Efficient single-copy HDR by 5’ modified long dsDNA donors
- Heidelberg University, Germany
Abstract
CRISPR/Cas9 efficiently induces targeted mutations via non-homologous-end-joining but for genome editing, precise, homology-directed repair (HDR) of endogenous DNA stretches is a prerequisite. To favor HDR, many approaches interfere with the repair machinery or manipulate Cas9 itself. Using Medaka we show that the modification of 5’ ends of long dsDNA donors strongly enhances HDR, favors efficient single-copy integration by retaining a monomeric donor conformation thus facilitating successful gene replacement or tagging.
https://doi.org/10.7554/eLife.39468.001eLife digest
CRISPR/Cas9 technology has revolutionized the ability of researchers to edit the DNA of any organism whose genome has already been sequenced. In the editing process, a section of RNA acts as a guide to match up to the location of the target DNA. The enzyme Cas9 then makes a cut in both strands of the DNA at this specific location. New segments of DNA can be introduced to the cell, incorporated into DNA ‘templates’. The cell uses the template to help it to heal the double-strand break, and in doing so adds the new DNA segment into the organism’s genome.
A drawback of CRISPR/Cas9 is that it often introduces multiple copies of the new DNA segment into the genome because the templates can bind to each other before being pasted into place. In addition, some parts of the new DNA segment can be missed off during the editing process. However, most applications of CRISPR/Cas9 – for example, to replace a defective gene with a working version – require exactly one whole copy of the desired DNA to be inserted into the genome.
In order to achieve more accurate CRISPR/Cas9 genome editing, Gutierrez-Triana, Tavhelidse, Thumberger et al. attached additional molecules to the end of the DNA template to shield the DNA from mistakes during editing. The modified template was used to couple a stem cell gene to a reporter that produces a green fluorescent protein into the genome of fish embryos. The fluorescent proteins made it easy to identify when the coupling was successful.
Gutierrez-Triana et al. found that the additional molecules prevented multiple templates from joining together end to end, and ensured the full DNA segment was inserted into the genome. Furthermore, the results of the experiments showed that only one copy of the template was inserted into the DNA of the fish. In the future, the new template will allow DNA to be edited in a more controlled way both in basic research and in therapeutic applications.
https://doi.org/10.7554/eLife.39468.002Introduction
The implementation of the bacterial CRISPR/Cas9 system in eukaryotes has triggered a quantum leap in targeted genome editing in literally any organism with a sequenced genome or targeting region (Cong et al., 2013; Jinek et al., 2012; Mali et al., 2013). Site-specific double-strand breaks (DSBs) are catalyzed by the Cas9 enzyme guided by a single RNA with a short complementary region to the target site. In response, the endogenous non-homologous end joining (NHEJ) DNA repair machinery seals the DSB. Since perfect repair will restore the CRISPR/Cas9 target site, mutations introduced by imprecise NHEJ are selected for.
To acquire precise genome editing the initial DSB should be fixed via the homology-directed repair (HDR) mechanism which is preferentially active during the late S/G2 phase of the cell cycle (Hustedt and Durocher, 2017). Donor DNA templates with flanking regions homologous to the target locus are used to introduce specific mutations or particular DNA sequences. Injected (linear) dsDNA rapidly multimerizes (Winkler et al., 1991), which likely also happens in CRISPR/Cas9 based approaches. Additionally, the high activity of NHEJ re-ligating CRISPR/Cas9 mediated DSBs can multimerize injected (linear) dsDNA donor templates. This poses a problem since consequently, the precise HDR-mediated recombination of single-copy donor templates is rather rare.
Several strategies have been followed to avoid NHEJ and/or favor HDR. NHEJ was interfered with by pharmacological inhibition of DNA ligase IV (Maruyama et al., 2015). Conversely, HDR was meant to be favored by fusing the HDR-mediating yeast protein Rad52 to Cas9 (Wang et al., 2017). Similarly, removal of Cas9 in the G1/S phase (Gutschner et al., 2016) by linking it to the N-terminal region of the DNA replication inhibitor Geminin, should restrict the introduction of double-strand cuts to the G2 phase, when HDR is most prominently occurring (Hustedt and Durocher, 2017). Those approaches improved HDR-mediated integration of the homology flanks, while they did not tackle reported integration of multimers (Auer et al., 2014) arising after injection of dsDNA templates such as plasmids or PCR products (Winkler et al., 1991).
Results
To enhance HDR without interfering with the endogenous DNA repair machinery, we aimed at establishing DNA donor templates that escape multimerization or NHEJ events. We thus blocked both 5´ends of PCR amplified long dsDNA donor cassettes using ‘bulky’ moieties like Biotin, Amino-dT (A-dT) and carbon spacers (e. g. Spacer C3, SpC3). This should shield the DNA donor from multimerization and integration via NHEJ, thus favoring precise and efficient single-copy integration via HDR (Figure 1A).
