
Utilization of Human Samples for Assessment of Mitochondrial Bioenergetics: Gold Standards, Limitations, and Future Perspectives

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Material and Reagents
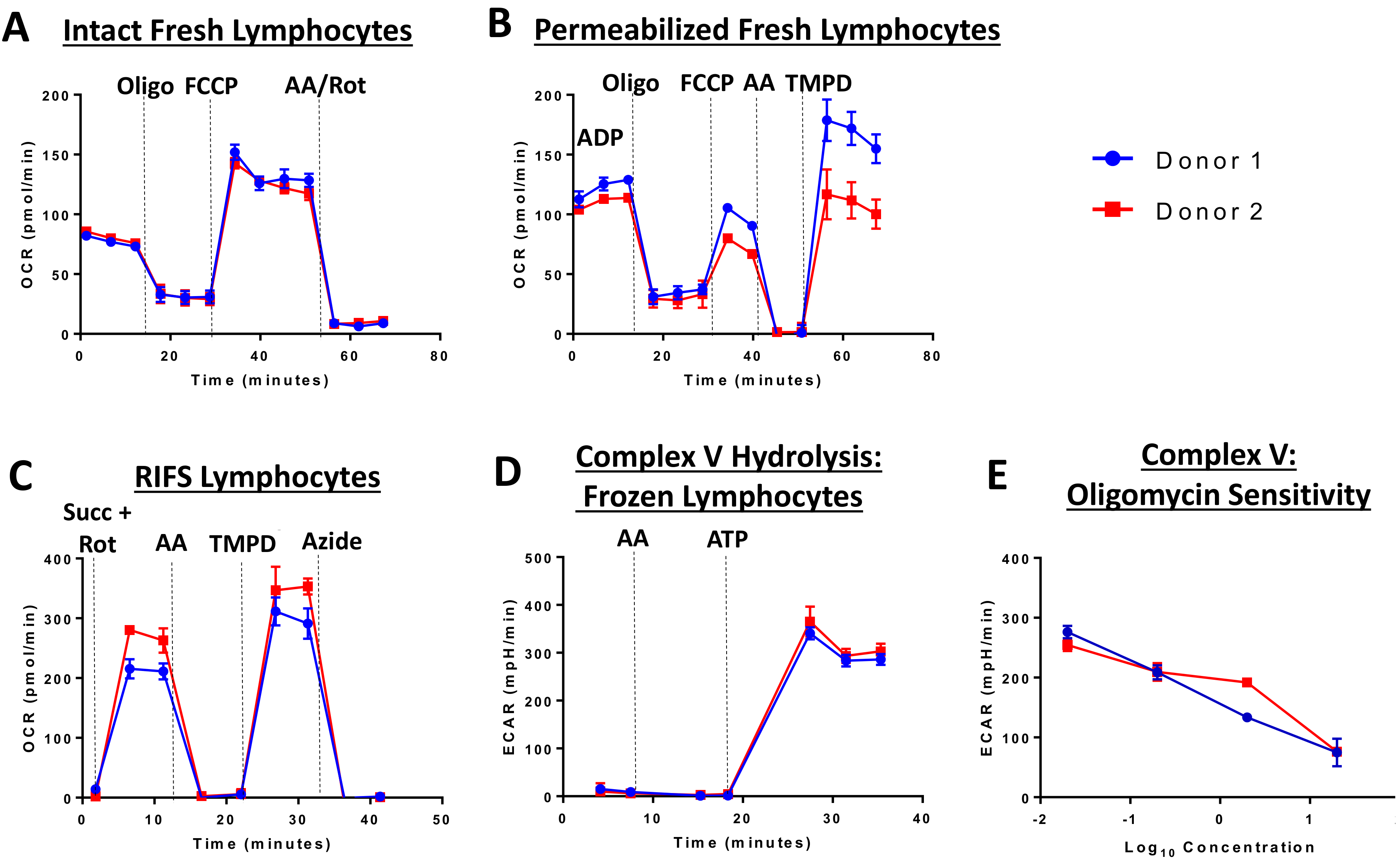
2.3. XF96 Extracellular Flux Analysis in Fresh and Frozen Cells
2.3.1. Intact Cell Respirometry
2.3.2. Permeabilized Cell Respirometry
2.3.3. RIFS
2.3.4. Complex V ATP Hydrolysis Assay
2.4. MitoTracker Deep Red in Frozen Cells
2.5. Enzymatic Assays in Frozen Cells
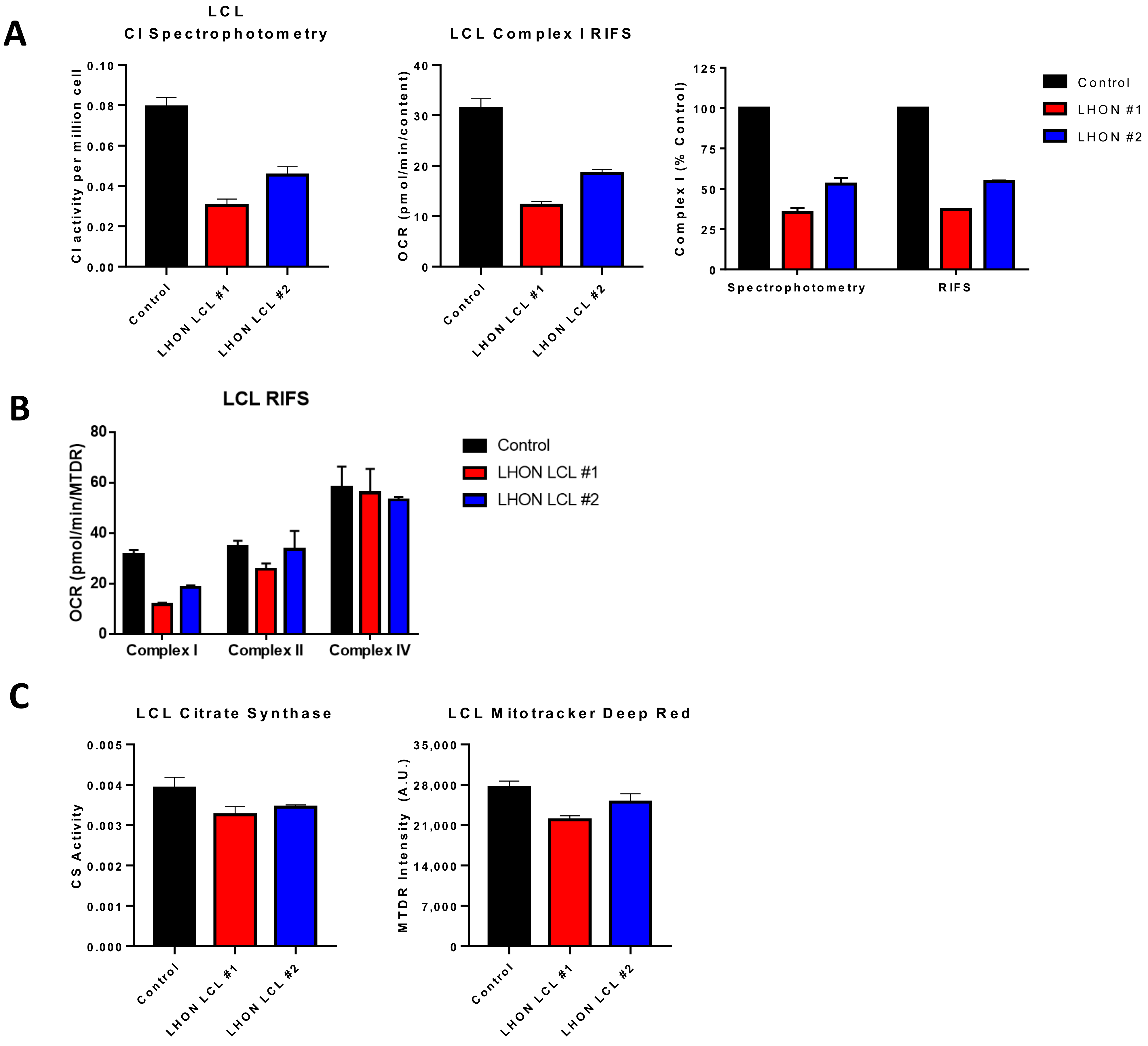
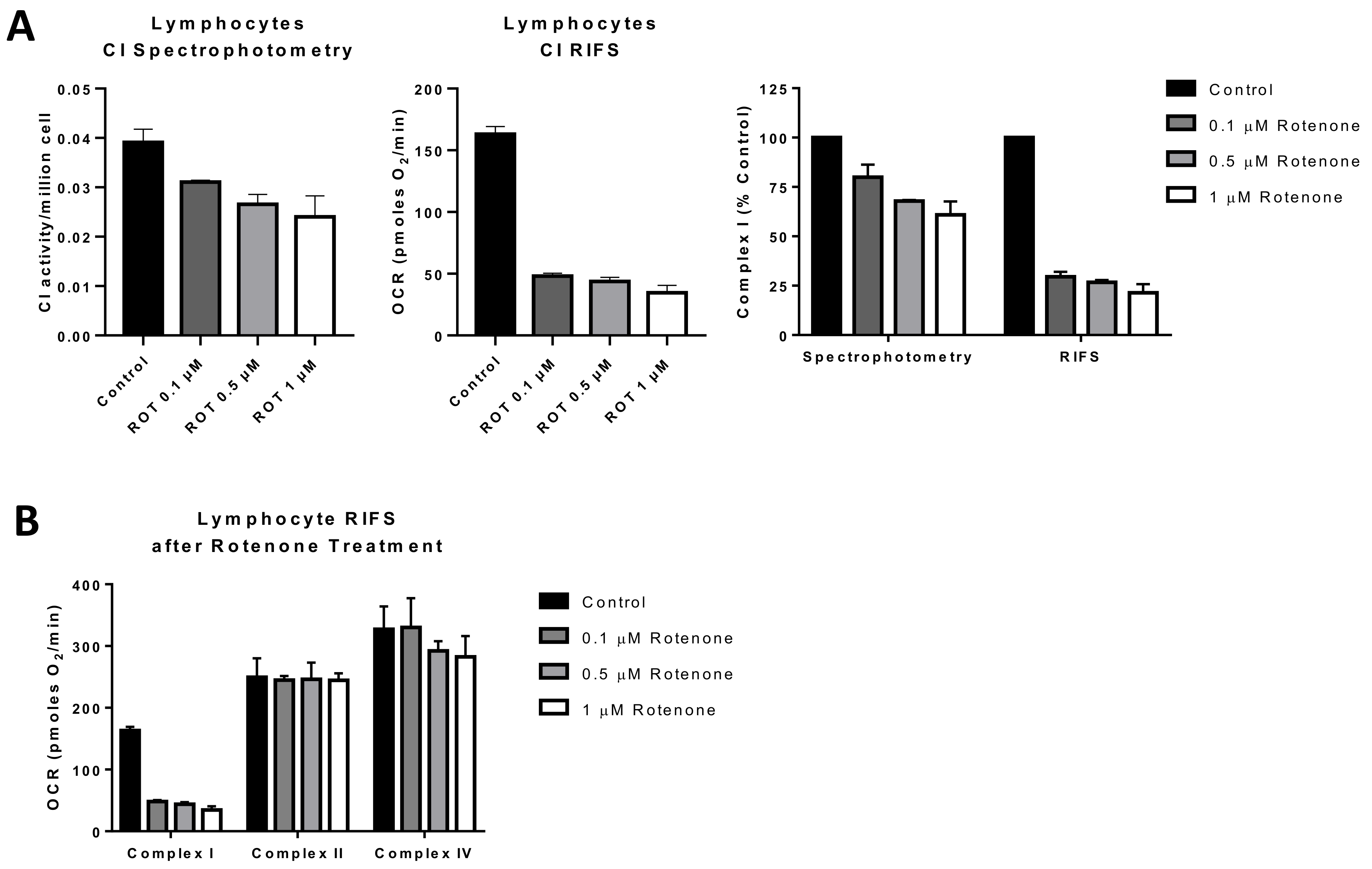
2.5.1. Complex I Activity
2.5.2. Citrate Synthase Activity
2.6. Statistical Analysis
3. Gold Standards: Human Samples for Assessment of Mitochondrial Bioenergetic Function
3.1. Bioenergetics Testing in Mitochondrial Disorders
3.2. Skeletal Muscle Biopsies
3.3. Human Fibroblasts
Considerations for Utilizing Fibroblasts for Respirometry Studies
3.4. Summary of Benchmark Human Samples to Study Mitochondrial Bioenergetics
4. Blood-Based Bioenergetics—Respirometry as a Systemic Biomarker of Mitochondrial Function
Considerations for Utilizing Circulating Blood Cells for Respirometry Studies
5. Mitochondrial Function in Previously Frozen Specimens: Overview and Commentary on an Updated Approach to Respirometry
5.1. Applications for Respirometry in Previously Frozen Samples
5.2. Limitations of RIFS
6. Future Outlook: Respirometry in Non-Invasive Samples
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional mitochondria in health and disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, E.; Enriquez, J.A.; Perez-Martos, A.; Montoya, J.; Fernandez-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 2011, 11, 207–213. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria bound to lipid droplets: Where mitochondrial dynamics regulate lipid storage and utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Herbers, E.; Kekalainen, N.J.; Hangas, A.; Pohjoismaki, J.L.; Goffart, S. Tissue specific differences in mitochondrial DNA maintenance and expression. Mitochondrion 2019, 44, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E. Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int. J. Biochem. Cell Biol. 2009, 41, 1837–1845. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and interpretation of microplate-based oxygen consumption and pH data. Methods Enzymol. 2014, 547, 309–354. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.S.; Rogers, G.W.; Murphy, A.N. Measuring mitochondrial function in permeabilized cells using the seahorse XF analyzer or a clark-type oxygen electrode. Curr. Protoc. Toxicol. 2014, 60, 25.2.1–25.2.16. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Ost, M.; Doerrier, C.; Gama-Perez, P.; Moreno-Gomez, S. Analysis of mitochondrial respiratory function in tissue biopsies and blood cells. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 336–342. [Google Scholar] [CrossRef]
- Horan, M.P.; Pichaud, N.; Ballard, J.W. Review: Quantifying mitochondrial dysfunction in complex diseases of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 1022–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djafarzadeh, S.; Jakob, S.M. High-resolution respirometry to assess mitochondrial function in permeabilized and intact cells. J. Vis. Exp. 2017, 120, e54985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Meszaros, A.T.; Gnaiger, E. High-resolution FluoRespirometry and OXPHOS protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. Methods Mol. Biol. 2018, 1782, 31–70. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef]
- Jones, A.E.; Sheng, L.; Acevedo, A.; Veliova, M.; Shirihai, O.S.; Stiles, L.; Divakaruni, A.S. Forces, fluxes, and fuels: Tracking mitochondrial metabolism by integrating measurements of membrane potential, respiration, and metabolites. Am. J. Physiol. Cell Physiol. 2021, 320, C80–C91. [Google Scholar] [CrossRef]
- Lanza, I.R.; Nair, K.S. Functional assessment of isolated mitochondria in vitro. Methods Enzymol. 2009, 457, 349–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Shiva, S.; Ballinger, S.; Zhang, J.; Darley-Usmar, V.M. Bioenergetics and translational metabolism: Implications for genetics, physiology and precision medicine. Biol. Chem. 2019, 401, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Polarographic oxygen sensors, the oxygraph, and high-resolution respirometry to assess mitochondrial function. In Drug-Induced Mitochondrial Dysfunction; Dykens, J.A., Will, Y., Eds.; Wiley: Hoboken, NJ, USA, 2008; pp. 327–348. [Google Scholar] [CrossRef]
