One-step efficient generation of dual-function conditional knockout and geno-tagging alleles in zebrafish
- Peking University, China
- Peking University Shenzhen Graduate School, China
- Gcrispr (Tianjin) Genetic Technology, China
Abstract
CRISPR/Cas systems are widely used to knock out genes by inducing indel mutations, which are prone to genetic compensation. Complex genome modifications such as knockin (KI) might bypass compensation, though difficult to practice due to low efficiency. Moreover, no ‘two-in-one’ KI strategy combining conditional knockout (CKO) with fluorescent gene-labeling or further allele-labeling has been reported. Here, we developed a dual-cassette-donor strategy and achieved one-step and efficient generation of dual-function KI alleles at tbx5a and kctd10 loci in zebrafish via targeted insertion. These alleles display fluorescent gene-tagging and CKO effects before and after Cre induction, respectively. By introducing a second fluorescent reporter, geno-tagging effects were achieved at tbx5a and sox10 loci, exhibiting CKO coupled with fluorescent reporter switch upon Cre induction, enabling tracing of three distinct genotypes. We found that LiCl purification of gRNA is critical for highly efficient KI, and preselection of founders allows the efficient germline recovery of KI events.
https://doi.org/10.7554/eLife.48081.001Introduction
Targeted genome modifications via customizable endonucleases such as TALENs or the CRISPR/Cas system have revolutionized the technology of genome editing, through which gene knockouts can be easily generated through nonhomologous end joining (NHEJ)-induced indel mutations in many species, including zebrafish (Danio rerio) (Huang et al., 2011; Miller et al., 2011; Sander et al., 2011; Tesson et al., 2011; Bedell et al., 2012; Huang et al., 2012; Chang et al., 2013; Cong et al., 2013; Hwang et al., 2013; Jao et al., 2013; Mali et al., 2013; Wang et al., 2013; Boch et al., 2014; Doudna and Charpentier, 2014; Bolukbasi et al., 2015; Anderson et al., 2017). However, indel mutations have been reported to be able to evoke transcriptional adaptation-derived genetic compensation, triggered by mutations bearing a premature termination codon (PTC), leading to complications and confusion in the study of gene functions (Rossi et al., 2015; El-Brolosy et al., 2019; Ma et al., 2019). Alternatively, advanced genome engineering, including conditional knockout (CKO) and gene tagging approaches, which rely on targeted knockin (KI) to introduce exogenous DNA sequences (e.g., loxP or fluorescent reporter genes) into a target site, might bypass the interference of the unwanted genetic compensation by adopting properly designed strategies (El-Brolosy and Stainier, 2017). However, these strategies are difficult in practice due to their low efficiency and the need for extensive screening, especially when dealing with multicellular organisms without developed embryonic stem (ES) cell technology. In addition, a ‘two-in-one’ dual-function KI strategy for achieving CKO coupled with gene tagging has rarely been reported (Su et al., 2017). Furthermore, the target gene is usually labeled by a single fluorescent reporter, and the concept and method for discriminating different alleles (normal vs defective) of the same target gene have not been established.
In zebrafish, KI is generally mediated by either homologous recombination (HR) or the NHEJ pathway for repairing DNA double-strand breaks (Zu et al., 2013; Auer et al., 2014; Irion et al., 2014; Shin et al., 2014; Li et al., 2015a; Li et al., 2015b; Carroll, 2016; Hoshijima et al., 2016; Luo et al., 2018), and NHEJ-based targeted insertion is more efficient, though less precise, than the HR-based strategy (Hagmann et al., 1998; Mao et al., 2008; Dai et al., 2010). However, current NHEJ-based KI methods are feasible for gene labeling but cannot be used to generate CKO alleles through a one-step single insertion since two loxP sequences have to be inserted at two separate locations (Burg et al., 2018). Alternatively, conditional gene inactivation or in combination with gene tagging can be achieved via various designs of gene-trap cassettes in zebrafish (Sivasubbu et al., 2006; Trinh et al., 2011; Ni et al., 2012; Sugimoto et al., 2017; Grajevskaja et al., 2018). Nevertheless, CKO and gene labeling are usually achieved through separate experiments, since they typically use different designs of donor DNA, and dual-function CKO alleles coupled with fluorescent gene labeling generated through targeted insertion have not been described. The combination of CKO and gene/cell labeling within the same allele or even CKO together with genotype/allele labeling, allowing researchers to easily distinguish defective alleles from normal ones by using different fluorescent reporters, could be particularly useful for mosaic analysis and cell fate tracing, especially for zebrafish research due to the superiority of the live imaging of embryogenesis in this animal model.
Here, we report the development of a highly efficient method for achieving one-step heritable generation of dual-function alleles in zebrafish through targeted insertion induced by the Cas9/gRNA system, based on the special design of a dual-cassette-donor. The feasibility of this strategy was first demonstrated at the tbx5a and kctd10 loci, resulting in a germline transmission efficiency as high as 56% after the preselection of F0 embryos showing correct expression of a fluorescent reporter. Under normal conditions, these KI alleles enable simultaneous expression of a fluorescent reporter together with the target gene, displaying a gene-labeling effect. After the injection of Cre mRNA, the expression and function of the target gene as well as the reporter gene were disrupted, and conditional knockout was successfully achieved. We further improved our strategy by introducing a second fluorescent reporter into the dual-cassette donor, which could be used to conditionally generate and distinguish different alleles (normal vs defective) by using different fluorescent reporters and, further, to distinguish different cells with different genotypes (homozygous normal, heterozygote, and homozygous mutant/defective), allowing real-time mosaic analysis within the same tissue. This allele-tagging or geno-tagging strategy was demonstrated at the tbx5a and sox10 loci.
Results
Rationale of our dual-cassette-donor strategy for the one-step generation of ‘two-in-one’ dual-function KI alleles through targeted insertion
To generate a dual-function allele through a single step of NHEJ-mediated KI, we developed a novel strategy based on the special design of a dual-cassette donor, in which CKO and gene labeling can be established simultaneously through one-step targeted insertion of the donor vector into an intron of the target gene via the NHEJ pathway activated by the CRISPR/Cas system. For this purpose, the initial donor was designed to contain two functional cassettes, a positive one and a negative one and is therefore called a PoNe (positive-negative) donor (Figure 1A). The positive cassette (Po-cassette) was designed to be able to maintain the correct expression/splicing and function of the target gene after the integration of the PoNe donor into the target site, and additionally to track target gene expression through the incorporation of a 2A-linked fluorescent reporter gene. For this purpose, the first part of the Po-cassette was designed to retain the integrity of the cDNA of the target gene by cloning the sequence of the intron, including the splice acceptor site, downstream of the target site as well as all the rest of the downstream coding sequence of the wild-type target gene into the donor. For gene labeling and the visualization of target gene expression, the second part of the Po-cassette was composed of a fluorescent reporter fused in-frame upstream of the stop codon of the target gene cDNA in the donor, separated by a 2A peptide coding sequence. The whole sequence described above was flanked by two loxP sites, so that it can be removed to generate conditional mutations in response to Cre recombinase. The negative cassette (Ne-cassette) was designed to ensure complete disruption of target gene expression and function after removal of the Po-cassette by Cre-induced recombination. This is achieved by two levels of interruption of gene expression, leading to the early termination of either transcription or translation. For this purpose, the initial Ne-cassette consisted of two transcription termination signals (TTSs), the SV40 polyA (PA) sequence followed by BGH PA, which were followed by the sequence of the same splice acceptor site as in the Po-cassette and the exon immediately after the target intron, but engineered to contain a nonsense mutation (mutExon).
Figure 1 with 1 supplement see all
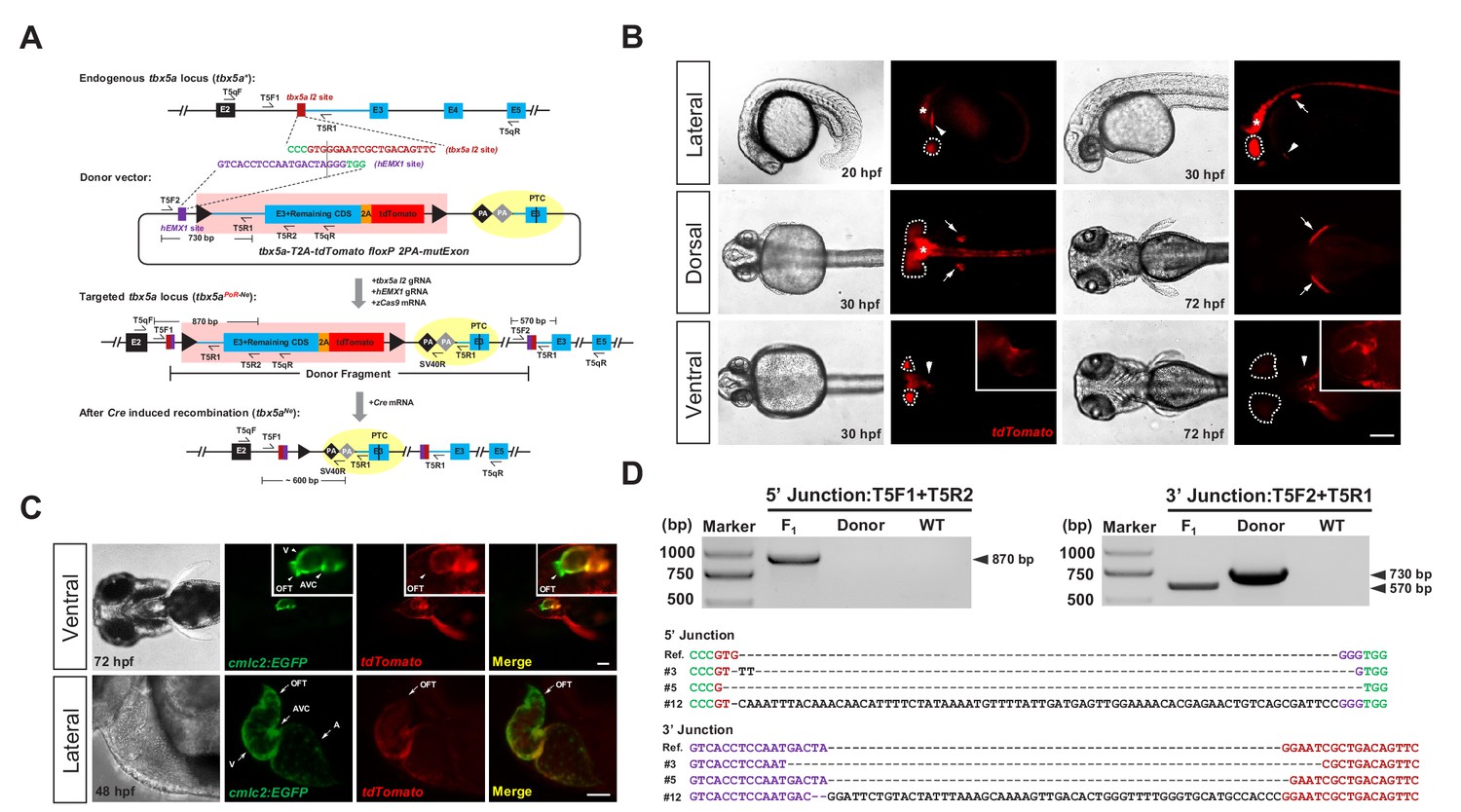
Generation of CKO coupled with gene labeling dual-function alleles through targeted insertion in zebrafish at the tbx5a locus.
