NCL1, A Highly Selective Lysine-Specific Demethylase 1 Inhibitor, Suppresses Castration-Resistant Prostate Cancer Growth via Regulation of Apoptosis and Autophagy

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. LSD1 Is Highly Expressed in CRPC
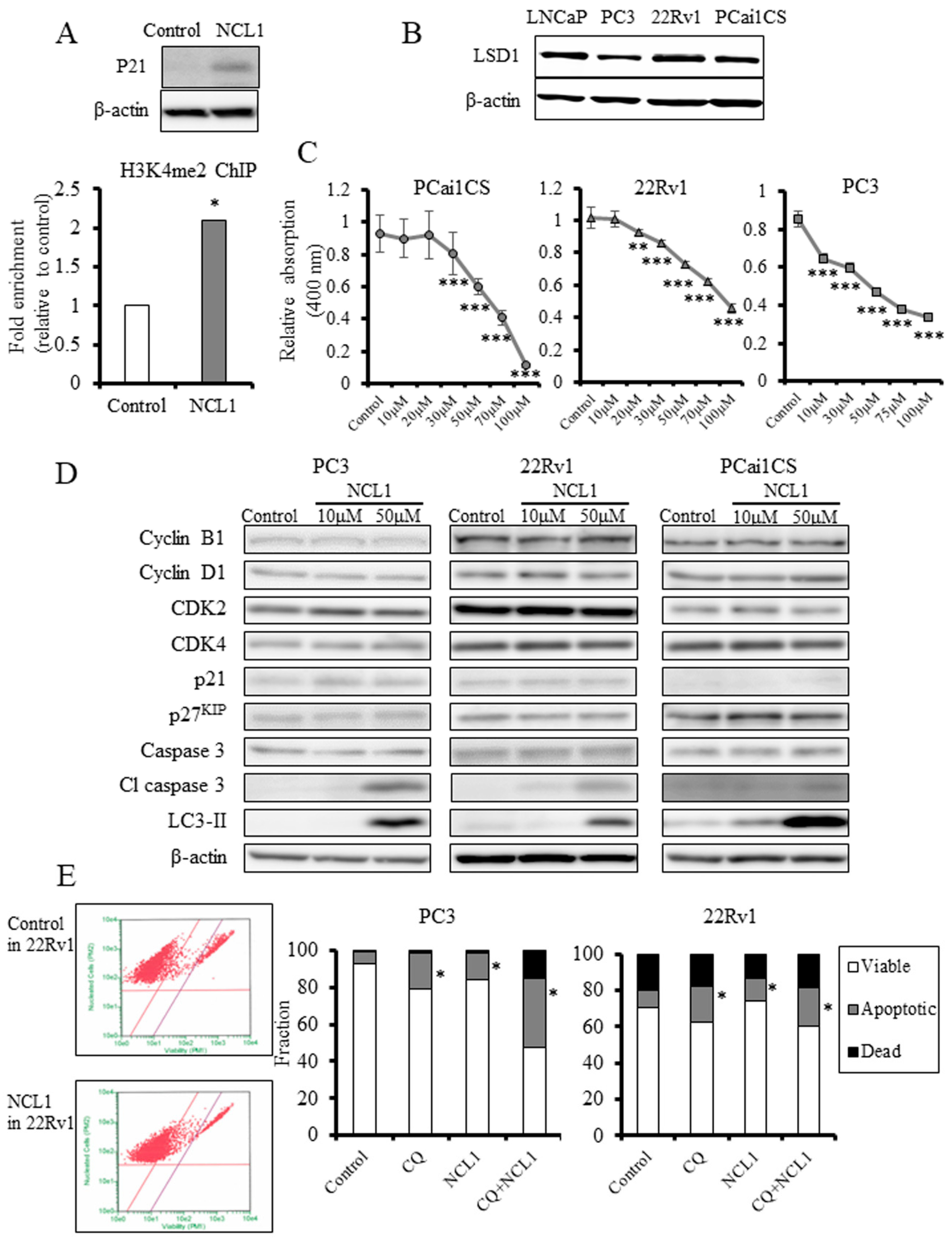
2.2. LSD1 Expression in Prostate Cancer Cell Lines and Suppression of Prostate Cancer Cell Proliferation by NCL1
2.3. NCL1 Inhibits CRPC Cell Growth by Apoptotic Mechanisms
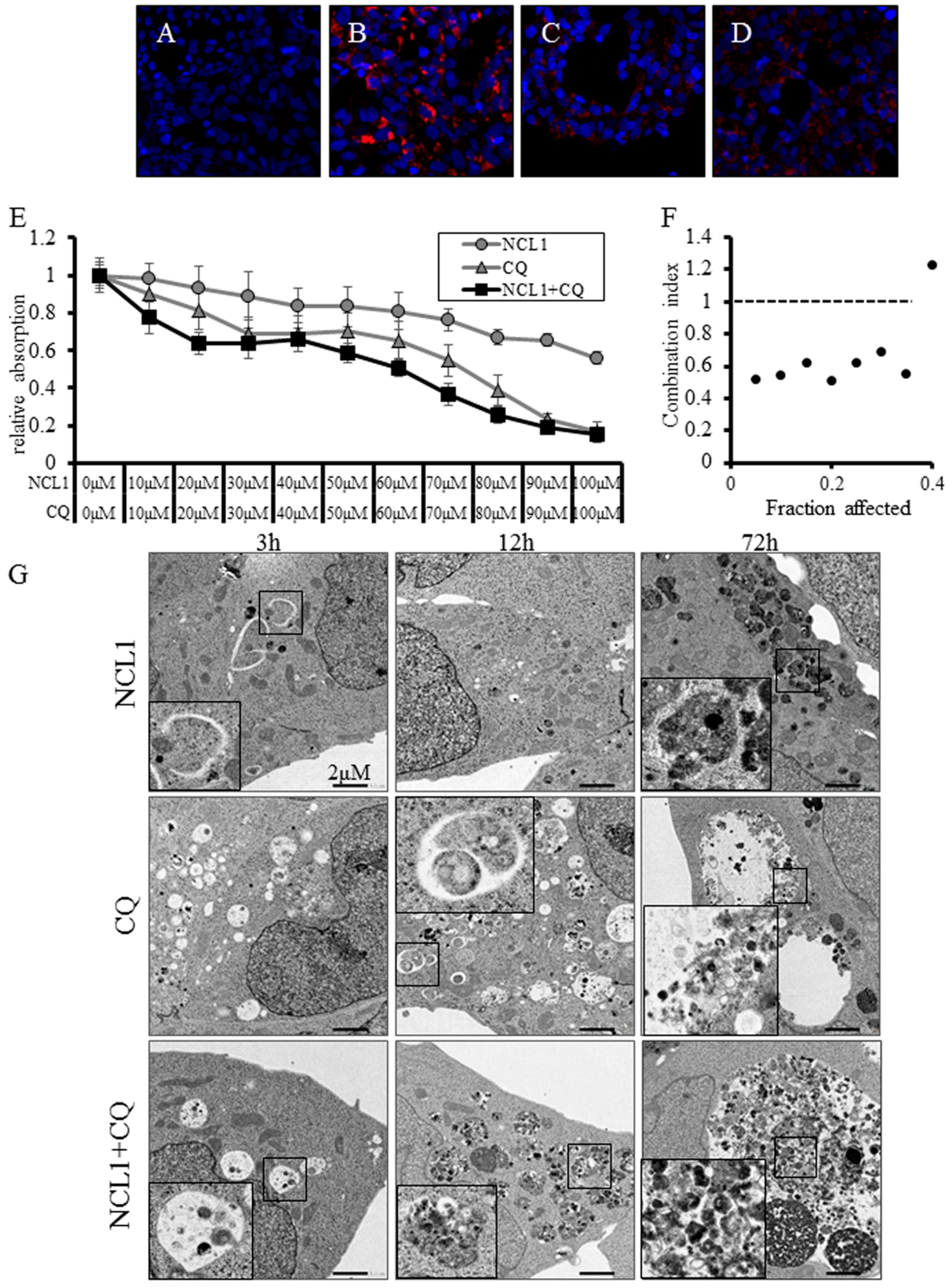
2.4. NCL1 Potentially Regulates Autophagy to Induce Cell Death in 22Rv1 Cells
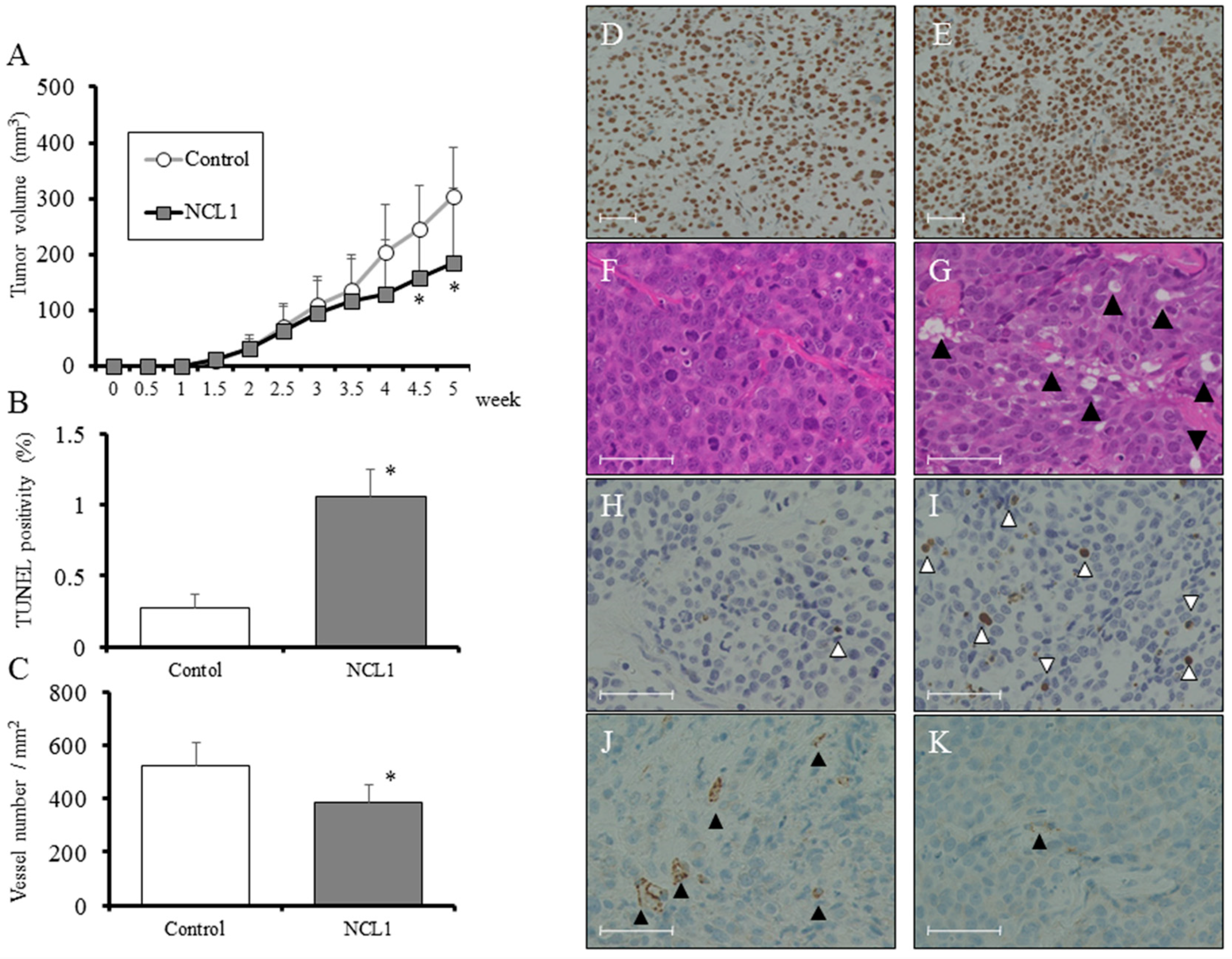
2.5. Ex Vivo Regulation of Tumorigenesis by NCL1
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Human Castration-Naïve and Castration-Resistant Prostate Cancer Specimens
5.2. Chemicals
5.3. Prostate Cancer Cell Lines
5.4. Cell Proliferation Assay
5.5. Analysis of the Cytotoxic Effect of NCL1 in Combination with Chloroquine
5.6. ChIP Assay
5.7. Western Blot Analysis
5.8. Flow Cytometry Analysis
5.9. Lysosome Localization and Activity Using LysoTracker® and LysoSensor™ Dyes
5.10. Transmission Electron Microscopy (TEM)
5.11. Ex Vivo Studies Using a Subcutaneous Castration-Resistant PCai1 Model
5.12. Immunohistochemical Analysis
5.13. TUNEL Assay
5.14. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Frydenberg, M.; Stricker, P.D.; Kaye, K.W. Prostate cancer diagnosis and management. Lancet 1997, 349, 1681–1687. [Google Scholar] [CrossRef]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B.; Saad, F., Jr.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogen and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Rossi, V.; Di Zasso, E.; Galasso, G.; De Rosa, C.; Abbondanza, C.; Sinisi, A.A.; Altucci, L.; Migliaccio, A.; Castoria, G. Estrogens Modulate Somatostatin Receptors Expression and Synergize With the Somatostatin Analog Pasireotide in Prostate Cells. Front. Pharmacol. 