
Role of EMT in Metastasis and Therapy Resistance

Department of Medicine, Cedars-Sinai Medical Center, 8750 Beverly Blvd., Atrium 103, Los Angeles, CA 90048, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2016, 5(2), 17; https://doi.org/10.3390/jcm5020017
Submission received: 20 October 2015
/
Revised: 22 December 2015
/
Accepted: 23 December 2015
/
Published: 27 January 2016
(This article belongs to the Special Issue Epithelial-Mesenchymal Transition)
{kind=link}
{kind=link}
Abstract
:Epithelial–mesenchymal transition (EMT) is a complex molecular program that regulates changes in cell morphology and function during embryogenesis and tissue development. EMT also contributes to tumor progression and metastasis. Cells undergoing EMT expand out of and degrade the surrounding microenvironment to subsequently migrate from the primary site. The mesenchymal phenotype observed in fibroblasts is specifically important based on the expression of smooth muscle actin (α-SMA), fibroblast growth factor (FGF), fibroblast-specific protein-1 (FSP1), and collagen to enhance EMT. Although EMT is not completely dependent on EMT regulators such as Snail, Twist, and Zeb-1/-2, analysis of upstream signaling (i.e., TGF-β, EGF, Wnt) is necessary to understand tumor EMT more comprehensively. Tumor epithelial–fibroblast interactions that regulate tumor progression have been identified during prostate cancer. The cellular crosstalk is significant because these events influence therapy response and patient outcome. This review addresses how canonical EMT signals originating from prostate cancer fibroblasts contribute to tumor metastasis and recurrence after therapy.
1. EMT Classifications
The steps of EMT include a combination of the microenvironment molecules adjusting to accommodate cellular expansion to either promote tissue development in physiological or pathological contexts [1,2,3,4,5]. These changes depend on spatial and temporal cues, characterized by the production of secretory enzymes to degrade the extracellular matrix (ECM), while cell expansion continues [5]. ECM degradation by matrix metalloproteinases (MMPs) is a clear example of this step of EMT. Furthermore, transforming growth factor-beta (TGF-β) regulates MMPs and downstream Smad signaling to enhance these degradation activities. Steps of EMT in cancer are comparable when cells intravasate into the circulatory and/or lymphatic systems, extravasate at the secondary site, and micrometastases develop during advanced disease [6,7,8]. These phenotypic changes illustrate the cancer cell plasticity required for tumor escape. To clearly discuss the molecules that control EMT and potential therapeutic approaches, EMT classifications as type-1, type-2, and type-3 are explained [9,10,11]. These types of EMT seem to be based on a combination of several factors, including components of the microenvironment, the context of the epithelial and stromal cells, and exogenous changes in host immunity. More specifically, stromal fibroblast “re-education” can be used to study adjacent epithelial cells. The transformation of the stromal compartment along with microenvironment changes provides a niche, where morphological signals influence tissue vitality and integrity (Figure 1A). Type-1 EMT is associated with embryogenesis and normal tissue development, type-2 with wound healing, and type-3 with tumor development and metastasis [12]. Type-1 EMT meets a terminal point during fetal development and after birth in contrast with type-2 and type-3 EMT. Embryo development requires the transformation of neural crest cells to generate three defined germ layers, responsible for physiological growth and expansion. Epithelial, mesenchymal, and mesodermal cells function during this process. Mesenchymal cells only exhibited migratory and invasive properties based on high expression of vimentin, N-cadherin, and low expression of E-cadherin. Wnt signaling orchestrates these steps of EMT based on findings where Wnt-deficient embryos lost their ability to undergo gastrulation. Similar results were observed when ectopic expression of Wnt8c in embryos induced primitive streak formation [13]. The epithelia also serve as the foundation for renal fibrosis where morphological changes define the clinical outcome [14]. The role of TGF-β during these steps control Wnt along with other regulators, such as Nodal and Vg1 [2]. Transcription factors related to type-1 EMT include mesodermal posterior 1 and 2 (Mesp1, Mesp2) during gastrulation [15], Snail [16,17], and eomesodermin (EOMES) [18] while downstream target gene transcription is either repressed or activated. Snail overexpression in ARCaPM cells influenced the ECM by decreasing the expression of integrins α5, α2, and β1 [16]. These effects were also similar when Snail transcriptional repressed maspin in 22Rv1 prostate cancer cells [17]. Snail knockdown reversed these effects to reduce EMT in prostate cancer cells. The common mesenchymal phenotypes at the initiation of embryogenesis generate diverse cell types based on these molecular interactions. Wnt signaling can regulate Snail activity to promote embryonic stem cell renewal or differentiation [19,20,21]. Snail conditional knockout experiments in embryonic stem cells had activated Wnt3a and Axin2 expression compared to control cells [19]. Wnt inhibition did decrease Snail activity, but Snail knockout did not affect EOMES expression. Traditional patterns of high Snail expression correlated with low E-cadherin expression were reversed after Snail knockout studies. These findings clearly demonstrate this molecular link during normal development as these cells also possessed cuboidal (epithelial) morphology. Furthermore, proliferation and cell death signals were unaffected after Snail knockout. A link to type-2 EMT can be seen when Snail did not affect embryonic stem cells migration after wound healing assays were performed [19]. Mesp1 induces mesoderm differentiation via Snail in embryonic stem cells, and have been studied in regards to miRNA regulation. miR-200 is downregulated when Snail is activated to enhance pluripotency (miR-294, miR-295, miR-292-3p) and repress the epithelial phenotype (miR-200c, miR-200b, miR-429), another feature of enhanced EMT [22,23]. TGF-β signaling inhibitor SB-431542 stopped activin receptor-like kinase 5 (activin) and miR-200 functions to promote EMT and differentiation. These features are important to note based on the function of activin during wound healing and skin carcinogenesis [12]. However, there is a balance of activin required to repair damaged tissues and prevent scar tissue formation that may lead to skin tumors.
