
SLCO1B1 Phenotype and CYP3A5 Polymorphism Significantly Affect Atorvastatin Bioavailability

 ,
, 
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Study Design and Procedures
2.3. Pharmacokinetic Analysis
2.4. Safety
2.5. Genotyping, Haplotyping and Phenotyping
2.6. Statistical Analysis
3. Results
3.1. Demographic Characteristics
3.2. Pharmacokinetics
3.3. Safety
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weng, T.-C.; Yang, Y.-H.K.; Lin, S.-J.; Tai, S.-H. A Systematic Review and Meta-Analysis on the Therapeutic Equivalence of Statins. J. Clin. Pharm. Ther. 2010, 35, 139–151. [Google Scholar] [CrossRef]
- Stancu, C.; Sima, A. Statins: Mechanism of Action and Effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, H.S.; Goa, K.L. Atorvastatin: An Updated Review of Its Pharmacological Properties and Use in Dyslipidaemia. Drugs 2001, 61, 1835–1881. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Pu, H.-H.; Liu, G.-Y.; Jia, J.-Y.; Weng, L.-P.; Xu, R.-J.; Li, G.-X.; Wang, W.; Zhang, M.-Q.; Lu, C.; et al. Pharmacokinetics and Bioequivalence Evaluation of Two Different Atorvastatin Calcium 10-Mg Tablets: A Single-Dose, Randomized-Sequence, Open-Label, Two-Period Crossover Study in Healthy Fasted Chinese Adult Males. Clin. Ther. 2010, 32, 1396–1407. [Google Scholar] [CrossRef]
- Agencia Española del Medicamento y Productos Sanitarios (AEMPS) Cardyl Comprimidos Recubiertos Con Película (Drug Label), Avda. de Europa, 20B, Parque Empresarial La Moraleja, 28108, Alcobendas, Madrid, Spain.
- García, M.J.; Reinoso, R.F.; Sánchez Navarro, A.; Prous, J.R. Clinical Pharmacokinetics of Statins. Methods Find. Exp. Clin. Pharmacol. 2003, 25, 457–481. [Google Scholar] [CrossRef]
- Schachter, M. Chemical, Pharmacokinetic and Pharmacodynamic Properties of Statins: An Update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of OATP Transporters on Pharmacokinetics: OATP Transporters and Pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGorter, M.K.; Tirona, R.G.; Schwarz, U.I.; Choi, Y.-H.; Dresser, G.K.; Suskin, N.; Myers, K.; Zou, G.; Iwuchukwu, O.; Wei, W.-Q.; et al. Clinical and Pharmacogenetic Predictors of Circulating Atorvastatin and Rosuvastatin Concentrations in Routine Clinical Care. Circ. Cardiovasc. Genet. 2013, 6, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.-H.; Bai, X.; Zhang, H.; Li, Y.; Hu, L.; Liu, L.; Mao, J.; Yang, X.; Dila, N. Gene Polymorphisms Affect the Effectiveness of Atorvastatin in Treating Ischemic Stroke Patients. Cell. Physiol. Biochem. 2016, 39, 630–638. [Google Scholar] [CrossRef]
- Dutch Pharmacogenetics Working Group Pharmacogenetic Recommendations. 2005.
