A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease


Department of Biology, Program in Cell and Molecular Biology, Boston University, Boston, MA 02215, USA
*
Author to whom correspondence should be addressed.
J. Cardiovasc. Dev. Dis. 2018, 5(3), 37; https://doi.org/10.3390/jcdd5030037
Submission received: 1 June 2018
/
Revised: 29 June 2018
/
Accepted: 4 July 2018
/
Published: 10 July 2018
(This article belongs to the Special Issue Non Coding RNAs in the Cardiovascular System)
Abstract
:The imprinted Dlk1-Dio3 genomic region harbors a noncoding RNA cluster encoding over fifty microRNAs (miRNAs), three long noncoding RNAs (lncRNAs), and a small nucleolar RNA (snoRNA) gene array. These distinct noncoding RNAs (ncRNAs) are thought to arise from a single polycistronic transcript that is subsequently processed into individual ncRNAs, each with important roles in diverse cellular contexts. Considering these ncRNAs are derived from a polycistron, it is possible that some coordinately regulate discrete biological processes in the heart. Here, we provide a comprehensive summary of Dlk1-Dio3 miRNAs and lncRNAs, as they are currently understood in the cellular and organ-level context of the cardiovascular system. Highlighted are expression profiles, mechanistic contributions, and functional roles of these ncRNAs in heart development and disease. Notably, a number of these ncRNAs are implicated in processes often perturbed in heart disease, including proliferation, differentiation, cell death, and fibrosis. However, most literature falls short of characterizing precise mechanisms for many of these ncRNAs, warranting further investigation. Taken together, the Dlk1-Dio3 locus represents a largely unexplored noncoding regulator of cardiac homeostasis, harboring numerous ncRNAs that may serve as therapeutic targets for cardiovascular disease.
Keywords:
cardiac; proliferation; hypertrophy; fibrosis; microRNA; long noncoding RNA; imprinting; epigenetics1. Introduction
Cardiovascular disease is associated with widespread perturbations in gene regulatory control that impact transcriptional, post-transcriptional, and epigenetic processes. Given the morphological and cellular complexity of the cardiovascular system, defining the gene regulatory networks required to form this vital physiological system is a formidable yet essential undertaking. This fundamental knowledge may yield insight into the genomic mechanisms through which insults, such as mutations, stress, and injury, trigger cellular abnormalities that ultimately drive disorders of the cardiovascular system.
Among various mediators of gene regulation, noncoding RNAs (ncRNAs) such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs) have emerged as core post-transcriptional and epigenetic regulators in the cardiovascular system [1,2,3,4,5]. During development, these classes of ncRNAs modulate gene programs controlling specification, proliferation, and differentiation in diverse cardiovascular cell types. The importance of ncRNAs in the cardiovascular system is exemplified by severe congenital defects that have emerged in gain/loss-of-function studies, their involvement in cardiac remodeling, and their dysregulated expression in a spectrum of cardiac disease phenotypes in human patients and animal model systems [6,7].
MiRNAs, one of the smallest classes of regulatory RNAs, inhibit gene expression through the degradation or translational block of protein-coding transcripts [8]. In the cardiovascular system, various miRNAs (miR-1/miR-133 clusters, miR-17–92 clusters) and the miRNA processing enzyme Dicer have been shown to play key regulatory roles in the differentiation of cardiomyocytes, smooth muscle cells, and neural crest derivatives in the developing heart [9,10,11,12,13,14]. MiRNAs also regulate cardiomyocyte proliferation and cardiac regeneration; for example, miR-590, miR-199a, and the miR-302–367 cluster can potently induce cardiomyocyte proliferation in vitro and in vivo [15,16]. In addition, the miR-15 family controls cell cycle withdrawal during early postnatal heart maturation in mice [17]. Given their central role in coordinating gene expression patterns that dictate cellular behavior within the cardiovascular system, these and many other miRNAs such as miR-208, miR-23a, and miR-29 have been implicated in modulating pathological remodeling in a spectrum of heart disease models [18,19,20].
LncRNAs, defined as polyadenylated RNAs greater than 200 nucleotides, exhibit complex stem-loop secondary structures, which along with their localization in the nucleus, cytoplasm, or both, enables them to regulate gene expression via multiple mechanisms [21,22]. Recent reports have described an important regulatory role for lncRNAs in cardiomyocyte specification and differentiation [7]. For example, the lncRNAs Fendrr and Bvht are important in cardiac development and cardiomyocyte lineage commitment, respectively [23,24]. Like their small ncRNA counterparts, differential expression of lncRNAs are observed in heart disease models, suggesting a role in pathological remodeling. Indeed the lncRNAs Mhrt, Chaer, MIAT, and CARL, to name a few, which are dysregulated in cardiac disease, confer epigenetic perturbations by modulating chromatin factor activity, or augment gene expression by sequestering miRNAs from their target transcripts [25,26,27,28].
Numerous reviews have described in great detail the function of aforementioned miRNAs and lncRNAs in cardiac development and disease. Whereas most miRNAs and lncRNAs identified to date are encoded as a single inter- or intragenic genes, or in some instances, expressed from a small cluster of several related RNAs, the mammalian Dlk1-Dio3 ncRNA locus is unusually massive: a >200 kilobase (kb) mega-cluster of distinct regulatory RNA functional categories. Specifically, more than 50 miRNAs, several lncRNAs, and a tandemly repeated array of C/D-box snoRNAs (small nucleolar RNAs) are expressed from this locus [29,30]. The Dlk1-Dio3 locus has been investigated extensively as a paradigm to understand the epigenetic mechanisms of genomic imprinting [31,32,33,34]. In this review, we highlight recent evidence that various ncRNAs expressed from this large, imprinted locus have key regulatory functions in developmental and disease-related processes pertaining to the cardiovascular system. We first explore a large body of findings that implicate many Dlk1-Dio3 miRNAs in cardiomyocyte proliferation and differentiation, heart development, and cardiac disease pathways. Then, we turn our attention toward Gtl2, a locus lncRNA with recently established roles in cardiac fibrosis and the vasculature. By highlighting the individual functions of the various Dlk1-Dio3 locus ncRNAs, a broader picture may emerge regarding a dynamic role for this remarkably complex locus in the cardiovascular system.