Figure 1 with 1 supplement see all
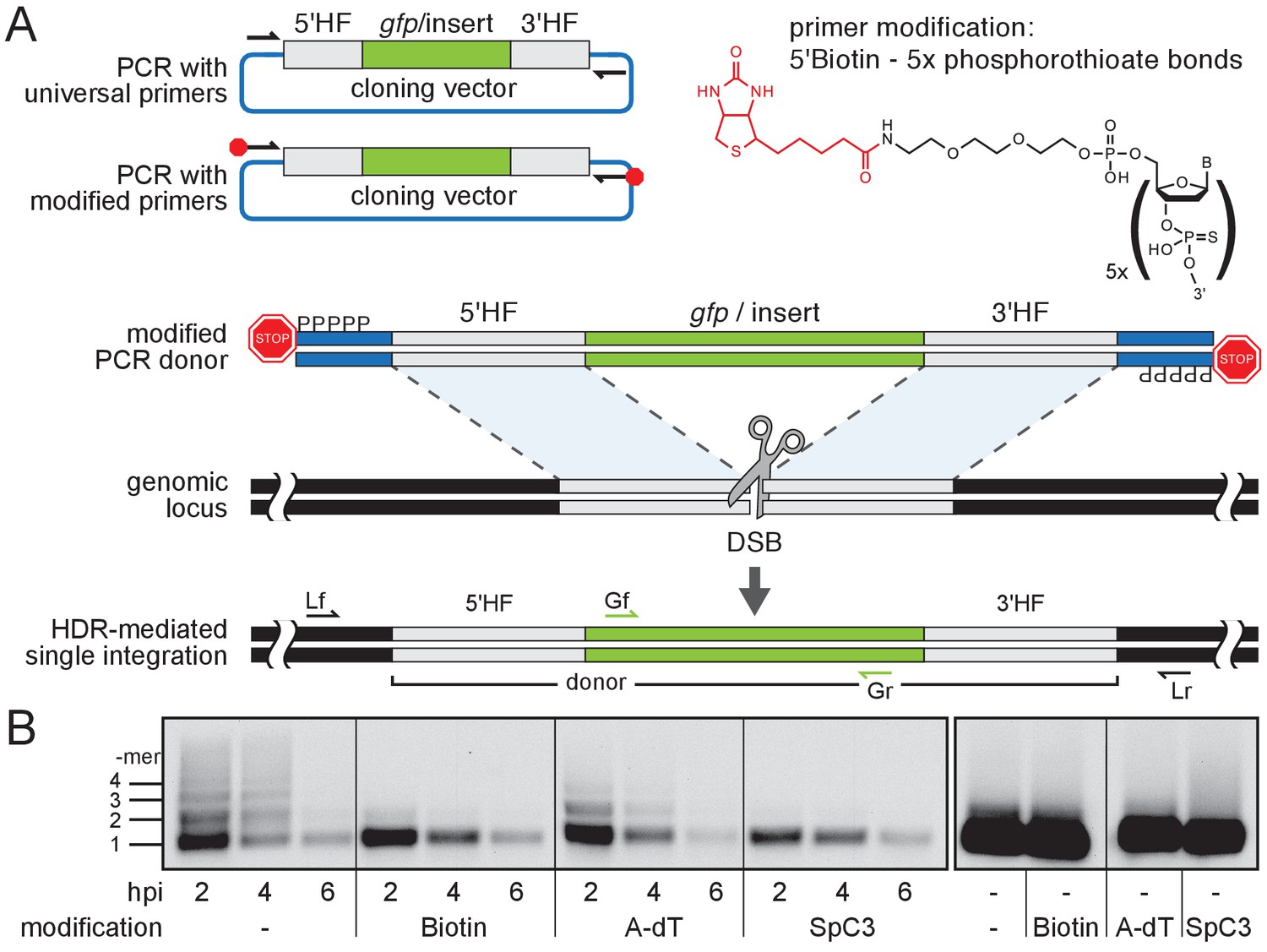
Modification of 5’ ends of long dsDNA fragments prevents in vivo multimerization.
(A) Schematic representation of long dsDNA donor cassette PCR amplification with universal primers (black arrows) complementary to the cloning vector backbone outside of the assembled donor cassette (e. g. gfp with homology flanks). Bulky moieties like Biotin at the 5’ ends of both modified primers (red octagon) prevent multimerization/NHEJ of dsDNA, providing optimal conditions for HDR-mediated single-copy integration following CRISPR/Cas9-introduced DSB at the target locus (grey scissors). Representation of locus (Lf/Lr) and internal gfp (Gf/Gr) primers for PCR genotyping of putative HDR-mediated gfp integration events. (B) Southern blot analysis reveals the monomeric state of injected dsDNA fragments in vivo for 5’ modification with Biotin or Spacer C3. Long dsDNAs generated with control unmodified primers or Amino-dT attached primers multimerize as indicated by a high molecular weight ladder apparent already within two hours post-injection (hpi). Note: 5’ moieties did not enhance the stability of injected DNA.
We first addressed the impact of the donor 5’ modification on the formation of multimers in vivo. We injected modified and unmodified dsDNA donors into one-cell stage medaka (Oryzias latipes) embryos and analyzed the conformational state of the injected material during zygotic development. dsDNA donors were generated by PCR employing 5’ modified and non-modified primers respectively, and by additionally providing traces of DIG-dUTP for labeling of the resulting PCR product. The conformation of the injected dsDNA donors was assayed in the extracted total DNA after 2, 4 and 6 hr post-injection, respectively. The DNA was size fractionated by gel electrophoresis and donor DNA conformation was detected after blotting the DNA to a nylon membrane by anti-DIG antibodies (Figure 1B). In unmodified DIG-labelled control donors, we uncovered multimerization already at 2 hr post-injection as evident by a ladder of labeled donor DNA representing different copy number multimers (Figure 1B). In contrast, Biotin and SpC3 modification of DIG-labelled donors prevented multimerization within six hours post-injection (Figure 1B). dsDNA donors established by A-dT modified primes, however, multimerized and produced similar results as unmodified DIG-labelled dsDNA donors (Figure 1B). Our results reveal that Biotin and SpC3 5’ modifications efficiently prevent donor multimerization in vivo. While strongly blocking multimerization, the 5’ modification of dsDNA did not apparently enhance the stability of the resulting dsDNA (compare modified and unmodified donors over time, Figure 1B).
To test whether 5’ modification not only reduces the degree of multimerization but also impacts on single-copy HDR-mediated integration of long dsDNA donors, we designed gfp containing donor cassettes for an immediate visual readout. We generated gfp in-frame fusion donors for four different genes: the retinal homeobox transcription factors rx2 and rx1 (Reinhardt et al., 2015), the non-muscle cytoskeletal beta-actin (actb) (Stemmer et al., 2015) and the DNA methyltransferase 1 (dnmt1). Donor cassettes contained the respective 5’ homology flank (HF) (462 bp for rx2, 430 bp for rx1, 429 bp for actb, 402 bp for dnmt1), followed by the in-frame gfp coding sequence, a flexible linker in case of rx2, rx1 and dnmt1, and the corresponding 3’ HF (414 bp for rx2, 508 bp for rx1, 368 bp for actb, 405 bp for dnmt1; schematic representation in Figure 1A, Figure 1—figure supplement 1 for detailed donor design). To amplify the long dsDNA donors we employed a pair of universal primers (5’ modified or unmodified as control) complementary to the backbone of the cloning vectors (pDestSC-ATG [Kirchmaier et al., 2013] or pCS2+ [Rupp et al., 1994]) encompassing the entire assembled donor cassette (Figure 1—figure supplement 1).
Modified or unmodified long dsDNA donors were subsequently co-injected together with Cas9 mRNA and the respective locus-specific sgRNA into medaka one-cell stage zygotes (Loosli et al., 1999; Rembold et al., 2006). For all four loci (and all 5’ modifications) tested we observed efficient targeting as apparent by the GFP expression within the expected expression domain (Figure 2A, Supplementary file 1, Figure 2—figure supplement 1).