- Salabei, J.K.; Gibb, A.A.; Hill, B.G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014, 9, 421–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaude, J.; Acin-Perez, R.; Martinez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villan, E.; Hervas-Stubbs, S.; Pelegrin, P.; Sander, L.E.; Enriquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Schlieben, L.D.; Prokisch, H. The dimensions of primary mitochondrial disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders—Past, present and future. Biochim. Biophys. Acta 2004, 1659, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, A.; Lim, A.; Gorman, G. Epidemiology of Mitochondrial Disease; Mancuso, M., Klopstock, T., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Loeffen, J.L.; Smeitink, J.A.; Trijbels, J.M.; Janssen, A.J.; Triepels, R.H.; Sengers, R.C.; van den Heuvel, L.P. Isolated complex I deficiency in children: Clinical, biochemical and genetic aspects. Hum. Mutat. 2000, 15, 123–134. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Kerr, D.S.; Dahms, B.; Roessmann, U. Deficiency of the reduced nicotinamide adenine dinucleotide dehydrogenase component of complex I of mitochondrial electron transport. Fatal infantile lactic acidosis and hypermetabolism with skeletal-cardiac myopathy and encephalopathy. J. Clin. Investig. 1987, 80, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Swalwell, H.; Kirby, D.M.; Blakely, E.L.; Mitchell, A.; Salemi, R.; Sugiana, C.; Compton, A.G.; Tucker, E.J.; Ke, B.X.; Lamont, P.J.; et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. Eur. J. Hum. Genet. 2011, 19, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, C.; Xiao, R.; Place, E.; Zhang, Z.; Sondheimer, N.; Bennett, M.; Yudkoff, M.; Falk, M.J. Mitochondrial respiratory chain disease discrimination by retrospective cohort analysis of blood metabolites. Mol. Genet. Metab. 2013, 110, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson Legault, J.; Strittmatter, L.; Tardif, J.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; Clish, C.B.; Cyr, D.; Daneault, C.; et al. A metabolic signature of mitochondrial dysfunction revealed through a monogenic form of leigh syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, K.R.; Ratnaike, T.; van den Ameele, J.; Horvath, R.; Chinnery, P.F. Mitochondrial diseases: A diagnostic revolution. Trends Genet. 2020, 36, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef]
- Watson, E.; Davis, R.; Sue, C.M. New diagnostic pathways for mitochondrial disease. J. Transl. Genet. Genom. 2020, 4, 188–202. [Google Scholar] [CrossRef]
- Chinnery, P.F. Mitochondrial disease in adults: What’s old and what’s new? EMBO Mol. Med. 2015, 7, 1503–1512. [Google Scholar] [CrossRef]
- Pfeffer, G.; Pyle, A.; Griffin, H.; Miller, J.; Wilson, V.; Turnbull, L.; Fawcett, K.; Sims, D.; Eglon, G.; Hadjivassiliou, M.; et al. SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 2015, 84, 1174–1176. [Google Scholar] [CrossRef] [Green Version]
- Muraresku, C.C.; McCormick, E.M.; Falk, M.J. Mitochondrial Disease: Advances in clinical diagnosis, management, therapeutic development, and preventative strategies. Curr. Genet. Med. Rep. 2018, 6, 62–72. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary mitochondrial disease and secondary mitochondrial dysfunction: Importance of distinction for diagnosis and treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, S.A.; Rani, Z.Z.M.; Radin, F.Z.M.; Murad, N.A. Advancement in the diagnosis of mitochondrial diseases. J. Transl. Genet. Genom. 2020, 4, 159–187. [Google Scholar] [CrossRef]
- Joyce, N.C.; Oskarsson, B.; Jin, L.W. Muscle biopsy evaluation in neuromuscular disorders. Phys. Med. Rehabil. Clin. N. Am. 2012, 23, 609–631. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Barrientos, A. In vivo and in organello assessment of OXPHOS activities. Methods 2002, 26, 307–316. [Google Scholar] [CrossRef]
- Barrientos, A.; Fontanesi, F.; Diaz, F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 2009, 63, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch-Machin, M.A.; Turnbull, D.M. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001, 65, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.C.; Boyle, K.E. Microplate assays for spectrophotometric measurement of mitochondrial enzyme activity. Methods Mol. Biol. 2019, 1978, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Hernansanz-Agustin, P.; Enriquez, J.A. Analyzing electron transport chain supercomplexes. Methods Cell Biol. 2020, 155, 181–197. [Google Scholar] [CrossRef]
- Haraux, F.; Lombes, A. Kinetic analysis of ATP hydrolysis by complex V in four murine tissues: Towards an assay suitable for clinical diagnosis. PLoS ONE 2019, 14, e0221886. [Google Scholar] [CrossRef]
- Morava, E.; Rodenburg, R.J.; Hol, F.; de Vries, M.; Janssen, A.; van den Heuvel, L.; Nijtmans, L.; Smeitink, J. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am. J. Med. Genet. A 2006, 140, 863–868. [Google Scholar] [CrossRef]
- Chi, M.M.; Hintz, C.S.; Coyle, E.F.; Martin, W.H., 3rd; Ivy, J.L.; Nemeth, P.M.; Holloszy, J.O.; Lowry, O.H. Effects of detraining on enzymes of energy metabolism in individual human muscle fibers. Am. J. Physiol. 1983, 244, C276–C287. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acin-Perez, R.; Fernandez-Silva, P.; Movilla, N.; Perez-Martos, A.; Rodriguez de Cordoba, S.; Gallardo, M.E.; Enriquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Iborra, S.; Marti-Mateos, Y.; Cook, E.C.L.; Conde-Garrosa, R.; Petcherski, A.; Munoz, M.D.M.; Martinez de Mena, R.; Krishnan, K.C.; Jimenez, C.; et al. Fgr kinase is required for proinflammatory macrophage activation during diet-induced obesity. Nat. Metab. 2020, 2, 974–988. [Google Scholar] [CrossRef]
- Gardner, P.R.; Nguyen, D.D.; White, C.W. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef] [Green Version]
- Oyanagui, Y. Reevaluation of assay methods and establishment of kit for superoxide dismutase activity. Anal. Biochem. 1984, 142, 290–296. [Google Scholar] [CrossRef]