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1 and Simvastatin-Induced Myopathy: 2014 Update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, C.; García, M. Causality assessment in reports on adverse drug reactions. Algorithm of Spanish pharmacovigilance system. Med. Clin. (Barc) 2016, 147, 461–464. [Google Scholar] [CrossRef]
- Belmonte, C.; Ochoa, D.; Román, M.; Saiz-Rodríguez, M.; Wojnicz, A.; Gómez-Sánchez, C.I.; Martín-Vílchez, S.; Abad-Santos, F. Influence of CYP2D6, CYP3A4, CYP3A5 and ABCB1 Polymorphisms on Pharmacokinetics and Safety of Aripiprazole in Healthy Volunteers. Basic Clin. Pharmacol. Toxicol. 2018, 122, 596–605. [Google Scholar] [CrossRef] [Green Version]
- Caudle, K.E.; Sangkuhl, K.; Whirl-Carrillo, M.; Swen, J.J.; Haidar, C.E.; Klein, T.E.; Gammal, R.S.; Relling, M.V.; Scott, S.A.; Hertz, D.L.; et al. Standardizing CYP 2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin. Transl. Sci. 2019. [Google Scholar] [CrossRef] [Green Version]
- Scott, S.A.; Sangkuhl, K.; Stein, C.M.; Hulot, J.-S.; Mega, J.L.; Roden, D.M.; Klein, T.E.; Sabatine, M.S.; Johnson, J.A.; Shuldiner, A.R. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C19 Genotype and Clopidogrel Therapy: 2013 Update. Clin. Pharmacol. Ther. 2013, 94, 317–323. [Google Scholar] [CrossRef]
- Caudle, K.E.; Rettie, A.E.; Whirl-Carrillo, M.; Smith, L.H.; Mintzer, S.; Lee, M.T.M.; Klein, T.E.; Callaghan, J.T. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing. Clin. Pharmacol. Ther. 2014, 96, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desta, Z.; Gammal, R.S.; Gong, L.; Whirl-Carrillo, M.; Gaur, A.H.; Sukasem, C.; Hockings, J.; Myers, A.; Swart, M.; Tyndale, R.F.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2B6 and Efavirenz-Containing Antiretroviral Therapy. Clin. Pharmacol. Ther. 2019, 106, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Birdwell, K.; Decker, B.; Barbarino, J.; Peterson, J.; Stein, C.; Sadee, W.; Wang, D.; Vinks, A.; He, Y.; Swen, J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubiaur, P.; Saiz-Rodríguez, M.; Ochoa, D.; Belmonte, C.; Román, M.; Mejía, G.; Martín-Vilchez, S.; Abad-Santos, F. Influence of CYP2B6 Activity Score on the Pharmacokinetics and Safety of Single Dose Efavirenz in Healthy Volunteers. Pharmacogenom. J. 2019. [Google Scholar] [CrossRef]