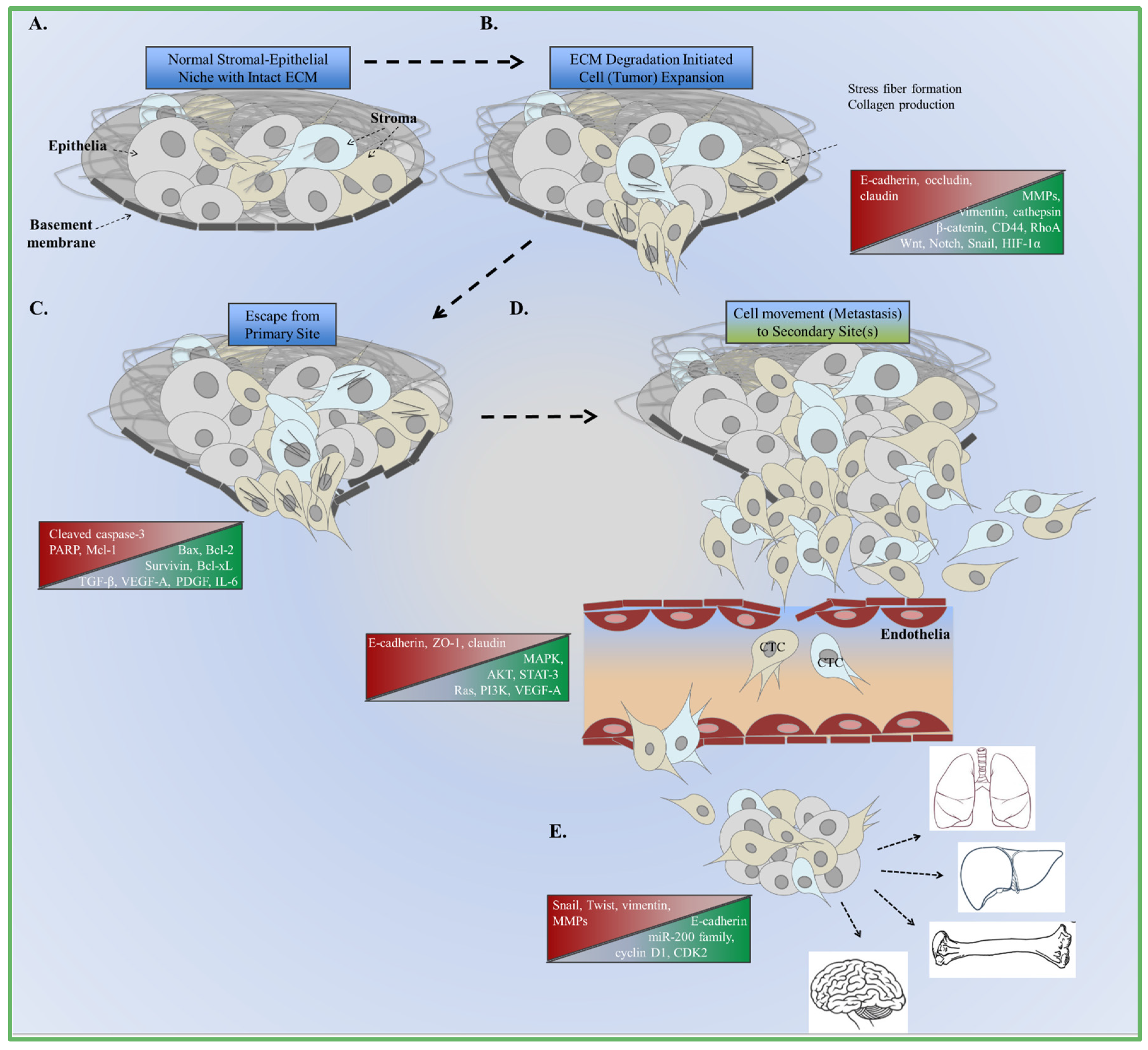
Cancer tissue remodeling associated with wound healing, described more than 150 years ago by Virchow, is a major component of the EMT mechanism [10]. Similarities between type-2 and type-3 EMT are found within the stroma of wound healing and transformed tumor cells. Cellular shape and extracellular matrix deposits from the stroma include collagen, which contributes to microenvironment remodeling (Figure 1B). Tenascin-C, fibronectin, and fibrinogen increase to accommodate these steps as cell shape changes [24,25]. The same markers used to monitor the changes of cell morphology and function is recognized within the circulatory system. Subsequently, the secretion of these proteins serves as the substrates to promote survival and migration [26,27] (Figure 1 and Figure 2). Claudins, occludins, and E-cadherin serve as these substrates during EMT to deregulate the ECM and microenvironment.
Development of cardiac tissue requires type-1 EMT in the generation of cardiac valves from endocardial cushions, whereas type-2 EMT is involved in adult cardiac disease and other diseases of the kidney and liver [14,28,29,30]. Disruption of normal myocardial tissues is regulated by adjacent fibroblasts. These fibroblasts recruit extracellular matrix (ECM) molecules and originate from surrounding endothelial (blood vessel) cells. These activities demonstrate a connection between type-1 EMT and endothelial EMT (endo-MT), another mechanism promoting support for expanding tissues [31,32]. TGF-β induced the development of fibroblast cells during type-1 and endo-MT. Conversely, bone morphogenetic protein 7 (BMP-7) maintained the endothelial phenotype, inhibited endo-MT and progression of cardiac fibrosis, renal injury, and colon cancer [33,34,35]. The expanding fibroblast cells involved in cardiac development invade and assist in epicardium formation, and maintain structural integrity observed in the adult mesodermal tissues [36,37]. Similar observations have been made in renal pathologies [9,33,38,39], liver fibrosis [30], and during gastrulation and neural crest formation [36].
Figure 1.
Summary of EMT and metastasus. (A) Normal interactions between cells and extracellular matrix (ECM) compobnents. Changes in canonical signaling and cellular function are the foundation of aberrant cell growth and expansion; (B) Increased expression of enzymes that degrade the ECM are involved in decreasing adhesion to the basement membrane; (C) Cells escape from the basement membrane as growth factor and cytokine signaling increases to accommodate changes in the microenvironment; (D) Cells intravasate and spread to the bloodstream as circulating tumor cells (CTC); (E) Extravasation at the secondary site promotes the formation of micrometastases and the re-induction of epithelial markers (MET). Sites of metastasis include the lungs, liver, bones, and brain.
Figure 1.
Summary of EMT and metastasus. (A) Normal interactions between cells and extracellular matrix (ECM) compobnents. Changes in canonical signaling and cellular function are the foundation of aberrant cell growth and expansion; (B) Increased expression of enzymes that degrade the ECM are involved in decreasing adhesion to the basement membrane; (C) Cells escape from the basement membrane as growth factor and cytokine signaling increases to accommodate changes in the microenvironment; (D) Cells intravasate and spread to the bloodstream as circulating tumor cells (CTC); (E) Extravasation at the secondary site promotes the formation of micrometastases and the re-induction of epithelial markers (MET). Sites of metastasis include the lungs, liver, bones, and brain.

Type-2 EMT is activated during injury, and has been studied during kidney fibrosis [40]. The mechanism of fibrosis can be monitored or predicted using specific fibroblast markers for cell morphology. Kidney fibrosis cells expressed fibroblast-specific protein 1 (FSP1) and conducted EMT, demonstrated by fibroblast phenotypes [41]. FSP1 is a filament-associated, calcium-binding protein specifically expressed at high levels in cells with the EMT phenotype. Findings also indicated that sometimes fibroblasts arise from the local conversion of epithelial cells [29]. NP-1 tubular epithelial cells submerged in type I collagen gels converged to a fibroblast phenotype accompanied with de novo FSP1 and vimentin expression. After developing a polyclonal FSP1 antibody, expression was also noted in kidney, lung, and spleen tissues. Embryonic analysis showed that FSP1 was not expressed in the embryo at day E8.5, although mRNA could be detected in decidual tissues from the uterus [3]. A monoclonal antibody to the β-subunit of the PDGF-receptor was used to determine expression in renal tissues [42]. Mesangial and peritubular interstitial cells within the glomerulus have high PDGF-receptor expression during glomerulonephritis, and cellular shape is similar to EMT. FSP1 is selective for cells exhibiting the fibroblast phenotype, unlike previous markers used to study injury and healing [43]. Platelet-derived growth factor (PDGF) [42], α-smooth muscle actin (α-SMA) [43], and ecto-5′-nucleotidase [44] expression was associated with myofibroblast accumulation during injury. The elevated expression of FSP-1 in kidney fibrosis helped support tubular epithelia cells EMT transformation in generating interstitial fibroblasts during kidney injury [28] and fibrosis [29]. FSP-1, for example, is regulated by a fibroblast transcription site (FTS-1) that exists in the promoters of EMT-promoting genes such as Twist, Snail, beta-catenin [42]. These activities correspond to fibroblast-regulated fibrosis in intestines and pulmonary tissues [45]. It is important to note other sources of myofibroblasts in tumor tissues include pericytes, resident fibroblasts, as well as bone marrow-derived mesenchymal stem cells [46,47]. Fibroblasts associated with lung cancer have a clear expression profile related to their involvement during tumor progression. The microarray gene expression of proteins regulated by TGF-β and MAPK signaling was higher in cancer-associate fibroblasts (CAF) cells compared to normal fibroblasts [46]. Similar to findings from Neal et al. [16], integrin α11 was higher in CAF cells following microarray analyses.
