Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
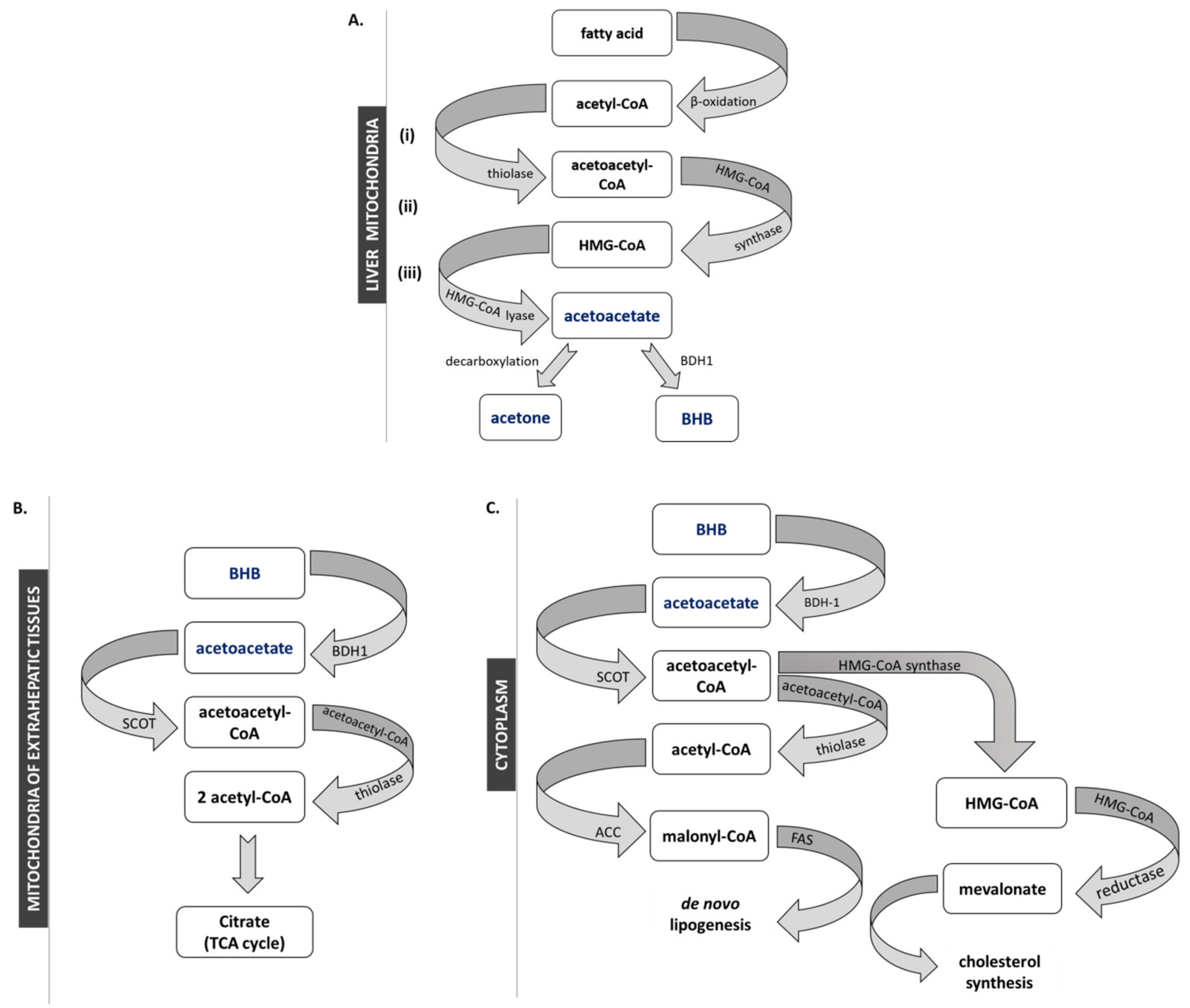
2. Anabolism and Catabolism of Ketone Bodies
3. Ketogenesis as A Physiological Response to Starving and Prolonged Physical Exercise and as A Pathological Phenomenon in Diabetes
4. Epigenetic Effects of Ketone Bodies
5. Signaling Pathways Linking Ketone Bodies to Protection from Oxidative Stress
5.1. Protection Against Oxidative Stress in Spinal Cord Injury
5.2. The Impact of Ketone Bodies/Ketogenic Diet on Alzheimer’s Disease
5.3. The Role of β-hydroxybutyrate in Ischemia/Reperfusion of Heart and Brain Injury
5.4. The Protective Role of β-hydroxybutyrate in Hypertension
6. The link between Ketone Bodies via Nutritional Intake and Physical Performance
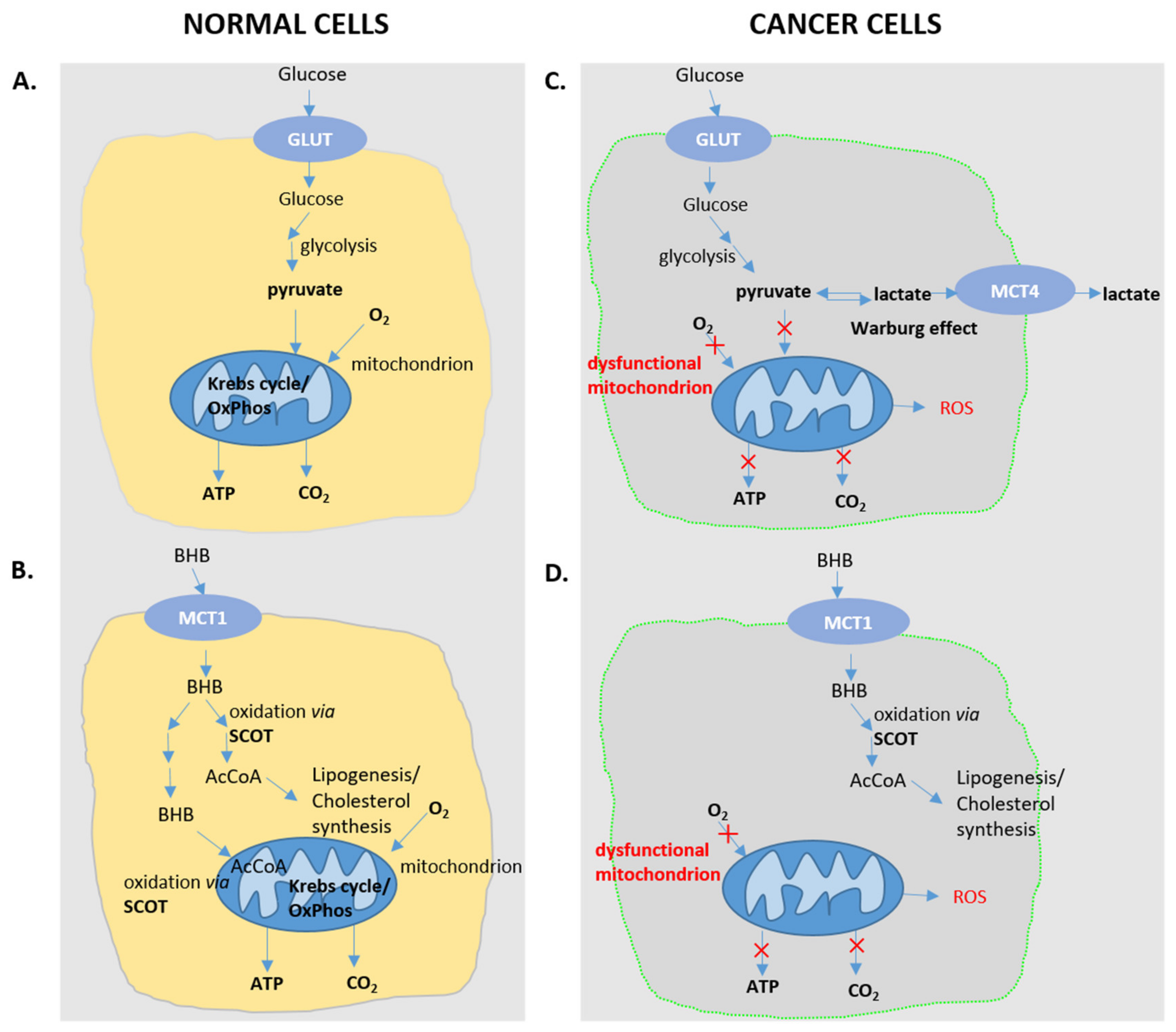
7. Curbing Cancer Progression with Ketone Bodies/Ketogenic Diet
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershuni, V.M.; Yan, S.L.; Medici, V. Nutritional Ketosis for Weight Management and Reversal of Metabolic Syndrome. Curr. Nutr. Rep. 2018, 7, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Hashim, S.A.; VanItallie, T.B. Ketone body therapy: From the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 2014, 55, 1818–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, B.; Raggi, P. The ketogenic diet: Pros and cons. Atherosclerosis 2019, 292, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, C.B.; Filloux, F.M.; Alder, S.C.; Lyon, J.L.; Caplin, D.A. Efficacy of the Ketogenic Diet as a Treatment Option for Epilepsy: Meta-Analysis. J. Child. Neurol. 2006, 21, 193–198. [Google Scholar] [PubMed]
- Castellana, M.; Conte, E.; Cignarelli, A.; Perrini, S.; Giustina, A.; Giovanella, L.; Giorgino, F.; Trimboli, P. Efficacy and safety of very low calorie ketogenic diet (VLCKD) in patients with overweight and obesity: A systematic review and meta-analysis. Rev. Endocr. Metab. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Westman, E.C.; Feinman, R.D.; Mavropoulos, J.C.; Vernon, M.C.; Volek, J.S.; Wortman, J.A.; Yancy, W.S.; Phinney, S.D. Low-carbohydrate nutrition and metabolism. Am. J. Clin. Nutr. 2007, 86, 276–284. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Anderson, A.J.; Dawes, E.A. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 1990, 54, 450–472. [Google Scholar] [CrossRef] [Green Version]
- Dedkova, E.N.; Blatter, L.A. Role of β-hydroxybutyrate, its polymer poly-β-hydroxybutyrate and inorganic polyphosphate in mammalian health and disease. Front. Physiol. 2014, 5, 260. [Google Scholar] [CrossRef] [Green Version]