(A) Schematic diagram of the KI strategy based on the dual-cassette PoNe donor tbx5a-T2A-tdTomato floxP 2PA-mutExon (the tbx5a PoR-Ne donor), consisting of a Po-cassette and a Ne-cassette (highlighted by pink and yellow shadows, respectively). The target sequences of hEMX1 and tbx5a are shown in purple and brown, respectively, and the PAMs are in shown green. Black triangles represent loxP. Black and gray diamonds indicate polyadenylation (PA) signals. The black bar in the third exon (E3) indicates the in-frame premature termination codon (PTC). Primers T5qF and T5qR are used for qRT-PCR in Figure 2—figure supplement 1E and F. (B) Images of F1 larvae from an outcross of a tbx5a PoR-Ne donor knockin founder (#12), showing tdTomato expression in the pectoral fins (white arrows), heart (white arrowheads), eyes (white dotted circle) and nervous system (white asterisk). Scale bar, 200 μm. (C) Images of F1 progeny from Tg(cmlc2:EGFP) transgenic zebrafish crossed with the tbx5a KI founder (#12), showing an antero-posterior gradient of tbx5a expression in the ventricle. Upper panel: Ventral view of a 72 hpf embryo. Lower panel: Z-stack confocal images of the heart region from a 48 hpf embryo. A: atrium. AVC: atrioventricular canal. OFT: outflow track. V: ventricle. Scale bar, 50 μm. (D) Junction PCR and direct sequencing results of individual positive F1 progeny from outcrosses of each of the three positive founders (#3, #5 and #12). Due to an extra copy of the T5R1 primer sequence in the PoR-Ne donor, PCR with the primer pair T5F2 and T5R1 targeting the donor plasmid results in a larger fragment than that of the F1 progeny. F1: an F1 embryo from F0 #12. Donor: tbx5a PoR-Ne donor plasmid. WT: pooled genomic DNA of five wild-type embryos.
After the insertion of the PoNe donor into the intron of the target gene, the targeted locus will acquire the properties of both a conditional allele and a gene-tagging allele. Under normal conditions, that is in the absence of Cre recombinase, this locus serves a gene-tagging function, and the expression and function of the target gene remain normal; a wild-type protein and a fluorescent reporter protein will be produced concurrently, with the fluorescent reporter recapitulating the expression pattern of the endogenous target gene. After Cre/loxP-mediated removal of the Po-cassette, the two polyA signals (2PA) in the Ne-cassette could prematurely stop the transcription of the target gene, resulting in a short transcript encoding a truncated and nonfunctional protein, thus achieving CKO. If there is leaky transcription after the polyA signals, the mutExon sequence in the Ne-cassette will be transcribed and spliced into the mature mRNA, again resulting in the disruption of the function of the target gene, due to the existence of the premature stop codon in this exon.
Efficient generation of a dual-function KI allele at the zebrafish tbx5a locus
The feasibility of this dual-cassette-donor strategy was first evaluated at the tbx5a locus (Basson et al., 1997; Garrity et al., 2002) in zebrafish. We identified a highly efficient guide RNA targeting the second intron of tbx5a (referred to the tbx5a I2 target site, Figure 1—figure supplement 1A–1C and Supplementary file 1, 2) and designed a PoNe donor containing an hEMX1 target site for in vivo linearization (Figure 1A; donor name: tbx5a-T2A-tdTomato floxP 2PA-mutExon, abbreviated as tbx5a PoR-Ne donor) (Lin et al., 2014). After the coinjection of this donor plasmid with the zCas9 mRNA, tbx5a I2 gRNA and hEMX1 gRNA into one-cell-stage zebrafish embryos, mosaic expression of tdTomato in the heart and pectoral fins, which are the expected tissues of endogenous tbx5a expression, was easily observed (Figure 1—figure supplement 1D and Supplementary file 2). Junction PCR results demonstrated correct integration of the PoNe donor into the tbx5a I2 target site (Figure 1—figure supplement 1E). Surprisingly, we found that the purification methods applied to the gRNAs after in vitro transcription, especially the gRNA targeting the donor plasmid (the hEMX1 target site), dramatically affected KI efficiency in our experimental settings. In our initial attempts, we employed ethanol precipitation to purify both gRNAs used for injection and found no tdTomato expression in any injected embryos. In contrast, when injected with LiCl-purified gRNAs, more than 10% of the embryos showed the expected red fluorescent signal (Figure 1—figure supplement 1D and Table 1). Furthermore, we could easily observe the expected fluorescent signals in the founder embryos when we injected the Cas9/gRNA system with the donor gRNA (targeting the hEMX1 site) purified by LiCl precipitation and the endogenous gene (tbx5a)-targeting gRNA purified by ethanol precipitation. However, no fluorescent signal could be detected with the other combination, that is the donor gRNA purified by ethanol precipitation and the tbx5a-targeting gRNA purified by LiCl precipitation (Table 1). These observations indicate that the gRNA targeting the donor plasmids is more sensitive to the purification method, and suggest that the purity or configuration of this gRNA may influence the efficiency of donor linearization in the zebrafish embryos, while the underlying mechanism of this phenomenon needs to be investigated in the future. Nevertheless, in the following experiments, we adopted all LiCl-purified gRNAs for injection.
Table 1
KI efficiency using different gRNA purification methods
https://doi.org/10.7554/eLife.48081.004| hEMX1 gRNA purification | tbx5a I2 gRNA purification | Ratio of fluorescence- positive F0 embryos |
|---|---|---|
| LiCl | LiCl | 18/119 (15.1%) |
| LiCl | Ethanol | 8/103 (7.8%) |
| Ethanol | LiCl | 0/94 (0.0%) |
| Ethanol | Ethanol | 0/43 (0.0%) |
Germline transmission and evaluation of the tbx5a KI allele
After the injection of the tbx5a PoR-Ne donor together with the Cas9/gRNA system into approximately 100 one-cell stage embryos, red fluorescent signals could be detected in 18 of the embryos. These founder (F0) embryos were separated into tdTomato-positive and tdTomato-negative groups and raised to adulthood for germline transmission analysis. Correct expression patterns of red fluorescent signals were detected in the F1 embryos derived from the outcrossing of 5 out of 9 F0 individuals grown from the tdTomato-positive embryos, with mosaicism ranging from 2.4% to 43.8% (Supplementary file 3), demonstrating successful and highly efficient germline transmission of the tbx5a PoR-Ne donor KI events among the embryos preselected for correct fluorescent reporter expression. Junction PCR and sequencing analysis confirmed the germline transmission of the KI alleles (Figure 1D). In contrast, no red fluorescent signal was detected in any F1 progeny from the outcrossing of 40 F0 adults raised from tdTomato-negative embryos, indicating a positive correlation of reporter gene expression in the founder embryos with successful and efficient germline transmission of the KI events. We also screened several batches of founders prepared from the injection of ethanol-purified gRNAs together with the tbx5a PoR-Ne donor and zCas9 mRNA, and no germline transmission of KI events was detected, demonstrating that LiCl purification of gRNA is critical for the successful generation of heritable KI alleles for our strategy.
Compared with the positive but mosaic founder embryos (Figure 1—figure supplement 1D), the F1 progeny from the germline-positive founders showed more specific and uniform expression of tdTomato in the heart, pectoral fins, eyes and nervous system (Figure 1B). After the introduction of the Tg(cmlc2:EGFP) transgenic background (Tong et al., 2014), we detected the colocalization of EGFP and tdTomato fluorescent signals in the progeny. Under higher magnification, we found a clear antero-posterior gradient of tbx5a expression in the ventricles, with no signal in the outflow track (OFT) and the highest expression in the atrioventricular canal (AVC) (Figure 1C). This phenomenon was scarcely detectable through in situ hybridization (Figure 1—figure supplement 1F), indicating the advantages of gene labeling for dissecting expression patterns in detail.