- Saiz-Rodríguez, M.; Ochoa, D.; Belmonte, C.; Román, M.; Vieira de Lara, D.; Zubiaur, P.; Koller, D.; Mejía, G.; Abad-Santos, F. Polymorphisms in CYP1A2, CYP2C9 and ABCB1 Affect Agomelatine Pharmacokinetics. J. Psychopharmacol. 2019, 33, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, B.; Tulsyan, S.; Mittal, R. The Effect of ABCB1 Polymorphisms on the Outcome of Breast Cancer Treatment. Pharmacogenom. Pers. Med. 2016, 9, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Graffelman, J.; Weir, B.S. Testing for Hardy–Weinberg Equilibrium at Biallelic Genetic Markers on the X Chromosome. Heredity 2016, 116, 558–568. [Google Scholar] [CrossRef] [Green Version]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity: Mechanistic Insights and Clinical Implications. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Zubiaur, P.; Soria-Chacartegui, P.; Koller, D.; Navares-Gómez, M.; Ochoa, D.; Almenara, S.; Saiz-Rodriguez, M.; Mejía-Abril, G.; Villapalos-García, G.; Román, M.; et al. Impact of Polymorphisms in Transporter and Metabolizing Enzyme Genes on Olanzapine Pharmacokinetics and Safety in Healthy Volunteer. Biomed. Pharmacother. 2021, 133, 111087. [Google Scholar] [CrossRef]
- Park, J.; Kim, C.O.; Jin, B.H.; Yang, S.; Park, M.S.; Hong, T. Pharmacokinetic Drug Interaction between Atorvastatin and Ezetimibe in Healthy Korean Volunteers. Transl. Clin. Pharmacol. 2017, 25, 202. [Google Scholar] [CrossRef] [Green Version]
- Gibson, D.M.; Bron, N.J.; Richens, M.A.; Hounslow, N.J.; Sedman, A.J.; Whitfield, L.R. Effect of Age and Gender on Pharmacokinetics of Atorvastatin in Humans. J. Clin. Pharmacol. 1996, 36, 242–246. [Google Scholar] [CrossRef]
- Romaine, S.P.R.; Bailey, K.M.; Hall, A.S.; Balmforth, A.J. The Influence of SLCO1B1 (OATP1B1) Gene Polymorphisms on Response to Statin Therapy. Pharmacogenom. J. 2010, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.-J.; Zhang, W.; Chen, Y.; Guo, D.; Tu, J.-H.; Xu, L.-Y.; Tan, Z.-R.; Chen, B.-L.; Li, Z.; Zhou, G.; et al. Rifampicin Alters Atorvastatin Plasma Concentration on the Basis of SLCO1B1 521T>C Polymorphism. Clin. Chim. Acta 2009, 405, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.; Perin, P.M.S.; Purim, S.G.; Silbiger, V.N.; Genvigir, F.D.V.; Willrich, M.A.V.; Arazi, S.S.; Luchessi, A.D.; Hirata, M.H.; Bernik, M.M.S.; et al. Pharmacogenetics of OATP Transporters Reveals That SLCO1B1 c.388A>G Variant Is Determinant of Increased Atorvastatin Response. Int. J. Mol. Sci. 2011, 12, 5815–5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-E.; Kim, K.