Figure 2 with 3 supplements see all
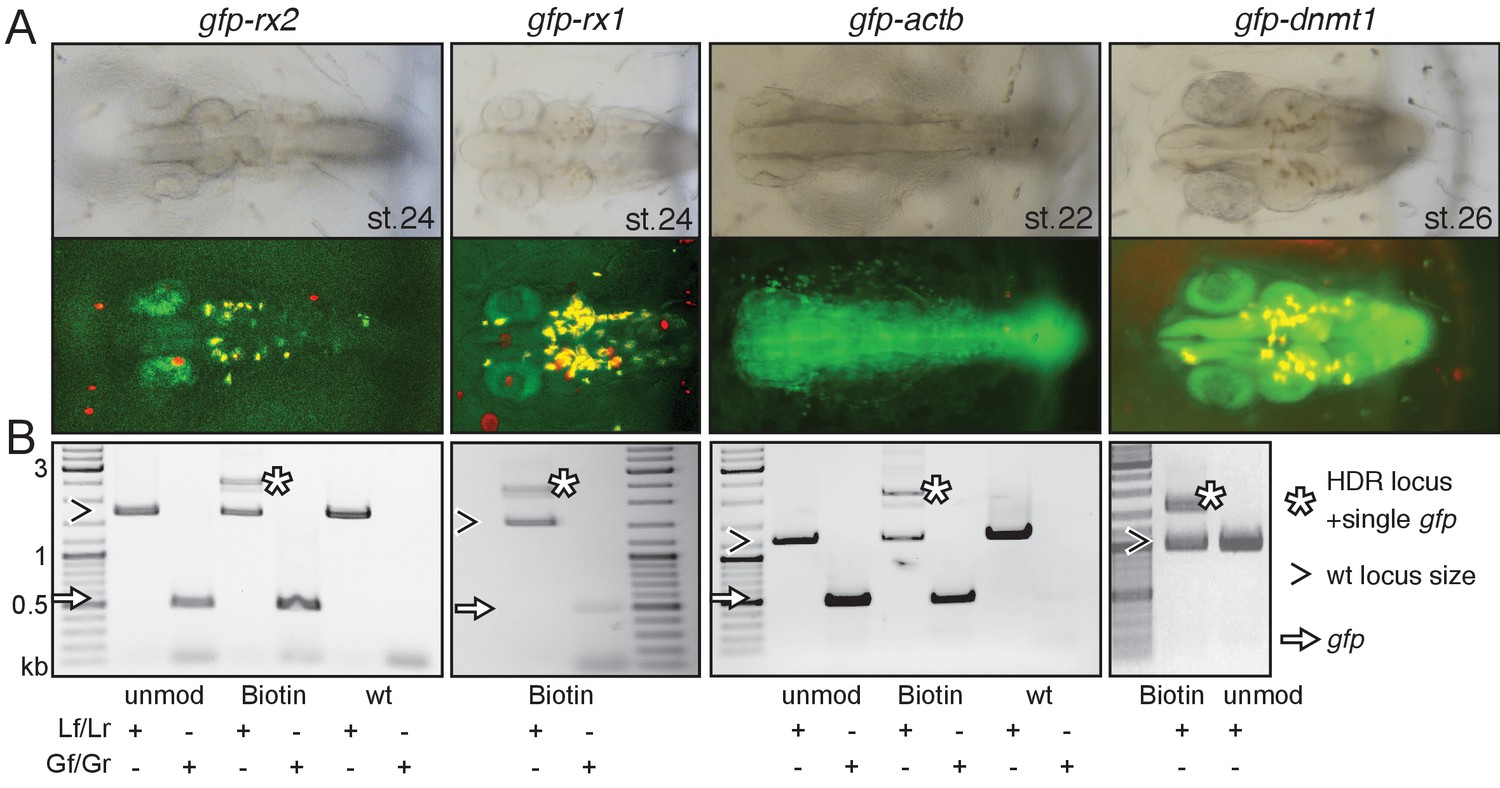
Modification of 5’ ends of long dsDNA fragments promotes HDR-mediated single-copy integration.
(A) GFP expression in the respective expression domain after HDR-mediated integration of modified dsDNA gfp donor cassettes into rx2, rx1, actb and dnmt1 ORFs in the injected generation. (B) Individual embryo PCR genotyping highlights efficient HDR-mediated single-copy integration of 5’Biotin modified long dsDNA donors, but not unmodified donor cassettes. Locus PCR reveals band size indicative of single-copy gfp integration (asterisk) besides alleles without gfp integration (open arrowhead). Amplification of gfp donor (white arrow) for control.
The survival rates of embryos injected with Biotin and SpC3 5’ modified dsDNA donors did not differ significantly from embryos injected with the unmodified dsDNA control donors (Supplementary file 1, Figure 2—figure supplement 1). In contrast, the injection of the A-dT 5’ modified dsDNA donors resulted in high embryonic lethality (Supplementary file 1, Figure 2—figure supplement 1).
We next analyzed the frequency of single-copy HDR events following a careful, limited cycle PCR approach on genomic DNA of GFP expressing embryos injected with unmodified, Biotin or SpC3 5’ modified dsDNA donors. Our approach allowed distinguishing alleles without gfp integration (i.e. size of wild-type locus) from those generated by HDR and NHEJ respectively and addressed the size of the integration by a locus spanning PCR with a reduced number of PCR cycles (<=30) to omit in vitro fusion-PCR artefacts (own data and [Won and Dawid, 2017]). To determine the predictive power of GFP expression for perfect integration, we genotyped randomly selected, GFP-expressing embryos using locus primers (Lf/Lr) located distal to the utilized HFs (Figure 1—figure supplement 1) and addressed the fusion of the gfp donor sequence to the target genes (Figure 2B, Figure 2—figure supplements 2 and 3).
Employing unmodified donors, the rate of HDR was very low as evidenced by the predominant amplification of the alleles without gfp integration (Figure 2B, Figure 2—figure supplement 2). In strong contrast, the 5’ modified long dsDNA donors resulted in efficient HDR already detectable in the injected generation for all targeted loci. For the gfp tagging of rx2, 6 out of 10 randomly selected, GFP-expressing embryos showed precise HDR-mediated single-copy integration in F0 (Figure 2—figure supplement 2) as sequence confirmed by the analysis of the locus (Figure 2—figure supplement 3). Thus, 9.5% of injected and surviving zygotes showed precise HDR-mediated single-copy integration (15.8% of the injected zygotes expressed GFP; 60% of those showed the precise single-copy integration; Supplementary file 1). For the gfp tagging of actb 46.5% of the injected zygotes expressed GFP, 35% of which (7 out of 20 randomly selected, GFP-expressing embryos) showed precise HDR-mediated single-copy integration in F0, accounting for 16.3% of the initially injected zygotes (Supplementary file 1).
In the case of dnmt1, the rate of precise HDR-mediated single-copy integration was even higher, since full gene functionality is required for the progression of development and embryonic survival. Here, strikingly, all GFP-expressing embryo showed the desired perfect integration.