- Francisco, A.; Ronchi, J.A.; Navarro, C.D.C.; Figueira, T.R.; Castilho, R.F. Nicotinamide nucleotide transhydrogenase is required for brain mitochondrial redox balance under hampered energy substrate metabolism and high-fat diet. J. Neurochem. 2018, 147, 663–677. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.; Vercesi, A.E.; Castilho, R.F. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic. Biol. Med. 2013, 63, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimomura, K.; Galvanovskis, J.; Goldsworthy, M.; Hugill, A.; Kaizak, S.; Lee, A.; Meadows, N.; Quwailid, M.M.; Rydstrom, J.; Teboul, L.; et al. Insulin secretion from beta-cells is affected by deletion of nicotinamide nucleotide transhydrogenase. Methods Enzymol. 2009, 457, 451–480. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Hoyos, B.; Zhao, F.; Vinogradov, V.; Fischman, D.A.; Harris, R.A.; Leitges, M.; Wongsiriroj, N.; Blaner, W.S.; Manfredi, G.; et al. Control of oxidative phosphorylation by vitamin A illuminates a fundamental role in mitochondrial energy homoeostasis. FASEB J. 2010, 24, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Hale, D.E.; Cornell, J.E.; Bennett, M.J. Stability of long-chain and short-chain 3-hydroxyacyl-CoA dehydrogenase activity in postmortem liver. Clin. Chem. 1997, 43, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Binstock, J.F.; Schulz, H. Fatty acid oxidation complex from Escherichia coli. Methods Enzymol. 1981, 71, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Grinblat, L.; Pacheco Bolanos, L.F.; Stoppani, A.O. Decreased rate of ketone-body oxidation and decreased activity of D-3-hydroxybutyrate dehydrogenase and succinyl-CoA:3-oxo-acid CoA-transferase in heart mitochondria of diabetic rats. Biochem. J. 1986, 240, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehninger, A.L.; Sudduth, H.C.; Wise, J.B. D-beta-Hydroxybutyric dehydrogenase of muitochondria. J. Biol. Chem. 1960, 235, 2450–2455. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Wong, K.E.; Koves, T.R.; Slentz, D.H.; Seiler, S.E.; Wittmann, A.H.; Ilkayeva, O.R.; Stevens, R.D.; Perry, C.G.; Lark, D.S.; et al. Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. J. Biol. Chem. 2014, 289, 8106–8120. [Google Scholar] [CrossRef] [Green Version]
- Scheer, W.D.; Lehmann, H.P.; Beeler, M.F. An improved assay for hexokinase activity in human tissue homogenates. Anal. Biochem. 1978, 91, 451–463. [Google Scholar] [CrossRef]
- Nuzum, C.T.; Snodgrass, P.J. Multiple assays of the five urea-cycle enzymes in human liver homogenates. In The Urea Cycle; Wiley: New York, NY, USA, 1976; pp. 325–349. [Google Scholar]
- Lawless, C.; Greaves, L.; Reeve, A.K.; Turnbull, D.M.; Vincent, A.E. The rise and rise of mitochondrial DNA mutations. Open Biol. 2020, 10, 200061. [Google Scholar] [CrossRef]
- Menzies, R.A.; Gold, P.H. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J. Biol. Chem. 1971, 246, 2425–2429. [Google Scholar] [CrossRef]
- Korr, H.; Kurz, C.; Seidler, T.O.; Sommer, D.; Schmitz, C. Mitochondrial DNA synthesis studied autoradiographically in various cell types in vivo. Braz. J. Med. Biol. Res. 1998, 31, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, J.M.; Tarnopolsky, M.A. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion 2004, 4, 441–452. [Google Scholar] [CrossRef]
- Shanely, R.A.; Zwetsloot, K.A.; Triplett, N.T.; Meaney, M.P.; Farris, G.E.; Nieman, D.C. Human skeletal muscle biopsy procedures using the modified Bergstrom technique. J. Vis. Exp. 2014, 51812. [Google Scholar] [CrossRef]
- Hayot, M.; Michaud, A.; Koechlin, C.; Caron, M.A.; Leblanc, P.; Prefaut, C.; Maltais, F. Skeletal muscle microbiopsy: A validation study of a minimally invasive technique. Eur. Respir. J. 2005, 25, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, T.E.; Brophy, P.; Lin, C.T.; Hickner, R.C.; Neufer, P.D. Assessment of in vivo skeletal muscle mitochondrial respiratory capacity in humans by near-infrared spectroscopy: A comparison with in situ measurements. J. Physiol. 2014, 592, 3231–3241. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saneto, R.P. Mitochondrial diseases: Expanding the diagnosis in the era of genetic testing. J. Transl. Genet. Genom. 2020, 4, 384–428. [Google Scholar] [CrossRef]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary skin fibroblasts as a model of ParkinsoN’s disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Huigsloot, M.; Janssen, A.J.; Kappen, A.J.; Smeitink, J.A.; Rodenburg, R.J. High-throughput assay to measure oxygen consumption in digitonin-permeabilized cells of patients with mitochondrial disorders. Clin. Chem. 2010, 56, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.; Bergquist, K.; Young, K.; Gnaiger, E.; Rao, R.R.; Bennett, J.P., Jr. Mitochondrial gene therapy improves respiration, biogenesis, and transcription in G11778A Leber’s hereditary optic neuropathy and T8993G Leigh’s syndrome cells. Hum. Gene Ther. 2012, 23, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Invernizzi, F.; D’Amato, I.; Jensen, P.B.; Ravaglia, S.; Zeviani, M.; Tiranti, V. Microscale oxygraphy reveals OXPHOS impairment in MRC mutant cells. Mitochondrion 2012, 12, 328–335. [Google Scholar] [CrossRef]
- Lahuerta, M.; Aguado, C.; Sanchez-Martin, P.; Sanz, P.; Knecht, E. Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 2018, 285, 2071–2090. [Google Scholar] [CrossRef] [Green Version]