1.1. The Dlk-Dio3 ncRNA Locus: Gene Organization, Imprinting and Human Disease
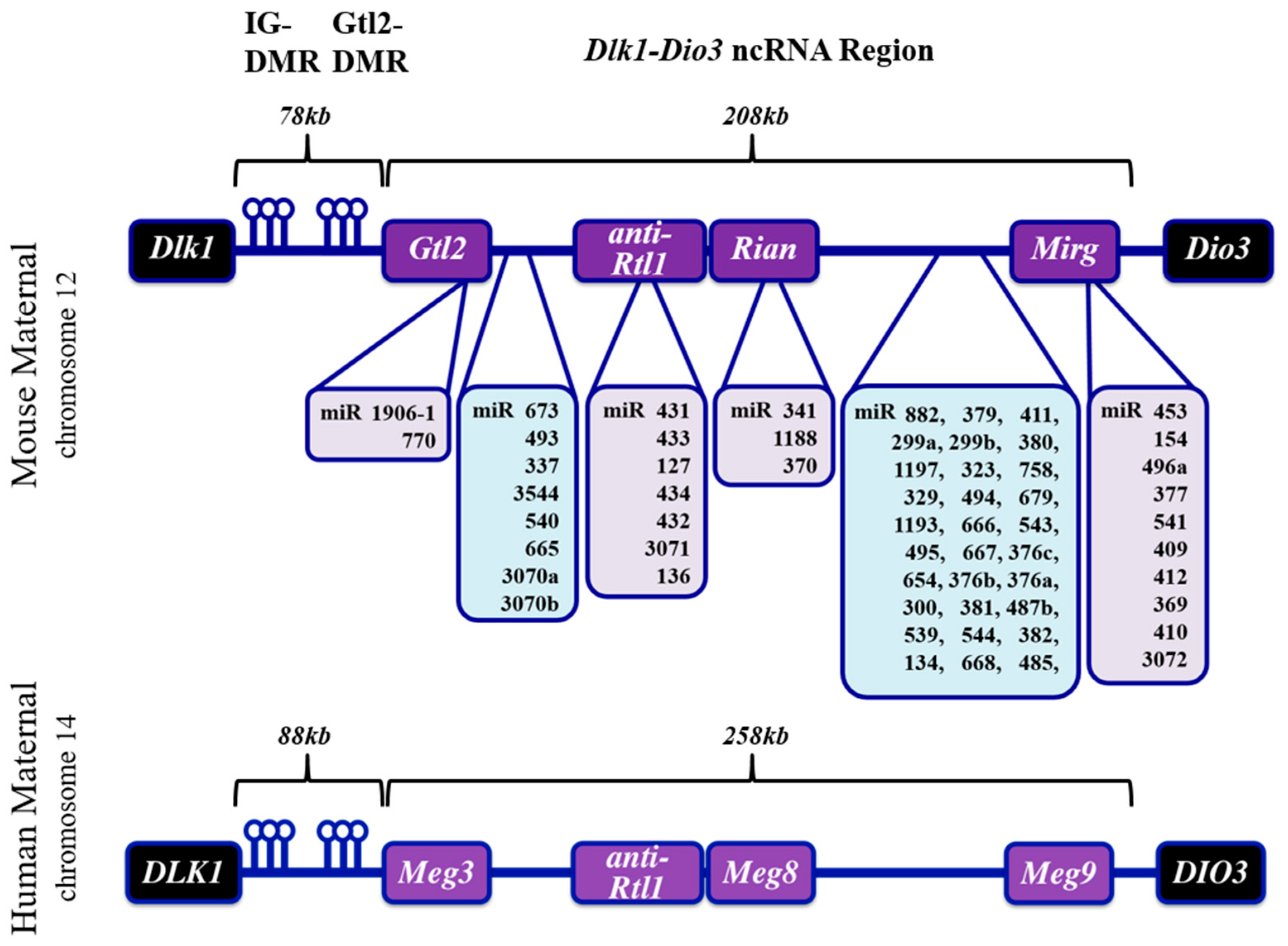
The mammalian Dlk1-Dio3 ncRNA locus resides on chromosomes 12 and 14 in mice and humans, respectively, and derives its name from the protein-coding genes that flank the ncRNA sequences (Figure 1). The organization of the locus is conserved in all mammals. The Dlk1 (Delta-like homolog 1) gene is located upstream of the ncRNA region and codes for a protein involved in the Notch signaling pathway [35]. Located at the 3′-end of the imprinted region is the Dio3 (type III iodothyronine deiodinase) gene which functions in thyroid hormone signaling [36]. Between the murine Dlk1 and Dio3 genes resides the >200 kb ncRNA mega-cluster: beginning with the lncRNA Gtl2 (MEG3 in humans) at the 5′ end and continuing through miR-3077 at the 3′ end [30]. The Gtl2 lncRNA is the most 5′ lncRNA transcript, exists in multiple splice variants, and interacts with chromatin remodeling proteins [37]. Anti-Rtl1, which harbors several miRNAs, is located immediately downstream of the Gtl2 gene. This region is followed by Rian, an annotated lncRNA which has no known function apart from harboring a tandemly repeated array of the C/D-box snoRNA family and three miRNAs. At the 3′ end of the locus is Mirg, which encodes a cluster of dozens of miRNAs (miR-379–410) that appear to fall into discrete subfamilies based on sequence similarities [29]. Moreover, although Mirg has no known lncRNA function, the transcript exhibits diverse tissue-specific expression in mouse embryogenesis [38].
Between the Dlk1 gene and the transcription start site of the ncRNA cluster are two intergenic (IG), differentially DNA-methylated regions (DMR): the IG-DMR and Gtl2-DMR [32]. The IG-DMR is located approximately 20 kb upstream of Gtl2, and its methylation state has been shown to dictate mono-allellic expression of the locus (i.e., protein-coding genes are exclusively expressed from the methylated paternal allele, whereas ncRNAs are expressed exclusively from the maternal allele). The Gtl2-DMR overlaps the proximal promoter and methylated CpG islands; in contrast, the IG-DMR is largely homomethylated, but it is currently unclear whether this epigenetic modification imparts any function [39]. A schematic depicting the organization of Dlk1-Dio3 locus ncRNAs is provided in Figure 1.
In humans, imprinting abnormalities at the Dlk1-Dio3 locus have been linked to syndromes of impaired fetal development and postnatal growth. Humans with uniparental disomy, that is, duplicate copies of either maternal or paternal chromosome 14q32 (MatUPD14 or PatUPD14), display severe growth retardation, skeletal malformations, and metabolic deficiencies [40]. Two known examples of chromosome 14q32 uniparental disomy are Temple and Kagami-Ogata syndromes [41,42]. Among the aforementioned clinical features in these patients, muscle hypotonia is a prominent symptom, and suggests a critical function for this locus in muscle development. Though not a prevalent feature, cardiac abnormalities such as septal defects and cardiomegaly have been described in a small number of patients with chromosome 14 uniparental disomy [43,44]. Mouse models of uniparental disomy of chromosome 14 (or mouse chromosome 12) display similar phenotypes to the human syndromes. Both paternal disomy (PatUPD12) and maternal disomy (MatUPD12) in mice display placental, bone, and skeletal muscle defects [45].
It is not entirely clear how the multiple ncRNAs are expressed from the Dlk1-Dio3 locus, since there are conflicting reports describing their transcriptional regulation. Multiple lines of evidence suggest mature Dlk-Dio3 ncRNAs are derived from post-transcriptional processing of a single, polycistronic transcript, synthesized from a promoter directly upstream of the first exon of the Gtl2 lncRNA. Supporting this notion, coordinate dysregulation of Dlk1-Dio3 miRNAs and lncRNAs has been reported in several experimental models indicating a common transcriptional start site for these ncRNAs [46,47,48,49,50,51]. However, other studies have reported discrete enhancers neighboring miRNA clusters within the ncRNA locus. For example, the miR-379–410 cluster is regulated by a MEF2-dependent enhancer immediately upstream of miR-379 [52]. Also, enhancers regulated by the estrogen receptor have been described upstream of the miR-433–127 cluster [53]. Regardless of the mechanism by which these ncRNAs are transcribed, while some of the miRNAs appear to target components belonging to a given pathway, many of the miRNAs expressed from this locus have distinct biological targets. Thus, much remains to be resolved regarding the regulation and activity of the Dlk1-Dio3 ncRNA locus.
1.2. The Dlk1-Dio3 ncRNA Locus in Striated Muscle
There is considerable evidence demonstrating an essential requirement for the ncRNAs in the Dlk1-Dio3 locus in mammalian striated muscle. In mice, a 10 kb deletion encompassing the Gtl2 lncRNA coding region (exons 1–5) and ~300 bp of the proximal promoter resulted in pronounced skeletal muscle defects in late fetal development [47]. Consistent with the genomic imprinting status and maternal expression of the ncRNAs, heterozygous mice with maternal inheritance of the deletion displayed the muscle phenotype whereas paternal inheritance did not affect growth or muscle development. Curiously, an independent line of knockout mice harboring a similar deletion as Zhou et al. described postnatal growth retardation with pulmonary and hepatic defects, but skeletal muscle defects were not reported [46]. It is worthwhile to note that both deletions caused the downregulation of all ncRNAs in the locus, reinforcing the notion of coordinate regulation.
Additional mouse models have linked the Dlk1-Dio3 ncRNA locus to skeletal muscle phenotypes. Homozygous mice harboring a deletion of the miR-379–544 cluster resulted in skeletal muscle hypertrophy, a phenotype likely attributable to altered regulation of the neighboring Dlk1 gene by one or more of the miRNAs embedded within this cluster [54]. In contrast, deletion of the miR-379–410 cluster, which removed additional miRNAs in the Mirg region downstream of miR-544, resulted in liver metabolic deficiencies and neonatal lethality. Although the authors measured significant downregulation of miRNAs in both heart and skeletal muscle, no further characterization was described [55]. Our group previously demonstrated that the Dlk1-Dio3 ncRNA locus is directly regulated by the MEF2 transcription factor and that this pathway is involved in muscle differentiation and regeneration [50]. Consistent with these findings, the MEF2-Dlk1-Dio3 pathway was recently demonstrated to function in muscle stem cell quiescence, metabolism, and differentiation [56,57]. Furthermore, genome-wide profiling of skeletal muscle in aged mice revealed coordinate downregulation of eight Dlk1-Dio3 miRNAs [58]. Finally, the Dlk1-Dio3 locus has been implicated in skeletal muscle hypertrophy. Callipyge sheep harbor a point mutation between the Dlk1 gene and the beginning of the ncRNA region (Gtl2/Meg3), and when inherited from the paternal allele, these animals develop muscle hypertrophy. This mutation presumably affects the activity of a distal enhancer, which alters protein-coding DLK1 and RTL/PEG11 genes expressed from the paternal allele [59,60]. There is also evidence that ncRNA expression is affected in callipyge sheep [61]. Taken together, it appears that the Dlk1-Dio3 locus and a subset of ncRNAs have biological relevance in skeletal muscle, although it remains unclear whether cardiac defects exist in callipyge sheep or any of the aforementioned mouse mutants.