TGF-β signaling was analyzed in NMuMG mouse mammary gland cells where TGF-β treatment induced epithelial differentiation [48]. Although these cells express elevated TGF-β type I receptor Tsk7L, they lack expression of ALK-5/R4 type I receptor. A shortened form of the Tsk7L type I receptor inhibited EMT in the presence of TGF-β, even with responsiveness. Cell growth and E-cadherin were reduced indicating a role for type I TGF-β receptors. Furthermore, these activities occur in a RhoA-dependent manner in NMuMG cells [49]. Type-3 EMT is identified during cancer development and metastasis—the primary focus of this review. TGF-β is a master regulator of cancer development, where decreased E-cadherin, ZO-1, and desmoplakin 1 expression in breast epithelial cells correlated with a fibroblast-like morphology [50]. Actin fiber reorganization occurred after TGF-β treatment, representing an early sign of EMT. This event was characterized by cellular differentiation [51], also indicated in another study where breast tumor EMT was regulated by TGF-β and RhoA [49]. These breast epithelial cells had the classical EMT morphology and expression of mesenchymal markers. This study also indicated that TGF-β-induced EMT was unchanged even when Smad signaling decreased, and stress fiber formation regulated by TGF-β was not Smad-dependent. TGF-β, p38MAPK [50], and AKT signaling interactions were found to be significant for tumor-specific EMT [52,53], as well as TGF-β ligand [54,55], and BMP-7 [34,39]. TGF-β signaling in the fibroblasts regulated adjacent tumor epithelia to promote prostate cancer [56]. These observations indicated a role of this cell type to regulate oncogenic potential in epithelial cells using a fibroblast-specific TGF-β receptor 2 knockout model (Tgfbr2fspKO). Alternatively, TGF-β ligand BMP-2 induces invasion and cancer stem features via STAT3 activation during colon cancer [35], and ERK1/2 and AKT during gastric cancer [57,58,59]. Tissue scarring depicted by fibrosis, followed by the administration of tumor necrosis factor alpha (TNF-α), leads to mesenchymal marker expression [60]. Migrating epithelial cells of human skin had increased vimentin and FSP1 expression. Cells contained within hypertrophic scars also had increased mesenchymal marker expression, combined with inflammatory cytokine expression. Expression of MMPs, FSP1 and vimentin were induced after TNF-α treatment antagonized by targeting BMP2/4. These findings demonstrated similarities during wound healing to promote EMT. Breast cancer EMT has been studied using fibroblast growth factor-10 (FGF-10) [61]. FGF-10 treatment increased tumorigenesis by increasing the MDA-MB-231 cell ability to migrate as indicated by wound healing assays, decreased apoptosis, and increased Wnt signaling. Cells lining the blood vessels supplying the tumors with nutrients, and a preferred environment to develop micrometastases characterize endo-MT. Endo-MT is observed to have a significant role in all three types of EMT. The endothelia serve as precursors for hematopoietic cells during embryonic development (type-1), fibrosis (type-2), and cell malignancy (type-3) in the adult [3,15,26]. Most of the cell types within the circulatory system develop according to the Endo-MT cues responding to factors like platelet-derived growth factor (PDGF), periostin, and several within the Notch signaling pathway. Studies focused on the signaling and cross-talk between different cell types during tumor progression have relied on the functionality and preference of tumor cells to respond to the fibroblast-specific cues, especially during later stages of tumorigenesis. Proliferative endothelia have been associated with the proliferations and motility of adjacent breast cancer epithelia [27,62]. Cancer cells associated with EMT promote expansion of mesenchymal cells where poor patient outcome was correlated with elevated Twist, Snail and CD44 expression in lung cancer [63]. Since tumor vasculature can be critical to the expansion of tumors, type-1 EMT in the form of endo-MT can play a paracrine role in tumorigenesis. Together, these can be a mechanism for the expansion of CAF.
2. EMT Mediators: Tumor and Microenvironment Crosstalk
Conserved zinc finger transcription factors (Snail, Zeb-1, Slug), regulated by upstream TGF-β activities, have been shown to drive EMT functions [64,65,66]. Snail also represses E-cadherin transcription at the promoter level by binding to its enhancer box sequence. These functions are related to the differentiated morphology to subsequently promote tumor metastasis. High expression of these types of molecules compared to non-tumor molecules are correlated with poorer prognosis in clinical analyses, as observed with Snail and Twist in lung adenocarcinoma [63], Slug in gastric cancer [64], Snail in prostate cancer [16,17] and breast cancer [67,68]. More specifically, the epigenetic events as described with zinc finger protein methylation of ZBTB20 [66], ZNF139 [69], and ZNF545 [70] are associated with tumor metastasis. Recent findings indicate that pancreatic tumor cells are able to conduct EMT in the absence of Snail and Twist transcription factors [71]. These cells were able to conduct metastasis even after Snail or Twist knockout. In vivo analysis of Snail- or Twist-induced EMT was not rate-limiting for invasion and metastasis in transgenic mouse models. Yet, in vitro overexpression of Snail regulates maspin tumor suppressor transcription in prostate cancer [17], and estrogen receptor alpha [67] and kinase activities in breast cancer cells [53]. The regulatory activities of Snail and Slug were also demonstrated when they activated TGF-β in breast cancer cells [68]. The data indicate a contrast between the physiologically relevant and cell culture conditions that are significant to EMT during cancer. Commonalities between the in vivo and in vitro conditions contributing to EMT are regulated by TGF-β, MAPK, and ECM component activities [72]. The fibroblast phenotype of these EMT tumor cells is critically important based on analyses of the surrounding microenvironment. CAF and tumor epithelial cells coordinate during EMT to promote vascular intravasation [73] (Figure 1). Circulating tumor cell (CTC) can escape from primary radiation or surgical intervention and their capture by current methods rarely indicate their potential to engraft and expand in a secondary metastatic site.
EMT is enhanced in part due to the maintenance of the fibroblast morphology and intricate crosstalk with the tumor microenvironment, also related to decreased cell adhesion [74]. Molecules that compose tight junctions, ECM components [75], and fibroblast-derived secretome [76] are examples of the crosstalk between the tumor and microenvironment. RhoA signaling is a traditional mediator of morphologic change. RhoA-ROCK signaling can serve as a switch to enable cell cycle arrest [49]. A general theme that has emerged is that proliferation and differentiation do not occur at the same time. There is controversy in the role of Smad proteins in the progression of EMT downstream of TGF-β. Smad2/3 may not be essential for morphologic changes of the epithelia, but it is a required component of cell cycle arrest for transdifferentiation steps to progress. These changes include cytoskeletal changes depicted by shifts in polarity due to actin reorganization in metastatic cancer cells [77], and increased motility [75,78]. Cytochalasin D (Cyt D) treatment of breast cancer cells increased E-cadherin and reduced RhoA expression. Remodeling of the cytoskeleton plays a crucial role in EMT to promote cancer. In vivo analysis indicated that vimentin was 3.7 times higher compared to E-cadherin in oral squamous cell carcinoma [78]. Wnt, Notch, and TGF-β signaling can additionally potentiate the expression of Zeb1, FoxC2, and Twist for mesenchymal morphologic changes mediated by the inhibition of E-cadherin expression and increased expression of vimentin and N-cadherin.
Transformation of mammary epithelial cells has been attributed to the functions of stroma, as compared to findings in prostate cancer [79]. The loss of stromal TGF-β responsiveness enables paracrine hepatocyte growth factor (HGF) signaling in adjacent epithelia tumors, characterized by the fibroblast phenotype [80,81]. Epithelial expression of the c-Met and Ron, HGF cognate receptors, support Stat5, JAK, and TAK1 to promote Snail expression. Platelet-derived growth factor (PDGF) is also identified as a functional determinant for CAF [4,82]. Following an orthotopic mouse model of colorectal cancer, the stroma cells required stanniocalcin-1 (STC1), a PDGF regulator, to promote intravasation of adjacent tumor cells [82]. Thus, epithelial tumors contained within invasive fronts may be dependent on paracrine signaling. Further, when associated with type 3 EMT, the morphologic changes can be associated with cell transformation [83,84]. There is clearly an intricate balance of the intracellular and extracellular activities that modulate EMT and in turn cancer initiation and progression. The cadherin and integrin switches during EMT involve relocation of the cells from the basement membrane environment to the fibrillary ECM zone [85]. Matrix metalloproteinases (MMPs) and integrins function with the tumor microenvironment to induce changes during EMT [62,86,87]. FSP1, matrilysin and stromelysin-1 are EMT-promoting genes that can downregulate E-cadherin to promote EMT [87,88]. MMP-3 and MMP-9 originating from the tumor microenvironment, tumor cells, and stromal cells adjacent to the tumor cells help to facilitate EMT via invasion and metastasis behaviors. The activation and expression of integrins also facilitate interaction with the tumor environment. Overexpression of αvβ6 integrin activated by MMP-3 in poorly differentiated squamous cell carcinomas gained a fibroblastic morphology to EMT during oral cancer [89] and melanoma [90]. Myosin filaments, restructured to accommodate changing cell shape by vimentin and beta-catenin signaling contribute to the invasive front of the tumor. These functions are specifically significant in head and neck squamous cell cancer (HNSCC), and oral squamous cell cancer (OSCC) [91]. Expression of a CD44high/CD24l0w protein signature, aldehyde dehydrogenase (ALDH1), Nanog, and stromal cell-derived factor-1/CXCR4 signaling regulate bone metastasis [92]. These activities regulate cellular plasticity to enhance the interactions between epithelial and fibroblast cell types.