- Hannibal, L.; Theimer, J.; Wingert, V.; Klotz, K.; Bierschenk, I.; Nitschke, R.; Spiekerkoetter, U.; Grunert, S.C. Metabolic profiling in human fibroblasts enables subtype clustering in glycogen storage disease. Front. Endocrinol. 2020, 11, 579981. [Google Scholar] [CrossRef]
- Tokuyama, T.; Hirai, A.; Shiiba, I.; Ito, N.; Matsuno, K.; Takeda, K.; Saito, K.; Mii, K.; Matsushita, N.; Fukuda, T.; et al. Mitochondrial dynamics regulation in skin fibroblasts from mitochondrial disease patients. Biomolecules 2020, 10, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.S.; Kao, S.H.; Ho, C.S.; Wei, Y.H.; Hung, P.L.; Hsu, M.H.; Wu, T.Y.; Wang, T.J.; Jian, Y.R.; Lee, T.H.; et al. Inflexibility of AMPK-mediated metabolic reprogramming in mitochondrial disease. Oncotarget 2017, 8, 73627–73639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gstraunthaler, G.; Seppi, T.; Pfaller, W. Impact of culture conditions, culture media volumes, and glucose content on metabolic properties of renal epithelial cell cultures. Are renal cells in tissue culture hypoxic? Cell Physiol. Biochem. 1999, 9, 150–172. [Google Scholar] [CrossRef] [PubMed]
- Van den Bogert, C.; Spelbrink, J.N.; Dekker, H.L. Relationship between culture conditions and the dependency on mitochondrial function of mammalian cell proliferation. J. Cell Physiol. 1992, 152, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Herzenberg, L.A.; Niemi, A.K.; Cowan, T.; Herzenberg, L.A. Importance of culturing primary lymphocytes at physiological oxygen levels. Proc. Natl. Acad. Sci. USA 2007, 104, 4547–4552. [Google Scholar] [CrossRef] [Green Version]
- Golpour, M.; Akhavan Niaki, H.; Khorasani, H.R.; Hajian, A.; Mehrasa, R.; Mostafazadeh, A. Human fibroblast switches to anaerobic metabolic pathway in response to serum starvation: A mimic of warburg effect. Int. J. Mol. Cell Med. 2014, 3, 74–80. [Google Scholar]
- Balin, A.K.; Fisher, A.J.; Anzelone, M.; Leong, I.; Allen, R.G. Effects of establishing cell cultures and cell culture conditions on the proliferative life span of human fibroblasts isolated from different tissues and donors of different ages. Exp. Cell Res. 2002, 274, 275–287. [Google Scholar] [CrossRef]
- Costa, C.F.; Pinho, S.A.; Pinho, S.L.C.; Miranda-Santos, I.; Bagshaw, O.; Stuart, J.; Oliveira, P.J.; Cunha-Oliveira, T. Mitochondrial and metabolic remodeling in human skin fibroblasts in response to glucose availability. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pereira, S.P.; Deus, C.M.; Serafim, T.L.; Cunha-Oliveira, T.; Oliveira, P.J. Metabolic and phenotypic characterization of human skin fibroblasts after forcing oxidative capacity. Toxicol. Sci. 2018, 164, 191–204. [Google Scholar] [CrossRef]
- Iannetti, E.F.; Smeitink, J.A.M.; Willems, P.; Beyrath, J.; Koopman, W.J.H. Rescue from galactose-induced death of Leigh Syndrome patient cells by pyruvate and NAD(.). Cell Death Dis. 2018, 9, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Fischer, A.; Reagan, J.D.; Yan, L.J.; Ames, B.N. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc. Natl. Acad. Sci. USA 1995, 92, 4337–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Tigges, J.; Krutmann, J.; Fritsche, E.; Haendeler, J.; Schaal, H.; Fischer, J.W.; Kalfalah, F.; Reinke, H.; Reifenberger, G.; Stuhler, K.; et al. The hallmarks of fibroblast ageing. Mech. Ageing Dev. 2014, 138, 26–44. [Google Scholar] [CrossRef]
- Tollefsbol, T.O. Techniques for analysis of biological aging. Methods Mol. Biol. 2007, 371, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Cristofalo, V.J.; Volker, C.; Allen, R.G. Use of the fibroblast model in the study of cellular senescence. In Aging Methods and Protocols (Methods in Molecular Medicine); Barnett, Y.A., Barnett, C.R., Eds.; Humana Press: Totowa, NJ, USA, 2000; Volume 38. [Google Scholar]
- Bittles, A.H.; Harper, N. Increased glycolysis in ageing cultured human diploid fibroblasts. Biosci. Rep. 1984, 4, 751–756. [Google Scholar] [CrossRef]
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871. [Google Scholar] [CrossRef]
- Hutter, E.; Renner, K.; Pfister, G.; Stockl, P.; Jansen-Durr, P.; Gnaiger, E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem. J. 2004, 380, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Latorre-Pellicer, A.; Lechuga-Vieco, A.V.; Johnston, I.G.; Hamalainen, R.H.; Pellico, J.; Justo-Mendez, R.; Fernandez-Toro, J.M.; Claveria, C.; Guaras, A.; Sierra, R.; et al. Regulation of mother-to-offspring transmission of mtDNA heteroplasmy. Cell Metab. 2019, 30, 1120–1130.e1125. [Google Scholar] [CrossRef]
- Schneider, E.L. Aging and cultured human skin fibroblasts. J. Investig. Dermatol. 1979, 73, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, M.; Villani, G.; Mazzucchelli, F.; Bresolin, N.; Papa, S.; Attardi, G. Marked aging-related decline in efficiency of oxidative phosphorylation in human skin fibroblasts. FASEB J. 2003, 17, 1706–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, S.; Moerman, E.J.; Porter, K. High-voltage electron microscopy of human diploid fibroblasts during ageing in vitro. Morphometric analysis of mitochondria. Exp. Cell Res. 1984, 154, 101–111. [Google Scholar] [CrossRef]