2. MicroRNAs Expressed from the Dlk1-Dio3 Locus in Cardiac Differentiation and Development
Despite the apparent lack of a cardiac phenotype in the various Dlk1-Dio3 mutant mouse lines, either because this organ has only been analyzed superficially or these mutations do not specifically cause cardiac abnormalities, there are numerous reports describing expression profiling, dysregulation, and functional roles for Dlk1-Dio3 ncRNAs in the cardiovascular system. In recent years, links between the Dkl-Dio3 locus and cardiac developmental processes, including proliferation and differentiation, have notably increased. Below, we summarize those Dlk1-Dio3 miRNAs and their connections to specific cardiac processes, as they are currently understood.
2.1. Cardiomyocyte and Endothelial Progenitors
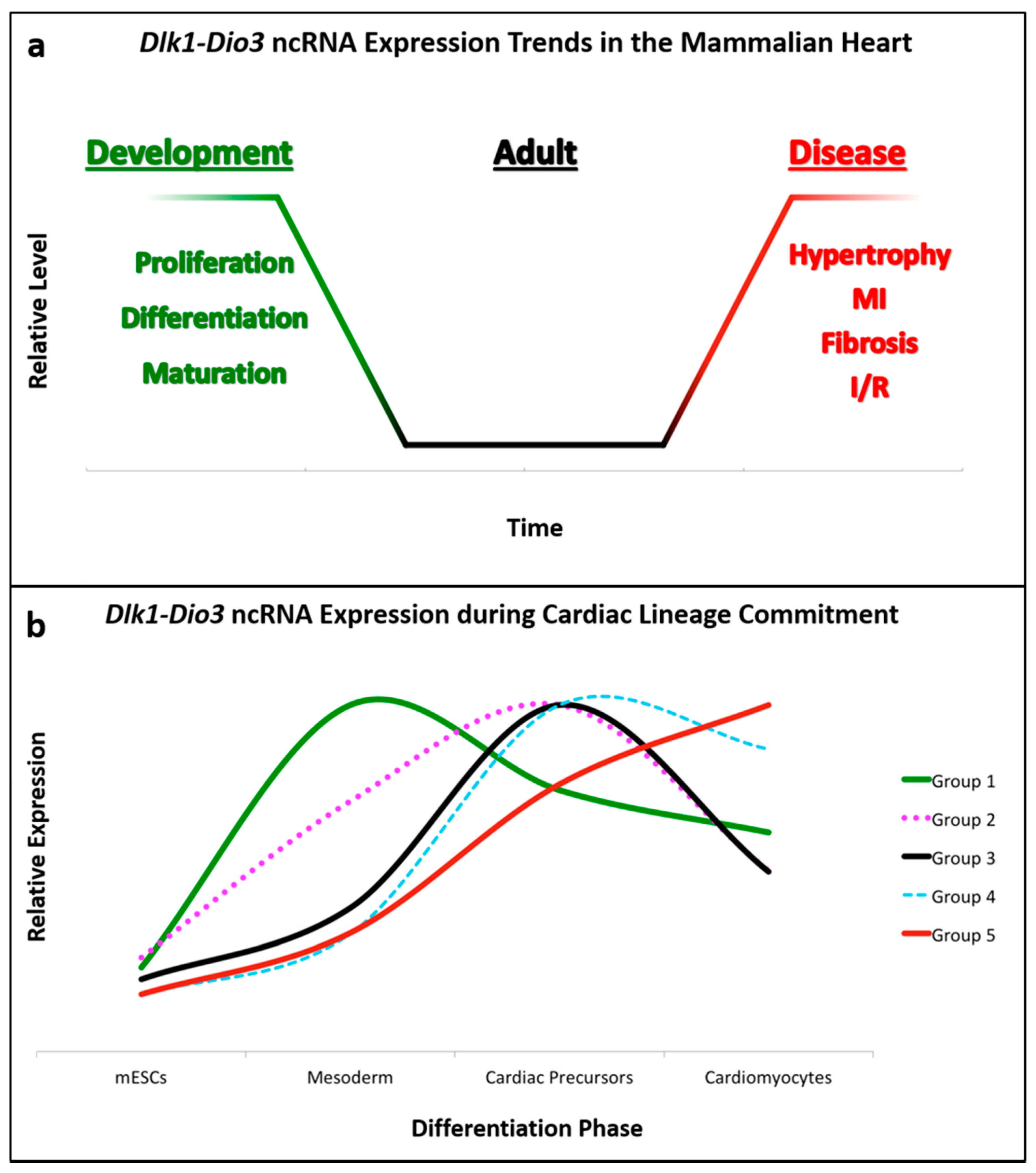
Temporal expression of Dlk1-Dio3 ncRNAs has been documented in directed cardiomyocyte differentiation from mouse embryonic stem cells. Although not the primary focus of the investigation, transcriptomic analysis revealed that ncRNA expression from this locus is highly upregulated in the transition from pluripotent stem cells to differentiated cardiomyocytes [62]. Interestingly, sub-groups of Dlk1-Dio3 miRNAs displayed differences in their temporal expression during phases of cardiomyocyte differentiation, which are summarized in Figure 2b. These observations suggest a function for one or more of Dlk1-Dio3 ncRNAs in cardiac lineage commitment, and may underscore dynamic roles for individual ncRNAs in heart development.
In mice, miR-300 has been shown to suppress cardiomyocyte progenitor differentiation in vitro. Specifically, miR-300 overexpression impaired cardiomyocyte differentiation marker expression and reduced the population of Sca1+ cardiac progenitor cells (CPCs) isolated from adult mouse hearts. Interestingly, overexpression of Bmi1, a member of the Polycomb Repressor Complex 1 (PRC1), appeared to significantly upregulate miR-300 in CPCs, but the treatment did not influence other miRNAs in the Dlk1-Dio3 locus [63].
Survival of CPCs from fetal human hearts is modulated by another distinct locus miRNA, miR-134. Overexpression of miR-134 in CPCs reduced proliferation, whereas inhibition enhanced proliferation. Numerous cell cycle genes, including cyclins A, B, and E, and CDK4, displayed dysregulated expression in these experiments. Moreover, miR-134 regulated cell cycle activity by targeting Meis2, a TALE homeobox transcription factor that regulates CPC proliferation and differentiation. Terminal differentiation of these progenitor cells was not affected by overexpression or inhibition of miR-134, but inhibition of this miRNA reduced levels of the cardiac transcription factors MEF2C, GATA4, and NKX2.5 [64].
Expression of the Dlk1-Dio3 miRNAs mir-495 and miR-543 was shown to correlate with the commitment of endothelial cells from mesodermal precursors derived from human induced pluripotent stem cells (iPSCs). Specifically, miR-495 and miR-543 were found to be significantly downregulated in CD31+ endothelial cells compared to non-endothelial cells (CD31-) and undifferentiated human iPSCs. Consistent with the expression analysis, stable human iPSC lines harboring antisense oligonucleotide against miR-495 enhanced endothelial cell differentiation. Moreover, miR-495 inhibition promoted angiogenesis (tube formation and migration) from human iPSC-endothelial cells in vitro. One target in this process is vascular endothelial zinc finger 1 (VEZF1), a regulator of endothelial cell differentiation and angiogenesis [65].
When considering temporal expression and several functional studies, it appears many Dlk1-Dio3 ncRNAs are involved with the commitment of mesoderm to a diverse subset of specialized cardiac cell lineages. The opposing roles of some Dlk1-Dio3 miRNAs in these contexts may reflect the versatility required for a single genetic locus to allocate diverse cell fates in early cardiac formation. Indeed, discrete subsets of miRNAs exhibit distinct expression patterns, and as demonstrated with Bmi1 manipulations, it is evident that mechanisms exist to enrich specific locus miRNAs over others. It therefore seems likely that transcriptional regulators interact with the locus polycistron during differentiation, dictating which locus miRNAs are primed to favor—or suppress—a particular cell fate.