- Smithen, M.; Elustondo, P.A.; Winkfein, R.; Zakharian, E.; Abramov, A.Y.; Pavlov, E. Role of polyhydroxybutyrate in mitochondrial calcium uptake. Cell Calcium 2013, 54, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elustondo, P.A.; Angelova, P.R.; Kawalec, M.; Michalak, M.; Kurcok, P.; Abramov, A.Y.; Pavlov, E.V. Polyhydroxybutyrate targets mammalian mitochondria and increases permeability of plasmalemmal and mitochondrial membranes. PLoS ONE 2013, 8, e75812. [Google Scholar] [CrossRef] [PubMed]
- Auestad, N.; Korsak, R.A.; Morrow, J.W.; Edmond, J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J. Neurochem. 1991, 56, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-W.; Biton, M.; Haber, A.L.; Gunduz, N.; Eng, G.; Gaynor, L.T.; Tripathi, S.; Calibasi-Kocal, G.; Rickelt, S.; Butty, V.L.; et al. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell 2019, 178, 1115–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Tang, K.; Ma, J.; Zhou, L.; Liu, J.; Zeng, L.; Zhu, L.; Xu, P.; Chen, J.; Wei, K.; et al. Ketogenesis-generated β-hydroxybutyrate is an epigenetic regulator of CD8+ T-cell memory development. Nat. Cell Biol. 2020, 22, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular mechanisms and clinical applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004, 447, 619–628. [Google Scholar] [CrossRef]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Saudubray, J.M.; Marsac, C.; Limal, J.M.; Dumurgier, E.; Charpentier, C.; Ogier, H.; Coudè, F.X. Variation in plasma ketone bodies during a 24-hour fast in normal and in hypoglycemic children: relationship to age. J. Pediatr. 1981, 98, 904–908. [Google Scholar] [CrossRef]
- Rehni, A.K.; Dave, K.R. Impact of Hypoglycemia on Brain Metabolism During Diabetes. Mol. Neurobiol. 2018, 55, 9075–9088. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Terada, S.; Banjo, M.; Seike, K.; Nakano, S.; Hatta, H. Effects of β-hydroxybutyrate treatment on glycogen repletion and its related signaling cascades in epitrochlearis muscle during 120 min of postexercise recovery. Appl. Physiol. Nutr. Metab. 2019, 44, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.-B.; Crawford, P.A. Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 260–266. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Miao, Z.; Xu, X. β-hydroxybutyrate alleviates depressive behaviors in mice possibly by increasing the histone3-lysine9-β-hydroxybutyrylation. Biochem. Biophys. Res. Commun. 2017, 490, 117–122. [Google Scholar] [CrossRef]