Conditional knockout could be achieved for the tbx5a KI allele
The tbx5a PoR-Ne donor KI alleles that we generated are not only gene-tagging alleles but also floxed conditional alleles. To evaluate whether conditional knockout could be achieved with these alleles, we isolated a tbx5a indel mutation with a 5 bp deletion in the third exon (tbx5aΔ5) (Figure 2—figure supplement 1A–1C). tbx5aΔ5/Δ5 homozygote embryos showed severe heart and fin defects at 48 hpf (hours post-fertilization) (Figure 2—figure supplement 1D), which is consistent with a previous report of a tbx5a loss-of-function mutation (Garrity et al., 2002). A tbx5a+/Δ5 heterozygote was crossed with the tbx5a KI founder showing the highest germline mosaicism (#12, 43.8%), and their progeny were injected with Cre mRNA at the one-cell stage. As expected, some of the injected F1 embryos (20 out of 78, 25.6%) showed distinct developmental defects of the heart and fins (Figure 2A). No red fluorescent signal was observed in any of the injected progeny, indicating that the Cre/loxP system was both functional and highly efficient in removing the Po-cassette. In contrast, 30 uninjected embryos developed normally, and 13 of them showed tdTomato expression, close to expectations. The genotyping results also showed that all the injected embryos with heart and fin defects were initially tbx5aΔ5/PoR-Ne compound heterozygotes before injection (we designated the original allele bearing the PoR-Ne donor insertion at the tbx5a locus from the #12 founder as tbx5aPoR-Ne, and the converted allele after Cre-induced loxP recombination was referred to as tbx5aNe), while the normal ones initially presented the tbx5a+/Δ5, tbx5a+/PoR-Ne, or tbx5a+/+ genotype (i.e., these embryos all harbored at least one wild-type tbx5a allele) (Figure 2B). To evaluate the expression of the tbx5aPoR-Ne KI allele in ‘on’ and ‘off’ states at the molecular level, quantitative RT-PCR (qRT-PCR) experiments were performed on 72 hpf embryos using primers T5qF and T5qR (Figure 1A, Supplementary file 7) in exons 2 and 5, respectively. The results obtained from outcrosses of tbx5aPoR-Ne/PoR-Ne homozygous fish with wild-type fish showed that the mRNA transcription level of the tbx5a locus in the tbx5a+/PoR-Ne heterozygous embryos without Cre mRNA injection was comparable with that in the wild-type control embryos, indicating normal tbx5a expression of the tbx5aPoR-Ne allele in the ‘on’ state (Figure 2—figure supplement 1E). No red fluorescence signal could be detected in the tbx5a+/PoR-Ne embryos after Cre mRNA injection, indicating efficient excision of the Po-cassette and that most of the tbx5aPoR-Ne alleles had been efficiently converted into tbx5aNe alleles. The tbx5a expression level in these tbx5a+/Ne embryos significantly decreased to approximately 50% of the level in the wild-type embryos, as expected (Figure 2—figure supplement 1E). To measure the expression of the tbx5aPoR-Ne allele in the ‘off’ state in homozygous mutants achieved by Cre-induced excision of the Po-cassette, we also performed qRT-PCR experiments on the embryos obtained from outcross of tbx5aPoR-Ne/PoR-Ne homozygous fish with a tbx5a+/PoR-Ne heterozygote after Cre mRNA injection. The results showed that the tbx5a transcripts decreased by ~15 fold in the tbx5aNe/Ne homozygous mutant embryos compared with wild-type ones, indicating highly efficient mutagenesis effect of the tbx5aNe allele (Figure 2—figure supplement 1F). Collectively, these results indicate that our PoNe donor knockin strategy can efficiently achieve a conditional gene knockout effect in addition to gene labeling in zebrafish.
Figure 2 with 2 supplements see all

The tbx5a and kctd10 conditional alleles are responsive to Cre recombinase.
(A) Images of the 72 hpf progeny (F1) from a tbx5a+/Δ5 heterozygote crossed with the F0 #12 (mosaic for the tbx5aPoR-Ne allele) against the Tg(cmlc2:EGFP) transgenic background with or without Cre mRNA injection. The fluorescent images were obtained in the lateral view. Black arrowheads indicate pectoral fins, and black or white arrows indicate the heart. A: atrium. V: ventricle. Scale bar, 200 μm. (B) Genotyping results of the individual Cre mRNA-injected embryos obtained from the cross in A. (C) Images of the 48 hpf progeny (F2) derived from the cross of two kctd10 KI heterozygotes (F1), each carrying a different KI allele (kctd10PoG-Ne-1 from #32 and kctd10PoG-Ne-2 from #5) with the Tg(cmlc2:EGFP) transgenic background, to reveal the morphology of the heart. The white dotted line indicates the outline of the heart. The hearts in the upper and middle panels developed normally, showing obvious heart looping. In contrast, the heart in the lower panel shows defective development, exhibiting AVC malformations and heart looping failure. Black arrowheads indicate the heart. Scale bar, 200 μm. (D) Genotyping by PCR amplification of the region flanking the loxP recombination site of the Cre mRNA-injected individual embryos obtained from the cross in C. (E) Representative junction PCR and direct sequencing results of the Cre mRNA-injected individual embryos showing normal or defective heart development obtained from the cross in C. As expected, the results indicate that the embryo showing the heart phenotype (labeled as ‘Single defective embryo’ in the figure) was a kctd10PoG-Ne-1/PoG-Ne-2 compound heterozygote (F2) before Cre mRNA injection since it showed overlapping peaks (red boxed region) in the sequencing results of the PCR products at both the 5’ and 3’ junctions (right panel), as the two alleles have different indel sequences at the junction sites. In contrast, the normal embryos (labeled as ‘Single normal embryos’ in the figure) were either kctd10+/PoG-Ne-1 or kctd10+/PoG-Ne-2 heterozygotes before Cre mRNA injection and therefore displayed uniform sequencing results corresponding to either the kctd10PoG-Ne-1 (or kctd10Ne-1) or kctd10PoG-Ne-2 (or kctd10Ne-2) allele, respectively. The expected corresponding sequences can be found in Figure 2—figure supplement 2J and K.
Generation and evaluation of dual-function alleles showing CKO with a gene labeling effect at the zebrafish kctd10 locus
We generated a second knockin allele according to the same strategy but replacing tdTomato with tdGFP as the reporter at another zebrafish locus, kctd10 (Figure 2—figure supplement 2A–2D), which has been reported to play an important role in heart development by suppressing Tbx5a activity, although its expression is somewhat ubiquitous (Tong et al., 2014). Similarly, we injected approximately 100 one-cell stage embryos and observed the mosaic expression of tdGFP in 22 embryos and we further verified the knockin events by junction PCR in the injected F0 embryos (Figure 2—figure supplement 2E–2F and Supplementary file 2). This time, we raised all the F0 embryos to adulthood without preselection, including those that were negative for the tdGFP fluorescent signals and found that 2 of the 42 F0 (4.8%) displayed germline transmission of the kctd10 PoG-Ne donor KI events (Figure 2—figure supplement 2G–2J and Supplementary file 4). kctd10 has been reported to show strong maternal expression in zebrafish (Tong et al., 2014), and this was confirmed with our kctd10 PoG-Ne donor KI alleles, since the expression of tdGFP could be detected as early as at one-cell stage in all the progeny from outcrosses of female KI heterozygotes but not in any of those from outcrosses of male KI heterozygotes. After the injection of the Cre mRNA into the progeny from a cross of two kctd10 KI heterozygotes (F1) derived from two F0 individuals harboring two slightly different PoG-Ne insertions (designated as kctd10PoG-Ne-1 and kctd10PoG-Ne-2, respectively, differing in the 5’ and 3’ junction sequences) (Figure 2—figure supplement 2J and K and Supplementary file 4), 25.9% (74/286) of the embryos exhibited the kctd10 mutant phenotype, and the rest of the injected embryos and all the uninjected embryos developed normally (Figure 2C), indicating that the Po-cassettes were efficiently excised from the kctd10 PoG-Ne KI alleles. The genotyping and junction PCR results confirmed that all the embryos displaying developmental defects showed loxP recombination and overlapping peaks in the sequencing results of the junction PCR products (Figure 2D–2E), indicating that they were kctd10PoG-Ne-1/PoG-Ne-2 compound heterozygotes before Cre mRNA injection. Similar to the tbx5a KI allele, qRT-PCR experiments were also performed to evaluate the mRNA expression level of the edited kctd10 allele with or without Cre-induced Po-cassette excision, using primers K10qF and K10qR (Figure 2—figure supplement 2D, Supplementary file 7) in exons 1 and 2, respectively. As expected, the results showed no significant difference in the expression of the kctd10 locus between the wild-type and kctd10+/PoG-Ne-1 heterozygous embryos, while its expression was significantly reduced by approximately 50% in the kctd10+/PoG-Ne-1 embryos after Cre mRNA injection due to the efficient conversion of the kctd10PoG-Ne-1 alleles into the kctd10Ne-1 (Figure 2—figure supplement 2L). The kctd10 transcription level further decreased to about 0.3% in the kctd10Ne-1/Ne-1 homozygous mutant embryos obtained by Cre mRNA injection of the progeny from the outcross of kctd10PoG-Ne-1/ PoG-Ne-1 homozygous fish with a kctd10+/PoG-Ne-1 heterozygote, demonstrating highly efficient mutagenesis effect of the kctd10Ne-1 allele (Figure 2—figure supplement 2M).
In the initial experiments, we used a donor with a single SV40 PA termination signal sequence without the modified exon (mutExon) in its Ne-cassette. Using such donors, we were able to isolate stable integrations at the kctd10 locus and observed the correct expression pattern of tdGFP. However, after Cre mRNA injection, all the injected embryos resulting from the incrosses of such heterozygous F1 fish developed normally, although the green fluorescent signal was no longer detectable in most of the embryos, indicating the successful and efficient removal of the Po-cassette. These results suggest that the single SV40 PA signal might not be strong enough to efficiently stop target gene transcription; thus, either multiple copies of the transcription termination sequences are necessary, or the modified exon (mutExon) following the PA sequence might be indispensable for efficiently disrupting target gene expression in our strategy (data not shown).
