Identification of Persuasive Antiviral Natural Compounds for COVID-19 by Targeting Endoribonuclease NSP15: A Structural-Bioinformatics Approach



Abstract
:1. Introduction
2. Results
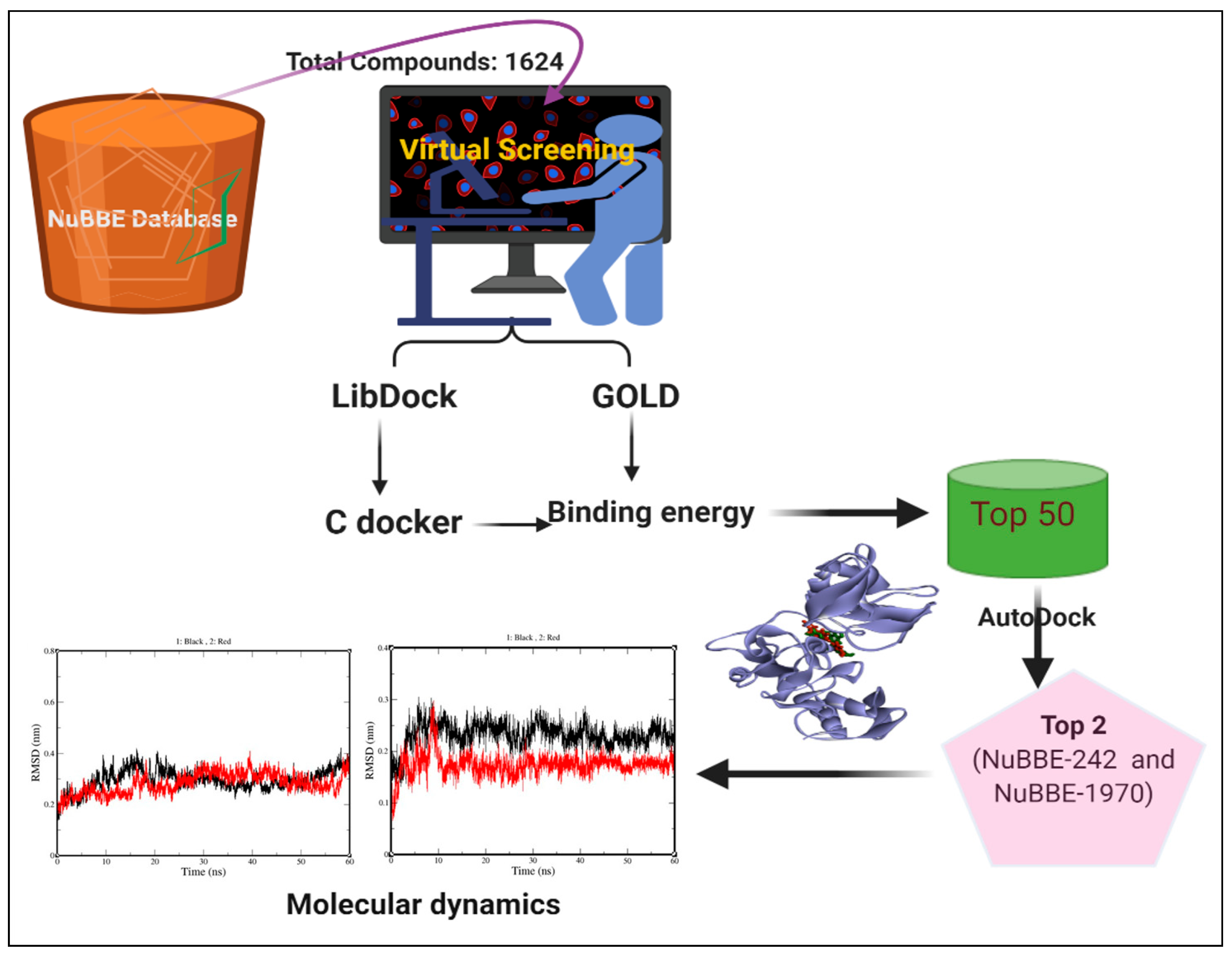
2.1. Virtual Screening
2.2. Pharmacokinetic Properties
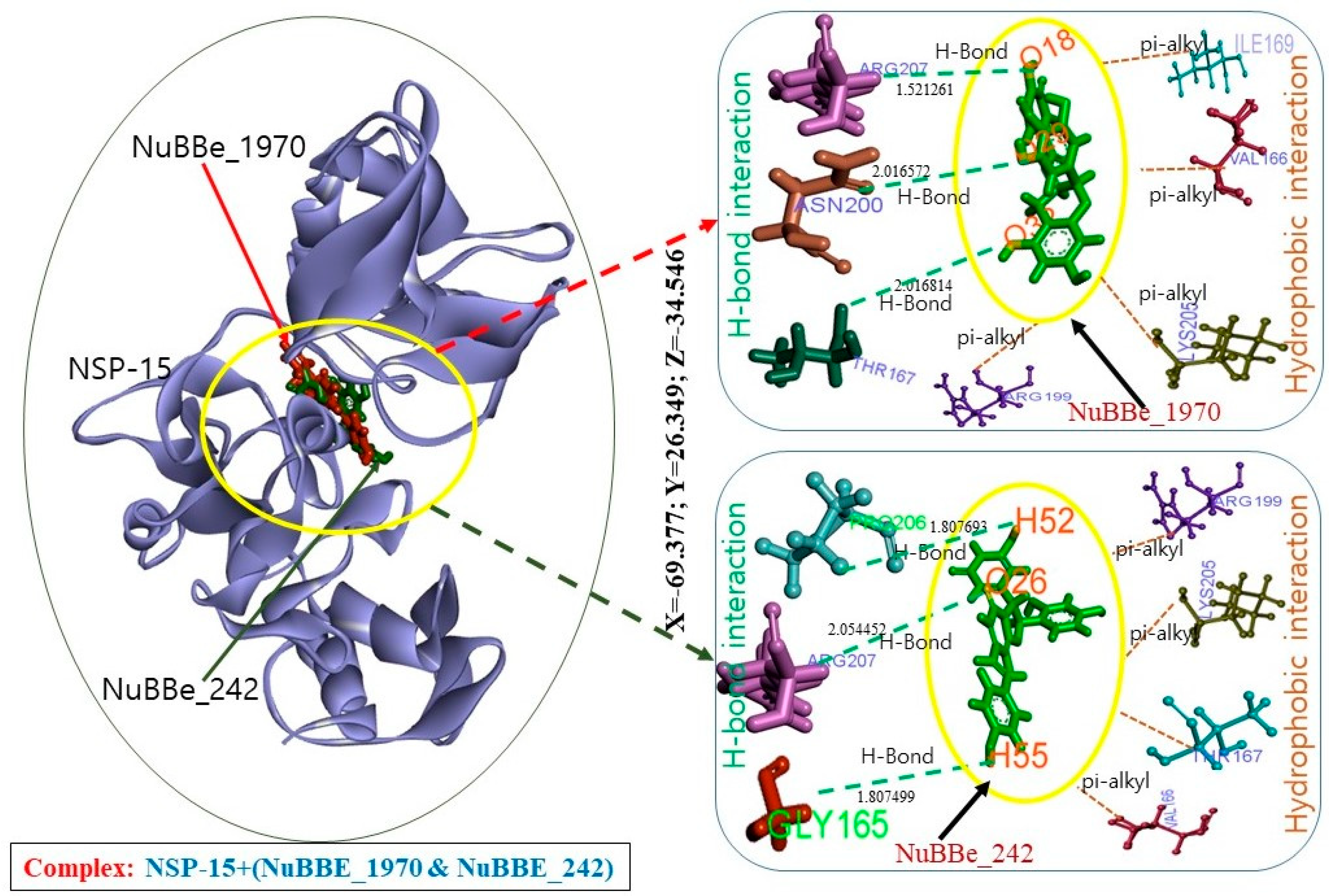
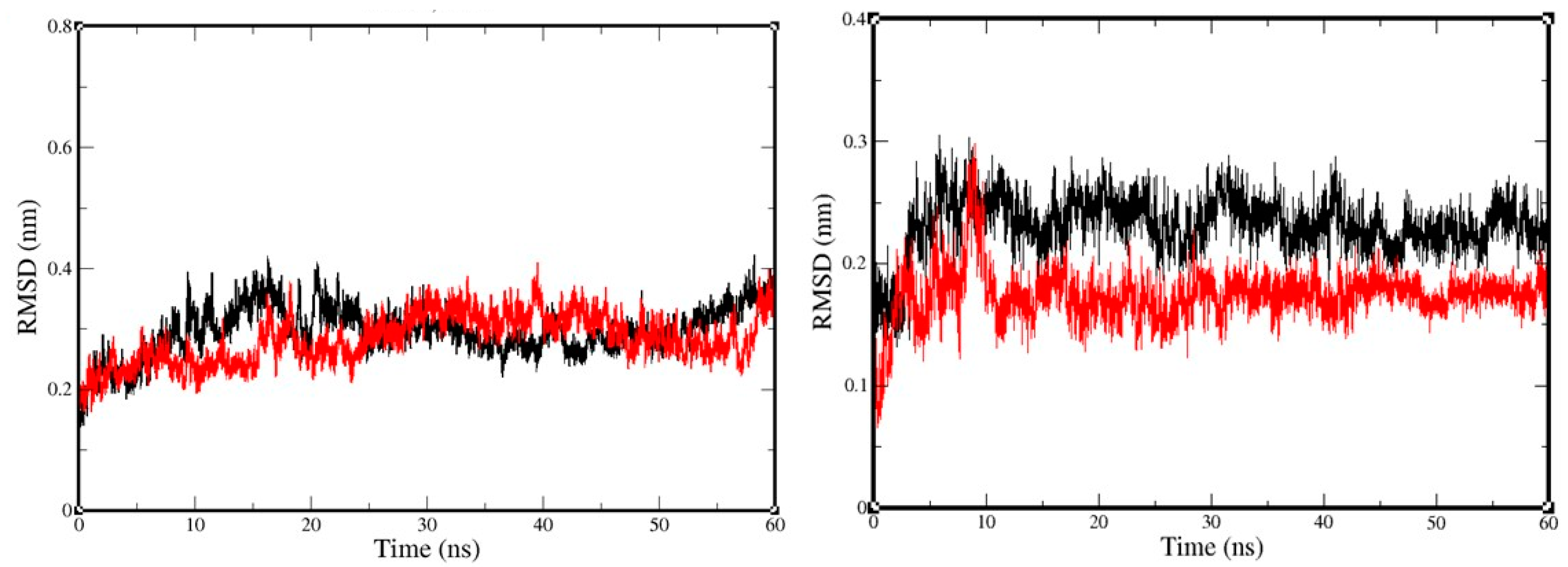
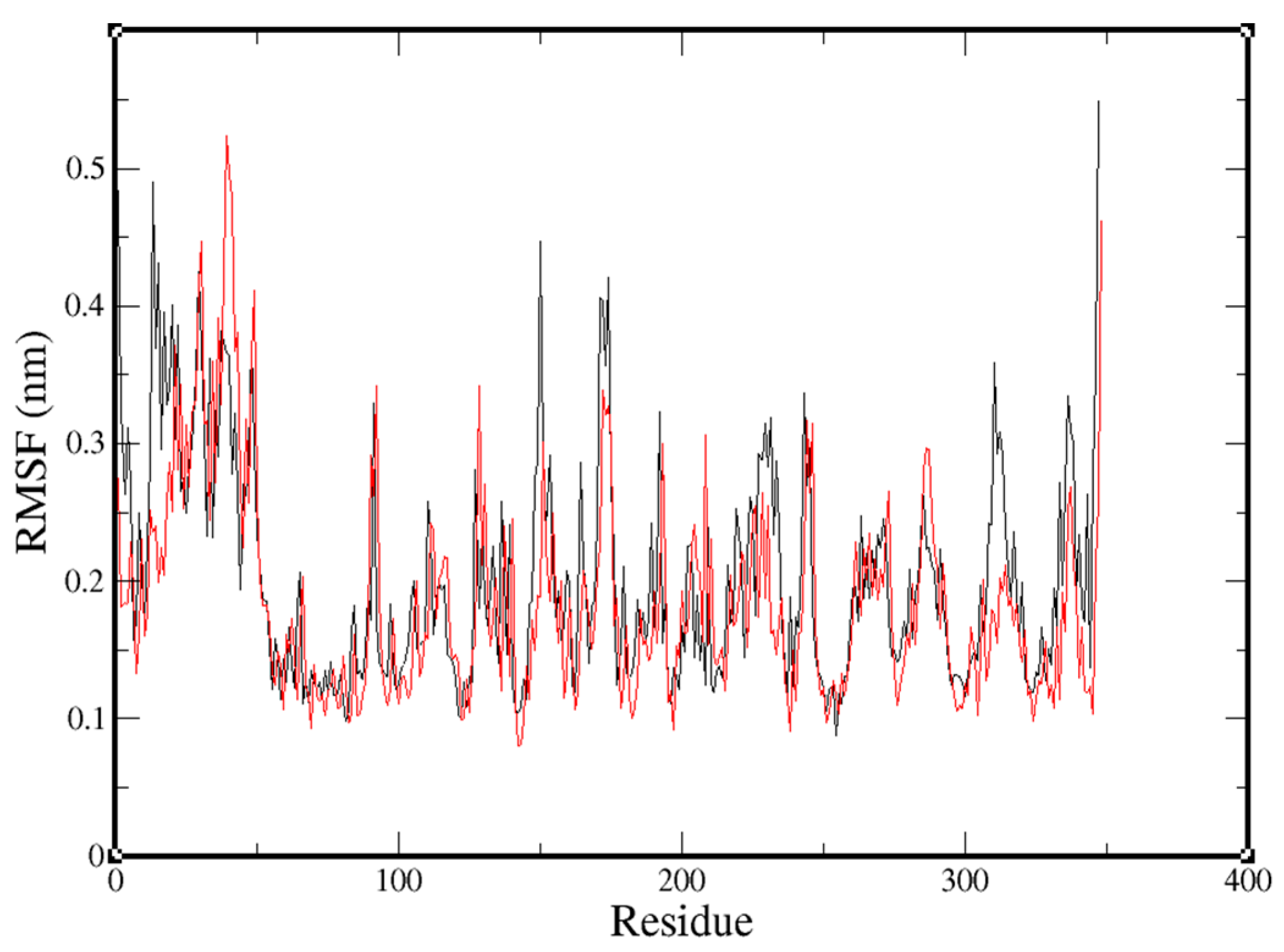
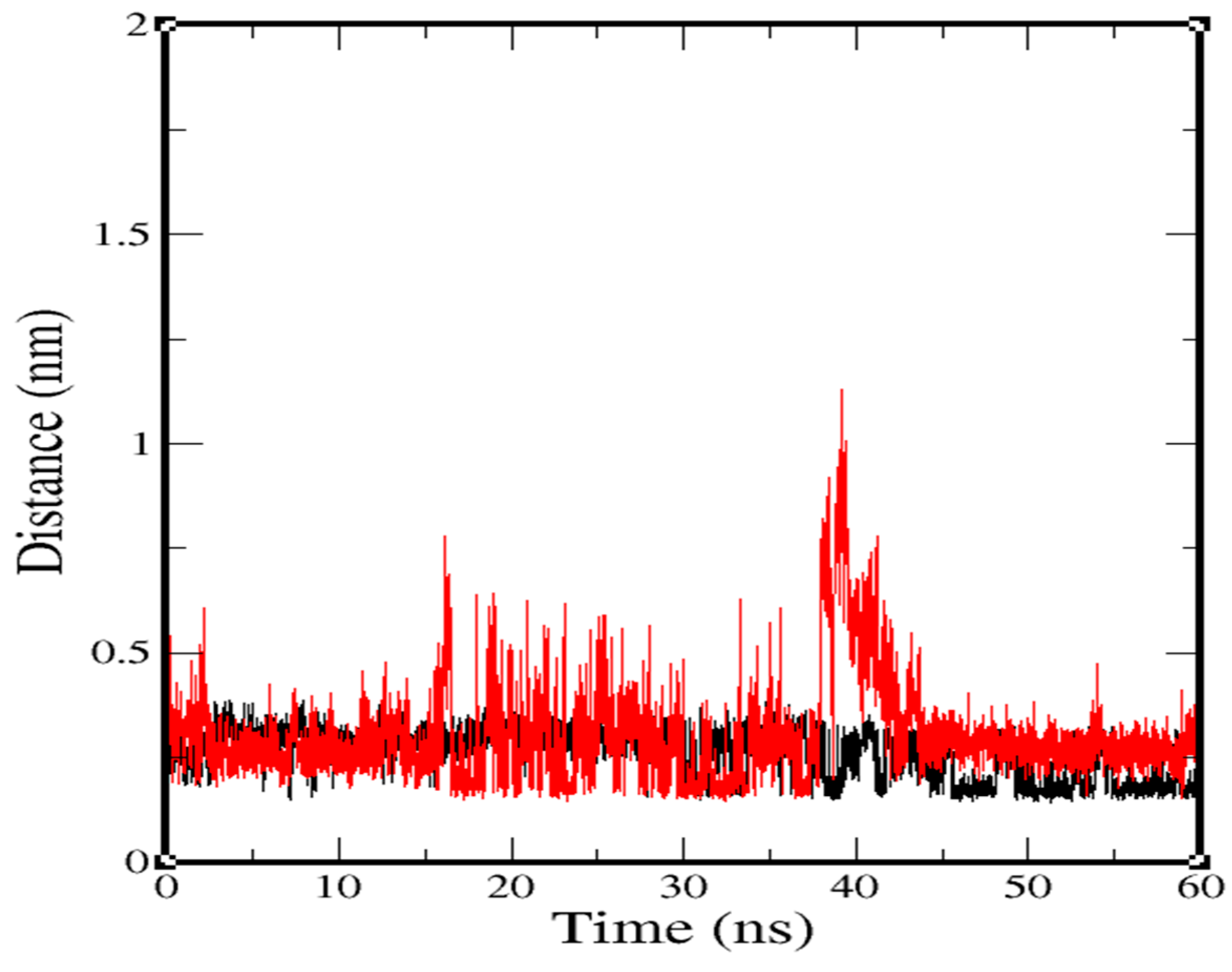
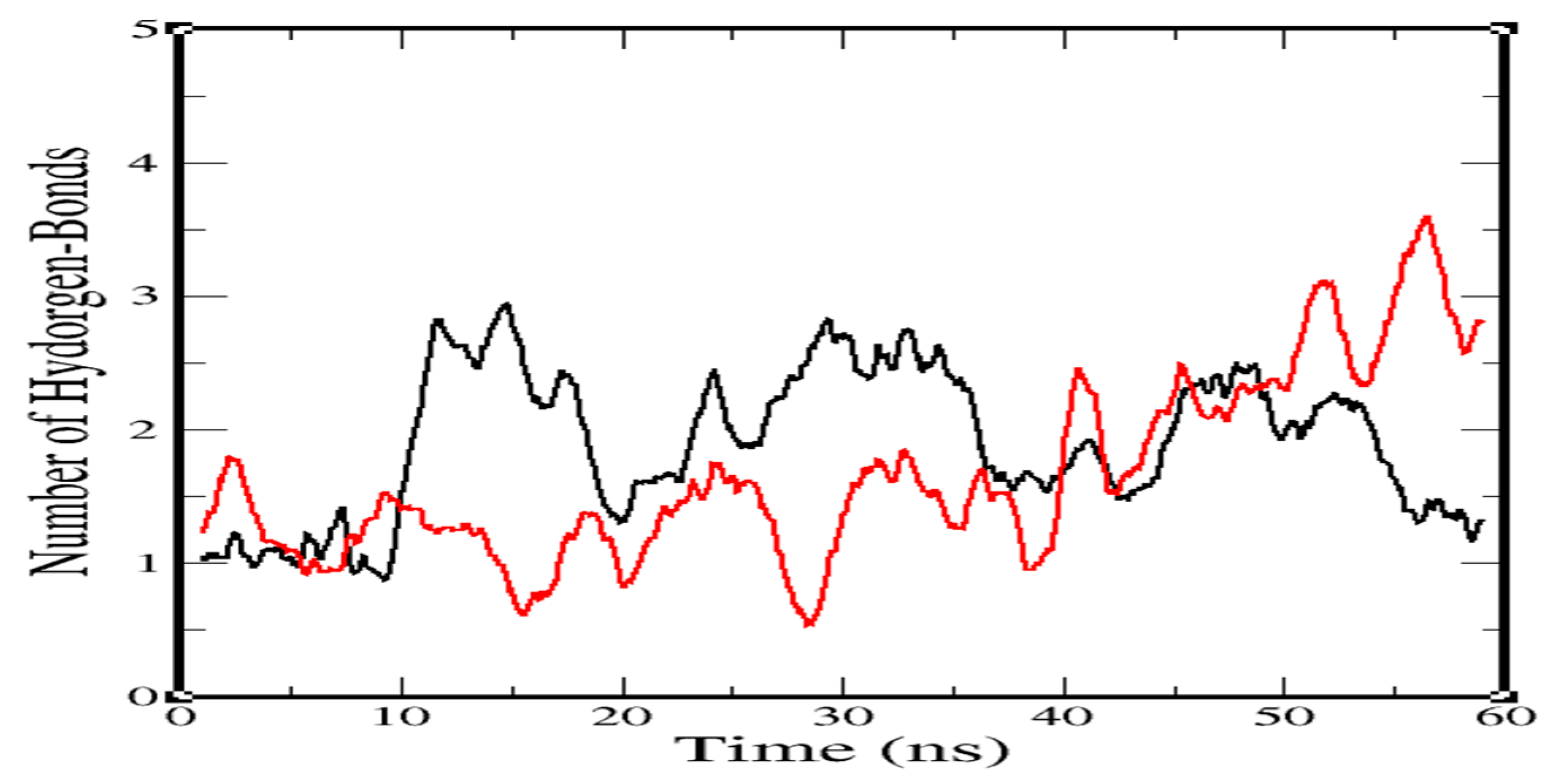
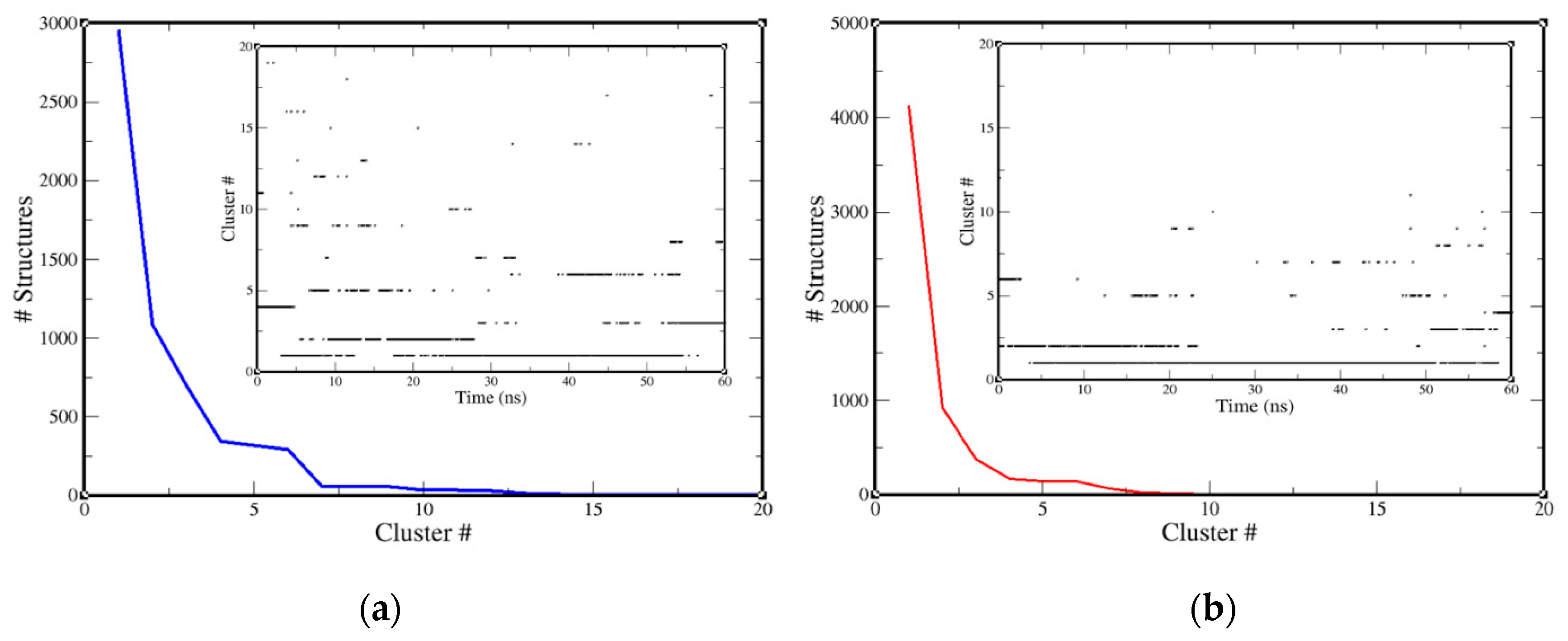
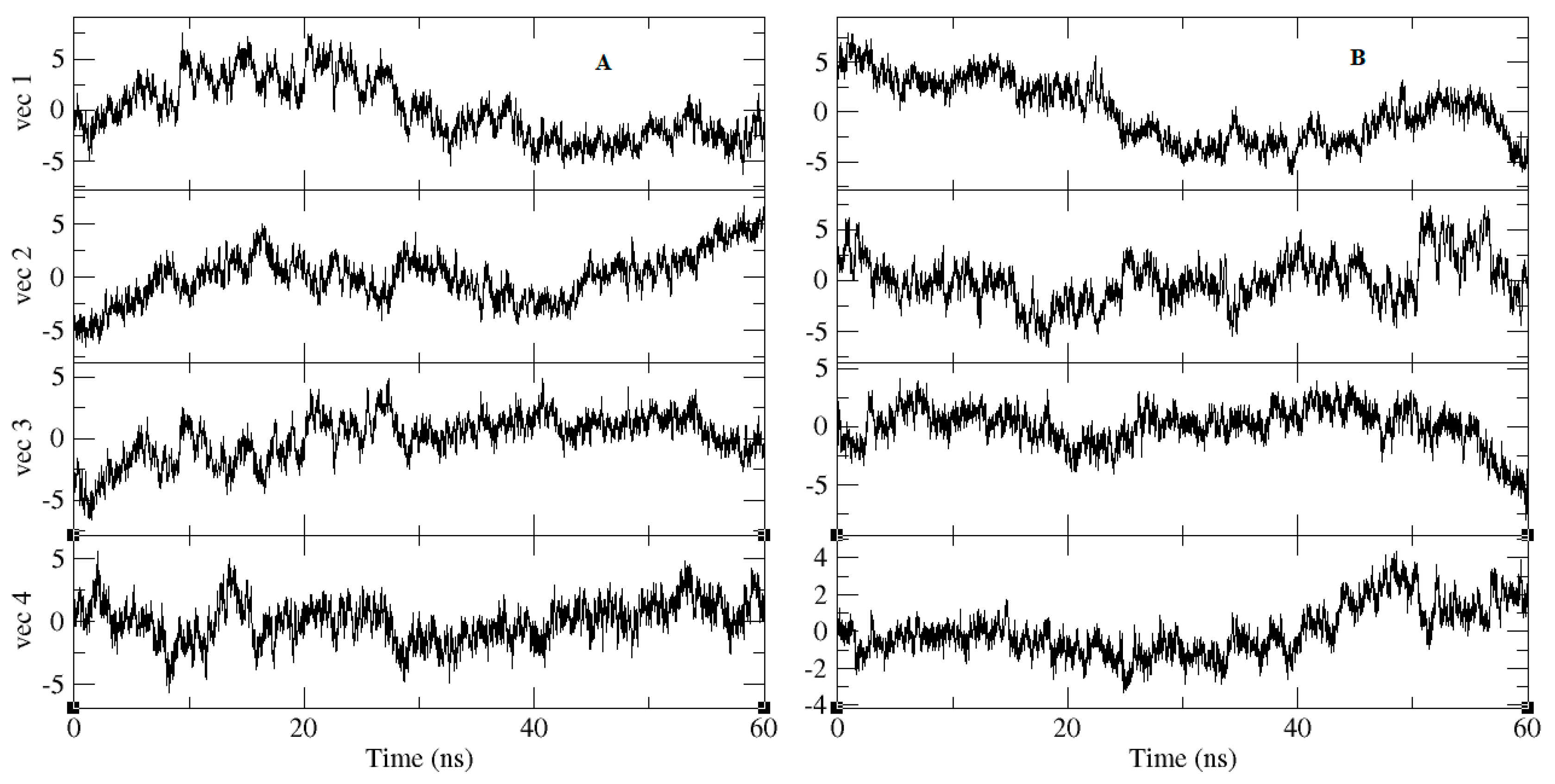
2.3. Molecular-Dynamics Simulation
3. Discussion
4. Materials and Methods
4.1. Data Sources and Preparation
4.2. Target Structure Preparation
4.3. Virtual Screening and Docking Experiment
4.4. Molecular-Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, N.; Viceconte, G.; Ergonul, O.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are they closely related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Lorusso, A. Novel human coronavirus (SARS-CoV-2): A lesson from animal coronaviruses. Vet. Microbiol. 2020, 244, 108693. [Google Scholar] [CrossRef] [PubMed]