- Fan, J.; Krautkramer, K.A.; Feldman, J.L.; Denu, J.M. Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 2015, 10, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.-P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Chriett, S.; Dąbek, A.; Wojtala, M.; Vidal, H.; Balcerczyk, A.; Pirola, L. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019, 9, 742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD+. Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A₁ receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusardi, T.A.; Akula, K.K.; Coffman, S.Q.; Ruskin, D.N.; Masino, S.A.; Boison, D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 2015, 99, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Kobow, K.; Kaspi, A.; Harikrishnan, K.N.; Kiese, K.; Ziemann, M.; Khurana, I.; Fritzsche, I.; Hauke, J.; Hahnen, E.; Coras, R.; et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013, 126, 741–756. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Locasale, J.W.; Lyssiotis, C.A.; Zheng, Y.; Teo, R.Y.; Ratanasirintrawoot, S.; Zhang, J.; Onder, T.; Unternaehrer, J.J.; Zhu, H.; et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013, 339, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Gietzen, D.W.; Lindström, S.H.; Sharp, J.W.; Teh, P.S.; Donovan, M.J. Indispensable Amino Acid-Deficient Diets Induce Seizures in Ketogenic Diet-Fed Rodents, Demonstrating a Role for Amino Acid Balance in Dietary Treatments for Epilepsy. J. Nutr. 2018, 148, 480–489. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.-P.; Li, S.-N.; Wang, J.-F.; Li, Y.; Xie, S.-S.; Xue, W.-J.; Liu, H.-M.; Huang, B.-X.; Lv, Q.-K.; Lei, L.-C.; et al. BHBA suppresses LPS-induced inflammation in BV-2 cells by inhibiting NF-κB activation. Mediators Inflamm. 2014, 2014, 983401. [Google Scholar] [CrossRef] [Green Version]
- Kong, G.; Huang, Z.; Ji, W.; Wang, X.; Liu, J.; Wu, X.; Huang, Z.; Li, R.; Zhu, Q. The Ketone Metabolite β-Hydroxybutyrate Attenuates Oxidative Stress in Spinal Cord Injury by Suppression of Class I Histone Deacetylases. J. Neurotrauma 2017, 34, 2645–2655. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 2011, 12, 1630–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, H.M.; Swerdlow, R.H. Amyloid precursor protein processing and bioenergetics. Brain Res. Bull. 2017, 133, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. Lond. 2005, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [Green Version]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. Lond. 2009, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef] [Green Version]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, Y.; Zhang, Y.; Zhang, Z.; An, W.; Zhao, X. Treatment with D-β-hydroxybutyrate protects heart from ischemia/reperfusion injury in mice. Eur. J. Pharmacol. 