3. EMT and Tumor Metastasis
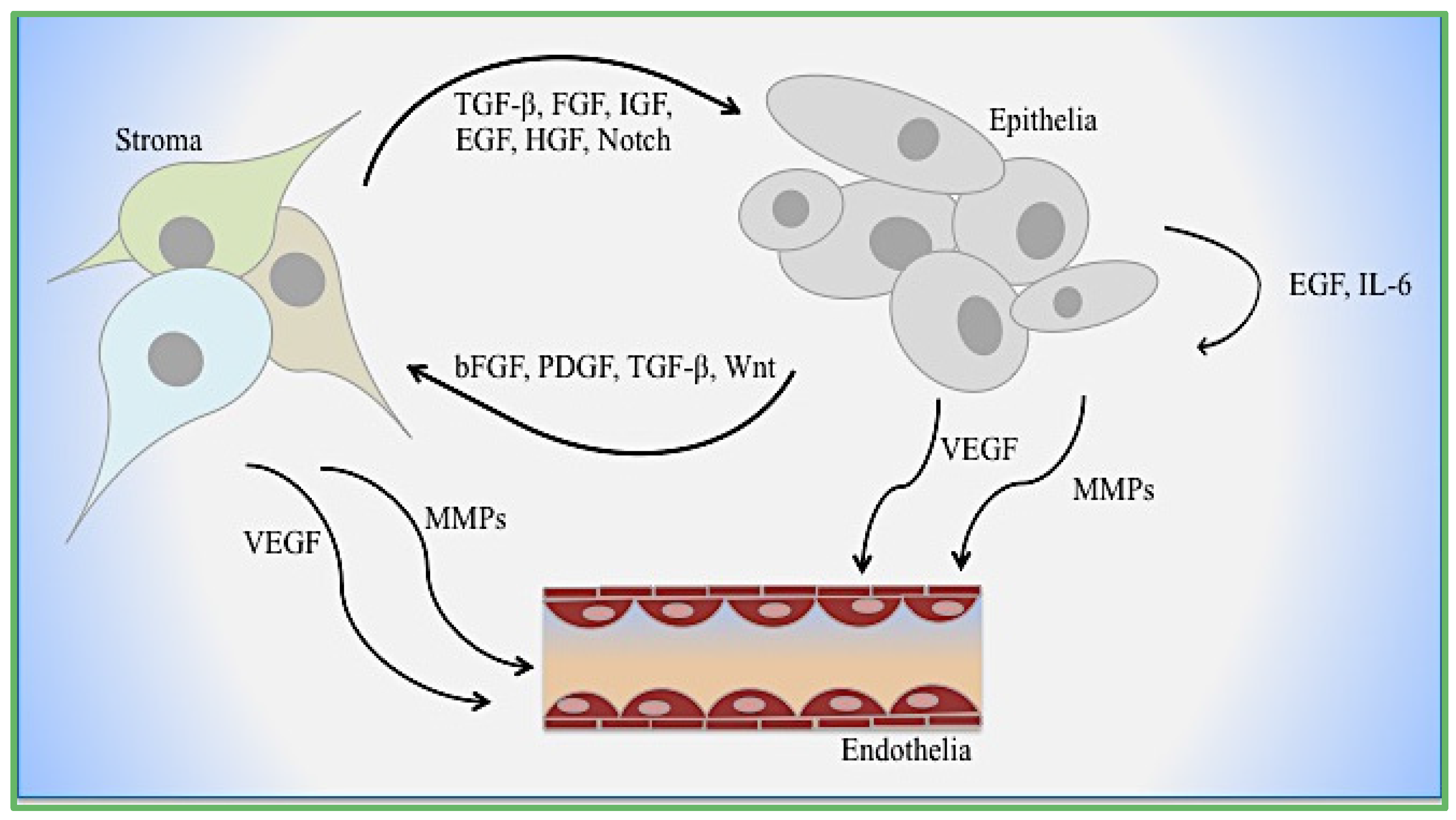
Although the function of EMT in development has not been disputed, its role in cancer metastasis has long been a point of controversy. EMT is a resultant of paracrine signaling by stromal-derived factors as well as a potential source of cells in the stromal compartment (Figure 2). The difficulty in proving that adult patient tissues unequivocally have cells undergoing EMT is the source of controversy. However, the process of EMT, either as a result of paracrine induction or inherent somatic mutations, can be associated with the gain of stem/progenitor cell differentiation. The gain of the fibroblastic phenotype is often associated with the gain of stem features, such as markers previously described, reduced proliferation, adaptability to secondary microenvironments during metastasis, and therapeutic resistance. The aberrant activation of Ras/mitogen-activated protein kinase (Ras/MAPK) is a feature of cancer stem cells in the pancreas and breast has long been recognized yet it is a difficult target to drug. In prostate cancer, epithelial expression of receptor activator of NF-κB ligand (RANKL) in an osteomimicry phenotype conveys the advantages of osteoclast activation in the bone microenvironment [93,94]. Further profile analysis of bone-specific proteins osteopontin (OPN), osteocalcin (OC), bone sialoprotein (BSP), and osteoprotegrin (OPG) demonstrated expression in prostate cancer epithelia [95,96,97]. Further, the ability of EMT cells to engraft in the bone marrow niche that also supports hematopoietic stem cells is reported. Survival within the skeleton during prostate cancer progression is supported by several growth factors related to TGF-β, ECM, and hormone-regulated receptors. The molecular crosstalk between the prostate tumor and bone stroma are targets of therapies to increase tumor cell death.
Figure 2.
Cellular interactions during EMT. Mechanisms of epithelial–stromal–endothelial crosstalk are depicted. Growth factors, cytokines, and enzymes function dynamically with and between each cell type.
Figure 2.
Cellular interactions during EMT. Mechanisms of epithelial–stromal–endothelial crosstalk are depicted. Growth factors, cytokines, and enzymes function dynamically with and between each cell type.

EMT was associated with RANKL activation in ARCaPM and LNCaP prostate cancer cells accompanied with Snail overexpression [94]. Similarly, Snail expression in the nucleus of epithelial ovarian tumors increased from precursor lesions to carcinomas that are especially resistant to traditional chemotherapy [98]. In this respect, the stem/progenitor state can mean the cancer epithelial may lose contact inhibition as well as become resistant to anoikis. This line of reasoning can help attribute EMT to these processes of metastasis while identifying transient cells in human primary tissues. The evidence of cells in CTC studies that simultaneously have epithelial and fibroblastic markers can support the role of EMT in the process of metastasis.
The controversial role of EMT has been noted during the regulation of the cytoskeleton protein complexes [99]. Here, it introduced the ability of cells to metastasize in the absence of an overt need to undergo EMT, through a process where a group of cells enter the leaky tumor vasculature in a raft-structure. Integrin outside–in signaling is evident in ECM remodeling by fibroblasts to provide an environment for invading tumor epithelia [100]. Cytoskeleton regulation of Rho GTPases Cdc42, Rac1, and RhoA were challenged using inhibitors. The findings from this study implicated that there are different modes of motility regulated by RhoA/ROCK-independent EMT mechanisms. Tumor cells were able to overcome inhibitors that target motility by switching to a different mode (Rac1 to RhoA and RhoC). Tracks of movement were detected as fibroblasts led epithelial tumors through the ECM. Subsequently, it is presumed that the group of cells could lodge in narrow blood vessels and expand without the need to extravasate. Histologic documentation of similar structures in the bone marrow and lung vasculature support this clinical metastatic possibility. In light of the plasticity of cancer epithelia, both EMT and collective migration methods are both likely modes of metastasis.