2019, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schule, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Sedukhina, A.S.; Nakagawa, Y.; Maeda, I.; Kubota, M.; Ohnuma, S.; Tsugawa, K.; Ohta, T.; Roche-Molina, M.; Bernal, J.A.; et al. LSD1 overexpression is associated with poor prognosis in basal-like breast cancer, and sensitivity to PARP inhibition. PLoS ONE 2015, 10, e0118002. [Google Scholar] [CrossRef] [PubMed]
- Etani, T.; Suzuki, T.; Naiki, T.; Naiki-Ito, A.; Ando, R.; Iida, K.; Kawai, N.; Tozawa, K.; Miyata, N.; Kohri, K.; et al. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses prostate cancer without adverse effect. Oncotarget 2015, 6, 2865–2878. [Google Scholar] [CrossRef] [PubMed]
- Ueda, R.; Suzuki, T.; Mino, K.; Tsumoto, H.; Nakagawa, H.; Hasegawa, M.; Sasaki, R.; Mizukami, T.; Miyata, N. Identification of cell-active lysine specific demethylase 1-selective inhibitors. J. Am. Chem. Soc. 2009, 131, 17536–17537. [Google Scholar] [CrossRef]
- Gurel, B.; Ali, T.Z.; Montgomery, E.A.; Begum, S.; Hicks, J.; Goggins, M.; Eberhart, C.G.; Clark, D.P.; Bieberich, C.J.; Epstein, J.I.; et al. NKX3.1 as a marker of prostatic origin in metastatic tumors. Am. J. Surg. Pathol. 2010, 34, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Culine, S.; El Demery, M.; Lamy, P.J.; Iborra, F.; Avances, C.; Pinguet, F. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J. Urol. 2007, 178, 844–848, discussion 848. [Google Scholar] [CrossRef]
- Conteduca, V.; Burgio, S.L.; Menna, C.; Carretta, E.; Rossi, L.; Bianchi, E.; Masini, C.; Amadori, D.; De Giorgi, U. Chromogranin A is a potential prognostic marker in prostate cancer patients treated with enzalutamide. Prostate 2014, 74, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Burgio, S.L.; Conteduca, V.; Menna, C.; Carretta, E.; Rossi, L.; Bianchi, E.; Kopf, B.; Fabbri, F.; Amadori, D.; De Giorgi, U. Chromogranin A predicts outcome in prostate cancer patients treated with abiraterone. Endocr. Relat. Cancer 2014, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, V.; Ahmad, S.; Nilsson, E.M.; Helczynski, L.; Kenna, S.