2.2. Cardiac Structures
Morphogenesis of specialized cardiac regions, such as the chambers, conduction system, and valves, requires precise spatio-temporal regulation of gene expression patterns that govern development of these structures. MiRNAs play an important role in fine-tuning such gene expression. To identify miRNAs potentially involved with development and/or function of specific cardiac structures, comparative transcriptomic analyses were performed on discrete cardiac regions from three mammalian species: rat, dog, and monkey. This ultimately identified miRNAs enriched in specific regions of the adult heart and conserved across the three mammalian species. One miRNA from the Dlk1-Dio3 locus, miR-127, was found to be enriched in cardiac valves in rat, dog, and monkey [66]. It is surprising that this particular miRNA appears to be the only Dlk1-Dio3 ncRNA enriched in this cardiac structure, suggesting region-specific post-transcriptional regulation of these miRNAs in the heart.
Ventricular septal defects are among the most common congenital cardiac abnormalities in humans [67]. A microarray screen to identify circulating miRNAs in ventricular-septal defect patients revealed Dlk1-Dio3 locus miRNAs-379, -654, -487b, -409, and -433 are significantly downregulated in plasma from these patients. Of these, all but miR-654 were independently validated using RT-PCR, and the authors used miR-targeting algorithms to model how miR-433 could regulate septal development: targeting NOTCH1 and/or the GATA3 transcription factor [68]. It is difficult to discern whether these circulating miRNAs are directly involved in ventricular septal defects, or a secondary consequence of this congenital disease. Along with the expression of miR-127 in the cardiac valves, these observations suggest a potential function for the Dlk1-Dio3 locus in cardiac morphogenesis, and warrant further investigation of these ncRNAs for pathophysiological roles in defective cardiac structures.
2.3. Cardiomyocyte Proliferation and Apoptosis
Understanding the mechanisms of cell cycle regulation in cardiomyocytes is a major driving force in cardiac regenerative medicine given the implications of repairing damaged tissue through myocyte proliferation. Studies have demonstrated that certain Dlk1-Dio3 miRNAs are capable of stimulating proliferation of post-mitotic neonatal cardiomyocytes. Although not the major focus of the study, a functional screen for pro-proliferative miRNAs in cardiomyocytes listed several Dlk1-Dio3 miRNAs, miR-411, -380, -495, -539, -668, -154, and -410 as capable of inducing DNA synthesis and cytokinesis in neonatal mouse and rat cardiomyocytes [15]. We have shown that overexpression of miR-410 or miR-495 induces robust proliferation of neonatal rat ventricular myocytes (NRVMs) in vitro. Both of these miRNAs directly targeted and inhibited Cited2, a transcriptional coactivator, resulting in the upregulation of pro-proliferative genes and inhibition of apoptotic genes [69].
MiRNAs that stimulate proliferation of post-mitotic cardiomyocytes may reflect their cell cycle regulatory activity in cardiac development. Along these lines, miR-410 and miR-495 and other miRNAs in the Dlk1-Dio3 locus such as miR-337 and miR-668 are expressed at higher levels in the neonatal mouse heart compared to the adult and first week post-natally, respectively [69,70]. Similarly, a miRNA screen comparing heart tissue before and after birth in sheep revealed higher expression of Dlk1-Dio3 miRNAs (-493, -127, -432, -379, -411, -380, -329, -543, -487b, -382, -485, -154, -409, and -410) in late fetal cardiac development compared to the post-natal heart [51]. The downregulation of Dlk1-Dio3 miRNAs coincides with a developmental timepoint where the regenerative capacity of the murine heart is lost, a consequence of cardiomyocyte cell cycle withdrawal.
Cardiomyocyte survival is also dependent on the Dlk1-Dio3 miRNA miR-377. The drug cyclosporin A (CsA) inhibits calcineurin, can block cytochrome c release, and also induces cardiomyocyte apoptosis. In NRVMs treated with CsA, miR-377 was the most upregulated miRNA. It was further characterized that miR-377 is a downstream mediator of CsA-induced cardiomyocyte apoptosis. Inhibition of miR-377 in CsA-treated NRVMs attenuated apoptosis, whereas miR-377 overexpression alone promoted cardiomyocyte apoptosis [71]. These results indicate a pro-apoptotic role for miR-377 in neonatal cardiomyocytes in vitro.
Data reported to date clearly indicate a function for Dlk1-Dio3 miRNAs in cardiomyocyte proliferation, whereas miR-377 functions in an entirely distinct manner: facilitating death. Continued investigation into transcriptional regulation of these miRNAs, as well as their therapeutic potential, is merited given their ability to modulate post-mitotic cardiomyocyte behaviors—specifically cardiomyocyte proliferation, which is extremely limited in the adult heart, and apoptotic cell death, which contributes to functional loss of heart muscle during disease.
3. MicroRNAs Expressed from the Dlk1-Dio3 Locus in Cardiovascular Pathology
In most tissues including the heart, post-natal expression of Dlk1-Dio3 ncRNAs is substantially downregulated. However, disease or exposure to various cardiac stressors upregulates these ncRNAs. Below, we summarize current research that implicates locus miRNAs in pathological remodeling of the heart, and highlight specific functions that may contribute to (or attenuate) heart disease.
3.1. Cardiac Hypertrophy
Hypertrophic myocyte growth is a hallmark of pathological cardiac remodeling, observed in numerous heart disease modalities. Because chronic hypertrophy inflicts deleterious functional consequences, its signaling and transcriptional mechanisms have been studied exhaustively. It is now firmly established that miRNAs impart important regulatory roles in pathological cardiac remodeling. Several miRNAs in the Dlk1-Dio3 locus have been implicated in cardiac hypertrophy, and their dysregulated expression has been described in various cardiac disease models associated with hypertrophy.
Our group reported that both miR-410 and miR-495 are upregulated in angiotensin II (Ang II)-induced hypertrophy in vivo and in neonatal rat ventricular myocytes (NRVMs) treated with phenylephrine (PE). AntimiRs that specifically inhibit miR-410 or miR-495 were shown to attenuate PE-induced hypertrophy in NRVMs. Moreover, we showed that the MEF2-dependent Gtl2 proximal promoter region is active in NRVMs, and this activity is amplified in response to PE, suggesting the ncRNA locus is sensitive to hypertrophic signals [72].
A microarray that surveyed miRNAs in serum from cats with spontaneous hypertrophic cardiomyopathy indicated that locus ncRNA miR-381 was upregulated. It was noted that in silico analyses revealed miR-381 has more predicted cardiac mRNA targets than most other miRNAs highlighted in the study, suggesting a pervasive role for this miRNA in the heart [73]. MiR-154 was also found to be upregulated in pressure overload and in hypertrophic cardiomyopathy in humans. Its inhibition in mice subjected to transverse aortic constriction (TAC) attenuated dysfunction and fibrosis, and it was determined that miR-154 directly targets p14, a cell cycle inhibitor [74]. Collectively, these data reveal the pro-hypertrophic activity attributed to a subset of Dlk1-Dio3 miRNAs.
Surprisingly, while the above Dlk1-Dio3 miRNAs have been shown to be upregulated in hypertrophic NRVMs, a subset of locus miRNAs display pathological downregulation, and unsuprisingly, these downregulated locus miRNAs functionally mitigate hypertrophy. For example, miR-485 was downregulated in PE-treated NRVMs, and unlike the aforementioned miRNAs, its overexpression attenuated cardiac hypertrophy in vivo [75]. MiR-541 has also been shown to be downregulated in hypertrophic cardiomyocytes induced by Ang II treatment. Consistent with these observations, transgenic mice that overexpress miR-541 in the heart displayed reduced hypertrophy in response to chronic Ang II treatment. This same study also described a promoter upstream of miR-541 and showed that it is negatively regulated by the MITF transcription factor [76].
Taken together, the dysregulation of the aforementioned miRNAs indicates a central role for these regulatory RNAs in facilitating cardiomyocyte hypertrophy. Moreover, the observation that some miRNAs display opposing patterns of dysregulation and antagonistic modulatory effects suggests complex post-transcriptional regulation of these transcripts, which appear to ultimately favor hypertrophic signaling in disease. If specific post-transcriptional mechanisms that dictate the balance between pro- and anti-hypertrophic Dlk1-Dio3 miRNAs were to be identified, they would constitute an appealing therapeutic target for modulating locus activity in hypertrophic remodeling.