-B.; Bae, S.K.; Moon, B.-S.; Liu, K.-H.; Shin, J.-G. Contribution of Cytochrome P450 3A4 and 3A5 to the Metabolism of Atorvastatin. Xenobiotica 2008, 38, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Pauly, D.F.; Pacanowski, M.A.; Langaee, T.; Frye, R.F.; Johnson, J.A. Effect of Cytochrome P450 3A5 Genotype on Atorvastatin Pharmacokinetics and Its Interaction with Clarithromycin. Pharmacotherapy 2011, 31, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Wilke, R.A.; Moore, J.H.; Burmester, J.K. Relative Impact of CYP3A Genotype and Concomitant Medication on the Severity of Atorvastatin-Induced Muscle Damage. Pharmacogenet. Genom. 2005, 15, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Willrich, M.A.V.; Hirata, M.H.; Genvigir, F.D.V.; Arazi, S.S.; Rebecchi, I.M.M.; Rodrigues, A.C.; Bernik, M.M.S.; Dorea, E.L.; Bertolami, M.C.; Faludi, A.A.; et al. CYP3A53A Allele Is Associated with Reduced Lowering-Lipid Response to Atorvastatin in Individuals with Hypercholesterolemia. Clin. Chim. Acta 2008, 398, 15–20. [Google Scholar] [CrossRef]
- Kivistö, K.T.; Niemi, M.; Schaeffeler, E.; Pitkälä, K.; Tilvis, R.; Fromm, M.F.; Schwab, M.; Eichelbaum, M.; Strandberg, T. Lipid-Lowering Response to Statins Is Affected by CYP3A5 Polymorphism. Pharmacogenetics 2004, 14, 523–525. [Google Scholar] [CrossRef]
- Thompson, J.F.; Man, M.; Johnson, K.J.; Wood, L.S.; Lira, M.E.; Lloyd, D.B.; Banerjee, P.; Milos, P.M.; Myrand, S.P.; Paulauskis, J.; et al. An Association Study of 43 SNPs in 16 Candidate Genes with Atorvastatin Response. Pharmacogenom. J. 2005, 5, 352–358. [Google Scholar] [CrossRef]
- Lennernäs, H. Clinical Pharmacokinetics of Atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef]
- Goswami, S.; Gong, L.; Giacomini, K.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Very Important Pharmacogene Information for SLC22A1. Pharmacogenet. Genom. 2014, 24, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.C.; Curi, R.; Genvigir, F.D.V.; Hirata, M.H.; Hirata, R.D.C. The Expression of Efflux and Uptake Transporters Are Regulated by Statins in Caco-2 and HepG2 Cells. Acta Pharmacol. Sin. 2009, 30, 956–964. [Google Scholar] [CrossRef]