- Wong, R.C.B.; Lim, S.Y.; Hung, S.S.C.; Jackson, S.; Khan, S.; Van Bergen, N.J.; De Smit, E.; Liang, H.H.; Kearns, L.S.; Clarke, L.; et al. Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging 2017, 9, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Herbst, A.; Lee, C.C.; Vandiver, A.R.; Aiken, J.M.; McKenzie, D.; Hoang, A.; Allison, D.; Liu, N.; Wanagat, J. Mitochondrial DNA deletion mutations increase exponentially with age in human skeletal muscle. Aging Clin. Exp. Res. 2020, 33, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed. Res. Int. 2014, 2014, 238463. [Google Scholar] [CrossRef] [Green Version]
- Keane, K.N.; Calton, E.K.; Cruzat, V.F.; Soares, M.J.; Newsholme, P. The impact of cryopreservation on human peripheral blood leucocyte bioenergetics. Clin. Sci. 2015, 128, 723–733. [Google Scholar] [CrossRef]
- Traba, J.; Miozzo, P.; Akkaya, B.; Pierce, S.K.; Akkaya, M. An optimized protocol to analyze glycolysis and mitochondrial respiration in lymphocytes. J. Vis. Exp. 2016, 117, 54918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, P.A.; Chacko, B.K.; Ravi, S.; Johnson, M.S.; Mitchell, T.; Darley-Usmar, V.M. Bioenergetics and the oxidative burst: Protocols for the isolation and evaluation of human leukocytes and platelets. J. Vis. Exp. 2014, 85, 51301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjovall, F.; Ehinger, J.K.; Marelsson, S.E.; Morota, S.; Frostner, E.A.; Uchino, H.; Lundgren, J.; Arnbjornsson, E.; Hansson, M.J.; Fellman, V.; et al. Mitochondrial respiration in human viable platelets—Methodology and influence of gender, age and storage. Mitochondrion 2013, 13, 7–14. [Google Scholar] [CrossRef]
- Rose, S.; Carvalho, E.; Diaz, E.C.; Cotter, M.; Bennuri, S.C.; Azhar, G.; Frye, R.E.; Adams, S.H.; Borsheim, E. A comparative study of mitochondrial respiration in circulating blood cells and skeletal muscle fibers in women. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E503–E512. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Bharadwaj, M.S.; Jorgensen, M.J.; Register, T.C.; Molina, A.J. Blood cell respirometry is associated with skeletal and cardiac muscle bioenergetics: Implications for a minimally invasive biomarker of mitochondrial health. Redox Biol. 2016, 10, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braganza, A.; Corey, C.G.; Santanasto, A.J.; Distefano, G.; Coen, P.M.; Glynn, N.W.; Nouraie, S.M.; Goodpaster, B.H.; Newman, A.B.; Shiva, S. Platelet bioenergetics correlate with muscle energetics and are altered in older adults. JCI Insight 2019, 5, e128248. [Google Scholar] [CrossRef] [Green Version]
- Pecina, P.; Houstkova, H.; Mracek, T.; Pecinova, A.; Nuskova, H.; Tesarova, M.; Hansikova, H.; Janota, J.; Zeman, J.; Houstek, J. Noninvasive diagnostics of mitochondrial disorders in isolated lymphocytes with high resolution respirometry. BBA Clin. 2014, 2, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, D.; Proctor, E.A.; Raval, F.M.; Ip, B.C.; Habib, C.; Ritou, E.; Grammatopoulos, T.N.; Steenkamp, D.; Dooms, H.; Apovian, C.M.; et al. Advances in the quantification of mitochondrial function in primary human immune cells through extracellular flux analysis. PLoS ONE 2017, 12, e0170975. [Google Scholar] [CrossRef]
- Avila, C.; Huang, R.J.; Stevens, M.V.; Aponte, A.M.; Tripodi, D.; Kim, K.Y.; Sack, M.N. Platelet mitochondrial dysfunction is evident in type 2 diabetes in association with modifications of mitochondrial anti-oxidant stress proteins. Exp. Clin. Endocrinol. Diabetes 2012, 120, 248–251. [Google Scholar] [CrossRef]
- Hartman, M.L.; Shirihai, O.S.; Holbrook, M.; Xu, G.; Kocherla, M.; Shah, A.; Fetterman, J.L.; Kluge, M.A.; Frame, A.A.; Hamburg, N.M.; et al. Relation of mitochondrial oxygen consumption in peripheral blood mononuclear cells to vascular function in type 2 diabetes mellitus. Vasc. Med. 2014, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Willig, A.L.; Kramer, P.A.; Chacko, B.K.; Darley-Usmar, V.M.; Heath, S.L.; Overton, E.T. Monocyte bioenergetic function is associated with body composition in virologically suppressed HIV-infected women. Redox Biol. 2017, 12, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Einsiedel, L.; Cherry, C.L.; Sheeran, F.L.; Friedhuber, A.; Wesselingh, S.L.; Pepe, S. Mitochondrial dysfunction in CD4+ lymphocytes from stavudine-treated HIV patients. Mitochondrion 2010, 10, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Lay, S.T.; De Piazza, S.W.; Sanislav, O.; Hammersley, E.; Allan, C.Y.; Francione, L.M.; Bui, M.Q.; Chen, Z.P.; Ngoei, K.R.; et al. Immortalized ParkinsoN’s disease lymphocytes have enhanced mitochondrial respiratory activity. Dis. Model. Mech. 2016, 9, 1295–1305. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.M.; Depp, C.; Ryan, B.J.; Johnston, G.I.; Alegre-Abarrategui, J.; Evetts, S.; Rolinski, M.; Baig, F.; Ruffmann, C.; Simon, A.K.; et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early ParkinsoN’s blood cells. Mov. Disord. 2018, 33, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Michalak, S.; Florczak-Wyspianska, J.; Rybacka-Mossakowska, J.; Ambrosius, W.; Osztynowicz, K.; Baszczuk, A.; Kozubski, W.; Wysocka, E. Mitochondrial respiration in intact peripheral blood mononuclear cells and sirtuin 3 activity in patients with movement disorders. Oxid. Med. Cell Longev. 2017, 2017, 9703574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Hejl, A.M.; Dinh, T.S.; Keijzers, G.; Hansen, A.M.; Desler, C.; Moreno-Villanueva, M.; Burkle, A.; Rasmussen, L.J.; Waldemar, G.; et al. Defective mitochondrial respiration, altered dNTP pools and reduced AP endonuclease 1 activity in peripheral blood mononuclear cells of Alzheimer’s disease patients. Aging 2015, 7, 793–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehinger, J.K.; Morota, S.; Hansson, M.J.; Paul, G.; Elmer, E. Mitochondrial respiratory function in peripheral blood cells from HuntingtoN’s disease patients. Mov. Disord. Clin. Pract. 2016, 3, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Tyrrell, D.J.; Bharadwaj, M.S.; Van Horn, C.G.; Marsh, A.P.; Nicklas, B.J.; Molina, A.J. Blood-cell bioenergetics are associated with physical function and inflammation in overweight/obese older adults. Exp. Gerontol. 2015, 70, 84–91. [Google Scholar] [CrossRef]