4. Cooperative EMT Signaling Mechanisms
A common signaling component of multiple paracrine factors including FGF, HGF, and PDGF signaling pathways is Ras activation. Even in the absence of somatic Ras mutations, paracrine Ras activation in the context of TGF-β is a long-recognized cooperation during tumor EMT. Oft et al. reported that EpH4 mammary epithelial cells transformed by mutant Ras signaling underwent EMT [101]. These cells possessed a fibroblast phenotype that directly corresponded to their metastatic behavior and resistance to TGF-β1 cell cycle inhibition. The cooperation of TGF-β and Ras signaling can be through multiple signaling proteins, including phosphatidylinositol-3 kinase (PI3K) and mitogen-activated protein kinase (MAPK), to result in the activation of Snail-downstream genes. Antagonizing the MAPK pathways with U0126 in ARCaP cells was able to partially reverse EMT through the downregulation of Snail [16]. TGF-β cooperates in this process by activating integrin signaling significant to epithelium, but not specific to lung tissue [102]. Forsyth et al. described EMT as a process that leads to the formation of stromal cells that progress in tandem with invasive carcinoma cells [103].
The various CTC capture technologies suggest, however, that cancer cells that have undergone EMT are in fact in circulation (Figure 1D). This observation can be explained by the gain of somatic mutations, bypassing the need for integrin signaling or additional mechanisms of EMT is the other obvious alternative. Integrin proteins are major players in the TGF-β-dependent tumor metastasis process [102,104,105]. Desmosomes and integrins relate consistently to TGF-β, Wnt, and Notch signaling by regulating not only cellular morphology and composition, but also increasing the potential for intravasation and extravasation during metastasis [106]. TGF-β potentiates integrin expression. However, integrin expression can in turn cooperate with TGF-β signaling. Studies in breast epithelia demonstrated that EMT progression by TGF-β can be interrupted by antagonizing integrin β1. Both integrin β1 and TGF-β have a common downstream signaling target, p38MAPK [50]. However, TGF-β activation of p38MAPK was lost if integrin β1 was neutralized. Since p38MAPK is required for TGF-β-mediated EMT to progress, these findings have interesting implications for epithelia that are non-adherent and possibly found in circulation. Presumably, the reduced outside–in integrin signaling by non-adherent cells could result in mesenchymal to epithelial transdifferentiation [49,105]. There are clearly multiple mechanisms of EMT initiation; however, the means of maintaining the mesenchymal state is not clear. Such understanding can support therapeutic limitations faced in treating cancer cells that undergo EMT.
5. Role of EMT in Therapeutic Resistance
EMT is associated with therapy resistance and tumor recurrence. Morphological changes observed during EMT are defined at distinct stages of differentiation. Invasive behavior carried out by tumor epithelia are regulated by the tumor-associated stroma [107]. The persistent accumulation of these cells during the recurrent phase following therapy presents an obstacle during clinical management of cancer. Research findings have long noted that tumor fibroblasts also contribute to therapy resistance. The role of EMT in this context during the reduction of therapy efficacy is evident in several tumors, including prostate and pancreas [108,109,110]. Neuroendocrine differentiation in prostate cancer cells was regulated during hormone treatments, but inadvertently resulted in resistance. These findings are particularly interesting, considering the role of Snail EMT marker during NED in LNCaP prostate cancer cells [111]. MAPK signaling and IL-6-induced neuroendocrine differentiation was increased in LNCaP prostate cancer cells, similar to activities observed in patients undergoing ADT [112,113,114]. It is important to note the fibroblastic and spindle-shaped cells proliferating during these studies. Canonical signaling regulated by MAPK and PI3K/Akt have also regulated EMT to promote resistance and tumor recurrence.
The stem features acquired in the specific EMT process can be of particular importance when considering therapeutic intervention. The activation of Notch signaling was associated with increased cell proliferation and survival in gemcitabine therapy resistance [111]. Similarly, Slug and Snail expression is associated with radio-resistance and chemo-resistance by reducing apoptosis while increasing stemness [99]. STAT-3 signaling (closely associated with IL-6) was antagonized using siRNA in metastatic PC3 prostate cancer cells [114]. The expression of anti-apoptotic gene Bcl-2, cyclin D1 and c-Myc were reduced, indicating a correlation between this signaling pathway and prostate cancer progression. Standard chemotherapy methods used to target cancer, can promote EMT where methods of reversing EMT had presumed benefit [115,116]. More specifically, resistance to therapies such as paclitaxel was associated with increased migration and invasion of tumor cells. Twist not only induces EMT, but increases therapy resistance in breast cancer cells via downregulation of estrogen receptor-α [117] and increased Akt [115]. Targeting the proteasome has also been shown to reduce chemoresistance via Snail signaling. Proteasome inhibitor NPI-0052 downregulated Snail and NF-kappaB expression while Raf-1 kinase inhibitory protein (RKIP) expression increased [116].
Tumor complexity is perpetuated by the communication between tumor cells and the microenvironment, contributing to therapy resistance [118]. Therapies targeting endothelial, cancer stem, and immune-evasive cells also present challenges when tumor signaling adapts. Doxorubicin therapy induced thymic endothelial cell expression of IL-6 and tissue inhibitor of metalloproteinase 1 (TIMP-1), providing tumor cells with a protective niche within the thymus tissue [118]. Following the administration of therapies, hypoxic tumor areas increase tumor growth and survival. The reason that a broader therapeutic strategy of targeting both the tumor epithelia and its microenvironment is starting to show promise may be because cells undergoing EMT are addressed as well as the source of paracrine mediators of tumor progression/differentiation. Tumor metastasis brings about another layer of complexity as the tumor microenvironment changes relative to the primary site. Tumor adaptation to the surrounding environment is influenced by selective pressure applied by drug therapy [119]. Claudin5 (CLDN5) is a potential target for antiangiogenic therapy because it was highly expressed in vascular endothelial cells [120]. Several other CLDN genes (1, 3, 4, 7, 10, 16) were highly expressed in several human tumors. Unfortunately, the long-term negative consequences tend to overshadow the short-lived efficiency of CLDN5 antagonism, as a result of upregulation of other tumor-promoting pathways [121]. Wide-spectrum therapies that are designed to impact more than one pathway or process have had greater efficacy in late-stage cancers. Taxane-based therapies in the context of other hormonal therapies or alone seem to have limited efficacy in breast and prostate cancers. However, in the case of ovarian cancer, where EMT and the gain of stem features by the cancer cells is common-place, large scale de-bulking of the tumor burden by surgical resection seems to have the greatest impact. Taxanes, platinum, and the like occupy the standard of care for late-stage cancer patients, but notoriously have short remission times. Thus, combining such traditional chemotherapy with targeted drugs or more recently immune checkpoint inhibition therapy seems to address the fast growing tumor cells, more quiescent, and those undergoing EMT.