- Syam Das, S.; Nair, S.S.; Indira, M. Atorvastatin Modulates Drug Transporters and Ameliorates Nicotine-Induced Testicular Toxicity. Andrologia 2018, 50, e13029. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of Genetic Variation in the Organic Cation Transporter 1 (OCT1) on Metformin Action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Sakaeda, T.; Fujino, H.; Komoto, C.; Kakumoto, M.; Jin, J.; Iwaki, K.; Nishiguchi, K.; Nakamura, T.; Okamura, N.; Okumura, K. Effects of Acid and Lactone Forms of Eight HMG-CoA Reductase Inhibitors on CYP-Mediated Metabolism and MDR1-Mediated Transport. Pharm. Res. 2006, 23, 506–512. [Google Scholar] [CrossRef] [Green Version]
- Haas, D.W.; Kwara, A.; Richardson, D.M.; Baker, P.; Papageorgiou, I.; Acosta, E.P.; Morse, G.D.; Court, M.H. Secondary Metabolism Pathway Polymorphisms and Plasma Efavirenz Concentrations in HIV-Infected Adults with CYP2B6 Slow Metabolizer Genotypes. J. Antimicrob. Chemother. 2014, 69, 2175–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarska, K.E.; Dekker, S.J.; Vermeulen, N.P.E.; Commandeur, J.N.M. Effect of UGT2B7*2 and CYP2C8*4 Polymorphisms on Diclofenac Metabolism. Toxicol. Lett. 2018, 284, 70–78. [Google Scholar] [CrossRef] [PubMed]


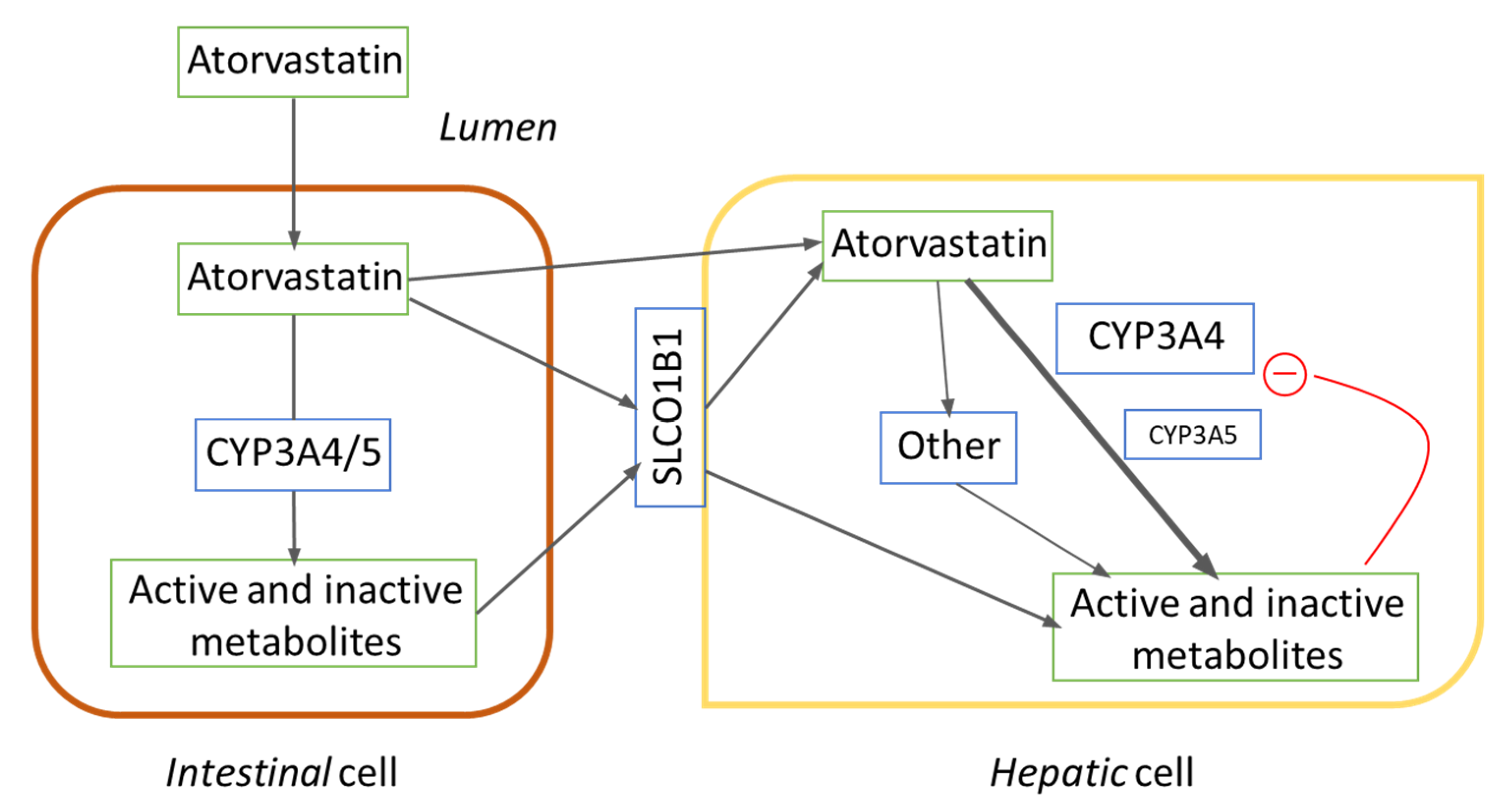{kind=link}
| Gene | Allele/SNP | Gene | Allele/SNP |
|---|---|---|---|
| CYP1A2 | *1C (rs2069514) | CYP3A4 | *22 (rs35599367) |
| *1F (rs762551) | rs55785340 | ||
| *1B (rs2470890) | rs4646438 | ||
| CYP2A6 | *9 (rs28399433) | CYP3A5 | *3 (rs776746) |
| CYP2B6 | *9 (rs3745274) | *6 (rs10264272) | |
| *5 (rs3211371) | ABCB1 | C3435T (rs1045642) | |
| *4 (rs2279343) | G2677T/A (rs2032582) | ||
| rs2279345 | C1236T (rs1128503) | ||
| rs4803419 | 1000-44G>T (rs10276036) | ||
| CYP2C8 | *2 (rs11572103) | 2895+3559C>T (rs7787082) | |
| *3 (rs10509681) | 330-3208C>T (rs4728709) | ||
| *4 (rs1058930) | 2481+788T>C (rs10248420) | ||
| CYP2C9 | *2 (rs1799853) | 2686-3393T>G (rs10280101) | |
| *3 (rs1057910) | 2320-695G>A (rs12720067) | ||
| CYP2C19 | *2 (rs4244285) | 2482-707A>G (rs11983225) | |
| *3 (rs4986893) | 2212-372A>G (rs4148737) | ||
| *4 (rs28399504) | rs3842 | ||
| *17 (rs12248560) | ABCC2 | c.1247G>A (rs2273697) | |
| CYP2D6 | *3 (rs35742686) | rs717620 | |
| *4 (rs3892097) | SLCO1B1 | *1B (rs2306283) | |
| *6 (rs5030655) | *5 (rs4149056) | ||
| *7 (rs5030867) | c.-910G>A (rs4149015) | ||
| *8 (rs5030865) | rs11045879 | ||
| *9 (rs5030656) | SLC22A1 | *2 (rs72552763) | |
| *10 (rs1065852) | *3 (rs12208357) | ||
| *14 (rs5030865) | *5 (rs34059508) | ||
| *17 (rs28371706) | UGT1A1 | *28 (rs887829) | |
| *41 (rs28371725) |
| Sex | n | Weight (kg) | CV% | BMI (kg/m2) | CV% | Height (m) | CV% | Age (years) | CV% |