- Braganza, A.; Annarapu, G.K.; Shiva, S. Blood-based bioenergetics: An emerging translational and clinical tool. Mol. Aspects Med. 2020, 71, 100835. [Google Scholar] [CrossRef]
- Silaidos, C.; Pilatus, U.; Grewal, R.; Matura, S.; Lienerth, B.; Pantel, J.; Eckert, G.P. Sex-associated differences in mitochondrial function in human peripheral blood mononuclear cells (PBMCs) and brain. Biol. Sex Differ. 2018, 9, 34. [Google Scholar] [CrossRef]
- Rausser, S.; Trumpff, C.; McGill, M.A.; Junker, A.; Wang, W.; Ho, S.H.; Mitchell, A.; Karan, K.R.; Monk, C.; Segerstrom, S.C.; et al. Mitochondrial phenotypes in purified human immune cell subtypes and cell mixtures. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Johnson, M.S.; Hardy, R.W.; Ballinger, S.W.; Darley-Usmar, V.M. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab. Investig. 2013, 93, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Banas, B.; Kost, B.P.; Goebel, F.D. Platelets, a typical source of error in real-time PCR quantification of mitochondrial DNA content in human peripheral blood cells. Eur. J. Med. Res. 2004, 9, 371–377. [Google Scholar] [PubMed]
- Urata, M.; Koga-Wada, Y.; Kayamori, Y.; Kang, D. Platelet contamination causes large variation as well as overestimation of mitochondrial DNA content of peripheral blood mononuclear cells. Ann. Clin. Biochem. 2008, 45, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Cronin, J.G.; Dolton, G.; Panetti, S.; Schauenburg, A.J.; Galloway, S.A.E.; Sewell, A.K.; Cole, D.K.; Thornton, C.A.; Francis, N.J. Metabolic adaptation of human CD4(+) and CD8(+) T-cells to T-cell receptor-mediated stimulation. Front. Immunol. 2017, 8, 1516. [Google Scholar] [CrossRef] [Green Version]
- Ravi, S.; Chacko, B.; Sawada, H.; Kramer, P.A.; Johnson, M.S.; Benavides, G.A.; O’Donnell, V.; Marques, M.B.; Darley-Usmar, V.M. Metabolic plasticity in resting and thrombin activated platelets. PLoS ONE 2015, 10, e0123597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer-Spurej, E.; Pfeiler, G.; Maurer, N.; Lindner, H.; Glatter, O.; Devine, D.V. Room temperature activates human blood platelets. Lab. Investig. 2001, 81, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Vollmar, B.; Slotta, J.E.; Nickels, R.M.; Wenzel, E.; Menger, M.D. Comparative analysis of platelet isolation techniques for the in vivo study of the microcirculation. Microcirculation 2003, 10, 143–152. [Google Scholar] [CrossRef]
- Garcia-Roche, M.; Casal, A.; Carriquiry, M.; Radi, R.; Quijano, C.; Cassina, A. Respiratory analysis of coupled mitochondria in cryopreserved liver biopsies. Redox Biol. 2018, 17, 207–212. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Kunz, W.S.; Saks, V.; Usson, Y.; Mazat, J.P.; Letellier, T.; Gellerich, F.N.; Margreiter, R. Cryopreservation of mitochondria and mitochondrial function in cardiac and skeletal muscle fibers. Anal. Biochem. 2003, 319, 296–303. [Google Scholar] [CrossRef]
- Larsen, S.; Wright-Paradis, C.; Gnaiger, E.; Helge, J.W.; Boushel, R. Cryopreservation of human skeletal muscle impairs mitochondrial function. Cryo Lett. 2012, 33, 170–176. [Google Scholar]
- Nukala, V.N.; Singh, I.N.; Davis, L.M.; Sullivan, P.G. Cryopreservation of brain mitochondria: A novel methodology for functional studies. J. Neurosci. Methods 2006, 152, 48–54. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Andreyev, A.; Murphy, A.N.; Perkins, G.A.; Ellisman, M.H.; Newmeyer, D.D. Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ. 2007, 14, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Acin-Perez, R.; Benador, I.Y.; Petcherski, A.; Veliova, M.; Benavides, G.A.; Lagarrigue, S.; Caudal, A.; Vergnes, L.; Murphy, A.N.; Karamanlidis, G.; et al. A novel approach to measure mitochondrial respiration in frozen biological samples. EMBO J. 2020, 39, e104073. [Google Scholar] [CrossRef] [PubMed]
- Osto, C.; Benador, I.Y.; Ngo, J.; Liesa, M.; Stiles, L.; Acin-Perez, R.; Shirihai, O.S. Measuring mitochondrial respiration in previously frozen biological samples. Curr. Protoc. Cell Biol. 2020, 89, e116. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Andreyev, A.Y.; Rogers, G.W.; Murphy, A.N. In situ measurements of mitochondrial matrix enzyme activities using plasma and mitochondrial membrane permeabilization agents. Anal. Biochem. 2018, 552, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Baracca, A.; Barogi, S.; Carelli, V.; Lenaz, G.; Solaini, G. Catalytic activities of mitochondrial ATP synthase in patients with mitochondrial DNA T8993G mutation in the ATPase 6 gene encoding subunit a. J. Biol. Chem. 2000, 275, 4177–4182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, R.A.; Diaz, V.; Meinild, A.K.; Gassmann, M.; Lundby, C. The C57Bl/6 mouse serves as a suitable model of human skeletal muscle mitochondrial function. Exp. Physiol. 2013, 98, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellerich, F.N.; Mayr, J.A.; Reuter, S.; Sperl, W.; Zierz, S. The problem of interlab variation in methods for mitochondrial disease diagnosis: Enzymatic measurement of respiratory chain complexes. Mitochondrion 2004, 4, 427–439. [Google Scholar] [CrossRef]