3.2. Myocardial Infarction
Myocardial infarction (MI) stems from reduced blood flow or complete blockage in vessels surrounding the heart, which ultimately results in extensive cardiomyocyte death and increased fibrosis. While the cellular pathophysiology of MI has been well documented, the post-transcriptional and epigenetic pathways that promote adverse myocardial remodeling remain poorly understood. Dysregulation of ncRNAs including the Dlk1-Dio3 ncRNA locus has been documented in animal models of MI, with a handful being functionally characterized.
One study examining miRNA profiles of mice subjected to MI revealed that nearly one-third of dysregulated miRNAs in this disease model belonged to the Dlk1-Dio3 locus, and all were found to be significantly upregulated though to varying levels [77]. In agreement with these observations, we found miR-410 and miR-495 to be dynamically upregulated in the infarcted mouse heart. The extent of upregulation in the infarct zone, where fibroblasts are abundant and myocytes are sparse, was considerably greater in the injured region relative to the remote area [72]. This may indicate roles for these specific miRNAs in fibroblast proliferation or activation, rather than surrounding cardiac muscle.
Additional MI studies independently demonstrate dysregulation of the Dlk1-Dio3 miRNAs miR-539, miR-433, miR-377, and miR-370. One such study focused on the potential metabolic role of miR-539 in MI. This Dlk1-Dio3 miRNA was shown to be upregulated in MI and to directly regulate O-linked GlcNAcase (OGA), a metabolic enzyme reduced in MI, thereby resulting in a pathological increase in O-linked GlcNAcylation of proteins [78]. Moreover, miR-539 contributes to MI-induced myocyte apoptosis and targets the mitochondrial prohibitin complex subunit PHB2 [28]. Another MI study performed in mice found miR-433 to be significantly upregulated in this cardiac injury model. Inhibition of miR-433 significantly reduced fibrosis, and preserved left ventricular ejection fraction and fractional shortening. In this same study, overexpression of miR-433 promoted cardiac fibroblast proliferation, and promoted pathological differentiation of fibroblasts into myofibroblasts [79]. Finally, miR-370 and miR-323 were upregulated in myocardial tissue from pigs subjected to coronary microembolization [80]. miR-370 was also expressed at significantly high levels in plasma from patients with coronary artery disease [81].
Taken together, we conclude that a large subset of Dlk1-Dio3 miRNAs are upregulated in MI models, and some have the capacity to functionally drive pathological cell behaviors. It seems a variety of locus miRNAs could serve as biomarkers, and targeting a subset of these miRNAs could mitigate the deleterious effects of fibrosis in cardiac injury.
3.3. Cardiac Fibroblasts
Many heart diseases, including MI, are associated with increased fibrosis. Fibrosis stems from cardiac fibroblast activation, proliferation, and increased extracellular matrix (ECM) production. In heart disease, fibrotic regions replace dead myocytes, but fail to restore heart function, which ultimately burdens surviving cardiomyocytes. Thus, understanding the mechanisms of fibroblast activity can help identify targets that mitigate the area of fibrosis and preserve contractile function in spite of disease.
To date, only two miRNAs expressed from the Dlk1-Dio3 locus have been explicitly characterized in cardiac fibrosis, each with opposing effects on fibroblast behavior. MiR-154 is dysregulated in pressure overloaded mice, and overexpression resulted in increased cardiac fibroblast proliferation via Wingless-related integrated site (WNT) signaling, whereas its inhibition attenuated fibrosis in the heart [82,83]. In contrast, miR-495 overexpression was found to inhibit myofibroblast inflammation, differentiation, and excess ECM accumulation in human cardiac fibroblasts exposed to high glucose-induced inflammation. MiR-495-mediated effects appear to be mediated via modulation of the NF-kB and TGFβ pathways [84]. Although these are only two miRNAs that have been characterized in cardiac fibroblasts, it strikingly illustrates how Dlk1-Dio3 locus miRNAs can mediate opposing cellular behaviours in the same cell context.
3.4. Ischemia/Reperfusion
Cardiac ischemia, i.e., reduced blood supply to the heart, precedes overt myocardial infarction. Reperfusion, i.e., reoxygenation of the heart, causes extensive tissue damage and oxidative stress, particularly in the vasculature. Several ischemia/reperfusion (I/R) models in mice have shown dysregulated Dlk1-Dio3 miRNA expression, suggesting these ncRNAs are involved in cardiac remodeling associated with this surgically-induced injury process.
Inhibition of the Dlk1-Dio3 miRNAs miR-329, miR-487b, miR-494, miR-495 promoted neovascularization after ischemia [85]. Similarly, inhibition of miR-377 in CD34+ cells which were transplanted in I/R-induced mice promoted neovascularization and reduced fibrosis post-I/R injury. Serine/threonine kinase 35 was shown to be regulated by miR-377 [86]. Consistent with this finding, miR-377 was downregulated in hypoxia-treated mesenchymal stem cells (MSCs), and injection of miR-377-deficient MSCs into the infarcted rat heart reduced fibrosis and improved cardiac function [87]. MiR-410 was found to be upregulated in cardiac I/R injury in mice and in hypoxia/reoxygenation stimulated human adult cardiac myocytes, and its inhibition attenuated mitophagy in I/R [88]. Contrary to the aforementioned findings, miR-494 was shown to be downregulated in I/R mice, and transgenic mice overexpressing miR-494 improved cardiac function and reduced infarct size [89].
Collectively, these studies describe functionally distinct Dlk1-Dio3 locus miRNAs upregulated in I/R, which broadly facilitates pathological remodeling. Similar to other instances, exceptions to the overall trend in coordinate locus-wide expression may reflect complex post-transcriptional modulation. Intriguingly, I/R appears to trigger pathological processing of the locus polycistron, favoring biologically detrimental miRNAs over functionally antagonistic ones.
3.5. Circulating miRNAs in Cardiovascular Disease
Many miRNAs circulate in blood plasma, and given their potential utility as non-invasive biomarkers, much work has been directed towards identifying circulating miRNAs that change with cardiovascular disease [90]. Several Dlk1-Dio3 miRNAs have been identified in studies surveying circulating RNAs from the sera of patients with congestive heart failure and other cardiovascular complications. A significant increase in miR-299 and mir-665 levels has also been detected in serum from human patients with chronic heart failure [91,92]. Another study demonstrated that miR-665 and miR-494 are downregulated and upregulated, respectively, in heart failure. This opposite effect on expression was thought to regulate the expression of cannabinoid receptor subtypes [93].
The Dlk1-Dio3 miRNAs miR-134 and miR-380 have been found to be enriched in plasma from patients with acute pulmonary embolism and acute myocardial infarction [38,94,95].
Upregulation of miR-494 and miR-495 expression are found in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC) and dogs with myxomatous mitral valve disease, respectively [96,97]. In other cardiovascular diseases, reduced levels of Dlk1-Dio3 miRNAs have been reported. For example, miR-382 and miR-432 are downregulated in plasma from patients with aortic stenosis and atrial fibrillation, respectively [98,99]. Although the physiological contributions of these circulating Dlk1-Dio3 locus miRNAs remain unclear, their dynamic expression may have utility as a panel of biomarkers for cardiovascular disease.
The opposing effects of the aforementioned miRNAs in a spectrum of cardiac disease models, where Dlk1-Dio3 locus miRNAs function antagonistically, reinforces the notion of a multifaceted role for this locus in modulating disease pathways. A summary of miRNA functions in development and disease can be found in Table 1.
4. Long noncoding RNAs Expressed from the Dlk1-Dio3 Locus in Cardiac Development and Disease
At the time of this review, the lncRNA Gtl2 (Meg3) remains the only functionally characterized lncRNA in the Dlk1-Dio3 locus. In non-cardiac cells, Gtl2/Meg3 has been hypothesized to function as a tumor suppressor given its reduced expression in a variety of tumors including pituitary adenomas [24]. Overexpression of Gtl2/Meg3 in various tumor cell lines caused a reduction in proliferation, lending support to its purported tumor suppressor function. Moreover, Gtl2/Meg3 has been shown to induce expression levels of the p53 tumor suppressor and enhance its transcriptional activity [105]. It is not yet known whether Gtl2/Meg3 lncRNA functions in an anti-proliferative capacity in cardiomyocytes. In embryonic stem cells, Gtl2/Meg3 interacts with the polycomb repressive complex (PRC2). The ability to regulate this chromatin modifier enzyme suggests that Gtl2/Meg3 plays an important role in pluripotency and epigenetics [37,106,107].