Improving the dual-cassette donor strategy to achieve a geno-tagging effect
The above results demonstrated the unique advantages of our dual-cassette donor strategy for efficiently generating dual-function alleles and achieving CKO coupled with gene labeling in zebrafish via one-step targeted insertion. To further improve our strategy and achieve more complex applications of genome editing, we developed a novel technique by designing a conditional dual-fluorescence reporter donor (Figure 3A), an advanced version of our original PoNe donor, in which Cre-mediated excision of the Po-cassette could simultaneously trigger conditional knockout and the fluorescent reporter switch, resulting in allele tagging instead of gene tagging, so that different alleles (i.e., normal/positive alleles vs defective/negative alleles) can be labeled with different fluorescent proteins. This goal was achieved by adding a different fluorescent reporter in the original Ne-cassette following the sequence containing the intron (including the splice acceptor site) plus a partial exon immediately downstream of the intron target site, and linked by a 2A peptide, as in the Po-cassette. After the removal of the Po-cassette containing the first fluorescent reporter by the Cre/loxP system, the second fluorescent reporter in the Ne-cassette will be transcribed together with the preceding intron and exon sequences and spliced into the incomplete mRNA of the target gene, thus achieving both target gene mutation (due to the incomplete coding sequence) and labeling of the mutated allele, simultaneously leading to the destruction of the target gene’s function and the allele-tagging switch. Under this strategy, the functionally normal and defective alleles can be easily distinguished by using two different fluorescent reporters. Note that the wild-type exon sequence was used in the Ne-cassette in this case since we would like to maintain normal transcription and translation of the fusion mRNA, which contains the incomplete target gene cDNA followed by a 2A-linked complete sequence encoding the fluorescent reporter (Figure 3A). In contrast to the previous strategy, which was based on a single fluorescent reporter, we designated this advanced strategy geno-tagging, since it can label different alleles with different fluorescent reporters, and the genotype of each cell is easily distinguishable by its fluorescent signal.
Figure 3 with 1 supplement see all
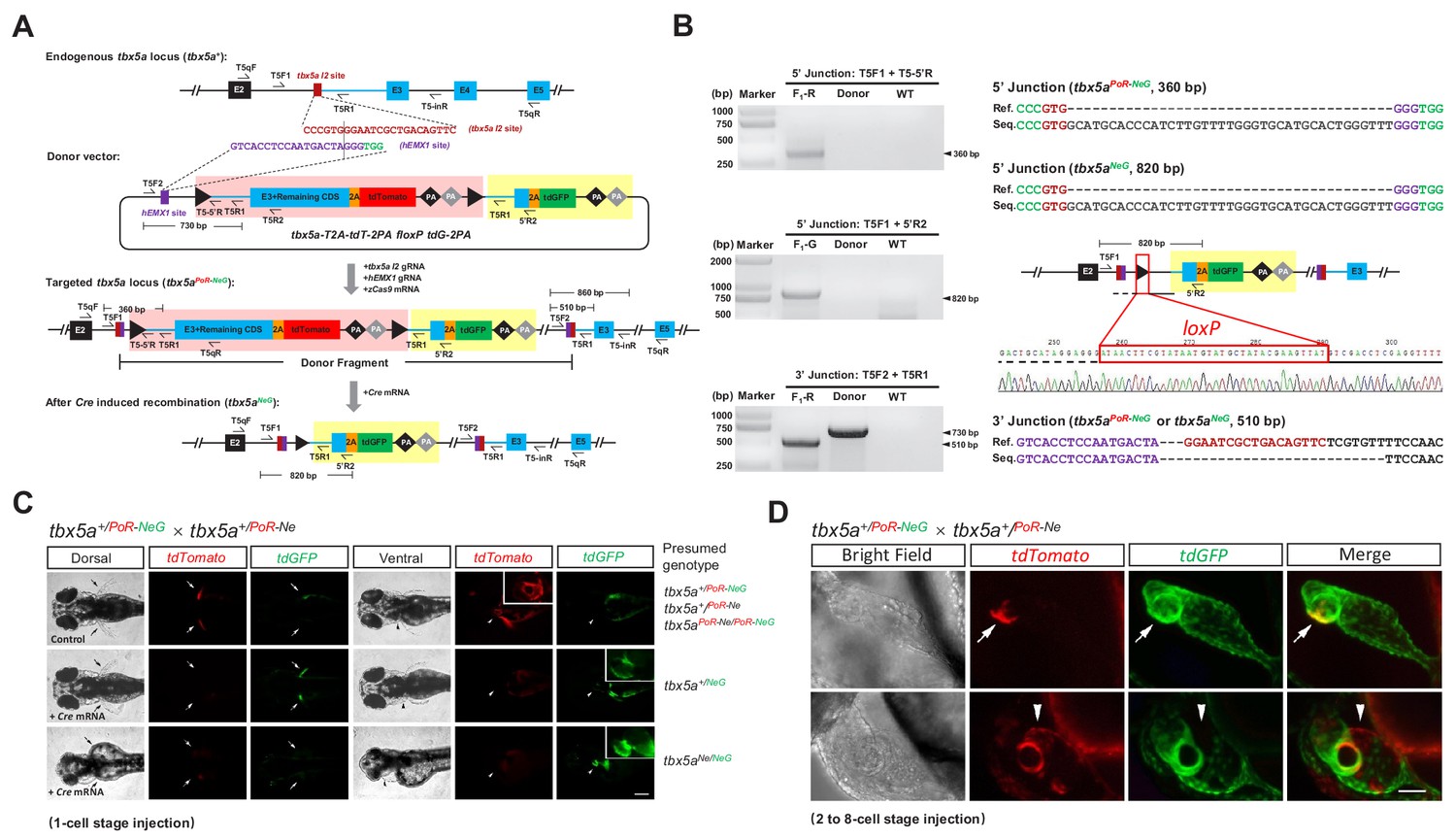
Generation of geno-tagging alleles by improving the dual-cassette donor strategy at the zebrafish tbx5a locus.
(A) Schematic diagram of the KI strategy for geno-tagging based on the tbx5a-T2A-tdT-2PA floxP tdG-2PA donor (or abbreviated as tbx5a PoR-NeG donor). Primers T5qF and T5qR are used for qRT-PCR in Figure 3—figure supplement 1E and F. (B) Results of junction PCR and direct sequencing to detect the tbx5a geno-tagging donor knockin and Cre-induced recombination events in 48 hpf embryos obtained from the cross in Figure 3—figure supplement 1B, that is the F1 embryos from the #42 positive F0 outcrossed with a wild-type zebrafish before or after Cre mRNA injection. Note that the sequences of both the T5F2 and T5R1 primers are also present in the donor vector, flanking the upstream loxP site (as shown in panel A); therefore, a 730 bp product could be amplified in the lane with the donor as the template. F1-R: an F1 embryo showing a red fluorescent signal (before Cre mRNA injection). F1-G: an F1 embryo showing a green fluorescent signal (after Cre mRNA injection). Donor: tbx5a PoR-NeG geno-tagging donor plasmid. WT: pooled genomic DNA of five wild-type embryos. (C) Phenotype analysis of the 72 hpf embryos from tbx5a+/PoR-NeG heterozygotes (derived from F0 #42 in Supplementary file 5) crossed with a tbx5a+/PoR-Ne heterozygote (derived from F0 #12 in Supplementary file 3) after the injection of Cre mRNA at the 1 cell stage. The upper panel represents an uninjected control embryo showing only a red fluorescent signal, whose genotype should be either tbx5a+/PoR-Ne, tbx5a+/PoR-NeG or tbx5a PoR-Ne/PoR-NeG. The middle panel represents a Cre mRNA-injected embryo showing normal development, whose genotype is expected to be tbx5a+/NeG. The lower panel represents a Cre mRNA-injected embryo showing a typical tbx5a mutant phenotype, including heart region defects and a lack of pectoral fins, whose genotype is expected to be tbx5aNe/NeG. Arrows indicate the pectoral fins, and arrowheads indicate the heart region. The boxed insets show a higher magnification of the corresponding heart region, for better comparison of heart morphology. Scale bar, 200 μm. (D) Z-stack confocal images of two representative 48 hpf embryos after the injection of Cre mRNA at the 2- to 8 cell stage from the same cross as in C. The white arrows indicate the colocalization of the tdGFP and tdTomato signals, and the white arrowheads indicate the mutually exclusive expression of the tdGFP and tdTomato signals. Scale bar, 50 μm.
Generation and evaluation of geno-tagging alleles at the zebrafish tbx5a locus
To test the feasibility of this geno-tagging strategy, we constructed a tbx5a geno-tagging donor based on the previous PoR-Ne donor, in which tdGFP was inserted into the negative cassette as the second fluorescent reporter followed by two additional polyA signals, and replaced the mutExon with the wild-type sequence (Figure 3A). This vector was referred to as the tbx5a PoR-NeG donor. In the generation of the previous tbx5aPoR-Ne KI allele, we showed that the preselection of F0 embryos according to the fluorescent signal largely ensured the successful identification of the germline transmission of the KI events. However, preselection of F0 embryos according to fluorescent signals is only feasible for certain genes and could be difficult to apply for many other genes, such as those showing late, low or little embryonic expression. To solve this problem, we aimed to establish a more universal method for the enrichment of the germline transmission of KI events. Therefore, during the generation of geno-tagging alleles at the tbx5a locus, we evaluated the potential correlation of KI events in germline transmission with the KI efficiency in F0 adult somatic tissues such as caudal fins. After the injection of the tbx5a geno-tagging donor (PoR-NeG donor) together with the zCas9 mRNA and corresponding gRNAs, red fluorescent signals could be detected in 16 of 124 normally developed F0 embryos. The ratio of embryos showing red fluorescent signals was comparable with that in the previous tbx5a PoR-Ne donor KI experiment (Supplementary file 2). However, we did not select the embryos according to their fluorescent signals and raised all the F0 embryos together to adulthood in this case. Then, junction PCR analysis was performed using genomic DNA isolated from caudal fin clips from 48 F0 adult fish, and the expected 5’ junction and/or 3’ junction amplicons were detected in six F0 adults (#1, #2, #9, #11, #24 and #42), indicating the successful integration of the donor in somatic cells (Figure 3—figure supplement 1A). F1 progeny were obtained from the outcrossing of five such F0 individually with wild-type zebrafish (F0 #24 failed to produce offspring), and germline transmission of the tbx5a PoR-NeG donor geno-tagging alleles was detected and confirmed in two F0 individuals (#2 and #42) (Figure 3B and Supplementary file 5). In contrast, no germline transmission events were detected in the F1 progeny from 20 F0 fish from the junction PCR-negative group. These results indicate a strong correlation between the germline transmission of KI events and adult somatic tissue integration of the donor, and provide an alternative efficient method for the enrichment of germline transmission events.