We observed the highest efficiency of HDR targeting by 5’ modified dsDNA for all loci tested for 5’Biotin modified donors (Figure 2—figure supplement 2). Already in the injected generation we prominently detected and validated the HDR-mediated fusion of the long dsDNA donors with the respective locus (Figure 2—figure supplement 2). While still giving rise to a high percentage of HDR events, SpC3 5’ modified dsDNA donors also resulted in elevated levels of additional bands indicative for the integration of higher order multimers and NHEJ events (Figure 2—figure supplement 2). Even though increasingly popular, in our hands the use of RNPs (Cas9 protein and respective sgRNA) did not even get close to the efficiency achieved by co-injection of Cas9 mRNA and the corresponding sgRNA assessed by gene targeting as described above.
The precise integration detected in the injected generation was successfully transmitted to the next generation (Figure 3, Figure 2—figure supplement 3, Figure 3—figure supplement 1). For gfp-rx2, 9% of fish originating from the initially injected embryos successfully transmitted the precisely modified locus to the next generation (15.8% of the injected embryos expressed GFP; 4 out of 7 GFP transmitting founder fish were also transmitting the precise single integration of the gfp donor cassette). For gfp-rx1, 3.9% of fish originating from the initially injected embryos were transmitting the precise single copy integrate to the next generation (13.5% of injected embryos expressed GFP; 2 out of 7 GFP transmitting founder fish were also transmitting the precise single integration of the gfp-rx1 donor cassette). For dnmt1, the high rate of precise HDR-mediated single-copy integration observed in the injected generation was fully maintained in the transmission to the next generation due to the absolute requirement of a functional/functionally tagged version of the locus. As we found in the course of our study, the precise integration of the gfp donor cassette into the actb locus results in late embryonic lethality. Consequently, stable transgenic lines could not be established.
Figure 3 with 1 supplement see all

Single-copy integration of long dsDNA donor establishes stably transmitted gfp-rx2 fusion gene.
(A) F2 homozygous embryos exhibit GFP-Rx2 fusion protein expression in the pattern of the endogenous gene in the retina. (B) Southern Blot analysis of F2 gfp-rx2 embryos reveals a single band for a digestion scheme cutting outside the donor cassette (BglII/HindIII) or within the 5’ donor cassette and in intron 2 (ScaI/HindIII) indicating precise single-copy donor integration. (B’) Schematic representation of the modified locus indicating the restriction sites and the domain complementary to the probe used in (B). (C) RT-PCR analysis on mRNA isolated from individual homozygous F3 embryos indicates the transcription of a single gfp-rx2 fusion mRNA in comparison to the shorter wild-type rx2 mRNA as schematically represented in (C’).
To estimate the timepoint of HDR events in the injected embryos, we investigated the actual rate of mosaicism in the germline as reflected by the germline transmission rate. For gfp-rx2, this rate ranged from 0.8% up to 12.9% indicating an HDR event earliest at the 4-cell stage (assuming that only a single event occurred per blastomere). In the case of gfp-rx1, HDR did not occur at the one-cell stage but rather later (2–8 cell stage as reflected by germline transmission rates of 23.9% and 5.8% respectively). Thus, for both cases, the transmission rates of the perfectly tagged locus reflect a level of mosaicism indicative for an HDR event between the 4- and 32-cell stage.
We addressed the nature of the insertion predicted to be single-copy by a combination of PCR and expression studies. We validated the genomic organization of the gfp-rx2 (Figure 3A) and gfp-rx1 knock-in in homozygous F2 animals by genomic sequencing (Figure 2—figure supplement 3C) and Southern Blot (Southern, 2006) analysis (Figure 3B and B’, Figure 3—figure supplement 1). In both cases, we detected a single band indicative for a single-copy HDR-mediated integration, when probing digested genomic DNA of F2 gfp-rx2+/+ and gfp-rx1+/+ knock-in fish respectively. We used enzyme combinations releasing the gfp-rx2 region including the HFs (BglII/HindIII, Figure 3B and B’) or cutting in the 5’ HF (ScaI/HindIII, Figure 3B,B’). For gfp-rx1 we released the 5’ flanking region including the donor cassette (HindIII/XmaI) or the respective 3’ flanking region and donor cassette (NcoI/EcoRI) (Figure 3—figure supplement 1).
This analyses crucially validated the PCR based predictions and sequencing results.
Furthermore, transcript analysis in gfp-rx2 homozygous F3 embryos exclusively uncovered a single fusion gfp-rx2 transcript (Figure 3C and C’). This molecular analysis confirmed that the 5’Biotin modification of the long dsDNA donor promoted precise single-copy HDR-mediated integration with high efficiency.
Taken together the simple 5’Biotin modification at both ends of long dsDNA donors by conventional PCR amplification presented here provides the means to favor HDR without interfering with the cellular DNA repair machinery.
Discussion
For efficient recombination the quality of the modified primers, that is the fraction of primers actually labeled with biotin and therefore the quality of the 5’ protected long ds DNA PCR product, is essential. The integration of 5’ modified PCR fragments larger than 2 kb does in principle not pose a problem. We already successfully integrated cassettes of up to 8.6 kb (data not shown) via HDR by in vivo linearization of the donor plasmid (Stemmer et al., 2015). In the approach presented here, the quality of the end-protected PCR product is likely to drop with higher length in part due to the (UV–) nicking in the extraction and purification process. Also, the rapid validation by locus spanning PCR is size limited but eventually Southern blot analysis will resolve the question of a single copy, perfect integration. Irrespective of the donor cassette size, the PCR cycles used to probe for the successful single integration via HDR must not exceed a total of 30 to avoid misleading in vitro fusion-PCR artifacts (own data and [Won and Dawid, 2017]).
As already mentioned earlier, HDR preferentially occurs during the late S/G2 phase of the cell cycle (Heyer et al., 2010; Hustedt and Durocher, 2017). Varying efficiencies of genome editing via HDR could be due to species-specific differences, in particular of the S/G2 phase of the cell cycle that likely crucially impacts on the rate of HDR. As reported extensively, there is no apparent G2 phase before mid-blastula-transition in amphibia (Xenopus) and fish (Danio rerio) with the marked exception of the first cleavage, where a G2 phase has been reported (Kimelman, 2014). The germline transmission rates obtained for the tagging of Rx1 and Rx2 indicated HDR events after the two-cell stage, predominantly at the 4-cell and subsequent stages. The slower cycling of medaka compared to zebrafish results in an extension of the G2 phase in the first cell cycle by more than 150% (total cycle zebrafish: 45 min [Kimmel et al., 1995], medaka: 65 min [Iwamatsu, 2004]). Assuming a comparable rate of DNA synthesis between the two species, there is more than double the time for S- and M-phase to replicate a genome encompassing roughly a third of the size of zebrafish. This extended cell cycle length is in particular apparent also in the subsequent cycles in medaka (15 min zebrafish [Kimmel et al., 1995], 40 min medaka [Iwamatsu, 2004]). Interestingly, phosphorylation of RNA polymerase, a prerequisite for the expression of the first zygotic transcripts has been reported to start at the 64cell stage in medaka (Kraeussling et al., 2011) and asynchronous divisions, a hallmark of MBT, are observed already at the transition of the 16- to 32-cell stage (Kraeussling et al., 2011), suggesting species-specific differences in the onset of zygotic transcription and consequently the lengthening of the cell cycle.