- Unchwaniwala, N.; Ahlquist, P. Coronavirus dons a new crown. Science 2020, 369, 1306–1307. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Lokugamage, K.G.; Makino, S. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. Adv. Virus Res. 2016, 96, 165–192. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Berretta, M.; Venanzi Rullo, E.; Nunnari, G.; Cacopardo, B. Differences and similarities between Severe Acute Respiratory Syndrome (SARS)-CoronaVirus (CoV) and SARS-CoV-2. Would a rose by another name smell as sweet? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2781–2783. [Google Scholar] [CrossRef]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases; ACS Publications: Washington, DC, USA, 2020. [Google Scholar]
- Sadegh, S.; Matschinske, J.; Blumenthal, D.B.; Galindez, G.; Kacprowski, T.; List, M.; Nasirigerdeh, R.; Oubounyt, M.; Pichlmair, A.; Rose, T.D.; et al. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 2020, 11, 3518. [Google Scholar] [CrossRef]
- Alazmi, M.; Motwalli, O. In silico virtual screening, characterization, docking and molecular dynamics studies of crucial SARS-CoV-2 proteins. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Quimque, M.T.J.; Notarte, K.I.R.; Fernandez, R.A.T.; Mendoza, M.A.O.; Liman, R.A.D.; Lim, J.A.K.; Pilapil, L.A.E.; Ong, J.K.H.; Pastrana, A.M.; Khan, A.; et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Salman, S.; Shah, F.H.; Idrees, J.; Idrees, F.; Velagala, S.; Ali, J.; Khan, A.A. Virtual screening of immunomodulatory medicinal compounds as promising anti-SARS-COV-2 inhibitors. Futur. Virol. 2020, 15, 267–275. [Google Scholar] [CrossRef]