Apart from its reported temporal expression in directed cardiomyocyte differentiation, a function for Gtl2/Meg3 has not been defined in cardiac development. Nevertheless, functions for Gtl2/Meg3 in pathological conditions relating to the cardiovasculature have been demonstrated. Recent studies in mice and cell culture systems suggest a role for this Dlk1-Dio3 lncRNA in regulating stress-response in the heart, and are detailed below.
4.1. Gtl2/Meg3 in Cardiomyocytes
Exposing H9c2 cells, a rat cardiomyocyte-like cell line, to hypoxic conditions resulted in upregulated expression of Gtl2/Meg3 [100]. Gong et al., showed that shRNA-mediated Gtl2/Meg3 knockdown greatly attenuated hallmarks of hypoxic injury and restored H9c2 viability and migration. Knockdown also reduced the overall percentage of apoptotic cells and markers, and taken together, this work indicates a robust role for Gtl2/Meg3 in facilitating hypoxic cell death Mechanistically, Gtl2/Meg3 appears to function as a molecular decoy via its interactions with miR-183 which has been shown to target the PI3K-Akt pathway. Gtl2/Meg3 expression levels were inversely proportional to miR-183 levels, and inhibition of miR-183 reversed the attenuation observed in Gtl2/Meg3 knockdown H9c2 cells [102].
4.2. Gtl2/Meg3 in Cardiac Fibroblasts and Pressure Overload
As described previously, cardiac fibroblasts are central players in pathological myocardial remodeling. Global lncRNA profiling of myocytes and non-myocytes of the adult mouse heart revealed that Gtl2/Meg3 is primarily enriched in cardiac fibroblasts. Piccoli and colleagues have shown that Gtl2/Meg3 transcripts are highly enriched in adult cardiac fibroblasts. In a mouse model of chronic pressure overload Gtl2/Meg3 transcripts were found to be significantly downregulated [103]. Curiously, further inhibition of Gtl2/Meg3 attenuated cardiac fibrosis and decreased matrix metalloproteinase 2 (MMP2), in mice subjected to TAC [103]. Overall, Gtl2/Meg3 appears to be an agonist of fibrosis, and its pathological downregulation in pressure overload may reflect mechanistic refinement of pathological fibrosis.
4.3. Gtl2/Meg3 in the Vasculature
Endothelial cell dysfunction in vasculature occurs during the human aging process, which can contribute to development of cardiovascular diseases Gtl2/Meg3 was among to highest expressed lncRNAs in endothelial cells from human umbilical vein. It was subsequently shown that Gtl2/Meg3 expression is higher in late passage versus early passage HUVECs, suggesting a role for this lncRNA in aging-mediated endothelial cell dysfunction. Inhibition of Gtl2/Meg3 enhanced sprouting of aged HUVECs and improved blood flow in ischemic hind limbs of aged mice [104].
Smooth muscle proliferation plays a role in hypoxia-induced pulmonary hypertension. Gtl2/Meg3 lncRNA is downregulated in pulmonary smooth muscle cells subjected to hypoxia [100]. Consistent with these findings, lung tissue and pulmonary arteries from patients with pulmonary arterial hypertension (PAH) displayed reduced Gtl2/Meg3 expression [80]. Inhibition of Gtl2/Meg3 has been shown to promote proliferation of human pulmonary artery smooth muscle cells via the PTEN and p53 pathways, respectively [100,101]. Taken together, Gtl2/Meg3 appears to generally suppress neovascularization, and modulating Gtl2/Meg3 activity could have utility in replenishing vasculature for diverse cardiovascular disease contexts.
5. Concluding Remarks
As highlighted here, the Dlk1-Dio3 ncRNA locus has emerged as a conserved, versatile mediator of cardiovascular development and disease. In the absence of disease, it appears that Dlk1-Dio3 ncRNAs function primarily in cardiac development, when their expression is higher compared to adult hearts (Figure 2a). While most ncRNAs in this locus are enriched during heart development, only a fraction have been functionally characterized for their specific contributions. Specifically, miRNAs expressed from this locus have been shown to influence cardiac lineage commitment (cardiomyocyte, endothelial) and embryonic/early-postnatal proliferative capacity (CPCs, fibroblasts, and cardiomyocytes). Furthermore, the observation that Dlk1-Dio3 ncRNAs are upregulated as pluripotent stem cells transition to mesodermal-derived lineages, and subsequently differentially expressed in cardiomyocyte differentiation reinforces the notion that they have a prominent regulatory role in cardiac development (Figure 2b). As additional individual ncRNAs are investigated, we anticipate their developmental functions will become evident.
Numerous models systems have demonstrated the coordinate expression of Dlk1-Dio3 ncRNAs, yet it remains ambiguous how these molecules–some with opposing effects–could faithfully orchestrate the proliferation, differentiation, and homeostasis of diverse cell types in the cardiovascular system. The dynamic expression patterns of Dlk1-Dio3 ncRNAs in different phases of cardiomyocyte differentiation (Figure 2b) suggest that levels of individual ncRNAs are subject to stringent, context-specific post-transcriptional regulation. Although more precise characterization must be performed to truly understand the locus as a whole, it is tempting to speculate that Dlk1-Dio3 ncRNA polycistron serves as an all-encompassing, but rudimentary template that can be refined to generate a complex array of functionalities.
Like many developmental regulatory factors in the heart, expression profiling strongly suggests that not only are these ncRNAs involved in cardiac development, but also function in pathological gene expression reprogramming in the diseased heart. At this juncture, it remains unclear whether pathological reactivation of locus ncRNAs is detrimental, cardioprotective, or some mixture of both. Once Dlk1-Dio3 noncoding RNAs are understood for their dynamic roles in cardiomyocyte differentiation, proliferation and hypertrophy, this knowledge can inform medical applications aimed at improved diagnostics, therapeutics, and regenerative strategies for treatment of cardiovascular disease.
Author Contributions
T.L.D. and F.J.N. wrote the paper.
Funding
This work was supported by funds provided by Boston University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, D.-Z. microRNAs in cardiovascular development. J. Mol. Cell. Cardiol. 2012, 52, 949–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaux, Y.; Zangrando, J.; Schroen, B.; Creemers, E.; Pedrazzini, T.; Chang, C.-P.; Dorn II, G.; Thum, T.; Heymans, S. Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 2015, 12, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Condorelli, G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat. Rev. Cardiol. 2015, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Olson, E.N. MicroRNA Regulatory Networks in Cardiovascular Development. Environ. Health 2010, 18, 510–525. [Google Scholar] [CrossRef] [PubMed]
- Hoelscher, S.C.; Doppler, S.A.; Dreßen, M.; Lahm, H.; Lange, R.; Krane, M. MicroRNAs: Pleiotropic players in congenital heart disease and regeneration. J. Thorac. Dis. 2017, 9, S64–S81. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, J.C.; Boyer, L.A. Getting to the heart of the matter: Long non-coding RNAs in cardiac development and disease. EMBO J. 2013, 32, 1805–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNA Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.; Tsuchihachi, T.; McManus, M.; Schwartz, R.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-F.; Murchison, E.P.; Tang, R.; Callis, T.; Tatsuguchi, M.; Deng, Z.; Rojas, M.; Hammond, S.; Schneider, M.; Selzman, C.; et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2008, 105, 2111–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Greene, S.B.; Bonilla-claudio, M.; Tao, Y.; Zhang, J.; Bai, Y.; Huang, Z.; Black, B.; Wang, F.; Martin, J. Bmp-signaling regulates myocardial differentiation from cardiac progenitors through a micro RNA-mediated mechanism. Dev. Cell 2010, 19, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-M.; Chen, H.-C.; Chen, S.-J.; Huang, C.-Y.; Wu, T.-W.; Feng, L.-Y.; Tsai, H.-C.; Lui, T.-N.; Hsueh, C.; Wei, K.-C. MicroRNA-495 inhibits proliferation of glioblastoma multiforme cells by downregulating cyclin-dependent kinase 6. World J. Surg. Oncol. 