Our approach facilitates the highly efficient detection of HDR events by GFP tagging even in non-essential genes. Other than in non-modified dsDNA donors, the locus-specific expression of GFP is an excellent predictor for precise, single-copy HDR-mediated integration already in the injected generation. This allows an easy selection and results in HDR rates in GFP positive embryos of up to 60% in the injected generation. This rate can even be higher in essential loci, such as dnmt1, where the full functionality of the (modified) gene under investigation is required for the survival of the affected cells, tissues, organs or organisms, thus providing the means for effective positive selection for single, fully functional integrates. It is interesting to note that approximately 400 bp flanking the insertion site in 5’ and 3’ direction are sufficient for HDR. The use of modified universal primers for the two donor template vectors employed allowed a flexible application of the procedure and ensured a high quality of the PCR generated 5’ modified long dsDNA donors. Strikingly, the primer-introduced non-homology regions in the very periphery of the long dsDNA donors did not negatively impact on the HDR efficiency.
Taken together, the simplicity and high reproducibility of our proof-of-concept analysis highlight that the presented protection of both 5’ ends of long dsDNA donors prevent multimerization and promote precise insertion/replacement of DNA elements, thus facilitating functional studies in basic research as well as therapeutic interventions.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Oryzias latipes) | Cab | other | medaka Southern wild-type population | |
| Strain, strain background (Oryzias latipes) | rx2-gfp | this paper | ||
| Strain, strain background (Oryzias latipes) | rx1-gfp | this paper | ||
| Strain, strain background (Oryzias latipes) | actb-gfp | this paper | ||
| Strain, strain background (Oryzias latipes) | dnmt1-gfp | this paper | ||
| Recombinant DNA reagent | rx2-gfp donor cassette | this paper | ||
| Recombinant DNA reagent | rx1-gfp donor cassette | this paper | ||
| Recombinant DNA reagent | actb-gfp donor cassette | this paper | ||
| Recombinant DNA reagent | dnmt1-gfp donor cassette | this paper | ||
| Sequence- based reagent | rx2 5'HF f | this paper | with BamHI restriction site: GCCGGATCCAAGCATGTCAAAACGTAGAAGCG | |
| Sequence- based reagent | rx2 5'HF r | this paper | with KpnI restriction site: GCCGGTACCCATTTGGCTGTGGACTTGCC | |
| Sequence- based reagent | rx2 3'HF f | this paper | with BamHI restriction site: GCCGGATCCCATTTGTCAATGGAC ACGCTTGGGATGGTGGACGAT | |
| Sequence- based reagent | rx2 3'HF r | this paper | with KnpI restriction site: GCCGGTACCTGGACTGGACTGGAAGTTATTT | |
| Sequence- based reagent | rx2 sgRNA f | this paper | substituted nucleotides to facilitate T7 in vitro transcription of the sgRNA oligonucleotides are shown in small letters TAgGCATTTGTCAATGGATACCC | |
| Sequence- based reagent | rx2 sgRNA r | this paper | AAACGGGTATCCATTGACAAATG | |
| Sequence- based reagent | rx2 Lf/5’UTRf | this paper | TGCATGTTCTGGTTGCAACG | |
| Sequence- based reagent | rx2 Lr | this paper | AGGGACCATACCTGACCCTC | |
| Sequence- based reagent | actb 5’HF f | this paper | with BamHI restriction site: GGGGATCCCAGCAACGACTTCGCACAAA | |
| Sequence- based reagent | actb 5’HF r | this paper | with KnpI restriction site: GGGGTACCGGCAATGTCATCATCCATGGC | |
| Sequence- based reagent | actb 3’HF f | this paper | with BamHI restriction site: GGGGATCCGACGACGATATAGCTG CACTGGTTGTTGACAACGGATCTG | |
| Sequence- based reagent | actb 3’HF r | this paper | with KnpI restriction site: GGGGTACCCAGGGGCAATTCTCAGCTCA | |
| Sequence- based reagent | actb sgRNA f | this paper | TAGGATGATGACATTGCCGCAC | |
| Sequence- based reagent | actb sgRNA r | this paper | AAACGTGCGGCAATGTCATCAT | |
| Sequence- based reagent | actb Lf | this paper | GTCCGAGTTGAGGGTGTCTG | |
| Sequence- based reagent | actb Lr | this paper | CATGTGCTCCACTGTGAGGT | |
| Sequence- based reagent | dnmt1 5’HF f | this paper | with SalI restriction site: AATTTGTCGACGCTTTGA CAGTTAACCTACACG | |
| Sequence- based reagent | dnmt1 5’HF r | this paper | with AgeI restriction site: AATTTACCGGTCGTAACTGCA AACTAAAAAATAAAAC | |
| Sequence -based reagent | dnmt1 3’HF f | this paper | with SpeI restriction site: AATTTACTAGTATGCCATCCAGAA CGTCCTTATCTCTACCAGACGATG TCAGAAAAAGGTAC | |
| Sequence- based reagent | dnmt1 3’HF r | this paper | with NotI restriction site: AATTTGCGGCCGCCTACACATA TTGTCTGTGATAC | |
| Sequence- based reagent | mgfpf | this paper | with AgeI restriction site: AATTTACCGGTACTAGTACCATG AGTAAAGGAGAAGAACTTTTCAC | |
| Sequence- based reagent | mgfpr | this paper | with SpeI restriction site: AATTTACTAGTCGCGGCTGCACTT CCACCGCCTCCCGATCCGCCACC GCCAGAGCCACCTCCGCCTGAAC CGCCTCCACCGCTCAGGCTAGCTT TGTATAGTTCATCCATGCCATG | |
| Sequence- based reagent | dnmt1 sgRNA f | this paper | substituted nucleotides to facilitate T7 in vitro transcription of the sgRNA oligonucleotides are shown in small letters TAgGACATCGTCTGGCAAAGAC | |
| Sequence- based reagent | dnmt1 sgRNA r | this paper | AAACGTCTTTGCCAGACGATGT | |
| Sequence- based reagent | dnmt1 Lf | this paper | CTCAATGTAAACACTTCGTGTCGCTTC | |
| Sequence -based reagent | dnmt1 Lr | this paper | TTGCATGCATATTCAAAGTTGTCAAAG | |
| Sequence- based reagent | rx1 5’HF f | this paper | with BamHI restriction site: GCCGGATCCGCATCCGAAAGG TAAGGACTGCAAACC | |