- Vergnes, L.; Davies, G.R.; Lin, J.Y.; Yeh, M.W.; Livhits, M.J.; Harari, A.; Symonds, M.E.; Sacks, H.S.; Reue, K. Adipocyte browning and higher mitochondrial function in periadrenal but not SC fat in pheochromocytoma. J. Clin. Endocrinol. Metab. 2016, 101, 4440–4448. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leung, M.C.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolkipli-Cunningham, Z.; Falk, M.J. Clinical effects of chemical exposures on mitochondrial function. Toxicology 2017, 391, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Bharadwaj, M.S.; Van Horn, C.G.; Kritchevsky, S.B.; Nicklas, B.J.; Molina, A.J. Respirometric profiling of muscle mitochondria and blood cells are associated with differences in gait speed among community-dwelling older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1394–1399. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Bharadwaj, M.S.; Jorgensen, M.J.; Register, T.C.; Shively, C.; Andrews, R.N.; Neth, B.; Keene, C.D.; Mintz, A.; Craft, S.; et al. Blood-based bioenergetic profiling reflects differences in brain bioenergetics and metabolism. Oxid. Med. Cell Longev. 2017, 2017, 7317251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winnica, D.; Corey, C.; Mullett, S.; Reynolds, M.; Hill, G.; Wendell, S.; Que, L.; Holguin, F.; Shiva, S. Bioenergetic differences in the airway epithelium of lean versus obese asthmatics are driven by nitric oxide and reflected in circulating platelets. Antioxid. Redox Signal. 2019, 31, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.; Murphy, M.P.; Darley-Usmar, V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013, 1, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Hedges, C.P.; Woodhead, J.S.T.; Wang, H.W.; Mitchell, C.J.; Cameron-Smith, D.; Hickey, A.J.R.; Merry, T.L. Peripheral blood mononuclear cells do not reflect skeletal muscle mitochondrial function or adaptation to high-intensity interval training in healthy young men. J. Appl. Physiol. 2019, 126, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Rodinova, M.; Trefilova, E.; Honzik, T.; Tesarova, M.; Zeman, J.; Hansikova, H. Non-invasive screening of cytochrome c oxidase deficiency in children using a dipstick immunocapture assay. Folia Biol. 2014, 60, 268–274. [Google Scholar]
- Frederiksen, A.L.; Andersen, P.H.; Kyvik, K.O.; Jeppesen, T.D.; Vissing, J.; Schwartz, M. Tissue specific distribution of the 3243A->G mtDNA mutation. J. Med. Genet. 2006, 43, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Yorns, W.R., Jr.; Valencia, I.; Jayaraman, A.; Sheth, S.; Legido, A.; Goldenthal, M.J. Buccal swab analysis of mitochondrial enzyme deficiency and DNA defects in a child with suspected myoclonic epilepsy and ragged red fibers (MERRF). J. Child. Neurol. 2012, 27, 398–401. [Google Scholar] [CrossRef]
- Goldenthal, M.J.; Kuruvilla, T.; Damle, S.; Salganicoff, L.; Sheth, S.; Shah, N.; Marks, H.; Khurana, D.; Valencia, I.; Legido, A. Non-invasive evaluation of buccal respiratory chain enzyme dysfunction in mitochondrial disease: Comparison with studies in muscle biopsy. Mol. Genet. Metab. 2012, 105, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Goldenthal, M.J.; Damle, S.; Sheth, S.; Shah, N.; Melvin, J.; Jethva, R.; Hardison, H.; Marks, H.; Legido, A. Mitochondrial enzyme dysfunction in autism spectrum disorders: A novel biomarker revealed from buccal swab analysis. Biomark. Med. 2015, 9, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Kolbasina, N.A.; Gureev, A.P.; Serzhantova, O.V.; Mikhailov, A.A.; Moshurov, I.P.; Starkov, A.A.; Popov, V.N. Lung cancer increases H2O2 concentration in the exhaled breath condensate, extent of mtDNA damage, and mtDNA copy number in buccal mucosa. Heliyon 2020, 6, e04303. [Google Scholar] [CrossRef]
- Delhey, L.M.; Nur Kilinc, E.; Yin, L.; Slattery, J.C.; Tippett, M.L.; Rose, S.; Bennuri, S.C.; Kahler, S.G.; Damle, S.; Legido, A.; et al. The effect of mitochondrial supplements on mitochondrial activity in children with autism spectrum disorder. J. Clin. Med. 2017, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Closas, M.; Egan, K.M.; Abruzzo, J.; Newcomb, P.A.; Titus-Ernstoff, L.; Franklin, T.; Bender, P.K.; Beck, J.C.; Le Marchand, L.; Lum, A.; et al. Collection of genomic DNA from adults in epidemiological studies by buccal cytobrush and mouthwash. Cancer Epidemiol. Biomark. Prev. 2001, 10, 687–696. [Google Scholar]
- Mahfuz, I.; Cheng, W.; White, S.J. Identification of Streptococcus parasanguinis DNA contamination in human buccal DNA samples. BMC Res. Notes 2013, 6, 481. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Supplier | Catalog Number |
|---|---|---|
| Acetyl-CoA | Sigma | A2181 |
| Adenosine 5′Diphosphate (ADP) | Sigma | A5285 |
| ATP | Sigma | A26209 |
| Alamethicin | Sigma | A4665 |
| Antimycin A (AA) | Sigma | A8674 |