- Joshi, G.; Poduri, R. Virtual screening enabled selection of antiviral agents against Covid-19 disease targeting coronavirus endoribonuclease NendoU: Plausible mechanistic interventions in the treatment of new virus strain. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Fatima, S.; Gupta, P.; Sharma, S.; Sharma, A.; Agarwal, S.M. ADMET profiling of geographically diverse phytochemical using chemoinformatic tools. Futur. Med. Chem. 2020, 12, 69–87. [Google Scholar] [CrossRef]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Devaux, C.A.; Rolain, J.M.; Colson, P.; Raoult, D. New insights on the antiviral effects of chloroquine against coronavirus: What to expect for COVID-19? Int. J. Antimicrob. Agents 2020, 55, 105938. [Google Scholar] [CrossRef]
- Maxwell, N.M.; Nevin, R.L.; Stahl, S.; Block, J.; Shugarts, S.; Wu, A.H.; Dominy, S.; Solano-Blanco, M.A.; Kappelman-Culver, S.; Lee-Messer, C.; et al. Prolonged neuropsychiatric effects following management of chloroquine intoxication with psychotropic polypharmacy. Clin. Case Rep. 2015, 3, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Sinha, M.; Ahmad, K.; Khalid, M.; Choi, I. Study of Caspase 8 Inhibition for the Management of Alzheimer’s Disease: A Molecular Docking and Dynamics Simulation. Molecules 2020, 25, 2071. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.; Koellner, G. Hydrogen bonds with pi-acceptors in proteins: Frequencies and role in stabilizing local 3D structures. J. Mol. Biol. 2001, 305, 535–557. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Khan, H.; Danish Rizvi, S.M.; Ansari, S.A.; Ullah, R.; Rastrelli, L.; Mahmood, H.M.; Siddiqui, M.H. Computational Study of Natural Compounds for the Clearance of Amyloid-Betaeta: A Potential Therapeutic Management Strategy for Alzheimer’s Disease. Molecules 2019, 24, 3233. [Google Scholar] [CrossRef] [Green Version]
- Abdelli, I.; Hassani, F.; Bekkel Brikci, S.; Ghalem, S. In silico study the inhibition of angiotensin converting enzyme 2 receptor of COVID-19 by Ammoides verticillata components harvested from Western Algeria. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Ottaviani, G.; Gosling, D.J.; Patissier, C.; Rodde, S.; Zhou, L.; Faller, B. What is modulating solubility in simulated intestinal fluids? Eur. J. Pharm. Sci. 2010, 41, 452–457. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Pilon, A.C.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 7215. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef] [PubMed]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Aided Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Toukan, K.; Rahman, A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B 1985, 31, 2643. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interactions | Target | NuBBE-1970 | NuBBE-242 |
|---|---|---|---|
| H Bond | NSP15 | THR167:HN—UNK0:O32 ASN200:HN—UNK0:O29 ARG207:HE—UNK0:O18 | UNK0:H52—PRO206:O UNK0:H55—GLY165:O ARG207:HE—UNK0:O26 |
| Hydrophobic interaction | VAL166—UNK0 UNK0—ILE169 UNK0—ARG199 UNK0—LYS205 | ARG199:HD1—UNK0:O26 LYS205:HE2—UNK0:O25 THR167:HN—UNK0 UNK0—VAL166 | |
| Electrostatic interaction | GLU203:OE1—UNK0. | — | |
| Total amino acid residues | GLY165, VAL166, THR167, LEU168, ILE169, ARG199, ASN200, GLU203, PHE204, LYS205, and ARG207, | LYS90, GLY165, VAL166, THR167, LEU168, ILE169, GLN197, SER198, ARG199, ASN200, LYS205, PRO206, and ARG207 |
| Compounds Name | Binding Energy (kcal/mol) | Inhibition Constant (µM) | Intermolecular Energy (kcal/mol) | Van der Waals, Hydrogen Bond, and Desolvation Energy(kcal/mol) | Electrostatic Energy (kcal/mol) |
|---|---|---|---|---|---|
| NuBBE-1970 | −6.28 | 24.98 | −8.52 | −8.30 | −0.22 |
| NuBBE-242 | −5.72 | 64.48 | −8.49 | −8.45 | −0.02 |
| Properties | NuBBE_242 | NuBBE_1970 | |