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. The lysine specific demethylase-1 (LSD1/KDM1A) regulates VEGF-A expression in prostate cancer. Mol. Oncol. 2013, 7, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A., 3rd; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2008, 115, E4179–E4188. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell death beyond apoptosis. Leukemia 2000, 14, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Demidenko, Z.N.; Blagosklonny, M.V. Flavopiridol induces p53 via initial inhibition of Mdm2 and p21 and, independently of p53, sensitizes apoptosis-reluctant cells to tumor necrosis factor. Cancer Res. 2004, 64, 3653–3660. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Jin, S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy 2006, 2, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.H.; Nguyen, T.L.; Delcroix, V.; Teres, S.; Bouchecareilh, M.; Salin, B.; Bodineau, C.; Vacher, P.; Priault, M.; Soubeyran, P.; et al. mTORC1 inhibition in cancer cells protects from glutaminolysis-mediated apoptosis during nutrient limitation. Nat. Commun. 2017, 8, 14124. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, F.; Thomas, C.; Crafter, C.; Kumano, M.; Zhang, F.; Davies, B.R.; Gleave, M.E.; Zoubeidi, A. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin. Cancer Res. 2013, 19, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.G.; Yang, J.C.; Kung, H.J.; Shi, X.B.; Tilki, D.; Lara, P.N., Jr.; DeVere White, R.W.; Gao, A.C.; Evans, C.P. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blessing, A.M.; Rajapakshe, K.; Reddy Bollu, L.; Shi, Y.; White, M.A.; Pham, A.H.; Lin, C.; Jonsson, P.; Cortes, C.J.; Cheung, E.; et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy 2017, 13, 506–521. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Weston, A.; Bearrs, J.; Thode, T.; Neiss, A.; Soldi, R.; Sharma, S. Reversible lysine-specific demethylase 1 antagonist HCI-2509 inhibits growth and decreases c-MYC in castration- and docetaxel-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2016, 19, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahm, E.R.; Singh, S.V. Honokiol causes G0-G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol. Cancer Ther. 2007, 6, 2686–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Kaur, M.; Agarwal, C.; Tecklenburg, M.; Sclafani, R.A.; Agarwal, R. p21 and p27 induction by silibinin is essential for its cell cycle arrest effect in prostate carcinoma cells. Mol. Cancer Ther. 2007, 6, 2696–2707. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, X.; Guo, J.; Weng, X.; Jiang, G.; Wang, Z.; He, L. Inhibition of LSD1 by Pargyline inhibited process of EMT and delayed progression of prostate cancer in vivo. Biochem. Biophys. Res. Commun. 2015, 467, 310–315. [Google Scholar] [CrossRef]
- Perillo, B.; Di Santi, A.; Cernera, G.; Ombra, M.N.; Castoria, G.; Migliaccio, A. Nuclear receptor-induced transcription is driven by spatially and timely resticted waves of ROS. The role of Akt, IKKα, and DNA damage repair enzymes. Nucleus 2014, 5, 482–491. [Google Scholar] [CrossRef]
- Perillo, B.; Di Santi, A.; Cernera, G.; Ombra, M.N.; Castoria, G.; Migliaccio, A. Phosphorylation of H3 serine 10 by IKKα governs cyclical production of ROS in estrogen-induced transcription and ensures DNA wholeness. Cell Death Differ. 2014, 9, 1503. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Chen, H.; Qiu, T.; Weng, X.D.; Guo, J.; Wang, L.; Liu, X.H. Effects of cisplatin on the LSD1-mediated invasion and metastasis of prostate cancer cells. Mol. Med. Rep. 2016, 14, 2511–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehm, S.M.; Schmidt, L.J.; Heemers, H.V.; Vessella, R.L.; Tindall, D.J. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008, 68, 5469–5477. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B.; et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Dai, B.; Ye, D.; Kong, Y.; Chang, K.; Jia, Z.; Yang, X.; Zhang, H.; Zhu, Y.; Shi, G. Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci. Rep. 2015, 5, 7654. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Fedor, H.L.; Lotan, T.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regufe da Mota, S.; Bailey, S.; Strivens, R.A.; Hayden, A.L.; Douglas, L.R.; Duriez, P.J.; Borrello, M.T.; Benelkebir, H.; Ganesan, A.; Packham, G.; et al. LSD1 inhibition attenuates androgen receptor V7 splice variant activation in castration resistant prostate cancer models. Cancer Cell Int. 2018, 18, 71. [Google Scholar] [CrossRef]
- Naiki, T.; Asamoto, M.; Toyoda-Hokaiwado, N.; Naiki-Ito, A.; Tozawa, K.; Kohri, K.; Takahashi, S.; Shirai, T. Organ specific Gst-pi expression of the metastatic androgen independent prostate cancer cells in nude mice. Prostate 2012, 72, 533–541. [Google Scholar] [CrossRef]
- Naiki, T.; Naiki-Ito, A.; Asamoto, M.; Kawai, N.; Tozawa, K.; Etani, T.; Sato, S.; Suzuki, S.; Shirai, T.; Kohri, K.; et al. GPX2 overexpression is involved in cell proliferation and prognosis of castration-resistant prostate cancer. Carcinogenesis 2014, 35, 1962–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, E.; Duigou-Osterndorf, R.; Krown, S.E.; Fanucchi, M.P.; Chou, J.; Hirsch, M.S.; Clarkson, B.D.; Chou, T.C. Synergistic cytotoxic effect of azidothymidine and recombinant interferon alpha on normal human bone marrow progenitor cells. Blood 1989, 74, 1281–1286. [Google Scholar] [PubMed]
- Schmukler, E.; Wolfson, E.; Haklai, R.; Elad-Sfadia, G.; Kloog, Y.; Pinkas-Kramarski, R. Chloroquine synergizes with FTS to enhance cell growth inhibition and cell death. Oncotarget 2014, 5, 173–184. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. of Mice | BW (g) | Liver (%) | R-Kidney (%) | L-Kidney (%) | |