2013, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.; Bassel-Duby, R.; Olson, E. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.-P.; Chen, J.-F.; Regan, J.; Maguire, C.; Tang, R.-H.; Dong, X.-R.; Majesky, M.; Wang, D.-Z. Loss of miRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, Y.; Wang, T.; Zhou, N.; Kong, J.; Chen, L.; Snitow, M.; Morley, M.; Li, D.; Petrenko, N.; et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Johnson, B.A.; Aurora, A.B.; Simpson, E.; Nam, Y.-J.; Matkovich, S.; Dorn II, G.; van Rooij, E.; Olson, E. The miR-15 Family Regulates Post-natal Mitotic Arrest of Cardiomyocytes Enzo. Circ. Res. 2011, 109, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.-F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, M.; Naseem, R.; Marshall, W.; Hill, J.; Olson, E. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werbner, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Lin, C.-H.; Yang, J.; Shang, C.; Nuernberg, S.; Jin, K.; Xu, W.; Lin, C.-Y.; Lin, C.-J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; Deng, K.-Q.; Gong, J.; Ren, S.; Wang, X.; Chen, I.; Wang, H.; et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat. Med. 2016, 22, 1131–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.-H.; Yuan, Y.-X.; Rao, S.-L.; Wang, P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3653–3660. [Google Scholar] [PubMed]
- Wang, K.; Long, B.; Zhou, L.-Y.; Liu, F.; Zhou, Q.-Y.; Liu, C.-Y.; Fan, Y.-Y.; Li, P.-F. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat. Commun. 2014, 5, 3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.; Royo, H.; Bortolin, M.-L.; Lin, S.-P.; Ferguson-Smith, A.C.; Cavaillé, J. A Large Imprinted microRNA Gene Cluster at the Mouse Dlk1-Gtl2 Domain. Genome Res. 2004, 14, 1741–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, J.P.; O’Neill, B.L.; Stewart, C.L.; Kozlov, S.V.; Croce, C.M. At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-P.; Coan, P.; da Rocha, S.T.; Seitz, H.; Cavaille, J.; Teng, P.-W.; Takada, S.; Ferguson-Smith, A. Differential regulation of imprinting in the murine embryo and placenta by the Dlk1-Dio3 imprinting control region. Development 2007, 134, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.S.; Yevtodiyenko, A.; Schmidt, C.; Schmidt, J.V. Allele-specific histone modifications regulate expression of the Dlk1-Gtl2 imprinted domain. Genomic 2007, 89, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 2014, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Falix, F.A.; Aronson, D.C.; Lamers, W.H.; Gaemers, I.C. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dentice, M.; Salvatore, D. Local impact of thyroid hormone inactivation. J. Endocrinol. 2011, 209, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, S.; Bonasio, R.; Saldaña-meyer, R.; Yoshida, T.; Son, J.; Nishino, K.; Umezawa, A.; Reinberg, D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol. Cell 2014, 53, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Jing, Z.C.; Ellinor, P.T.; Liang, D.; Zhang, H.; Liu, Y.; Chen, X.; Pan, L.; Lyon, R.; Liu, Y.; et al. MicroRNA-134 as a potential plasma biomarker for the diagnosis of acute pulmonary embolism. J. Transl. Med. 2011, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Guntrum, M.; Vlasova, E.; Davis, T.L. Asymmetric DNA methylation of CpG dyads is a feature of secondary DMRs associated with the Dlk1/Gtl2 imprinting cluster in mouse. Epigenet. Chromatin 2017, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Kagami, M. Molecular Mechanisms Leading to the Phenotypic Development in Paternal and Maternal Uniparental Disomy for Chromosome 14. Clin. Pediatr. Endocrinol. 2008, 17, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannides, Y.; Lokulo-Sodipe, K.; Mackay, D.J.G.; Davies, J.H.; Temple, I.K. Temple syndrome: Improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: An analysis of 51 published cases. J. Med. Genet. 2014, 51, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Kagami, M. Kagami-Ogata syndrome: A clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet. 2016, 61, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, D.A.; Brothman, A.R.; Chen, Z.; Bayrak-Toydemir, P.; Longo, N. Paternal uniparental disomy of chromosome 14: Confirmation of a clinically-recognizable phenotype. Am. J. Med. Genet. A 2004, 130A, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Schwartz, S.; McPherson, E. Paternal uniparental isodisomy for chromosome 14 in a patient with a normal 46, XY karyotype. Am. J. Med. Genet. A 2004, 127A, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, P.; Watkins, M.; Surani, M.A.; Ferguson-Smith, A.C. Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development 2000, 127, 4719–4728. [Google Scholar] [PubMed]
- Takahashi, N.; Okamoto, A.; Kobayashi, R.; Kono, T. Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum. Mol. Genet. 2009, 18, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.; Zhong, Y.; Rice, K.; Zhang, L.; Zhang, X.; Gordon, F.; Lidov, H.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, C.; Longmire, T.A.; Shen, S.S.; Bourdon, A.; Sommer, A.; Gadue, P.; Spira, A.; Gouon-Evans, V.; Murphy, G.; Mostoslavsky, G.; et al. Mouse ES and iPS cells can form similar definitive endoderm despite differences in imprinted genes. J. Clin. Investig. 2011, 121, 2313–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdmanis, P.N.; Roy-chaudhuri, B.; Kim, H.K.; Sayles, L.; Zheng, Y.; Chuang, C.-H.; Caswell, D.; Chu, K.; Winslow, M.; Sweet-Cordero, E.; et al. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene 2015, 34, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Rice, A.L.; Estrella, N.L.; Held, A.; Kandarian, S.C.; Naya, F.J. MEF2A regulates the Gtl2-Dio3 microRNA mega-cluster to modulate WNT signaling in skeletal muscle regeneration. Development 2013, 140, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.L.; Zhang, S.; Tellam, R.L.; Brooks, D.; McMillen, I.; Porrello, E.; Botting, K. Regulation of microRNA during cardiomyocyte maturation in sheep. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fiore, R.; Khudayberdiev, S.; Christensen, M.; Siegel, G.; Flavell, S.; Kim, T.-K.; Greenberg, M.; Schratt, G. Mef2-mediated transcription of the miR379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009, 28, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, G.; Wang, L. Transcriptional mechanism for the paired miR-433 and miR-127 genes by nuclear receptors SHP and ERRγ. Nucleic Acids Res. 2008, 36, 5727–5735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.-Q.; Chen, X.; Wang, P.; Lu, L.; Zhao, W.; Chen, C.; Chen, C.-P.; Tao, T.; Sun, J.; Zheng, Y.-Y.; et al. Regulation of DLK1 by the maternally expressed miR-379/miR-544 cluster may underlie callipyge polar overdominance inheritance. Proc. Natl. Acad. Sci. USA 2015, 112, 13627–13632. [Google Scholar] [CrossRef] [PubMed]
- Labialle, S.; Marty, V.; Bortolin-Cavaillé, M.-L.; Hoareau-Osman, M.; Pradere, J.-P.; Valet, P.; Martin, P.; Cavaille, J. The miR-379/miR-410 cluster at the imprinted Dlk 1 -Dio 3 domain controls neonatal metabolic adaptation. EMBO J. 2014, 33, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Wüst, S.; Dröse, S.; Heidler, J.; Wittig, I.; Klockner, I.; Franko, A.; Bonke, E.; Gunther, S.; Garner, U.; Boettger, T.; et al. Metabolic Maturation during Muscle Stem Cell Differentiation Is Achieved by miR-1/133a-Mediated Inhibition of the Dlk1-Dio3 Mega Gene Cluster. Cell Metab. 2018, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Baghdadi, M.B.; Mella, S.; Gayraud-Morel, B.; Marty, V.; Cavaille, J.; Antoniewski, C.; Tajbaksh, S. Small-RNA sequencing identifies dynamic microRNA deregulation during skeletal muscle lineage progression. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Park, Y.-K.; Lee, K.-P.; Lee, S.-M.; Kong, T.-W.; Kim, H.-J.; Dho, S.; Kim, S.-Y.; Kwon, K.-S. Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging (Albany NY) 2014, 6, 524–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, C.; Segers, K.; Karim, L.; Shay, T.; Gyapay, G.; Cockett, N.; Georges, M. The callipyge mutation enhances the expression of coregulated imprinted genes in cis without affecting their imprinting status. Nat. Genet. 2001, 27, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, C.A.; Waddell, J.N.; Taxis, T.M.; Yu, H.; Tellam, R.; Neary, M.; Cockett, N. New insights into polar overdominance in callipyge sheep. Anim. Genet. 2014, 45 (Suppl. 1), 51–61. [Google Scholar] [CrossRef] [PubMed]