|---|---|---|---|
| Physicochemical | Formula | C22H18O11 | C28H22O7 |
| Molecular weight | 458.37 g/mol | 470.47 g/mol | |
| Number of heavy atoms | 33 | 35 | |
| Number of aromatic heavy atoms | 18 | 24 | |
| Number of rotatable bonds | 4 | 4 | |
| Number of H-bond acceptors | 11 | 7 | |
| Number of H-bond donors | 8 | 6 | |
| Molar refractivity | 112.06 | 132.27 | |
| TPSA | 197.37 Å2 | 130.61 Å2 | |
| Lipophilicity (Log Po/w) | iLOGP | 1.83 | 2.40 |
| XLOGP3 | 1.17 | 5.05 | |
| WLOGP | 1.91 | 4.81 | |
| MLOGP | −0.18 | 2.33 | |
| SILICOS-IT | 0.57 | 3.98 | |
| Consensus Log Po/w | 1.06 | 3.71 | |
| Water solubility | Log S (ESOL) | −3.56 (soluble) | −6.18 (poorly soluble) |
| Log S (Ali) | −4.91 (moderately soluble) | −7.53 (poorly soluble) | |
| Log S (SILICOS-IT) | −2.50 (Soluble) | −6.27 (poorly soluble) | |
| Pharmacokinetics | Log Kp (skin permeation) | −8.27 cm/s | −5.58 cm/s |
| GI absorption | Low | Low | |
| BBB permeant | No | No | |
| CYP2C9 inhibitor | No | Yes | |
| AMES toxicity | No | No | |
| hERG I inhibitor | No | No | |
| Hepatotoxicity | No | No | |
| Skin sensitization | No | No | |
| Minnow toxicity | 5.305 (log mM) | 1.927 (log mM) | |
| Druglikeness | Lipinski | 2 violations: NorO > 10, NHorOH > 5 | 1 violation: NHorOH > 5 |
| Ghose | Yes | 1 violation: MR > 130 | |