2018, 829, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Suzuki, M.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn. J. Pharmacol. 2001, 87, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Sasaguri, S.; Rajesh, K.G.; Suzuki, R. dl-3-Hydroxybutyrate administration prevents myocardial damage after coronary occlusion in rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1968–H1974. [Google Scholar] [CrossRef] [PubMed]
- Godar, R.J.; Ma, X.; Liu, H.; Murphy, J.T.; Weinheimer, C.J.; Kovacs, A.; Crosby, S.D.; Saftig, P.; Diwan, A. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy 2015, 11, 1537–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelton, P.K.; Carey, R.M.; Aronow, W.S.; Casey, D.E.; Collins, K.J.; Dennison Himmelfarb, C.; DePalma, S.M.; Gidding, S.; Jamerson, K.A.; Jones, D.W.; et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 2018, 71, e13–e115. [Google Scholar]
- Kjeldsen, S.E. Hypertension and cardiovascular risk: General aspects. Pharmacol. Res. 2018, 129, 95–99. [Google Scholar] [CrossRef]
- Walkowska, A.; Kuczeriszka, M.; Sadowski, J.; Olszyñski, K.H.; Dobrowolski, L.; Červenka, L.; Hammock, B.D.; Kompanowska-Jezierska, E. High salt intake increases blood pressure in normal rats: putative role of 20-HETE and no evidence on changes in renal vascular reactivity. Kidney Blood Press. Res. 2015, 40, 323–334. [Google Scholar] [CrossRef]
- Chakraborty, S.; Galla, S.; Cheng, X.; Yeo, J.-Y.; Mell, B.; Singh, V.; Yeoh, B.; Saha, P.; Mathew, A.V.; Vijay-Kumar, M.; et al. Salt-Responsive Metabolite, β-Hydroxybutyrate, Attenuates Hypertension. Cell Rep. 2018, 25, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Cox, P.J.; Clarke, K. Acute nutritional ketosis: Implications for exercise performance and metabolism. Extrem. Physiol. Med. 2014, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Castellano, C.A.; Cuenoud, B.; Cunnane, S.C. Medium Chain Triglycerides Modulate the Ketogenic Effect of a Metabolic Switch. Front. Nutr. 2020, 7, 3. [Google Scholar] [CrossRef]
- Kiens, B.; Astrup, A. Ketogenic Diets for Fat Loss and Exercise Performance: Benefits and Safety? Exerc. Sport Sci. Rev. 2015, 43, 109. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.; Sharman, M.J.; Avery, N.G.; Love, D.M.; Gómez, A.L.; Scheett, T.P.; Kraemer, W.J.; Volek, J.S. Endurance capacity and high-intensity exercise performance responses to a high fat diet. Int. J. Sport Nutr. Exerc. Metab. 2003, 13, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.J.; Kirk, T.; Ashmore, T.; Willerton, K.; Evans, R.; Smith, A.; Murray, A.J.; Stubbs, B.; West, J.; McLure, S.W.; et al. Nutritional Ketosis Alters Fuel Preference and Thereby Endurance Performance in Athletes. Cell Metab. 