Management of the anticancer drugs for patients based on neurotoxicity, the effects on canonical MAPK and EGFR signaling have been the focus of several studies. Phase II clinical trial analysis indicated that although toleration of CI-1040 was considerable, it failed to demonstrate sufficient antitumor activity [122]. The second generation MEK PD 0325901 showed significantly higher response in various tumors tested, including: breast, colon, pancreatic, and non-small-cell lung cancer. These findings were also related to breast cancer cell resistance to gefitinib, a EGFR tyrosine kinase inhibitor. Gefitinib successfully inhibited EGFR activation in SK-Br-3, MDA-MB-261, and MDA-MB-468 breast cancer cells, although p42/p44-MAPK and AKT phosphorylation was not reduced in MDA-MB-468 compared to the other cells [123]. PI3K inhibitor LY294002 or MEK inhibitor PD 98059 were more effective at reducing cells growth. Combination therapy resulted in apoptosis that was higher compared to single treatments. Overexpression of activated MAPK in normal mammary MCF-10A cells increased resistance, once again indicating the role of this signaling pathway for tumor progression. Epigenetics and transcriptional regulation also play a role in therapeutic efficacy. Promoter hypermethylation and silencing of a Wnt antagonist, secreted frizzled related protein 5 (SFRP5), was associated with ovarian cancer cell chemoresistance to cisplatin [124]. SFRP5 methylation was directly related to poor prognosis in patients who were treated with platinum-based chemotherapy. Cancer-promoting genes were activated based on the epigenetic silencing of Wnt-associated gene SFRP5. These findings link to the Twist- and AKT2-mediated events of EMT where cancer cell invasion, colony formation, and tumor growth were increased. Plant-based natural products such as Genistein, targets Akt/glycogen synthase kinase-3 (GSK-3) signaling [125]. The transgenic adenocarcinoma mouse prostate model (TRAMP/FVB) treated with Genistein had reduce prostate size and poorly defined tissue. Prostatic intra-epithelial neoplasia (PIN) was reduced using Genistein, and there was decreased Snail and migratory potential. Genistein is effective for treatment of various tumors and alters apoptosis, cell cycle and angiogenesis – further inhibiting metastasis. Specifically, caspases, Bcl-2, MAPK, Wnt, and AKT signaling are targeted with Genistein, while possessing synergy with other chemotherapy (i.e., adriamycin, docetaxel, tamoxifen). Quercetin (EGCG) is reported to inhibit EMT, migration, and stem features [126]. The CD44+/CD133+ expressed prostate cancer cells decreased following EGCG treatment, indicating its ability to inhibit self-renewal. Caspase-3/7 activation was coupled with inhibition of Bcl-2, survivin, and Snail. Molecules responsible for invasion and migration were also inhibited and this agent may be useful in future therapy.
Natural products often have more than one target, which comes with advantages and disadvantages. However, the lack of rigorous studies on effective doses of such natural products has been the greatest obstacle to their wide adoption. The heterogeneous nature of the tumor and its microenvironment defines therapeutic efficacy. Mechanisms of inducing synthetic lethality targeting both compartments consider the plasticity of the disease. Ultimately, combination therapies that balance the blocking of resistance mechanisms of first-line therapies without compromising systemic functions are the obvious goal.
6. Conclusions
Studies discussed here indicate important mechanisms of cancer progression that are intimately linked to EMT progression in multiple solid tumors. Although the linking of EMT to the process of metastasis is not clear, its role in therapeutic resistance is evident. This is the result of the stem-like properties acquired by cells undergoing EMT as well as it being a means of potentiating tumor heterogeneity. The study of EMT benefits the strategies of cancer prevention, therapy, and prognosis.
Acknowledgments
This work was supported by T325T32DK007770-14 (to BNS) and RO1 CA108646 (to NAB)
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| EMT | Epithelial mesenchymal transition |
| TGF-β | Transforming growth factor β |
| EGF | Epidermal growth factor |
| MET | Mesenchymal-epithelial transition |
| MAPK | Mitogen activated protein kinase |
| ECM | Extracellular matrix |
| VEGF-A | Vascular endothelial growth factor-A |
| NED | Neuroendocrine differentiation |
| RANKL | Receptor activator of NFκB ligand |
| TME | Tumor microenvironment |
| PDGF | Platelet-derived growth factor |
| CAF | Cancer-associated fibroblast |
| NAF | Normal-associated fibroblast |
| FSP-1 | Fibroblast-specific protein-1 |
| LNCaP | Prostate cancer cells from left supraclavicular lymph node |
| PI3K | Phosphatidylinositol-3 kinase |
| IL-6 | Pro-inflammatory cytokine interleukin-6 |
| STC-1 | Stanniocalcin-1 |
| AR | Androgen receptor |
| ZEB-1 | Zinc finger E-box-binding homeobox 1 |
| ARCaPM | Androgen Repressed Metastatic Human Prostate Cancer Cell Line |
| UO126 | MEK1 and MEK2 selective inhibitor |
| MMP | Matrix metalloproteinase |
| BMP | Bone morphogenetic protein |
| HGF | Hepatocyte growth factor |
| FGF | Fibroblast growth factor |
| CTC | Circulating tumor cells |
| PAR1 | Protease-activated receptor-1 |
| α-SMA | α-smooth muscle actin |
| MSC | Mesenchymal stem cells |
| ROS | Reactive oxygen species |
| HNSCC | Head and neck squamous cell cancer |
| OSCC | Oral squamous cell cancer |
| FAK | Focal adhesion kinase |
| CSC | Cancer stem cells |
| ALDH1 | Aldehyde dehydrogenase 1 |
| CXCR4 | chemokine (C-X-C motif) receptor 4 |
| uPA | Urokinase plasminogen activator |
| IGF | Insulin growth factor |
| OC | Osteocalcin |
| BSP | Bone sialoprotein |
| OPN | Osteopontin |
| OPG | Osteoprotegrin |
| PTEN | Phosphatase and tensin homolog |
| NSCLC | Non-small-cell lung cancer |
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition. From: Cardiovascular Development to Disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Invest. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Werner, S.; Grose, R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003, 83, 835–870. [Google Scholar] [PubMed]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Die Cellular Pathologie in Ibrer Begruendung auf Physiologische und Pathologische Gewebelebre; August Hirschwald: Berlin, Germany, 1858. [Google Scholar]
- Arnoux, V.; Come, C.; Kusewitt, D.F.; Hudson, L.G.; Savagner, P. Cutaneous wound reepithelialization: A partial and reversible EMT. In Rise and Fall of Epithelial Phenotype: Concepts of Epithelial-Mesenchymal Transition; Savagner, P., Ed.; Springer: New York, NY, USA, 2005; pp. 111–134. [Google Scholar]
- Antsiferova, M.; Werner, S. The bright and dark sides of activin in wound healing and cancer. J. Cell Sci. 2012, 125, 3929–3937. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, V.; Schoenwolf, G.C. Primitive-streak origin of the cardiovascular system in avian embryos. Dev. Biol. 1993, 159, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Kaissling, B.; le Hir, M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: Fact or fantasy? J. Clin. Invest. 2011, 121, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.; Acloque, H.; Huang, R.Y.J.; Angela Nieto, M. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Neal, C.L.; Mckeithen, D.; Odero-Marah, V.A. Snail negatively regulates cell adhesion to extracellular matrix and integrin expression via the MAPK pathway in prostate cancer cells. Cell Adh. Migr. 2015, 5, 249–257. [Google Scholar] [CrossRef]
- Neal, C.L.; Henderson, V.; Smith, B.N.; Mckeithen, D.; Graham, T.; Vo, B.T.; Odero-Marah, V.A. Snail transcription factor negatively regulates maspin tumor suppressor in human prostate cancer cells. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Baala, L.; Brialt, S.; Etchevers, H.C.; Laumonnier, F.; Natiq, A.; Amiel, J.; Boddaert, N.; Picard, C.; Sbiti, A.; Asermouh, A.; et al. Homozygous silencing T-box transcription factor EOMES leads to microcephaly with polymicrogyria and corpus callosum agenesis. Nat. Genet. 2007, 39, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Li, X.-Y.; Willis, A.L.; Liu, C.; Chen, G.; Weiss, S.J. Snail1-dependent control of embryonic stem cell pluripotency and lineage commitment. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Sokol, S.Y. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 2011, 138, 4341–4350. [Google Scholar] [CrossRef] [PubMed]