| Veber | 1 violation: TPSA > 140 | Yes | |
| Egan | 1 violation: TPSA > 131.6 | Yes | |
| Muegge | 3 violations: TPSA > 150, H-acc > 10, H-don > 5 | 2 violations: XLOGP3 > 5, H-don > 5 | |
| Bioavailability score | 0.17 | 0.55 | |
| Medicinal chemistry | PAINS | 1 alert | 0 alert |
| Brenk | 1 alert | 1 alert | |
| Leadlikeness | 1 violation: MW > 350 | 2 violations: MW > 350, XLOGP3 > 3.5 | |
| Synthetic accessibility | 4.20 | 4.48 | |
| Compound | Binding Energy | Ligand Energy | Protein Energy | Complex Energy | Complex Entropy | Protein Entropy | Ligand Entropy | Entropic Energy | C Docker Energy | C Docker Interaction Energy |
|---|---|---|---|---|---|---|---|---|---|---|
| NuBBE_242 | −483.68 | 162.69 | −14,112.7 | −14,433.7 | −31.45 | −31.43 | −20.34 | 20.33 | 45.56 | 57.05 |
| NuBBE_1970 | −305.87 | 78.97 | −14,088.5 | −14,315.4 | −31.45 | −31.43 | −20.81 | 20.80 | 22.42 | 55.35 |
| NuBBE_1485 | −296.971 | 122.49 | −14,120.4 | −14,294.9 | −31.45 | −31.43 | −20.06 | 20.05 | 8.61 | 51.05 |
| NuBBE_1966 | −295.89 | 97.28 | −14,122.8 | −14,321.4 | −31.45 | −31.43 | −20.77 | 20.76 | 31.35 | 58.23 |
| NuBBE_1078 | −287.46 | 77.71 | −14,129.4 | −14,339.1 | −31.44 | −31.43 | −19.47 | 19.46 | 6.98 | 37.12 |
| NuBBE_1969 | −280.30 | 99.14 | −14,130.7 | −14,311.9 | −31.45 | −31.43 | −20.43 | 20.41 | −10.80 | 44.28 |
