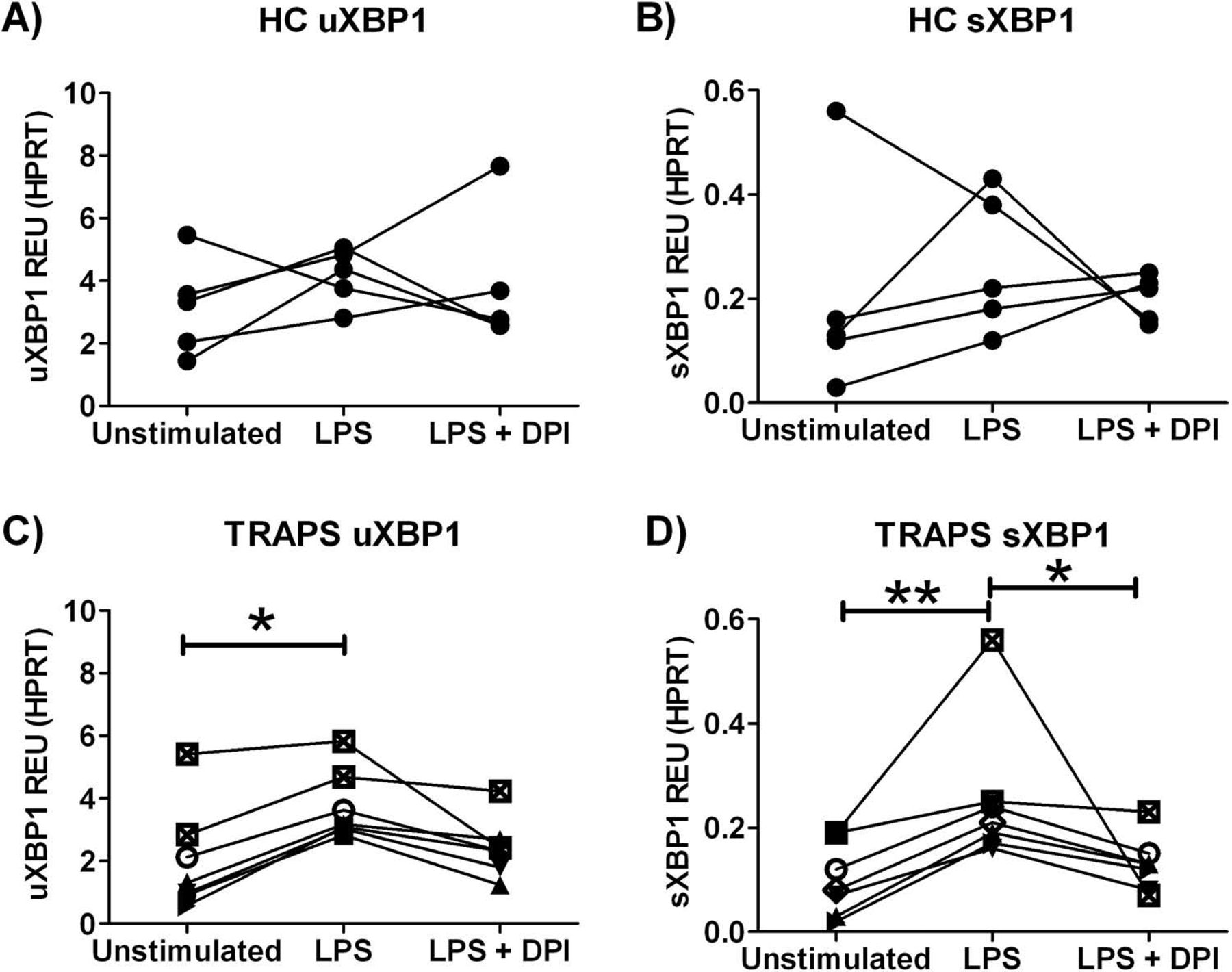
LPS increases sXBP1 transcript levels in PBMCs from patients with TRAPS, an effect abolished by antioxidant treatment
So far, we have demonstrated activation of the XBP1 pathway in TRAPS, and XBP1 has been described to play an important role in enhancement of LPS signalling.24 Taken together with previous reports of LPS hyper-responsiveness in TRAPS,15 we investigated whether XBP1 activation could help explain this phenomenon. PBMCs from HCs (n=5) and from patients with TRAPS (n=7) were cultured for 6 h in the presence of LPS or LPS plus DPI antioxidant, to investigate whether LPS could induce XBP1 splicing, and whether this was influenced by high ROS levels in cells of patients with TRAPS. Interestingly, we noticed that cultured TRAPS PBMCs no longer had increased sXBP1 transcripts, evident when analysing the same cells immediately after PBMC isolation (figures 2C and 5D). In response to LPS stimulation, HC PBMCs showed no significant increases in uXBP1 or sXBP1 (figure 5A,B). However, significantly increased uXBP1 and sXBP1 transcripts were observed with LPS stimulation in the PBMCs of patients with TRAPS (figure 5C,D; p<0.05 and p<0.01 respectively). Co-treatment with LPS and antioxidant significantly reduced sXBP1 levels in patients with TRAPS by 40% (figure 5D, p<0.05), suggesting an association between ER stress due to mutant TNFR1 accumulation and enhanced ROS levels mediating LPS hyper-responsiveness through the XBP1 pathway.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patients with tumour necrosis factor-receptor associated periodic syndrome (TRAPS) demonstrate LPS-induced XBP1 splicing which is abolished by antioxidant treatment. XBP1 splicing was measured using quantitative PCR. Peripheral blood mononuclear cells from healthy controls (HCs) (n=5) and patients with TRAPS (n=7) were cultured for 6 h (lipopolysaccharide (LPS) 10 ng/ml, diphenyleneiodonium chloride (DPI) 5 μM), and levels of uXBP1 and sXBP1 measured. Data are plotted as relative expression units (REU); expression of the transcript relative to hypoxanthine phosphoribosyltransferase 1 (HPRT) using the 2(–ΔCT) method. (A) HC uXBP1; (B) HC sXBP1; (C) TRAPS uXBP1 and (D) TRAPS sXBP1. A Kruskal–Wallis test with Dunn's multiple comparison correction was carried out to analyse significant differences between the groups (*p<0.05, **p<0.01).