| Sequence- based reagent | rx1 5’HF r | this paper | with KpnI restriction site: GCCGGTACCCATGAGAGCG TCTGGGCTCTGACC | |
| Sequence- based reagent | rx1 3’HF f | this paper | with BamHI restriction site: GGCGGATCCCATTTATCAC TCGATACCATGAGCA | |
| Sequence- based reagent | rx1 3’HF r | this paper | with KpnI restriction site: GGCGGTACCTTCCAGTTTA AGAACATCCCCTCT | |
| Sequence- based reagent | rx1 sgRNA1 f | this paper | substituted nucleotides to facilitate T7 in vitro transcription of the sgRNA oligonucleotides are shown in small letters TAggAAATGCATGAGAGCGTCT | |
| Sequence- based reagent | rx1 sgRNA1 r | this paper | AAACAGACGCTCTCATGCATTT | |
| Sequence- based reagent | rx1 sgRNA2 f | this paper | substituted nucleotides to facilitate T7 in vitro transcription of the sgRNA oligonucleotides are shown in small letters TAggCTCTCATGCATTTATCAC | |
| Sequence- based reagent | rx1 sgRNA2 r | this paper | AAACGTGATAAATGCATGAGAG | |
| Sequence- based reagent | rx1 Lf | this paper | CTTTGCTGTTTTGAGAATTGCACC | |
| Sequence- based reagent | rx1 Lr | this paper | GAGACCGAACGATGACAATAACAC | |
| Sequence- based reagent | pDest f (control) | this paper | CGAGCGCAGCGAGTCAGTGAG | |
| Sequence- based reagent | pDest r (control) | this paper | CATGTAATACGACTCACTATAG | |
| Sequence- based reagent | pDest f mod | this paper | Asterisks indicate phosphorothioate bonds, ‘5’moiety’ was either 5’Biotin, Amino-dT or Spacer C3. 5’moiety-C*G*A*G*C*GCAGCGAGTCAGTGAG | |
| Sequence- based reagent | pDest r mod | this paper | Asterisks indicate phosphorothioate bonds, ‘5’moiety’ was either 5’Biotin , Amino-dT or Spacer C3. 5’moiety-C*A*T*G*T*AATACGACTCACTATAG | |
| Sequence- based reagent | pCS2 f | this paper | CCATTCAGGCTGCGCAACTG | |
| Sequence- based reagent | pCS2 r | this paper | CACACAGGAAACAGCTATGAC | |
| Sequence -based reagent | pCS2 f mod | this paper | Asterisks indicate phosphorothioate bonds, ‘5’moiety’ was either 5’Biotin, Amino-dT or Spacer C3. 5’moiety-C*C*A*T*T*CAGGCTG CGCAACTG | |
| Sequence- based reagent | pCS2 r mod | this paper | Asterisks indicate phosphorothioate bonds, ‘5’moiety’ was either 5’Biotin, Amino-dT or Spacer C3. 5’moiety-C*A*C*A*C*AGGAAACAGCTATGAC | |
| Sequence- based reagent | Gf | this paper | ATGGCAAGCTGACCCTGAAGTTCAT CTGCACCACCGGCAAGC | |
| Sequence- based reagent | Gr | this paper | CTCAGGTAGTGGTTGTCG | |
| Sequence- based reagent | gfpf | this paper | GCTCGACCAGGATGGGCA | |
| Sequence- based reagent | gfpr | this paper | CTGAGCAAAGACCCCAACGAGA AGCGCGATCACATG | |
| Sequence- based reagent | gfp probe f | this paper | GTGAGCAAGGGCGAGGAGCT | |
| Sequence- based reagent | gfp probe r | this paper | CTTGTACAGCTCGTCCATG |
Fish maintenance
Request a detailed protocolAll fish are maintained in closed stocks at Heidelberg University. Medaka (Oryzias latipes) husbandry (permit number 35–9185.64/BH Wittbrodt) and experiments (permit number 35–9185.81/G-145/15 Wittbrodt) were performed according to local animal welfare standards (Tierschutzgesetz §11, Abs. 1, Nr. 1) and in accordance with European Union animal welfare guidelines (Bert et al., 2016). The fish facility is under the supervision of the local representative of the animal welfare agency. Embryos of medaka of the wild-type Cab strain were used at stages prior to hatching. Medaka was raised and maintained as described previously (Koster et al., 1997).
Donor plasmids
Request a detailed protocolRx2 and actb template plasmids for gfp donor cassette amplification are described in Stemmer et al. (2015) and were generated by GoldenGATE assembly into the pGGDestSC-ATG destination vector (addgene #49322) according to Kirchmaier et al. (2013). See Supplementary file 2 for primers used to amplify respective homology flanks. The dnmt1 gfp plasmid was cloned with homology flanks (5’ HF 402 bp, primers dnmt1 5’HF f/dnmt1 5’HF r; 3’ HF 405 bp, dnmt1 3’HF f/dnmt1 3’HF r) that were PCR amplified with Q5 polymerase (New England Biolabs, 30 cycles) from wild-type medaka genomic DNA. mgfp-flexible linker was amplified with primers mgfpf/mgfpr. The respective restriction enzyme was used to digest the amplicons (5’HF: SalI HF (New England Biolabs), AgeI HF (New England Biolabs); mgfp-flexible linker: AgeI HF (New England Biolabs), SpeI HF (New England Biolabs); 3’HF: SpeI HF (New England Biolabs), NotI HF (New England Biolabs)) followed by gel purification (Analytik Jena) and ligation into pCS2+ (Rupp et al., 1994) (digested with SalI HF (New England Biolabs), NotI HF (New England Biolabs)). The rx1 gfp plasmid was cloned with homology flanks (5’HF 430 bp, primers rx1 5’HF f/rx1 5’HF r; 3’ HF 508 bp, rx1 3’HF f/rx1 3’HF r) that were PCR amplified with Q5 polymerase (New England Biolabs, 30 cycles) from wild-type medaka genomic DNA. All primers were obtained from Eurofins Genomics.
Donor amplification
Request a detailed protocolWe designed universal primers that match the pGGDestSC-ATG (Kirchmaier et al., 2013) (addgene #49322) or pCS2+ (Rupp et al., 1994) backbone encompassing the assembled inserts (i.e. the gfp donor cassette). Unmodified control primers (pDest f, pDest r, pCS2 f, pCS2 r) were ordered from Eurofins Genomics. Modified primers obtained from Sigma-Aldrich (pDest f mod, pDest r mod, pCS2 f mod, pCS2 r mod) consist of the same sequences with phosphorothioate bonds in the first five nucleotides and 5’moiety extension: 5’Biotin, 5’Amino-dT or 5’Spacer C3.