- Ten Berge, D.; Kurek, D.; Blauwkamp, T.; Koole, W.; Maas, A.; Eroglu, E.; Siu, R.K.; Nusse, R. Embryonic stem cells require Wnt proteins to prevent differentation to epiblast stem cells. Nat. Cell Biol. 2011, 13, 1070–1075. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the microRNA-200 family act in opposition to regulate epithelial-to-mesenchymal transition and germ layer fate restriciton in differentiating ESCs. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Samatov, T.R.; Tonevitsky, A.G.; Schumacher, U. Epithelial-mesenchymal transition: Focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Flier, S.N.; Tanjore, H.; Kokkotou, E.G.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J. Biol. Chem. 2010, 285, 20202–20212. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Hanai, J.; Sugimoto, H.; Mammoto, T.; Charytan, D.; Strutz, F.; Kalluri, R. BMP-7 counteracts TGF-beta1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 2003, 9, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Duangkumpha, K.; Techasen, A.; Loilome, W.; Namwat, N.; Thanan, R.; Khuntikeo, N.; Yongvanit, P. BMP-7 blocks the effects of TGF-β-induced EMT in cholangiocarcinoma. Tumour Biol. 2014, 35, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.R.; Oh, S.C.; Lee, D.H.; Kim, J.L.; Lee, S.Y.; Kang, M.H.; Lee, S.I.; Kang, S.; Joung, S.Y.; Min, B.W. BMP-2 induces motility and invasiveness by promoting colon cancer stemness through STAT3 activation. Tumour Biol. 2015, 36, 9475–9486. [Google Scholar] [CrossRef] [PubMed]
- Tam, P.P.; Parameswaran, M.; Kinder, S.J.; Weinberger, R.P. The allocation of epiblast cells to the embryonic heart and other mesodermal lineages: The role of ingression and tissue movement during gastrulation. Development 1997, 124, 1631–1642. [Google Scholar] [PubMed]
- Kissa, K.; Herbomel, P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 2010, 464, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Rastaldi, M.P.; Ferrario, F.; Giardino, L.; Dell’Antonio, G.; Grillo, C.; Grillo, P.; Strutz, F.; Muller, G.A.; Colasanti, G.; D’Amico, G. Epithelial-mesenchymal transition of tubular epithelial cells in human renal biopsies. Kidney Int. 2002, 62, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Shah, A.A.; Kalluri, R. Bone morphogenic protein-7 induces mesenchymal to epithelial transition in adult renal fibroblasts and facilitates regeneration of injured kidney. J. Biol. Chem. 2005, 280, 8094–8100. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Seifert, R.A.; Hudkins, K.L.; Johnson, R.J.; Bowen-Pope, D.F. PDGF-receptor localizes to mesangial, parietal epithelial, and interstitial cells in human and primate kidneys. Kidney Int. 1993, 43, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Darby, I.; Skalli, O.; Gabbiani, G. α-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab. Invest. 1990, 63, 21–30. [Google Scholar] [PubMed]
- Dawson, T.P.; Gandhi, R.; Le Hir, M.; Kaissling, B. Ecto-5′-nucleotidase: Localization in rat kidney by light microscopic histochemical methods. J. Histochem. Cytochem. 1989, 37, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Liau, G.; Yamada, Y.; de Crombrugghe, B. Coordinate regulation of the levels of type III and type I collagen mRNA in most but not all mouse fibroblasts. J. Biol. Chem. 1985, 260, 531–536. [Google Scholar] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Venkov, C.; Link, A.J.; Jennings, J.L.; Plieth, D.; Inoue, T.; Nagai, K.; Xu, C.; Dimitrova, Y.N.; Rauscher, F.J.; Neilson, E.G. A proximal activator of transcription in epithelial-mesenchymal transition. J. Clin. Invest. 2007, 117, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Zent, R.; Ghiassi, M.; McDonnell, M.; Moses, H.L. Integrin beta 1 signaling is necessary for transforming growth factor-β activation of p38MAPK and epithelial plasticity. J. Biol. Chem. 2001, 276, 46707–46713. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Nabi, I.R. Actin cytoskeletal regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M.; et al. Snail promotes epithelial mesenchymal transition in breast cancer cells in part via activation of nuclear ERK2. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, M.; Pozzi, A.; Bhowmick, N.; Moses, H.L.; Zent, R. Transforming growth factor-beta (TGF-β) and TGF-β-associated kinase 1 are required for R-Ras-mediated transformation of mammary epithelial cells. Cancer Res. 2008, 68, 6224–6231. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Begai, S.; Jakkaraju, S.; Mattingly, R.R.; Pan, D.; Schuger, L. High RhoA activity maintains the undifferentiated mesenchymal cell phenotype, whereas RhoA down-regulation by laminin-2 induces smooth muscle myogenesis. JCB 2002, 156, 893–903. [Google Scholar]
- Yang, Y.; Yang, C.; Zhang, J. C23 protein meditates bone morphogenetic protein-2-mediated EMT via up-regulation of Erk1/2 and Akt in gastric cancer. Med. Oncol. 2015, 32. [Google Scholar] [CrossRef] [PubMed]
- Liao, A.; Wang, W.; Sun, D.; Jiang, Y.; Tian, S.; Li, J.; Yang, X.; Shi, R. Bone morphogenetic protein 2 mediates epithelial-mesenchymal transition via AKT and ERK signaling pathways in gastric cancer. Tumour Biol. 2015, 36, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Grimm, W.A.; Garner, W.L.; Qin, L.; Travis, T.; Tan, N.; Han, Y.P. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-α through bone morphogenic protein-2. Am. J. Pathol. 2010, 176, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, A.; Riazi, G.H.; Azizi, E.; Amanpour, S.; Muhammadnejad, S.; Haddadi, M.; Zekri, A.; Shirkoohi, R. FGF10: Type III epithelial mesenchymal transition and invasion in breast cancer cell lines. J. Cancer 2014, 5, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wu, H.; Zhang, M.; Ding, L.; Meng, F.; Fan, X. Expression of the epithelial-mesenchymal transition-related proteins and their clinical significance in lung adenocarcinoma. Diagn. Pathol. 2013, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Uchikado, Y.; Okumura, H.; Ishigami, S.; Setoyama, T.; Matsumoto, M.; Owaki, T.; Kita, Y.; Natsugoe, S. Increased Slug and decreased E-cadherin expression is related to poor prognosis in patients with gastric cancer. Gastric Cancer. 2011, 14, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Deep, G.; Jain, A.K.; Ramteke, A.; Ting, H.; Vijendra, K.C.; Gangar, S.C.; Agarwal, C.; Agarwal, R. Snai1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol. Cancer. 2014, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tan, Y.; Ren, Y.; Dong, L.; Xie, Z.; Tang, L.; Cao, D.; Zhang, W.; Hu, H.; Wang, H. Zinc finger protein ZBTB20 expression is increased in hepatocellular carcinoma and associated with poor prognosis. BMC Cancer 2011, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, D.; Kajita, M.; Wade, P.A. The transcription factor Snail mediates epithelial to mesenchymal transition by repression of estrogen receptor-alpha. Mol. Endocrinol. 2007, 21, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Dhasarathy, A.; Phadke, D.; Mav, D.; Shah, R.R.; Wade, P.A. The transcription factors of Snail and Slug activate the transforming growth factor-beta signaling pathway in breast cancer. PLoS ONE 2011, 6, e26514. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Q.; Fan, L.Q.; Wang, L.L.; Tan, B.B.; Leng, Y.L.; Liu, Y.; Wang, D. Zinc finger protein 139 expression in gastric cancer and its clinical significance. World J. Gastroenterol. 2014, 20, 18346–18353. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Jiang, H.; Ying, G.; Dong, Q.; Zhang, R.; Yu, J.; Fan, D.; Hao, X. Poor survival is associated with the methylated degree of zing-finger protein 545 (ZNF545) DNA promoter in gastric cancer. Oncotarget 2015, 6, 4482–4495. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Hirohashi, S.; Kanai, Y. Cell adhesion system and human cancer morphogenesis. Cancer Sci. 2003, 94, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Pantel, K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastab. Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Valles, A.M.; Jouanneau, J.; Yamada, K.M.; Thiery, J.P. Alternative splicing fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol. Biol. Cell 1994, 5, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Nabi, I.R. Actin cytoskeletal regulation of epithelial mesenchymal transition in metastatic cancer cells. PLoS ONE 2015, 10, e0119954. [Google Scholar]