| NuBBE_1263 | −277.71 | 29.814 | −14,110.4 | −14,358.3 | −31.44 | −31.43 | −19.77 | 19.76 | 22.55 | 40.49 |
| NuBBE_1969 | −277.03 | 95.89 | −14,122.6 | −14,303.7 | −31.45 | −31.43 | −20.48 | 20.46 | −6.83 | 45.60 |
| NuBBE_1770 | −270.24 | 35.044 | −14,080 | −14,315.2 | −31.44 | −31.43 | −19.44 | 19.43 | −4.63 | 35.99 |
| NuBBE_2205 | −268.32 | 230.91 | −14,113.1 | −14,150.5 | −31.45 | −31.43 | −20.23 | 20.22 | −94.30 | 59.87 |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, M.; Saeed, A.; Alam, M.J.; Alreshidi, M. Identification of Persuasive Antiviral Natural Compounds for COVID-19 by Targeting Endoribonuclease NSP15: A Structural-Bioinformatics Approach. Molecules 2020, 25, 5657. https://doi.org/10.3390/molecules25235657
Saeed M, Saeed A, Alam MJ, Alreshidi M. Identification of Persuasive Antiviral Natural Compounds for COVID-19 by Targeting Endoribonuclease NSP15: A Structural-Bioinformatics Approach. Molecules. 2020; 25(23):5657. https://doi.org/10.3390/molecules25235657
Chicago/Turabian StyleSaeed, Mohd, Amir Saeed, Md Jahoor Alam, and Mousa Alreshidi. 2020. "Identification of Persuasive Antiviral Natural Compounds for COVID-19 by Targeting Endoribonuclease NSP15: A Structural-Bioinformatics Approach" Molecules 25, no. 23: 5657. https://doi.org/10.3390/molecules25235657