The dsDNA donor cassettes were amplified by PCR using 1x Q5 reaction buffer, 200 µM dNTPs, 200 µM primer forward and reverse and 0.6 U/µl Q5 polymerase (New England Biolabs). Conditions used: initial denaturation at 98°C 30 s, followed by 35 cycles of: denaturation at 98°C 10 s, annealing at 62°C 20 s and extension at 72°C 30 s per kb and a final extension step of 2 min at 72°C. The PCR reaction was treated with 20 units of DpnI (New England Biolabs) to remove any plasmid template following gel purified using the QIAquick Gel Extraction Kit (Qiagen, 28706) and elution with 20 µl nuclease-free water.
The LacZ cassette of the pGGDestSC-ATG (Kirchmaier et al., 2013) (addgene #49322) which served as DIG labelled dsDNA fragment to test in vivo multimerization was amplified via Q5-PCR as above using a mixture of 200 µM dATP, dCTP, dGTP, 170 µM dTTP and 30 µM DIG-dUTP and purified as detailed.
sgRNA target site selection
Request a detailed protocolDnmt1 sgRNAs were designed with CCTop as described in Stemmer et al. (2015). sgRNAs for rx2 and actb were the same as in Stemmer et al. (2015). The following target sites close to the translational start codons were used (PAM in brackets): rx2 (GCATTTGTCAATGGATACCC[TGG]), actb (GGATGATGACATTGCCGCAC[TGG]), dnmt1 (TGACATCGTCTGGCAAAGAC[AGG]) and rx1 (AAATGCATGAGAGCGTCT[GGG] and CTCTCATGCATTTATCAC[TGG]). Cloning of sgRNA templates and in vitro transcription was performed as detailed in Stemmer et al. (2015).
In vitro transcription of mRNA
Request a detailed protocolThe pCS2 +Cas9 plasmid was linearized using NotI and the mRNA was transcribed in vitro using the mMessage_mMachine SP6 kit (ThermoFisher Scientific, AM1340).
Microinjection and screening
Request a detailed protocolMedaka zygotes were injected with 10 ng of DIG-labelled donors and were allowed to develop until 2, 4 and 6 hr post injection. For the CRISPR/Cas9 experiments, medaka zygotes were injected with 5 ng/µl of either unmodified and modified long dsDNA donors together with 150 ng/µl of Cas9 mRNA and 15–30 ng/µl of the gene-specific sgRNAs. Injected embryos were maintained at 28°C in embryo rearing medium (ERM, 17 mM NaCl, 40 mM KCl, 0.27 mM CaCl2, 0.66 mM MgSO4, 17 mM Hepes). One day post-injection (dpi) embryos were screened for survival, GFP expression was scored at two dpi.
Southern blot
Request a detailed protocolIn order to check for multimerization of unmodified and modified donors, we used a modified Southern Blot approach. In brief, embryos were injected with DIG-labelled donors which were PCR-amplified from pGGDestSC-ATG (addgene #49322) using primers pDest f/pDest r (LacZ cassette) harboring either no 5’ moiety or one of the following: 5’Biotin, Amino-dT or Spacer C3. 2, 4 and 6 hr post injection, 30 embryos were lysed in TEN buffer plus proteinase K (10 mM Tris pH 8, 1 mM EDTA, 100 mM NaCl, 1 mg/ml proteinase K) at 60°C overnight. DNA was ethanol precipitated after removal of lipids and proteins by phenol-chloroform extraction. Total DNA was resuspended in TE buffer (10 mM Tris HCl pH 8.0, 1 mM EDTA pH 8.0). 200 ng of each sample was run on a 0.8% agarose gel. As a control, 100 pg of uninjected donor PCR product were loaded. The agarose gel was transferred to a nylon membrane overnight using 10x SSC (1.5 M NaCl, 0.15 M C6H5Na3O7) as transfer solution. The cross-linked membrane was directly blocked in 1% w/v blocking reagent (Roche) in 1x DIG1 solution (0.1 M maleic acid, 0.15 M NaCl, pH 7.5) and the labeled DNA was detected using CDP star (Roche) following the manufacturer’s instructions.
In order to check for copy number insertions in the gfp-rx2 and gfp-rx1 transgenic lines, genomic DNA was isolated as described above from F2 embryos expressing GFP. 10 µg digested genomic DNA were loaded per lane on a 0.8% agarose gel and size fractionated by electrophoresis. The gel was depurinated in 0.25 N HCl for 30 min at room temperature, rinsed with H2O, denatured in 0.5 N NaOH, 1.5 M NaCl solution for 30 min at room temperature and neutralized in 0.5 M Tris HCl, 1.5 M NaCl, pH 7.2 before it was transferred overnight at room temperature onto a Hybond membrane (Amersham). The membrane was washed with 50 mM NaPi for 5 min at room temperature, then crosslinked and pre-hybridized in Church hybridization buffer (0.5 M NaPi, 7% SDS, 1 mM EDTA pH 8.0) at 65°C for at least 30 min. The probe was synthesized from the donor plasmid with primers gfp probe f and gfp probe r using the PCR DIG Probe Synthesis Kit (Roche, 11636090910) and the following PCR protocol: initial denaturation at 95°C for 2 min, 35 cycles of 95°C 30 s, 60°C 30 s, 72°C 40 s and final extension at 72°C 7 min. The probe was boiled in hybridization buffer for 10 min at 95°C and the membrane was hybridized overnight at 65°C. The membrane was washed with pre-heated (65°C) Church washing buffer (40 mM NaPi, 1% SDS) at 65°C for 10 min, then at room temperature for 10 min and with 1x DIG1% and 0.3% Tween for 5 min at room temperature. The membrane was blocked in 1% w/v blocking reagent (Roche) in 1x DIG1 solution at room temperature for at least 30 min. The membrane was incubated with 1:10,000 anti-digoxigenin-AP Fab fragments (Roche) for 30 min at room temperature in 1% w/v blocking reagent (Roche) in 1x DIG1 solution. Two washing steps with 1x DIG1% and 0.3% Tween were performed for 20 min at room temperature, followed by a 5 min washing step in 1x DIG3 (0.1 M Tris pH 9.5, 0.1 M NaCl) at room temperature. Detection was performed using 6 µl/ml CDP star (Roche).