We chose positive founder #42 for the following experiments and designated the corresponding geno-tagging allele tbx5aPoR-NeG. We injected Cre mRNA into the F1 progeny from F0 #42 outcrossed with wild-type zebrafish and observed successful switching of the fluorescent signals (Figure 3—figure supplement 1B). Junction PCR and sequencing results further confirmed the correct KI events and excision of the PoR-cassette. Note that the 3’ junction does not change after Cre mRNA injection (Figure 3B). To evaluate the geno-tagging effect, we crossed a tbx5a+/PoR-NeG heterozygote with a previously identified tbx5a+/PoR-Ne heterozygote and divided the embryos into three groups: Group I was injected with 100 pg of Cre mRNA at the one-cell stage, Group II was injected with 25 pg of Cre mRNA in a single cell at the 2–8 cell stage, and Group III remained untreated as a control (Figure 3—figure supplement 1C). No tdTomato signal could be detected in any of the Group I embryos, indicating efficient switching of the fluorescent reporters in all the geno-tagging alleles. Among these embryos, 27.4% (29/106) showed the typical tbx5a mutant phenotype (Figure 3—figure supplement 1C), and all of these defective embryos were positive for the green fluorescent signal, as expected (Figure 3C). Mosaic expression of tdTomato and tdGFP could be observed in some embryos from Group II, indicating partial switching of the geno-tagging alleles (Figure 3D). In some embryos, double fluorescent signals could be observed in the same cell, suggesting that the genotype of these cells was most likely to be tbx5aPoR-Ne/NeG (Figure 3D). Among the Group II embryos, 11.6% (8/69) also showed obvious developmental defects in the heart and fins (Figure 3—figure supplement 1C), and not surprisingly, all of these defective embryos showed dominant or exclusive green fluorescent signals compared with the red fluorescent signal (Figure 3D). In contrast, all the embryos from Group III appeared normal during development (Figure 3—figure supplement 1C), and only tdTomato expression was observed, as expected (Figure 3C).
To verify that tissue and temporal control of Cre-induced recombination events can be achieved for this geno-tagging allele, we generated a transgenic line, Tg(cmlc2:zCreERT2-2A-ECFP), showing heart-specific expression of both ECFP and 4-hydroxyltamoxifen (4-HT)-inducible zebrafish codon-optimized CreERT2. After crossing this transgenic fish with tbx5aPoR-NeG/PoR-NeG adults, the embryos were collected and separated into two groups. Then 4-HT was added to one group from 5 hpf to 48 hpf. Green fluorescent signals were detected in the hearts of 56 hpf ECFP-positive embryos, indicating the successful switching of tdTomato to tdGFP expression in some cells (Figure 3—figure supplement 1D). In contrast, only red fluorescent signals were observed in the control group without chemical treatment (data not shown). qRT-PCR experiments were performed to evaluate the expression of the tbx5a geno-tagging allele with a similar strategy to that used for the previous tbx5aPoR-Ne allele. As expected, the expression of tbx5a in the tbx5a+/PoR-NeG heterozygous embryos was comparable with the wild-type, but reduced to ~50% after the Cre-induced excision of the Po-cassette (Figure 3—figure supplement 1E). Furthermore, mRNA quantification results of the tbx5aNeG/NeG homozygous mutant embryos derived from the Cre mRNA-injected tbx5aPoR-NeG/PoR-NeG embryos showed ~12.5 fold reduction of the tbx5a transcripts, confirmed the high mutagenesis efficiency of the tbx5aNeG allele (Figure 3—figure supplement 1F). These results together demonstrated that our geno-tagging technique successfully achieved switching of fluorescent reporters and CKO simultaneously, which provides unique opportunities for mosaic analysis and lineage tracing of cells with different genotypes.
Geno-tagging effect achieved for the zebrafish sox10 gene
To further prove the feasibility of our geno-tagging technique, we also applied this strategy at the sox10 locus (Figure 4—figure supplement 1A–1C), which is mainly expressed in neural crest cells in zebrafish (Dutton et al., 2001a; Dutton et al., 2001b). It has been reported that the efficiency of transcription termination correlates with the number of polyA signals (Ni et al., 2012). We began sox10 geno-tagging experiments before the work on tbx5a and kctd10 was completed. Because of concern that the double SV40/BGH polyA signals may be insufficient to achieve complete transcriptional termination (Ni et al., 2012), we constructed a vector with eight tandem repeats of SV40 pA (8xPA) (Figure 4—figure supplement 1D). In addition, since the hEMX1 gRNA targeting the previous donors needs to be purified by LiCl precipitation for efficient KI, we used another highly efficient gRNA target site known as lamGolden (Zu et al., 2016) for the linearization of the sox10 geno-tagging donor. However, similar to the case of hEMX1, the gRNA targeting the lamGolden site also needed to be purified with LiCl for successful knockin; otherwise, knockin events were scarcely detectable in F0 embryos (data not shown). We first coinjected the sox10 geno-tagging donor with the zCas9 system into one-cell-stage zebrafish embryos, after which approximately half of the embryos were injected again with 25 pg of Cre mRNA in a single cell at the 4 cell stage. As expected, we detected the expression of two different fluorescent reporters in otic vesicles as well as in cranial and trunk neural crest cells in some embryos (29/126) injected with Cre mRNA (Figure 4A). In some cells, colocalization of the tdGFP and tdTomato signals could be observed (Figure 4A), indicating that these cells were most likely to be sox10PoR-NeG/NeG heterozygous. In contrast, only the tdTomato signal was observed in the embryos injected without Cre mRNA (103/152) (Supplementary file 2). NHEJ-mediated knockin and Cre-induced recombination events were further confirmed by junction PCR and sequencing (Figure 4B). The 103 founder embryos showing red fluorescent signals were raised to adulthood, and germline screening was conducted outcrossed with wild-type zebrafish. Correct red fluorescent patterns in F1 embryos were observed in 10 out of 21 founders, and the proportion of mosaicism varied from 8.2% to 50.0% (Supplementary file 6). Positive F1 embryos from F0 #6 were raised as sox10+/PoR-NeG heterozygotes for further study. To test the functionality of the sox10PoR-NeG allele, the F2 progeny from an incross of the F1 heterozygotes were injected with 100 pg Cre mRNA per embryo at the one-cell stage. An efficient switch of the fluorescent signals from red (tdTomato) to green (tdGFP) was observed in all the injected fluorescent embryos (Figure 4—figure supplement 1E), indicating successful high-effciency deletion of the positive cassettes of the sox10PoR-NeG alleles in response to Cre recombinase. Furthermore, 24.6% (56/228) of the injected embryos exhibited extensive loss of pigmentation at 48 hpf, recapitulating the phenotype reported previously for the sox10 mutant colourless (Dutton et al., 2001a; Dutton et al., 2001b), and all these defective embryos exclusively showed green fluorescent signals, indicating that they were most likely homozygous for the sox10NeG allele (Figure 4—figure supplement 1E). Genotyping results confirmed that all the abnormal embryos were sox10PoR-NeG/PoR-NeG homozygotes before Cre injection and that all the normal embryos contained at least one wild-type sox10 allele (Figure 4—figure supplement 1F). qRT-PCR experiments were performed to further evaluate the expression of the sox10 geno-tagging allele. As expected, there was no significant difference in mRNA levels between the sox10 wild-type allele and the geno-tagging allele without Cre mRNA injection, while the expression of the sox10 geno-tagging allele was significantly decreased after Cre-induced excision of the Po-cassette (Figure 4—figure supplement 1G). The transcription level of sox10 in the sox10NeG/NeG homozygous mutant embryos obtained by Cre-induced Po-cassette excision in the sox10PoR-NeG/PoR-NeG embryos decreased by ~25 fold compared with wild-type embryos, further proved the strong mutagenesis effect of the sox10NeG allele (Figure 4—figure supplement 1H). Taken together, these results demonstrated the universality and high efficiency of our geno-tagging strategy.