|---|---|---|---|---|---|---|---|---|---|
| Women | 85 | 61.5 | 13.8 | 23.1 | 13.4 | 1.63 | 3.7 | 30.1 | 28.2 |
| Men | 71 | 75.3 * | 12.7 | 24.4 * | 10.2 | 1.75 * | 4.0 | 27.8 | 27.0 |
| Race | |||||||||
| Caucasian | 81 | 65.5 | 16.8 | 22.6 *2 | 10.6 | 1.7 | 5.9 | 25.5 *2 | 22.4 |
| Latin-American | 70 | 69.2 | 15.0 | 24.8 | 11.7 | 1.67 | 6.0 | 32.7 | 26.9 |
| Black or Arabic | 5 | 85.3 *1 | 15.0 | 27.0 | 7.4 | 1.77 *3 | 5.6 | 33.7 | 18.7 |
| Total | 156 | 67.7 | 16.8 | 23.7 | 12.2 | 1.69 | 5.9 | 29.0 | 27.9 |
| N | AUC/DW (kg*h*ng/mL*mg) | CV% | Cmax/DW (kg*ng/mL*mg) | CV% | tmax (h) | CV% | t1/2 (h) | CV% | Vd/F (l/kg) | CV% | Cl/F (L/h*kg) | CV% | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Female | 85 | 142.6 | 56.7 | 34.3 | 61.2 | 1.4 | 57.1 | 9.3 | 31.2 | 124.0 | 66.3 | 9023.4 | 49.6 |
| Male | 71 | 136.6 | 58.4 | 29.9 | 74.9 | 1.4 | 57.1 | 8.7 | 25.3 | 117.0 | 52.3 | 9248.5 | 42.7 | |
| Clinical trial | A | 14 | 129.2 | 35.9 | 22.7 | 36.1 | 1.7 | 88.2 | 8.1 | 23.5 | 103.5 | 45.1 | 8745 | 35.5 |
| B | 30 | 174.9 | 63.0 | 36.1 | 58.2 | 1.5 | 53.3 | 9.1 | 23.1 | 107.3 | 70.7 | 7836.7 | 61.5 | |
| C | 39 | 116.3 *1 | 59.3 | 26.6 *2 | 60.2 | 1.3 | 61.5 | 9.4 | 28.7 | 147.1 *1*2 | 53.0 | 11,036.7 *1 | 45.7 | |
| D | 37 | 149.4 | 46.3 | 37.4 | 61.0 | 1.4 | 42.9 | 8.7 | 33.3 | 103.9 | 71.9 | 8247.0 | 45.9 | |
| E | 36 | 130.5 | 57.5 | 33.7 | 82.2 | 1.4 | 42.9 | 9.5 | 30.5 | 127.7 | 52.2 | 9181.5 | 30.4 | |
| Ezetimibe | No | 119 | 136.9 | 60.9 | 30.7 | 69.1 | 1.4 | 64.3 | 9.2 | 27.2 | 126.1 | 57.3 | 9399.1 | 46.2 |
| Yes | 37 | 149.4 ! | 46.3 | 37.4 *! | 61.0 | 1.4 | 42.9 | 8.7 | 33.3 | 103.9 | 71.9 | 8247.0 | 45.9 | |
| Race | Caucasian | 81 | 132.5 | 61.1 | 33.5 | 71.6 | 1.4 | 50.0 | 9.5 | 29.5 | 138.1 *3! | 63.8 | 9938.9 | 49.4 |
| Latin-American | 70 | 147.8 | 54.2 | 31.0 | 62.9 | 1.5 | 60.0 | 8.7 | 27.6 | 103.4 | 45.9 | 8279.3 | 38.7 | |
| Black or Arabic | 5 | 148.7 | 48.4 | 31.8 | 34.3 | 1.6 | 43.8 | 7.6 | 9.2 | 83.8 | 33.4 | 7808.3 | 36.8 |
| N | AUC/DW (kg*h*ng/mL*mg) | CV% | Cmax (kg*ng/mL*mg) | CV% | tmax (h) | CV% | t1/2 (h) | CV% | Vd/F (l/kg) | CV% | Cl/F (L/h*kg) | CV% | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLCO1B1 | NF | 86 | 122.6 *1! | 44.9 | 28.5 *1! | 59.3 | 1.4 | 64.3 | 9 | 30.0 | 127.8 *1! | 57.8 | 9827.4 *1! | 43.1 |
| DF | 30 | 181.5 | 59.3 | 37.5 | 60.0 | 1.4 | 57.1 | 8.7 | 25.3 | 99.7 | 78.0 | 7680.4 | 67.0 | |