- Chaw, S.Y.; Majeed, A.; Dalley, A.J.; Chan, A.; Stein, S.; Farah, C.S. Epithelial to mesenchymal transition (EMT) biomarkers—E-cadherin, beta-catenin, APC and Vimentin—In oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012, 48, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, M.; Pozzi, A.; Bhowmick, N.; Moses, H.L.; Zent, R. Transforming growth factor-beta (TGF-β) and TGF-β-associated kinase I are required for R-Ras-mediated transformation of mammary epithelial cells. Cancer Res. 2008, 68, 6224–6231. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Sheng, T.; Stelter, A.A.; Li, C.; Zhang, X.; Sinha, M.; Luxon, B.A.; Xie, J. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J. Biol. Chem. 2006, 281, 35598–33602. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; Cespedes, M.V.; Lindh, M.B.; Kiflemariam, S.; Mezheyeuski, A.; Edgvist, P.H.; Hägglöf, C.; Birgisson, H.; Bojmar, L.; Jirström, K.; et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013, 73, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Sneddon, J.B.; Alizadeh, A.A.; Sood, R.; West, R.B.; Montgomery, K.; Chi, J.-T.; van de Rijn, M.; Botstein, D.; Brown, P.O. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004, 2, E7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression profiles clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, J.; Chanreshekar, C.; Radharkrishnan, R. Critical biomarkers of epithelial-mesenchymal transition in the head and neck cancers. J. Cancer Res. Ther. 2014, 10, 512–518. [Google Scholar] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Keirsebilck, A.; Bonne, S.; Bruyneel, E.; Vermassen, P.; Lukanidin, E.; Mareel, M.; van Roy, F. E-cadherin and metastasin (mts-1/S100A4) expression levels are inversely regulated in two tumor cell families. Cancer Res. 1998, 58, 4587–4591. [Google Scholar] [PubMed]
- Noe, V.; Fingleton, B.; Jacobs, K.; Crawford, H.C.; Vermeulen, S.; Steelant, W.; Bruyneel, E.; Matrisian, L.M.; Mareel, M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 2001, 114, 111–118. [Google Scholar] [PubMed]
- Ramos, D.M.; Dang, D.; Sadler, S. The role of the integrin αvβ6 in regulating the epithelial to mesenchymal transition in oral cancer. Anticancer Res. 2009, 29, 125–130. [Google Scholar] [PubMed]
- Dang, D.; Bamburg, J.R.; Ramos, D.M. αvβ3 integrin and cofilin molecule modulate K1735 melanoma cell invasion. Exp. Cell Res. 2006, 312, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Balasundaram, P.; Singh, M.K.; Dinda, A.K.; Thakar, A.; Yadav, R. Study of β-catenin, E-cadherin and vimentin in oral squamous cell carcinoma with and without lymph node metastases. Diagn. Pathol. 2014, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegrin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA. 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Odero-Marah, V.A.; Wang, R.; Chu, G.; Zayzafoon, M.; Xu, J.; Shi, C.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Receptor activator of NF-κB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res. 2008, 18, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [PubMed]
- Fizazi, K.; Yang, J.; Peleg, S.; Sikes, C.R.; Kreimann, E.L.; Daliani, D.; Olive, M.; Raymond, K.A.; Janus, T.J.; Logothetis, C.J.; et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegrin in osteoblasts. Clin. Cancer Res. 2003, 9, 2587–2597. [Google Scholar] [PubMed]
- Koeneman, K.S.; Yeung, F.; Chung, L.W. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate 1999, 39, 246–261. [Google Scholar] [CrossRef]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and Slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Oft, M.; Peli, J.; Rudaz, C.; Schwarz, H.; Beug, H.; Reichmann, E. TGF-β1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996, 10, 2462–2477. [Google Scholar] [CrossRef] [PubMed]
- Aluwihare, P.; Munger, J.S. What the lung has taught us about latent TGF-beta activation. Am. J. Respir. Cell Mol. Biol. 2008, 39, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelial-mesenchymal transition in colon and breast cancer cells. Alcohol. Clin. Exp. Res. 2010, 34, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Pasche, B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat. Rev. Rheumatol. 2009, 5, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.L. Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am. J. Pathol. 2009, 175, 1362–1370. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial mesenchymal to epithelial reverting transition in breast and prostate cancer metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Jiborn, T.; Biartell, A.; Abrahamsson, P.A. Neuroendocrine differentiation in prostatic carcinoma during hormonal treatment. Urology 1998, 51, 585–589. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Chio, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Mckeithen, D.; Graham, T.; Chung, L.W.; Odero-Marah, V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate 2010, 70, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Bruchovsky, N.; Sadar, M.D. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J. Biol. Chem. 2002, 277, 7076–7085. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Murakami-Mori, K.; Bonavida, B. Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem. Biophys. Res. Commun. 1999, 257, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, L.; Hu, J.; Li, F.; Shao, Y.; Kalvakolanu, D.V.; Kopecko, D.J.; Zhao, X.; Xu, D.-Q. Down-regulation of signal transducer and activator of transcription 3 expression using vector-based small interfering RNA suppressing growth of human prostate tumor in vivo. Clin. Cancer Res. 2005, 11, 6333–6341. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Yeung, K.; Palladino, M.; Berenson, J.; Bonavida, B. Pivotal roles of snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cells chemoimmunosensitization. Cancer Res. 2009, 69, 8376–8385. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; Lisok, A.; Kimble, B.; Domek, J.; Kato, Y.; van der Groep, P.; Artemov, D.; Kowalski, J.; Carraway, H.; van Diest, P.; et al. Twist contributes to hormone resistance in breast cancer by down-regulating estrogen receptor alpha. Oncogene 2012, 31, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour microenvironment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Alberti, P.; Cavaletti, G. Management of side effects in the personalized medicine era: Chemotherapy-induced peripheral neuropathy. Pharmacogn. Drug Discov. Dev. Methods Mol. Biol. 2014, 1175, 301–322. [Google Scholar]
- Hewitt, K.J.; Agarwal, R.; Morin, P.J. The claudin gene family: expression in normal and neoplastic tissues. BMC Cancer 2006, 6, 186. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J. Claudin proteins in human cancer: promising new targets for diagnosis and therapy. Cancer Res. 2005, 65, 9603–9606. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.; Adjei, A.A.; Lorusso, P.M.; Waterhouse, D.; Hecht, J.R.; Natale, R.B.; Hamid, O.; Varterasian, M.; Asbury, P.; Kaldjian, E.P.; et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004, 22, 4456–4462. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Maiello, M.R.; Campiglio, M.; Napolitano, M.; Mancino, M.; Carotenuto, A.; Viglietto, G.; Menard, S. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell. Physiol. 2006, 207, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Su, H.Y.; Lai, H.C.; Lin, Y.W.; Liu, C.Y.; Chen, C.K.; Chou, Y.C.; Lin, S.P.; Lin, W.C.; Lee, H.Y.; Yu, M.H. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through wnt signaling pathway. Int. J. Cancer 2010, 127, 555–567. [Google Scholar] [CrossRef] [PubMed]
- El Touny, L.H.; Banerjee, P.P. Akt GSK-3 pathway as a target in Genistein-induced inhibition of TRAMP prostate cancer progression toward a poorly differentiated phenotype. Carcinogenesis 2007, 28, 1710. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Singh, C.; Nall, D.; Meeker, D.; Shankar, S.; Srivastava, R.K. The dietary bioflavonoid Quercetin synergizes with epigallocathechin gallate (EGCG) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial-mesenchymal transition. J. Mol. Signal. 2010, 5, 14. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. https://doi.org/10.3390/jcm5020017
AMA Style
Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. Journal of Clinical Medicine. 2016; 5(2):17. https://doi.org/10.3390/jcm5020017
Chicago/Turabian StyleSmith, Bethany N., and Neil A. Bhowmick. 2016. "Role of EMT in Metastasis and Therapy Resistance" Journal of Clinical Medicine 5, no. 2: 17. https://doi.org/10.3390/jcm5020017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.