Figure 4 with 1 supplement see all
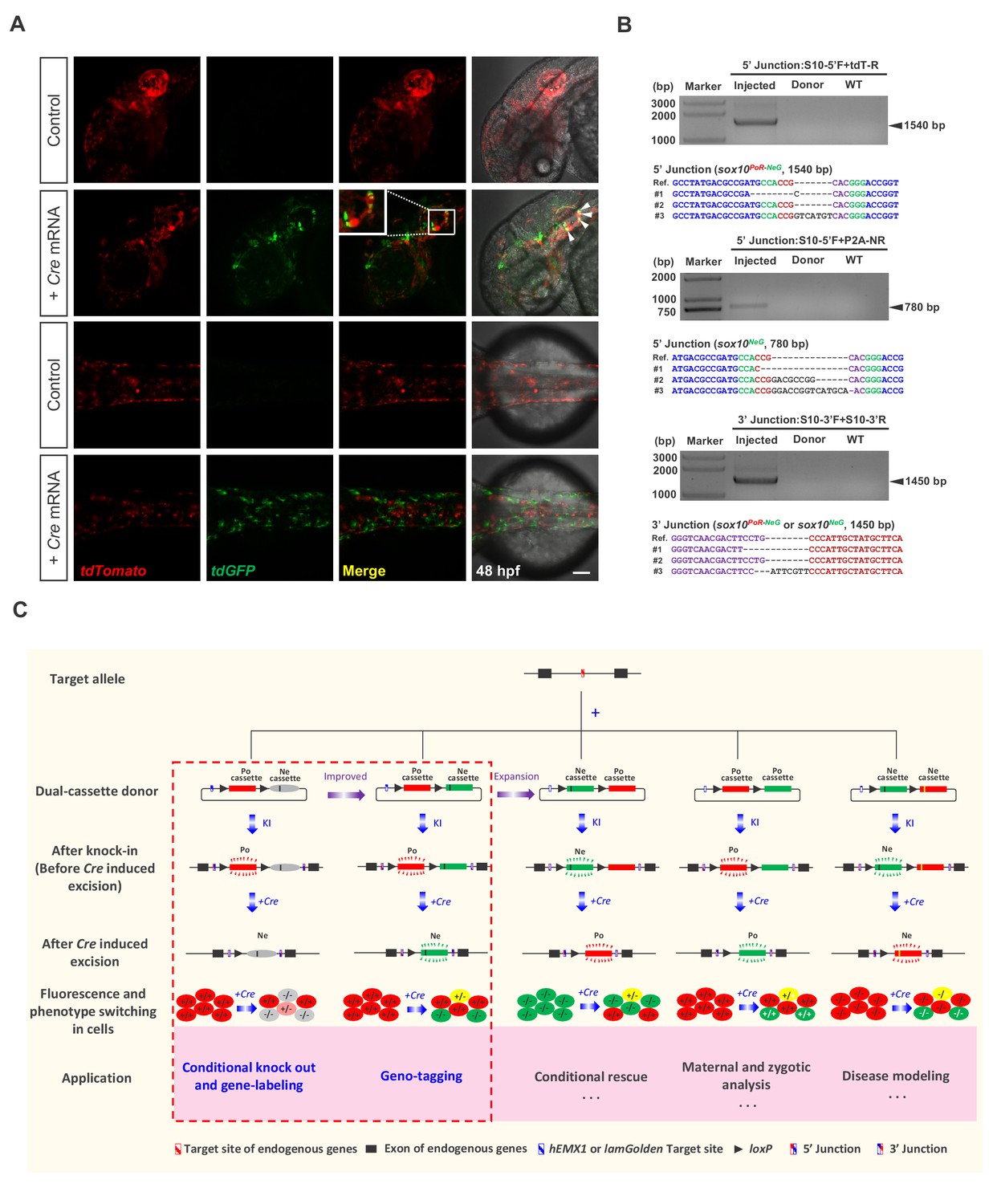
Generation of geno-tagging at the sox10 locus and summary of our dual-cassette donor KI strategy.
(A) Z-stack confocal images of 48 hpf zebrafish embryos after the injection of the sox10 geno-tagging donor knockin system at the one-cell stage (Control) followed by the further injection of 25 pg Cre mRNA into a single cell at the 4 cell stage (+Cre mRNA). White arrowheads indicate the colocalization of the tdGFP and tdTomato signals. Scale bar, 100 μm. (B) Junction PCR and clonal sequencing to detect the knockin and Cre-induced recombination events in the injected embryos showing mosaic double-fluorescence signals. Injected: Embryos injected with the donor, Cas9/gRNA system and Cre mRNA. Donor: sox10 geno-tagging donor plasmid. Uninjected: Uninjected embryos. (C) Summary of the applications and potential expansion of our dual-cassette donor KI strategy.
Discussion
In summary, we reported a highly efficient knockin strategy based on the special design of multipurpose dual-cassette PoNe donors and NHEJ-mediated targeted insertion, facilitated by the CRISPR/Cas system. Three levels of complex genome modifications could be achieved in zebrafish by using this method: (i) one-step generation of conditional knockout alleles through targeted insertion; (ii) simultaneous fluorescent labeling of CKO alleles to achieve a gene tagging effect as well as labeling of the cells expressing the target gene, through the incorporation of an in-frame fluorescent reporter gene (tdTomato or tdGFP) in the positive-cassette of the donor (the feasibility of this dual-function ‘two-in-one’ KI allele strategy was demonstrated at zebrafish tbx5a and kctd10 loci); (iii) differential labeling of two different alleles (normal vs defective) to achieve a geno-tagging effect via the differential labeling of the two cassettes (Po- vs Ne-cassette) in the donor with two different fluorescent reporters. This advanced strategy enables the conditional switching of the fluorescent reporters and, thus, the discrimination of cells with three different genotypes (homozygous normal, heterozygote, and homozygous mutant/defective). We successfully generated such conditional coupled geno-tagging alleles at the tbx5a and sox10 loci in zebrafish and demonstrated the fluorescent reporter switch in response to Cre recombinase treatment. As the activity of Cre recombinase can easily be manipulated (e.g., by using inducible CreERT2), genetic mosaicism of target tissues could be generated through conditional knockout. However, previously reported KI techniques usually cannot distinguish cells or tissues with different genotypes, which limits the in-depth study of gene functions. With simple modifications of our dual-cassette donor system, we have established a novel geno-tagging technique that allows conditional knockout and geno-tagging switching to occur simultaneously in the same allele. Fluorescence label switching, together with conditional gene knockout, not only allows the real-time labeling and discrimination of knockout cells or tissues from normal ones to trace and compare their fates in vivo but can even go further, potentially labeling different alleles with different fluorescent reporters. In this manner, one can easily determine and follow the genotype of each allele and each cell.
In addition, there is another potential advantage of the geno-tagging strategy due to the special design of the Ne-cassette in the donor. While the original single-fluorescent-reporter version of the dual-cassette donor might trigger genetic compensation since it contains a mutExon bearing a premature stop codon in its Ne-cassette, the mutant alleles produced through the geno-tagging KI strategy, which does not contain a premature stop codon to disrupt the target gene, are likely exempted from the disturbance caused by the genetic compensation response, thus simplifying as well as consolidating subsequent phenotype analyses. Therefore, we recommend the use of the geno-tagging donor for the generation of CKO alleles coupled with gene or allele labeling effects. Regarding polyA signals, in principle, more polyA signals seem to be more effective for efficient termination of transcription (Ni et al., 2012). Although 2PA was satisfactory for the generation of the tbx5aPoR-NeG geno-tagging allele in the present study, we adopted eight tandem repeats of SV40 polyA signals (8xPA) for stronger transcriptional termination for the sox10 locus. However, there are potential disadvantages of employing two 8xPA sequences, each containing eight tandem repeats of the ~160 bp polyA signal sequence, concerning stability. The repeats within each 8xPA sequence might be reduced during expansion and/or passaging to the next generations of the sox10PoR-NeG allele; on the other hand, the sequence flanked by the two 8xPA sequences (containing the downstream loxP and Ne-cassette) might be lost due to occasional recombination of the two 8xPA sequences, leading to the abolition of the CKO and fluorescence-switch effects. Practically, since different target genes may have very different expression patterns and regulation properties, for each particular target gene, we recommend beginning with the construction of the donor with 2PA, then testing its effect in founder embryos to determine whether this is sufficient to stop the expression of the second fluorescent reporter, and introducing more polyA signals when necessary.
Germline transmission is crucial for the successful establishment of stable KI transgenic fish lines. Interestingly, our results indicate a strong correlation between the efficient germline transmission and correct insertion of KI donors in somatic tissues. We observed high-efficiency germline recovery of the KI events after preselection for mosaic founders that acquired the correct donor insertions in somatic tissues by selecting either embryos on the basis of fluorescent reporter signals or adult fish on the basis of fin clip junction PCR. During the construction of the tbx5a PoR-Ne KI fish line, approximately 15% of F0 embryos showed the expected tdTomato expression after the injection of the donor together with the Cas9/gRNA system. More than 50% (5/9) of these fluorescent reporter-positive F0 fish successfully transmitted the KI alleles to their progeny. In contrast, no germline transmission events were identified in the 40 founders negative for the somatic cell expression of tdTomato. Similarly, approximately half (10/21) of the embryonic stage preselected founders showed germline transmission of the sox10 geno-tagging KI alleles. For the screening of tbx5a PoR-NeG geno-tagging KI fish, we preselected adult fish for somatic cell integration by junction PCR of fin clips and found that 6 out of 48 founders were positive for donor insertions in this somatic tissue. Among these founders, 2 out of 5 positive founders were also positive for germline transmission. Not surprisingly, we also screened for germline transmission in 20 founders negative by fin clip junction PCR and found that none of them transmitted a KI donor to their offspring. In summary, to achieve efficient recovery of germline-transmissible KI alleles, we strongly recommend the preselection of F0 embryos according to fluorescent signals or adults on the basis of fin clip junction PCR and then screening only these founders in germline transmission analysis.
Compared with traditional KI strategies, which generally adopt donors containing a single functional cassette, our dual-cassette donor design provides more flexibility to achieve complex genome manipulation, enabling a variety of applications. In addition to coupling conditional knockout with gene-labeling or geno-tagging, this dual-cassette donor system could theoretically be expanded to other applications after further modification (Figure 4C), such as to simulate precise genome modifications, achieve conditional gene rescue or differentiate and analyze maternal, zygotic and paternal gene expression and activity. For example, the Ne-cassette can be engineered to contain a coding sequence with a precise mutation (such as a disease-causing point mutation) so that the deletion of the Po-cassette could conditionally simulate the precise mutation. On the other hand, conditional gene rescue can be achieved by swapping the Po-cassette and Ne-cassette. This strategy may also be utilized to study cell-autonomous and cell-non-autonomous gene functions. Furthermore, our dual-cassette donor knockin strategy could theoretically be customized for use in other model systems, such as mice and Drosophila, or adapted for medical research, including the establishment of disease models for drug screening and mechanism dissection or the tracing of cancer cell transformation and metastasis to provide new insights into carcinogenesis and cancer therapy.