Genotyping
Request a detailed protocolSingle injected GFP positive embryos were lysed in DNA extraction buffer (0.4 M Tris/HCl pH 8.0, 0.15 M NaCl, 0.1% SDS, 5 mM EDTA pH 8.0, 1 mg/ml proteinase K) at 60°C overnight. Proteinase K was inactivated at 95°C for 10 min and the solution was diluted 1:2 with H2O. Genotyping was performed in 1x Q5 reaction buffer, 200 µM dNTPs, 200 µM primer forward and reverse and 0.012 U/µl Q5 polymerase and 2 µl of diluted DNA sample and the respective locus primers. The conditions were: 98°C 30 s, 30 cycles of 98°C 10 s, annealing for 20 s and 72°C 30 s per kb (extension time used would allow for detecting potential NHEJ events on both ends of the donor) (rx2 Lf/rx2 Lr: 68°C annealing, 90 s extension time; rx1 Lf/rx1 Lr: 66°C annealing, 90 s extension time; actb Lf/actb Lr: 66°C annealing, 84 s extension time; dnmt1 Lf/dnmt1 Lr: 65°C annealing, 90 s extension time) and a final extension of 2 min at 72°C. PCR products were analyzed on a 1% agarose gel.
Diagnostic GFP PCR here: 63°C annealing, 500 bp, 15 s extension
RT-PCR
Request a detailed protocolTotal RNA was isolated from 60 homozygous embryos (stage 32) by lysis in TRIzol (Ambion) and chloroform extraction according to the manufacturer’s protocol. RNA was precipitated using isopropanol and resuspended in H2O. cDNA was reverse transcribed with Revert Aid Kit (Thermo Fisher Scientific) after DNAse digestion and inactivation following the manufacturer’s instructions. PCR was performed using 5’UTRf, 3’UTRr and Q5 polymerase (New England Biolabs): 98°C 30 s, 35 cycles of 98°C 10 s, annealing 65°C for 20 s and 72°C 210 s and a final extension of 2 min at 72°C. PCR products were analyzed on a 1.5% agarose gel.
Sequencing
Request a detailed protocolPlasmids and PCR fragments were sequenced with the indicated primers by a commercial service (Eurofins Genomics).
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files. Source data files have been provided for Figure 2-figure supplement 1.
References
-
Regulation of homologous recombination in eukaryotesAnnual Review of Genetics 44:113–139.https://doi.org/10.1146/annurev-genet-051710-150955
-
The control of DNA repair by the cell cycleNature Cell Biology 19:1–9.https://doi.org/10.1038/ncb3452
-
Stages of normal development in the medaka Oryzias latipesMechanisms of Development 121:605–618.https://doi.org/10.1016/j.mod.2004.03.012
-
Stages of embryonic development of the zebrafishDevelopmental Dynamics 203:253–310.https://doi.org/10.1002/aja.1002030302
-
Medaka Spalt acts as a target gene of hedgehog signalingDevelopment 124:3147–3156.
-
Six3 overexpression initiates the formation of ectopic retinaGenes & Development 13:649–654.https://doi.org/10.1101/gad.13.6.649
-
Cas9 as a versatile tool for engineering biologyNature Methods 10:957–963.https://doi.org/10.1038/nmeth.2649
-
Xenopus embryos regulate the nuclear localization of XMyoDGenes & Development 8:1311–1323.https://doi.org/10.1101/gad.8.11.1311
-
Transient expression of foreign DNA during embryonic and larval development of the medaka fish (Oryzias latipes)MGG Molecular & General Genetics 226:129–140.https://doi.org/10.1007/BF00273596
Article and author information
Author details
Tinatini Tavhelidse

Funding
Deutsche Forschungsgemeinschaft (CRC 873,TP A3)
- Joachim Wittbrodt
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This research was funded through the German Science funding agency (DFG, CRC 873, Project A3 to JW). TTa and ET are members of HBIGS, the Heidelberg Biosciences International Graduate School. We are grateful to M Majewsky, E Leist and A Saraceno for fish husbandry. We thank Oliver Gruss (Bonn) and Daigo Inoue (Yokohama) and all members of the Wittbrodt lab for their critical, constructive feedback on the procedure and the manuscript.
Version history
- Received: June 22, 2018
- Accepted: August 14, 2018
- Accepted Manuscript published: August 29, 2018 (version 1)
- Version of Record published: September 5, 2018 (version 2)
Copyright
© 2018, Gutierrez-Triana et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 7,190
- views
-
- 1,263
- downloads
-
- 84
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Efficient single-copy HDR by 5’ modified long dsDNA donors
eLife 7:e39468.
https://doi.org/10.7554/eLife.39468
Further reading
-
- Computational and Systems Biology
- Developmental Biology
Organisms utilize gene regulatory networks (GRN) to make fate decisions, but the regulatory mechanisms of transcription factors (TF) in GRNs are exceedingly intricate. A longstanding question in this field is how these tangled interactions synergistically contribute to decision-making procedures. To comprehensively understand the role of regulatory logic in cell fate decisions, we constructed a logic-incorporated GRN model and examined its behavior under two distinct driving forces (noise-driven and signal-driven). Under the noise-driven mode, we distilled the relationship among fate bias, regulatory logic, and noise profile. Under the signal-driven mode, we bridged regulatory logic and progression-accuracy trade-off, and uncovered distinctive trajectories of reprogramming influenced by logic motifs. In differentiation, we characterized a special logic-dependent priming stage by the solution landscape. Finally, we applied our findings to decipher three biological instances: hematopoiesis, embryogenesis, and trans-differentiation. Orthogonal to the classical analysis of expression profile, we harnessed noise patterns to construct the GRN corresponding to fate transition. Our work presents a generalizable framework for top-down fate-decision studies and a practical approach to the taxonomy of cell fate decisions.
-
- Developmental Biology
- Evolutionary Biology
Despite rapid evolution across eutherian mammals, the X-linked MIR-506 family miRNAs are located in a region flanked by two highly conserved protein-coding genes (SLITRK2 and FMR1) on the X chromosome. Intriguingly, these miRNAs are predominantly expressed in the testis, suggesting a potential role in spermatogenesis and male fertility. Here, we report that the X-linked MIR-506 family miRNAs were derived from the MER91C DNA transposons. Selective inactivation of individual miRNAs or clusters caused no discernible defects, but simultaneous ablation of five clusters containing 19 members of the MIR-506 family led to reduced male fertility in mice. Despite normal sperm counts, motility, and morphology, the KO sperm were less competitive than wild-type sperm when subjected to a polyandrous mating scheme. Transcriptomic and bioinformatic analyses revealed that these X-linked MIR-506 family miRNAs, in addition to targeting a set of conserved genes, have more targets that are critical for spermatogenesis and embryonic development during evolution. Our data suggest that the MIR-506 family miRNAs function to enhance sperm competitiveness and reproductive fitness of the male by finetuning gene expression during spermatogenesis.




{kind=link}
{kind=link}
{kind=link}