| Ascorbic Acid (Asc) | Fisher | A61-100 |
| Azide | Sigma | S8032 |
| Coenzyme Q1 | Sigma | C7956 |
| Cytochrome c | Sigma | C2506 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Sigma | D5030 |
| 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) | Sigma | D218200 |
| Ethylene glycol-bis(2-aminoethylether)- N,N,N’,N’-tetraacetic acid (EGTA) | Sigma | E4378 |
| FBS | Corning | 35010CV |
| Carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) | Enzo | BML-CM120 |
| Glucose | Sigma | G5767 |
| Glutamine | Gibco | 25030081 |
| 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) | Gibco | 15630080 |
| Reduced nicotinamide adenine dinucleotide (NADH) | Sigma | N8129 |
| Magnesium Chloride | Sigma | M0250 |
| Mannitol | Sigma | M9546 |
| MitoTracker Deep Red (MTDR) | Invitrogen | M22426 |
| Oligomycin (Oligo) | Sigma | 495455 |
| Oxaloacetic acid | Sigma | O4126 |
| Phosphate Buffered Saline (PBS) | Gibco | 14190250 |
| Poly-D-Lysine | Sigma | P6407 |
| Potassium cyanide | Sigma | 60178 |
| Potassium phosphate | Sigma | P5655 |
| RPMI-1640 | Gibco | 11875119 |
| Rotenone (Rot) | Sigma | R8875 |
| Sodium Pyruvate | Gibco | 11360070 |
| Succinic Acid (Succ) | Sigma | S9512 |
| Sucrose | Sigma | S0389 |
| N, N, N’, N’-tetramethyl-p-phenylenediamine (TMPD) | Sigma | 87890 |
| XF plasma membrane permeabilizer (XF PMP) | Agilent | 102504-100 |
| Mitochondrial Compound/Substrate | Function |
|---|---|
| ADP | Induce State 3, ADP-stimulated respiration |
| ATP | Induce Complex V (CV) hydrolysis in uncoupled mitochondria |
| Alamethicin | Permeabilize the mitochondrial inner membrane |
| Antimycin A | Complex III (CIII) inhibitor |
| Ascorbate | Maintains TMPD in the reduced state |
| Azide | Complex IV (CIV) inhibitor |
| Cytochrome c | Soluble component of ETC; added to replace the cytochrome c that is lost with freezing |
| FCCP | Chemical uncoupler of OXPHOS to measure maximal respiration |
| NADH | Complex I (CI) substrate |
| MitoTracker Deep Red | Fluorescent dye to measure mitochondrial content |
| Oligomycin | CV inhibitor |
| Rotenone | CI inhibitor |
| recombinant perfringolysin O (rPFO) or XF PMP | Permeabilize the plasma membrane |
| Succinate | Complex II (CII) substrate |
| TMPD | Electron donor to cytochrome c/CIV |
| Enzyme | Metabolic Pathway | References |
|---|---|---|
| CI: NADH/CoQ1 Oxidoreductase | ETC, OXPHOS | [50,51] |
| CII: Succinate DH | ETC, OXPHOS, TCA | [25,50,51] |
| CIII: Coenzyme Q: Cytochrome c—oxidoreductase | ETC, OXPHOS | [50,51] |
| CIV: Cytochrome c oxidoreductase | ETC, OXPHOS | [50,51] |
| Combined CI + CIII | ETC, OXPHOS | [51] |
| Combined CII + CIII | ETC, OXPHOS | [51] |
| Combined CI + CIII + CIV | ETC, OXPHOS | [52] |
| CV: ATP hydrolysis | ETC, OXPHOS | [53,54] |
| Creatine kinase | ATP homeostasis | [55] |
| Adenylate kinase | ATP homeostasis | [55] |
| Citrate synthase | TCA | [50,51] |
| α-ketoglutarate DH | TCA | [56,57] |
| Isocitrate DH | TCA | [58] |
| Malate DH | TCA | [55,56,57] |
| Aconitase | TCA, Redox balance | [50,56,59] |
| Manganese Superoxide dismutase (MnSOD) | Redox balance | [60] |
| Nicotinamide nucleotide transhydrogenase | Redox balance | [61,62,63] |
| Catalase | Redox balance | [56] |
| Glycerol-3 phosphate DH | Glycerophosphate shuttle | [25,51] |
| Pyruvate DH | Glucose and Fatty acid oxidation | [64] |
| β-Hydroxyacyl CoA DH | Fatty acid oxidation | [50,55] |
| Short-chain hydroxyl-acyl-CoA DH | Fatty acid oxidation | [58,65,66] |
| β-hydroxybutyrate DH | Ketone body | [67,68,69] |
| Hexokinase | Glucose metabolism | [55,70] |
| Arginase | Urea cycle | [71] |
| Intact | Permeabilized | Isolated Mitochondria | |
|---|---|---|---|
| Advantages |
|
|
|
| Drawbacks |
|
|
|
| Drawbacks | • Cell culture conditions, confluence, and experimental media influence outcomes • Less mechanistic insight • May be more difficult to interpret since so many processes can alter OCR • Changes in OCR could reflect changes in mitochondrial content • Normalization is required | • No cellular control of metabolism/nutrient preference • Careful titration of permeabilizing agent required • Changes in OCR could reflect changes in mitochondrial content • Normalization is required | • No cellular control of metabolism/nutrient preference • Loss of intracellular environment • Loss of mitochondrial morphology • Isolation procedure can damage mitochondria • Selection bias • More starting material required |
| Fresh | Frozen | |
|---|---|---|
| Advantages |
|
|
| Drawbacks |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acin-Perez, R.; Benincá, C.; Shabane, B.; Shirihai, O.S.; Stiles, L. Utilization of Human Samples for Assessment of Mitochondrial Bioenergetics: Gold Standards, Limitations, and Future Perspectives. Life 2021, 11, 949. https://doi.org/10.3390/life11090949
Acin-Perez R, Benincá C, Shabane B, Shirihai OS, Stiles L. Utilization of Human Samples for Assessment of Mitochondrial Bioenergetics: Gold Standards, Limitations, and Future Perspectives. Life. 2021; 11(9):949. https://doi.org/10.3390/life11090949
Chicago/Turabian StyleAcin-Perez, Rebeca, Cristiane Benincá, Byourak Shabane, Orian S. Shirihai, and Linsey Stiles. 2021. "Utilization of Human Samples for Assessment of Mitochondrial Bioenergetics: Gold Standards, Limitations, and Future Perspectives" Life 11, no. 9: 949. https://doi.org/10.3390/life11090949