Nevertheless, our current dual-functional donor KI strategy has certain limitations and is open to further improvement. To simplify donor design and construction and reduce the size of the donor plasmids as much as possible, we have omitted most intron sequences downstream of the Cas9/gRNA target sites as well as the endogenous 3’-UTRs, and mainly relied on the downstream coding sequence from the cDNA to rescue the correct expression of the target gene. This approach may alter the expression pattern and/or regulation of the target gene, so we suggest careful evaluation of the reporter gene expression and comparison of the result with the endogenous gene characteristics before performing further experiments, such as conditional knockout or lineage tracing. Of course, one can easily include introns and 3’-UTRs in the donor design when necessary. On the other hand, since only one form of the downstream intronless cDNA sequence is inserted into the final KI allele to complement the disruption of endogenous genes, this approach may not be applicable to genes with multiple alternative splicing isoforms or genes regulated by RNA splicing. However, this limitation may be avoided by including all the downstream introns in the Po-cassette of the donor. In addition, this limitation sometimes might be considered a unique advantage, through which KI alleles with different alternative splicing isoforms could be purposely designed and generated separately, facilitating specific and detailed investigation as well as the comparison of different isoforms. In addition to the disadvantages in the design of the rescue sequences in the Po-cassette, another concern is that all the current KI alleles retain the plasmid backbone sequence at the insertion site in the genome, which might induce DNA methylation and silencing of transcription, as previously reported (Suzuki et al., 2016). In the future, we will try to improve the donor by introducing another recombination system, such as Flp/FRT, attB/attP, or Dre/Rox to flank the unwanted backbone and then delete it after successful knock in Lu et al. (2011); Wong et al. (2011); Park and Leach, 2013; Carney and Mosimann (2018), or by introducing two Cas/gRNA target sites in the donor, flanking the unwanted backbone to separate it from the functional cassettes during targeted insertion, or by using minicircle DNA to minimize the unwanted backbone sequence.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Danio rerio) | tbx5a | Gene: 30071 | ENSDARG00000024894 | |
| Gene (Danio rerio) | kctd10 | Gene: 406787 | ENSDARG00000017115 | |
| Gene (Danio rerio) | sox10 | Gene: 140616 | ENSDARG00000077467 | |
| Strain, strain background (Danio rerio) | Tübingen (TU) | Our lab | A commonly used wild-type zebrafish strain | |
| Strain, strain background (Danio rerio) | Tg(cmlc2:EGFP) | Tong et al., 2014 | PMID: 24430697 | |
| Strain, strain background (Danio rerio) | Tg(cmlc2:zCreERT2-T2A-ECFP) | This paper | A transgenic zebrafish expressing zebrafish codon-optimized CreERT2 and ECFP driven by the heart-specific cmlc2 promoter | |
| Recombinant DNA reagent | pMD18-T vector (plasmid) | TAKARA | Cat#: 6011 | |
| Recombinant DNA reagent | pMD19-T simple vector (plasmid) | TAKARA | Cat#: 3271 | |
| Recombinant DNA reagent | pminiTol2 (plasmid) | Balciunas et al., 2006 | PMID: 17096595 | |
| Recombinant DNA reagent | pGH-T7-zCas9 (plasmid) | Our lab | PMID: 24480746 | Liu et al., 2014 |
| Recombinant DNA reagent | pMD18T-tdGFP (plasmid) | Dr. Yulong Li | ||
| Recombinant DNA reagent | pUC19-scaffold (plasmid) | Dr. Jingwei Xiong | PMID: 23528705 | Chang et al., 2013 |
| Recombinant DNA reagent | bait otx2 CreERT2 (plasmid) | Dr. Michael Brand | PMID: 29435650 | Kesavan et al., 2018 |
| Recombinant DNA reagent | pX-T7-Cre (plasmid) | Dr. Yao Zu | ||
| Commercial assay or kit | Gibson assembly | NEB | E5510S | |
| Commercial assay or kit | mMessage mMachine T7 kit | Ambion | AM1344 | |
| Chemical compound, drug | 4-HT (Hydroxyltamoxifen) | Sigma | H6278-10MG | |
| Chemical compound, drug | Tricaine (ethyl 3-aminobenzoate methanesulfonate salt) | Sigma | E10521 | |
| Software, algorithm | AxioVision Rel.4.8 | Zeiss | RRID: SCR_002677 | |
| Software, algorithm | ZEN 2009 | Zeiss | RRID: SCR_013672 | |
| Other | TRIzol reagent | Invitrogen | Cat#: 10296028 | |
| Other | 5x All-In-One RT MasterMix | abm | G485 | |
| Other | EvaGreen 2x qRT-PCR Mastermix | abm | Mastermix-S |
Zebrafish husbandry
Request a detailed protocolAll the zebrafish used in this study were raised in groups and maintained at 28.5°C in the zebrafish facility of Peking University with a 14 hr/10 hr light/dark cycle. The wild-type strain used was Tübingen (TU). All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Peking University. The reference from IACUC of Peking University is LSC-ZhangB-2.
Donor construction
Request a detailed protocolTo construct the PoNe donor for the tbx5a locus targeting the second intron (i.e., the tbx5a-T2A-tdTomato floxP 2PA-mutExon donor) as an example, the hEMX1 target site and the first loxP site were linked by a 111 bp DNA fragment cloned from the pMD18-T vector (TAKARA, 6011) to avoid the disruption of the loxP sequence due to NHEJ-mediated DSB repair induced by the cleavage of the hEMX1 sequence, and this hEMX1-linker-loxP1st sequence was cloned into the pMD19-T simple vector (TAKARA, 3271). Then, the intron two sequence downstream of the tbx5a I2 target site (including the splice acceptor) of the tbx5a gene was cloned downstream of this first loxP site. The tbx5a partial CDS (referred to as ‘E3+Remaining CDS’ in Figure 1A) (except for exon 1, exon two and the stop codon) was fused with the T2A-tdTomato sequence. Finally, the second loxP site, SV40 PA (polyA signal), BGH PA and a modified exon 3 of tbx5a that contained an in-frame stop codon and included its upstream splice acceptor sequence, were successively cloned downstream of the tdTomato coding sequence. The tbx5a-T2A-tdTomato floxP 2PA-mutExon donor (abbreviated as tbx5a PoR-Ne donor) was used as the basic vector to construct the PoNe donor for the kctd10 gene and for the geno-tagging donors for the tbx5a and sox10 loci. To construct the tbx5a geno-tagging PoR-NeG donor as an example, we improved the initial tbx5a PoR-Ne donor by cloning the sequence around the splice acceptor of the second intron of the tbx5a gene and fused it in-frame with a tdGFP coding sequence, which was separated by a 2A peptide sequence, and we subsequently inserted the whole sequence between the second loxP site and the 2PA; then, another copy of 2PA was inserted upstream of the second loxP site, and the mutExon was removed (Figure 3A).
Preparation of the gRNAs, zCas9 mRNA and Cre mRNA
Request a detailed protocolThe gRNAs were designed with the CasOT program (http://casot.cbi.pku.edu.cn/) (Xiao et al., 2014). Forward oligonucleotides containing a T7 promoter, gRNA target site and partial gRNA scaffold sequences were designed for gRNA template synthesis through PCR amplification by using the pUC19-scaffold as the template (Chang et al., 2013), together with a universal reverse primer (5’-AAAAAAAGCACCGACTCGGTGCCAC-3’). Then, gRNAs were synthesized by in vitro transcription with T7 RNA polymerase (TAKARA) and purified by LiCl or ethanol precipitation. The gRNA target sequences are shown in Supplementary file 1. The zebrafish codon-optimized Cas9 expression vector pGH-T7-zCas9 was linearized by XbaI and used as the template for generating zCas9 mRNA through in vitro transcription using the mMessage mMachine T7 kit (Ambion) (Liu et al., 2014). Then, the zCas9 mRNA was purified by LiCl precipitation. The Cre expression vector pX-T7-Cre was linearized with XbaI digestion and used as the template for synthesizing Cre mRNA through in vitro transcription by using the mMessage mMachine T7 kit (Ambion). Then, the Cre mRNA was purified by LiCl precipitation.
Microinjection of zebrafish embryos
Request a detailed protocolThe zCas9 mRNA and gRNAs were coinjected into one-cell-stage zebrafish embryos with or without donor plasmids. Each embryo was injected with 1–2 nL of a solution containing 700 ng/μL zCas9 mRNA, 100 ng/μL gRNA and 15 ng/μL donor plasmid; 100 pg or 25 pg of Cre mRNA was injected into 1 cell stage or a single cell of 4 cell stage zebrafish embryos, respectively.
Restriction endonuclease assay to evaluate the indel efficiency of the Cas9/gRNA system
Request a detailed protocolFor indel efficiency evaluation, one-cell stage zebrafish embryos were injected with the zCas9 mRNA and the corresponding gRNA. To extract genomic DNA, at 24 hpf, fifteen injected embryos were divided into three groups and lysed with 50 mM NaOH solution (10 μL each embryo) at 95°C for 15 min, followed by neutralization with 1 M Tris-HCl (pH = 8.0, 1 μL of each embryo). Then, 1 μL of the genomic DNA extract was used as the template to amplify the target site regions with the corresponding primers (Supplementary file 7). The PCR products were subsequently digested by using the corresponding restriction endonucleases and analyzed by agarose gel electrophoresis and Sanger sequencing after TA cloning.
Junction PCR and sequencing
Request a detailed protocolGenomic DNA was extracted with lysis buffer (10 mM Tris-HCl, pH 8.2, 200 mM NaCl, 5% SDS solution, 200 μg/mL proteinase K and 10 mM EDTA) from either individual or pools of 72 hpf zebrafish embryos and then used to PCR amplify the 5’ and 3’ junction fragments of target genes or the region flanking the loxP site using the appropriate primers (Supplementary file 7). The PCR products were either directly sent for sequencing (F1 and F2 embryos) or cloned into pMD18-T (TAKARA) for clonal sequencing (F0 embryos).
Generation of transgenic fish and chemical treatment
View detailed protocolTo construct the plasmid for the generation of Tg(cmlc2:zCreERT2-T2A-ECFP) zebrafish, the zebrafish cmlc2 (myl7) promoter sequence and the SV40 polyadenylation site were cloned and ligated into the pminiTol2 vector (Balciunas et al., 2006) by the Gibson assembly method according to the manufacturer’s instructions (NEB). Then, the coding regions of both zebrafish codon-optimized CreERT2 from the bait otx2 CreERT2 (Kesavan et al., 2018) and ECFP were cloned and ligated into this Tol2 vector via the Gibson assembly method. Approximately 35 pg of the plasmid and 80 pg of Tol2 transposase mRNA were coinjected into one-cell-stage fertilized zebrafish eggs to generate F0 transgenic fish. F0 adults were screened for germline transmission of zCreERT2-T2A-ECFP expression vector integration by the observation of correct ECFP fluorescent signals in the progeny. To activate CreERT2, zebrafish embryos were treated with 5 μM 4-HT (Sigma) from 5 hpf to 48 hpf and the embryo medium was changed once at 24 hpf.