2016, 24, 256–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phinney, S.D.; Bistrian, B.R.; Evans, W.J.; Gervino, E.; Blackburn, G.L. The human metabolic response to chronic ketosis without caloric restriction: Preservation of submaximal exercise capability with reduced carbohydrate oxidation. Metab. Clin. Exp. 1983, 32, 769–776. [Google Scholar] [CrossRef]
- Volek, J.S.; Freidenreich, D.J.; Saenz, C.; Kunces, L.J.; Creighton, B.C.; Bartley, J.M.; Davitt, P.M.; Munoz, C.X.; Anderson, J.M.; Maresh, C.M.; et al. Metabolic characteristics of keto-adapted ultra-endurance runners. Metab. Clin. Exp. 2016, 65, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.C.; Bryce, G.R.; Conlee, R.K. Adaptations to a high-fat diet that increase exercise endurance in male rats. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 78–83. [Google Scholar] [CrossRef]
- Goedecke, J.H.; Christie, C.; Wilson, G.; Dennis, S.C.; Noakes, T.D.; Hopkins, W.G.; Lambert, E.V. Metabolic adaptations to a high-fat diet in endurance cyclists. Metab. Clin. Exp. 1999, 48, 1509–1517. [Google Scholar] [CrossRef]
- Gibson, A.A.; Seimon, R.V.; Lee, C.M.Y.; Ayre, J.; Franklin, J.; Markovic, T.P.; Caterson, I.D.; Sainsbury, A. Do ketogenic diets really suppress appetite? A systematic review and meta-analysis. Obes. Rev. 2015, 16, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Lambert, E.V.; Speechly, D.P.; Dennis, S.C.; Noakes, T.D. Enhanced endurance in trained cyclists during moderate intensity exercise following 2 weeks adaptation to a high fat diet. Eur. J. Appl. Physiol. Occup. Physiol. 1994, 69, 287–293. [Google Scholar] [CrossRef]
- Zajac, A.; Poprzecki, S.; Maszczyk, A.; Czuba, M.; Michalczyk, M.; Zydek, G. The effects of a ketogenic diet on exercise metabolism and physical performance in off-road cyclists. Nutrients 2014, 6, 2493–2508. [Google Scholar] [CrossRef]
- Sansone, M.; Sansone, A.; Borrione, P.; Romanelli, F.; Di Luigi, L.; Sgrò, P. Effects of Ketone Bodies on Endurance Exercise. Curr. Sports Med. Rep. 2018, 17, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Khodadadi, S.; Sobhani, N.; Mirshekar, S.; Ghiasvand, R.; Pourmasoumi, M.; Miraghajani, M.; Dehsoukhteh, S.S. Tumor Cells Growth and Survival Time with the Ketogenic Diet in Animal Models: A Systematic Review. Int. J. Prev. Med. 2017, 8, 35. [Google Scholar]