Discussion
This study was undertaken to investigate convergence of ER stress pathways and enhanced production of ROS in patients with TRAPS. Accumulation of intracellular mutant TNFR1 is proposed as an explanation for the pro-inflammatory TRAPS phenotype.18 ,26 Results presented here point to activation of a select minority of UPR-signalling mediators in patients with TRAPS, with significantly increased sXBP1 transcript and elevated PERK and p-PERK levels. It has been reported that activated IRE1α degrades its own mRNA using endoribonuclease activity, which may explain our observation of increased sXBP1, yet decreased IRE1α levels.35
Treatment of patients' PBMCs with LPS significantly increased the levels of sXBP1, an effect not observed in the PBMCs of HCs, which complements previous reports of hypersensitivity to LPS in TRAPS.15 TLR/XBP1 cooperation was described in mouse macrophages,24 but the results presented here are, to our knowledge, the first to demonstrate activation of this pathway in humans and, crucially, with higher induction seen in the PBMCs of patients with TRAPS. It is possible that mild ER stress, through accumulation of mutant TNFR1, results in XBP1 splicing but not a full classical UPR. Subsequently, during bacterial challenge, even at low doses which would not usually induce signalling in healthy individuals, activation of both XBP1 and LPS signalling pathways cooperate to enhance the inflammatory response. An interesting observation was that after culturing, TRAPS PBMCs lost their increased sXBP1, indicating potential recovery after being removed from an inflammatory environment.
Enhanced activation of JNK and p38 have been reported in TNFR1 mutant mice.15 In addition, a direct link between p38 signalling pathways and sXBP1 has been described, whereby, p38 directly phosphorylates sXBP1 leading to enhanced nuclear translocation and transcriptional activity.36 This may provide a further potential feedback loop in TRAPS, whereby upregulated p38 activation may enhance XBP1 signalling. Investigations into the phosphorylation status of sXBP1 and cellular distribution, whether cytoplasmic or nuclear, would be an interesting future step in the investigation of TRAPS.