|---|---|---|---|---|---|
| Control | 11 | 23.4 ± 1.3 | 5.14 ± 0.22 | 0.78 ± 0.02 | 0.80 ± 0.05 |
| NCL1 1.0 mg/kg | 10 | 23.5 ± 1.5 | 5.30 ± 0.26 | 0.81 ± 0.03 | 0.82 ± 0.04 |
| Control | NCL1 1.0 mg/kg | |
|---|---|---|
| AST | 56.2 ± 10.3 | 55.9 ± 10.6 |
| ALT | 28.2 ± 6.3 | 27.1 ± 3.6 |
| ALP | 240.7 ± 20.6 | 250.2 ± 30.9 |
| T-Bil | 0.04 ± 0.01 | 0.04 ± 0.01 |
| T-Chol | 83.5 ± 8.3 | 85.1 ± 8.3 |
| Crea | 0.09 ± 0.01 | 0.10 ± 0.01 |
| BUN | 23.4 ± 3.2 | 23.5 ± 2.7 |
| Na | 146.3 ± 3.8 | 146.8 ± 2.7 |
| K | 6.7 ± 1.0 | 5.9 ± 0.7 |
| Cl | 110.4 ± 6.5 | 109.7 ± 5.3 |
| Ca | 8.5 ± 0.2 | 8.4 ± 0.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etani, T.; Naiki, T.; Naiki-Ito, A.; Suzuki, T.; Iida, K.; Nozaki, S.; Kato, H.; Nagayasu, Y.; Suzuki, S.; Kawai, N.; et al. NCL1, A Highly Selective Lysine-Specific Demethylase 1 Inhibitor, Suppresses Castration-Resistant Prostate Cancer Growth via Regulation of Apoptosis and Autophagy. J. Clin. Med. 2019, 8, 442. https://doi.org/10.3390/jcm8040442
Etani T, Naiki T, Naiki-Ito A, Suzuki T, Iida K, Nozaki S, Kato H, Nagayasu Y, Suzuki S, Kawai N, et al. NCL1, A Highly Selective Lysine-Specific Demethylase 1 Inhibitor, Suppresses Castration-Resistant Prostate Cancer Growth via Regulation of Apoptosis and Autophagy. Journal of Clinical Medicine. 2019; 8(4):442. https://doi.org/10.3390/jcm8040442
Chicago/Turabian StyleEtani, Toshiki, Taku Naiki, Aya Naiki-Ito, Takayoshi Suzuki, Keitaro Iida, Satoshi Nozaki, Hiroyuki Kato, Yuko Nagayasu, Shugo Suzuki, Noriyasu Kawai, and et al. 2019. "NCL1, A Highly Selective Lysine-Specific Demethylase 1 Inhibitor, Suppresses Castration-Resistant Prostate Cancer Growth via Regulation of Apoptosis and Autophagy" Journal of Clinical Medicine 8, no. 4: 442. https://doi.org/10.3390/jcm8040442