- Aminzadeh-Gohari, S.; Feichtinger, R.G.; Vidali, S.; Locker, F.; Rutherford, T.; O’Donnel, M.; Stöger-Kleiber, A.; Mayr, J.A.; Sperl, W.; Kofler, B. A ketogenic diet supplemented with medium-chain triglycerides enhances the anti-tumor and anti-angiogenic efficacy of chemotherapy on neuroblastoma xenografts in a CD1-nu mouse model. Oncotarget 2017, 8, 64728–64744. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.D.; Aminzadeh-Gohari, S.; Tulipan, J.; Catalano, L.; Feichtinger, R.G.; Kofler, B. Ketogenic diet in the treatment of cancer—Where do we stand? Mol. Metab. 2020, 33, 102–121. [Google Scholar] [CrossRef]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Lin, R.; Jin, L.; Zhao, L.; Kang, H.-B.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Konstantakou, E.; et al. Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth. Cell Metab. 2017, 25, 358–373. [Google Scholar] [CrossRef] [Green Version]
- Klement, R.J. Beneficial effects of ketogenic diets for cancer patients: A realist review with focus on evidence and confirmation. Med. Oncol. 2017, 34, 132. [Google Scholar] [CrossRef]
- Buchhalter, J.R.; D’Alfonso, S.; Connolly, M.; Fung, E.; Michoulas, A.; Sinasac, D.; Singer, R.; Smith, J.; Singh, N.; Rho, J.M. The relationship between d-beta-hydroxybutyrate blood concentrations and seizure control in children treated with the ketogenic diet for medically intractable epilepsy. Epilepsia Open 2017, 2, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Lambrechts, D.A.; de Kinderen, R.J.; Vles, J.S.; de Louw, A.J.; Aldenkamp, A.P.; Majoie, H.J. A randomized controlled trial of the ketogenic diet in refractory childhood epilepsy. Acta Neurol. Scand. 2017, 135, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.C.; Kim, Y.J.; Kim, D.W.; Kim, H.D. Efficacy and safety of the ketogenic diet for intractable childhood epilepsy: Korean multicentric experience. Epilepsia 2005, 46, 272–279. [Google Scholar] [CrossRef]
- Dressler, A.; Stöcklin, B.; Reithofer, E.; Benninger, F.; Freilinger, M.; Hauser, E.; Reiter-Fink, E.; Seidl, R.; Trimmel-Schwahofer, P.; Feucht, M. Long-term outcome and tolerability of the ketogenic diet in drug-resistant childhood epilepsy—the Austrian experience. Seizure 2010, 19, 404–408. [Google Scholar] [CrossRef]
- De Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dąbek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. https://doi.org/10.3390/nu12030788
Dąbek A, Wojtala M, Pirola L, Balcerczyk A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients. 2020; 12(3):788. https://doi.org/10.3390/nu12030788
Chicago/Turabian StyleDąbek, Arkadiusz, Martyna Wojtala, Luciano Pirola, and Aneta Balcerczyk. 2020. "Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States" Nutrients 12, no. 3: 788. https://doi.org/10.3390/nu12030788