Although ER and oxidative stresses are closely linked events, the molecular pathways that couple these processes are poorly understood. It is known that UPR can upregulate antioxidant defence mechanisms,37 ,38 and a number of XBP1 gene targets are known to have roles in redox homeostasis and cell survival during hypoxia.39 Liu et al described how uXBP1 induced expression of antioxidant genes (catalase, superoxide dismutase and thioredoxin), independently of sXBP1.40 Interestingly, uXBP1 transcript levels were not significantly elevated in patients with TRAPS compared with HC, despite increased ROS levels, suggesting possible defective antioxidant responses. A recent paper described the involvement of NLRP3 inflammasome in ER stress responses, with NLRP3 inflammasome-dependent IL-1β secretion, which was dependent on ROS and independent of UPR signalling pathways.41 This finding could help explain why many patients with TRAPS respond to the interleukin-1-receptor antagonist, anakinra.7
Levels of ROS production were measured in nine patients with TRAPS, with common (T50M) and rare (c.472+1G>A) mutations. The results confirm previous findings that mitochondria may be the source.16 ROS have already been implicated in TNF signalling pathways,42 including cell death,30 MAPK activation,16 and TNFR1 shedding.43 Therefore, excessive ROS can affect many essential mechanisms involved in the pathogenesis of TRAPS, including self-association of TNFRs.44
A further effect of ROS might involve IL-6 signalling; however, this has not been thoroughly studied and, to our knowledge, only one report has demonstrated a link between these two mechanisms.45 Our data show that IL-6 induced the production of ROS in monocytes of TRAPS. The previous study suggested that IL-6 induced reduction of mitochondrial membrane potential and decreased cellular ATP production. Overall, the observed high IL-6 levels in sera of patients with TRAPS, which could increase ROS production, provides a rationale for IL-6 antagonism as a therapy for TRAPS.8
To further investigate the connection between enhanced sXBP1 and ROS production in TRAPS, an antioxidant was used to analyse the effect on LPS responses. We found that high sXBP1 levels in TRAPS PBMCs, caused by LPS, could be significantly decreased when co-treated with DPI antioxidant, whereas, there was no such effect in cells of HCs. So, as proposed by Martinon et al, involvement of sXBP1 in TLR signalling pathways may be dependent on ROS24 and, therefore, high ROS levels in TRAPS could contribute to enhanced LPS responses by promoting sXBP1 signalling.15 However, it is also possible that the effects of DPI may not purely be ROS inhibition, as DPI has been suggested to have other, albeit less specific effects, including effects on mRNA transcription.16 ,46 Treatment of patients with TRAPS with antioxidants designed to decrease levels of ROS may be a useful adjunct treatment.47
To conclude, we describe a novel XBP1 pathway activated in patients with TRAPS that, together with high production of ROS, can lower the threshold of innate immune activation, thereby causing inappropriate cellular responses leading to an autoinflammatory phenotype.
Acknowledgments
The authors thank the patients and controls for contributing blood samples. The authors thank Dr Sarah Churchman for assistance with real-time PCR and Dr Thomas Baboolal for assistance with producing flow cytometry histogram overlays (both from LIMM). The authors also thank Dr Nasim Yousaf from the William Harvey Research Institute for providing the T37I-TNFR1 construct, and Dr Gina Doody and Dr Reuben Tooze (both LIMM) for providing initial XBP1 RT-PCR primers.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding This work was supported in part by a University of Leeds and Leeds Institute of Molecular Medicine (LIMM) Research Scholarship, an FP7-HEALTH-2007-2.4.4-1 Grant (Number 200923), and the NIHR-Leeds Musculoskeletal Biomedical Research Unit.
-
Competing interests None.
-
Ethics approval Ethics approval was granted by the Leeds (East) Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Correction notice This article has been corrected since it was published Online First.