| PF | 4 | 283.7 | 41.4 | 62.4 | 35.9 | 1.3 | 38.5 | 10.5 | 9.5 | 66.4 | 58.9 | 4382.7 | 60.2 | |
| CYP3A5*3 | *1/*1 | 5 | 244.2 *1 | 21.5 | 57.1 *1 | 55.2 | 1.9 *1 | 42.1 | 9.8 | 26.5 | 65.2 | 41.9 | 4405.1 *1 | 23.7 |
| *1/*3 | 32 | 153.7 | 70.3 | 35.1 | 75.5 | 1.4 | 71.4 | 8.8 | 21.6 | 114.3 | 60.6 | 9181.8 | 55.0 | |
| *3/*3 | 119 | 131.8 | 52.0 | 30.5 | 63.0 | 1.4 | 50.0 | 9.1 | 30.8 | 124.9 | 59.9 | 9309.2 | 42.7 | |
| SLC22A1*2 | *1/*1 | 71 | 131.8 | 48.7 | 28.9 | 53.6 | 1.4 | 57.1 | 8.9 | 29.2 | 121.4 | 57.5 | 9393.2 | 47.7 |
| *1/*2 | 41 | 151.2 | 66.2 | 33.5 | 64.5 | 1.4 | 64.3 | 9 | 28.9 | 121.1 | 72.0 | 9136.4 | 54.0 | |
| *2/*2 | 8 | 195.7 | 51.8 | 49.8 *2 | 64.7 | 1.6 | 68.8 | 8.9 | 19.1 | 83 | 59.5 | 6449.1 | 47.9 | |
| SLC22A1*5 | *1/*1 | 114 | 144.6 | 57.0 | 32.3 | 61.9 | 1.4 | 64.3 | 8.9 | 28.1 | 114.8 | 59.7 | 8933.1 | 49.3 |
| *1/*5 | 6 | 105.6 | 58.4 | 24.2 | 34.3 | 1.6 | 43.8 | 10.8 | 30.6 | 193.9 * | 75.6 | 12,455.2 | 56.3 | |
| UGT2B7 rs7439366 | *1/*1 | 9 | 159.6 | 80.6 | 44.1 | 103.9 | 1.8 *3 | 44.4 | 8.2 | 24.4 | 106.9 | 50.4 | 8821 | 43.8 |
| *1/*2 | 12 | 108.9 | 29.5 | 26.6 | 35.3 | 1.1 | 36.4 | 9.5 | 22.1 | 137.8 | 35.6 | 9990.2 | 22.7 | |
| *2/*2 | 15 | 130.4 | 41.1 | 33.2 | 69.3 | 1.4 | 28.6 | 10.1 | 36.6 | 132 | 64.2 | 8751 | 28.3 | |
| Total | 156 | 139.9 | 57.3 | 32.3 | 67.2 | 1.4 | 57.1 | 9.1 | 28.6 | 120.8 | 60.6 | 9125.9 | 46.4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubiaur, P.; Benedicto, M.D.; Villapalos-García, G.; Navares-Gómez, M.; Mejía-Abril, G.; Román, M.; Martín-Vílchez, S.; Ochoa, D.; Abad-Santos, F. SLCO1B1 Phenotype and CYP3A5 Polymorphism Significantly Affect Atorvastatin Bioavailability. J. Pers. Med. 2021, 11, 204. https://doi.org/10.3390/jpm11030204
Zubiaur P, Benedicto MD, Villapalos-García G, Navares-Gómez M, Mejía-Abril G, Román M, Martín-Vílchez S, Ochoa D, Abad-Santos F. SLCO1B1 Phenotype and CYP3A5 Polymorphism Significantly Affect Atorvastatin Bioavailability. Journal of Personalized Medicine. 2021; 11(3):204. https://doi.org/10.3390/jpm11030204
Chicago/Turabian StyleZubiaur, Pablo, Maria Dolores Benedicto, Gonzalo Villapalos-García, Marcos Navares-Gómez, Gina Mejía-Abril, Manuel Román, Samuel Martín-Vílchez, Dolores Ochoa, and Francisco Abad-Santos. 2021. "SLCO1B1 Phenotype and CYP3A5 Polymorphism Significantly Affect Atorvastatin Bioavailability" Journal of Personalized Medicine 11, no. 3: 204. https://doi.org/10.3390/jpm11030204