Imaging and processing
Request a detailed protocolFor general imaging, zebrafish embryos were anesthetized with 0.02% tricaine (ethyl 3-aminobenzoate methanesulfonate salt, 1 M Tris-HCl, pH 9.0), then placed in 3% methylcellulose (Sigma), and imaged under a microscope (AXIO Imager Z1; Zeiss) equipped with AxioCam MRm (Zeiss). Confocal imaging was performed using an LSM 710 confocal microscope (Zeiss) with a 20X water immersion objective. Z-stack images were acquired with a 5- to 7 μm resolution. The images were processed with AxioVision Rel.4.8 and ZEN 2009 imaging software.
qRT-PCR analysis
Request a detailed protocolRNA was isolated using TRIzol reagent (Invitrogen) according to the standard protocol from 10 randomly picked 72 hpf sibling embryos with or without the injection of Cre mRNA. All the embryos were collected from crosses of the zebrafish carrying the corresponding homozygous KI alleles with either a wild-type or a heterozygote zebrafish. Embryos of different genotypes were distinguished and grouped by fluorescent signals and corresponding phenotypes. At least 500 ng of total RNA was employed for reverse transcription using the 5x All-In-One RT MasterMix (Applied Biological Materials). All reactions were performed in at least technical triplicates, and the results represent biological triplicates. Each primer pair was designed to span the KI donor integration site, and gapdh was used as the internal reference gene. qRT-PCR primer information is listed in Supplementary file 7. qRT-PCR experiments were performed in a Roche LightCycler 96 instrument or BioRad CFX96 using EvaGreen 2x qRT-PCR Mastermix (Applied Biological Materials) and a 500 nM final primer concentration. Fold changes were calculated using the 2−ΔΔCt method, and p<0.05 was accepted as statistically significant.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
TAL effectors--pathogen strategies and plant resistance engineeringNew Phytologist 204:823–832.https://doi.org/10.1111/nph.13015
-
DNA-binding-domain fusions enhance the targeting range and precision of Cas9Nature Methods 12:1150–1156.https://doi.org/10.1038/nmeth.3624
-
Switch and trace: recombinase genetics in zebrafishTrends in Genetics 34:362–378.https://doi.org/10.1016/j.tig.2018.01.004
-
Non-Homologous end joining plays a key role in transgene concatemer formation in transgenic zebrafish embryosInternational Journal of Biological Sciences 6:756–768.https://doi.org/10.7150/ijbs.6.756
-
Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fatesDevelopment 128:4113–4125.
-
The heartstrings mutation in zebrafish causes heart/fin Tbx5 deficiency syndromeDevelopment 129:4635–4645.
-
Precise Editing of the Zebrafish Genome Made Simple and EfficientDevelopmental Cell 36:654–667.https://doi.org/10.1016/j.devcel.2016.02.015
-
Heritable gene targeting in zebrafish using customized TALENsNature Biotechnology 29:699–700.https://doi.org/10.1038/nbt.1939
-
Reverse Genetic Approaches in ZebrafishJournal of Genetics and Genomics 39:421–433.https://doi.org/10.1016/j.jgg.2012.07.004
-
Efficient genome editing in zebrafish using a CRISPR-Cas systemNature Biotechnology 31:227–229.https://doi.org/10.1038/nbt.2501
-
Targeted knock-in of CreER T2 in zebrafish using CRISPR/Cas9Cell and Tissue Research 372:41–50.https://doi.org/10.1007/s00441-018-2798-x
-
Efficient gene targeting in zebrafish mediated by a zebrafish-codon-optimized cas9 and evaluation of off-targeting effectJournal of Genetics and Genomics 41:43–46.https://doi.org/10.1016/j.jgg.2013.11.004
-
PhiC31 integrase induces efficient site-specific excision in zebrafishTransgenic Research 20:183–189.https://doi.org/10.1007/s11248-010-9394-5
-
A TALE nuclease architecture for efficient genome editingNature Biotechnology 29:143–148.https://doi.org/10.1038/nbt.1755
-
Targeted gene disruption in somatic zebrafish cells using engineered TALENsNature Biotechnology 29:697–698.https://doi.org/10.1038/nbt.1934
-
Knockout rats generated by embryo microinjection of TALENsNature Biotechnology 29:695–696.https://doi.org/10.1038/nbt.1940
-
A versatile gene trap to visualize and interrogate the function of the vertebrate proteomeGenes & Development 25:2306–2320.https://doi.org/10.1101/gad.174037.111
-
FLPe functions in zebrafish embryosTransgenic Research 20:409–415.https://doi.org/10.1007/s11248-010-9410-9
-
CasOT: a genome-wide Cas9/gRNA off-target searching toolBioinformatics 30:1180–1182.https://doi.org/10.1093/bioinformatics/btt764
-
Biallelic editing of a lamprey genome using the CRISPR/Cas9 systemScientific Reports 6:23496.https://doi.org/10.1038/srep23496
Article and author information
Author details
Funding
National Key Research and Development Program of China (2018YFA0801000)
- Bo Zhang
National Key Research and Development Program of China (2016YFA0100500)
- Bo Zhang
National Key Basic Research Program of China (2015CB942803)
- Bo Zhang
National Natural Science Foundation of China (31671500)
- Bo Zhang
National Natural Science Foundation of China (31871458)
- Bo Zhang
National Natural Science Foundation of China (81371264)
- Bo Zhang
Peking University (Qidong-SLS Innovation Fund)
- Bo Zhang
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Yulong Li for providing us with the pMD18T-tdGFP vector. We thank Jingwei Xiong for providing us with the pUC19-scaffold plasmid. We thank Yao Zu for providing us with the pX-T7-Cre vector. We thank Dr. Michael Brand for kindly providing the bait otx2 CreERT2 plasmid. We thank Christopher J Krueger for language editing, Yuying Gao and Yan Shen for lab management, and Yingdi Jia and Jingliang Chen for zebrafish husbandry. We also thank Xiaochen Li and Xuemei Hao from the Core Facilities at the School of Life Sciences of Peking University in Beijing, China, for assistance with confocal microscopy. This work was partially supported by grants from the National Key Research and Development Program of China [2018YFA0801000, 2016YFA0100500]; the National Key Basic Research Program of China [2015CB942803]; the National Natural Science Foundation of China (NSFC) [31671500, 31871458 and 81371264]; and the PKU Qidong-SLS Innovation Fund.
Ethics
Animal experimentation: All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) of Peking University. The reference from IACUC of Peking University is LSC-ZhangB-2.
Version history
- Received: April 30, 2019
- Accepted: October 30, 2019
- Accepted Manuscript published: October 30, 2019 (version 1)
- Version of Record published: November 11, 2019 (version 2)
Copyright
© 2019, Li et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 8,207
- views
-
- 1,131
- downloads
-
- 30
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
One-step efficient generation of dual-function conditional knockout and geno-tagging alleles in zebrafish
eLife 8:e48081.
https://doi.org/10.7554/eLife.48081
Further reading
-
- Genetics and Genomics
- Immunology and Inflammation
Transposable elements (TEs) are repetitive sequences representing ~45% of the human and mouse genomes and are highly expressed by medullary thymic epithelial cells (mTECs). In this study, we investigated the role of TEs on T-cell development in the thymus. We performed multiomic analyses of TEs in human and mouse thymic cells to elucidate their role in T-cell development. We report that TE expression in the human thymus is high and shows extensive age- and cell lineage-related variations. TE expression correlates with multiple transcription factors in all cell types of the human thymus. Two cell types express particularly broad TE repertoires: mTECs and plasmacytoid dendritic cells (pDCs). In mTECs, transcriptomic data suggest that TEs interact with transcription factors essential for mTEC development and function (e.g., PAX1 and REL), and immunopeptidomic data showed that TEs generate MHC-I-associated peptides implicated in thymocyte education. Notably, AIRE, FEZF2, and CHD4 regulate small yet non-redundant sets of TEs in murine mTECs. Human thymic pDCs homogenously express large numbers of TEs that likely form dsRNA, which can activate innate immune receptors, potentially explaining why thymic pDCs constitutively secrete IFN ɑ/β. This study highlights the diversity of interactions between TEs and the adaptive immune system. TEs are genetic parasites, and the two thymic cell types most affected by TEs (mTEcs and pDCs) are essential to establishing central T-cell tolerance. Therefore, we propose that orchestrating TE expression in thymic cells is critical to prevent autoimmunity in vertebrates.
-
- Genetics and Genomics
The ‘diabetic bone paradox’ suggested that type 2 diabetes (T2D) patients would have higher areal bone mineral density (BMD) but higher fracture risk than individuals without T2D. In this study, we found that the genetically predicted T2D was associated with higher BMD and lower risk of fracture in both weighted genetic risk score (wGRS) and two-sample Mendelian randomization (MR) analyses. We also identified ten genomic loci shared between T2D and fracture, with the top signal at SNP rs4580892 in the intron of gene RSPO3. And the higher expression in adipose subcutaneous and higher protein level in plasma of RSPO3 were associated with increased risk of T2D, but decreased risk of fracture. In the prospective study, T2D was observed to be associated with higher risk of fracture, but BMI mediated 30.2% of the protective effect. However, when stratified by the T2D-related risk factors for fracture, we observed that the effect of T2D on the risk of fracture decreased when the number of T2D-related risk factors decreased, and the association became non-significant if the T2D patients carried none of the risk factors. In conclusion, the genetically determined T2D might not be associated with higher risk of fracture. And the shared genetic architecture between T2D and fracture suggested a top signal around RSPO3 gene. The observed effect size of T2D on fracture risk decreased if the T2D-related risk factors could be eliminated. Therefore, it is important to manage the complications of T2D to prevent the risk of fracture.




{kind=link}
{kind=link}
{kind=link}
{kind=link}