- Caiment, F.; Charlier, C.; Hadfield, T.; Cockett, N.; Georges, M.; Baurain, D. Assessing the effect of the CLPG mutation on the microRNA catalogue of skeletal muscle using high throughput sequencing. Genome Res. 2010, 20, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.; Ding, H.; Wylie, J.; Pico, A.; Capra, J.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.M.; Tomé, M.; Bernal, J.A.; Bernad, A. MIR-300 mediates Bmi1 function and regulates differentiation in primitive cardiac progenitors. Cell Death Dis. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Zhao, H.; Zhou, L.P.; Zhao, C.-X.; Wu, Y.-F.; Zhen, L.-X.; Li, J.; Ge, D.-X.; Xu, L.; Lin, L.; et al. miR-134 modulates the proliferation of human cardiomyocyte progenitor cells by targeting Meis2. Int. J. Mol. Sci. 2015, 16, 25199–25213. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Huang, W.; Cai, W.; Wang, L.; Guo, L.; Paul, C.; Yu, X.-Y.; Wang, Y. Inhibition of microRNA-495 Enhances Therapeutic Angiogenesis of Human Induced Pluripotent Stem Cells. Stem. Cells 2016, 35, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Vacchi-Suzzi, C.; Hahne, F.; Scheubel, P.; Marcellin, M.; Dubost, V.; Westphal, M.; Boeglen, C.; Buchmann-Moller, S.; Chueng, M.; Cordier, A.; et al. Heart Structure-Specific Transcriptomic Atlas Reveals Conserved microRNA-mRNA Interactions. PLoS ONE 2013, 8, e52442. [Google Scholar] [CrossRef] [PubMed]
- Van der Bom, T.; Zomer, A.C.; Zwinderman, A.H.; Meijboom, F.J.; Bouma, B.J.; Mulder, B.J.M. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 2010, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ji, L.; Liu, L.; Liu, Y.; Hou, H.; Yu, K.; Sun, Q.; Zhao, Z. Characterization of circulating microRNA expression in patients with a ventricular septal defect. PLoS ONE 2014, 9, e106318. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Naya, F.J. MicroRNAs in the myocyte enhancer factor 2 (MEF2)-regulated Gtl2-Dio3 noncoding RNA locus promote cardiomyocyte proliferation by targeting the transcriptional coactivator Cited2. J. Biol. Chem. 2015, 290, 23162–23172. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Zhu, J.G.; Liu, Y.Q. Identification of the microRNA Expression Profile in the Regenerative Neonatal Mouse Heart by Deep Sequencing. Cell Biochem. Biophys. 2014, 70, 635. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.D.; Chi, J.Y.; Liang, H.H.; Huangfu, L.-T.; Guo, Z.-D.; Zou, H.; Yin, X.-H. MicroRNA-377 Mediates Cardiomyocyte Apoptosis Induced by Cyclosporin A. Can. J. Cardiol. 2016, 32, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.L.; Maruyama, S.; Sano, S.; Accorsi, A.; Girgenrath, M.; Walsh, K.; Naya, F. miR-410 and miR-495 are dynamically regulated in diverse cardiomyopathies and their inhibition attenuates pathological hypertrophy. PLoS ONE 2016, 11, e0151515. [Google Scholar] [CrossRef]
- Weber, K.; Rostert, N.; Bauersachs, S.; Wess, G. Serum microRNA profiles in cats with hypertrophic cardiomyopathy. Mol. Cell. Biochem. 2015, 402, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Nguyen, S.S.; Gao, X.M.; Tham, Y.-K.; Ooi, J.; Patterson, N.; Kiriazis, H.; Su, Y.; Thomas, C.; Lin, R.; et al. Inhibition of miR-154 Protects Against Cardiac Dysfunction and Fibrosis in a Mouse Model of Pressure Overload. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ponnusamy, M.; Liu, C.; Tian, J.; Dong, Y.; Gao, J.; Wang, C.; Zhang, Y.; Zhang, L.; Wang, K.; et al. MiR-485-5p modulates mitochondrial fission through targeting mitochondrial anchored protein ligase in cardiac hypertrophy. Biochim. Biophys. Acta-Mol. Basis Dis. 2017, 1863, 2871–2881. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, N.; Long, B.; Fan, Y.-Y.; Liu, C.-Y.; Zhou, Q.-Y.; Murtaza, I.; Wang, K.; Li, P.-F. Cardiac hypertrophy is negatively regulated by miR-541. Cell Death Dis. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Janssen, R.; Zuidwijk, M.; Muller, A.; Mulders, J.; Oudejans, C.B.M.; Simonides, W.S. Cardiac expression of deiodinase type 3 (Dio3) following myocardial infarction is associated with the induction of a pluripotency microRNA signature from the Dlk1-Dio3 genomic region. Endocrinology 2013, 154, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, S.; DeMartino, A.M.; Watson, L.J.; Brittian, K.; Zafir, A.; Dassanyaka, S.; Hong, K.; Jones, S. MicroRNA-539 is up-regulated in failing heart, and suppresses O-GlcNAcase expression. J. Biol. Chem. 2014, 289, 29665–29676. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Bei, Y.; Chen, P.; Lei, Z.; Fu, S.; Zhang, H.; Xu, J.; Che, L.; Chen, X.; Sluijter, J.; et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics 2016, 6, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, L.; Zhao, J.; Sun, Y.; Yang, H. MiRNA Expression Profile of the Myocardial Tissue of Pigs with Coronary Microembolization. Cell. Physiol. Biochem. 2017, 43, 1012–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Yang, N.; Fei, Z.; Qiu, J.; Ma, D.; Liu, X.; Cai, G.; Li, S. Analysis of plasma miR-208a and miR-370 expression levels for early diagnosis of coronary artery disease. Biomed. Rep. 2016, 5, 332–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.Y.; Bie, Z.D.; Zhang, C.H.; Li, H.; Li, L.D.; Yang, J. MiR-154 directly suppresses DKK2 to activate Wnt signaling pathway and enhance activation of cardiac fibroblasts. Cell Biol. Int. 2016, 40, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Liu, W.; Wang, D.-Z. MiR-154 promotes myocardial fibrosis through β-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jin, H.; Jiang, S.; Xu, Y. MicroRNA-495 inhibits the high glucose- induced inflammation, differentiation and extracellular matrix accumulation of cardiac fibroblasts through downregulation of NOD1. Cell. Mol. Biol. Lett. 2018, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Welten, S.M.J.; Bastiaansen, A.J.N.M.; De Jong, R.C.M.; de Vries, M.; Peters, E.; Boonstra, M.; Sheikh, S.; Monica, N.; Kandimalla, E.; Quax, P.; et al. Inhibition of 14q32 MicroRNAs miR-329, miR-487b, miR-494, and miR-495 increases neovascularization and blood flow recovery after ischemia. Circ. Res. 2014, 115, 696–708. [Google Scholar] [CrossRef] [PubMed]
- Joladarashi, D.; Garikipati, V.N.S. Enhanced Cardiac Regenerative Ability of Stem Cells After Ischemia-Reperfusion Injury: Role of Human CD34+ Cells Deficient in MicroRNA-377. J. Am. Coll. Cardiol. 2015, 66, 2214–2226. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Huang, W.; Feng, Y.; Cai, W.; Wang, Y.; Wang, X.; Liang, J.; Wani, M.; Chen, J.; Zhu, P.; et al. MicroRNA-377 regulates mesenchymal stem cell-induced angiogenesis in ischemic hearts by targeting VEGF. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, T.; Dong, Z.; Mi, R. MicroRNA-410 is involved in mitophagy after cardiac ischemia/reperfusion injury by targeting high-mobility group box 1 protein. J. Cell. Biochem. 2018, 119, 2427–2439. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Ren, X.-P.; Chen, J.; Liu, H.; Yang, J.; Medvedovic, M.; Hu, Z.; Fan, G.-C. MicroRNA-494 Targeting both Pro-apoptotic and Anti-apoptotic Proteins Protects against Ischemia/Reperfusion-Induced Cardiac Injury. Circulation 2010, 122, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Creemers, E.E.; Tijsen, A.J.; Pinto, Y.M. Circulating MicroRNAs: Novel biomarkers and extracellular communicators in cardiovascular disease? Circ. Res. 2012, 110, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, H.A.; Coskunpinar, E.; Ikitimur, B. The prognostic value of circulating microRNAs in heart failure: Preliminary results from a genome-wide expression study. J. Cardiovasc. Med. 2015, 16, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, J.; Yin, Z.; Wang, F.; Chen, C.; Wang, D.-W. Identification of cardiac-related circulating microRNA profile in human chronic heart failure. Oncotarget 2015, 7, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möhnle, P.; Schütz, S.V.; Schmidt, M.; Hinske, C.; Hubner, M.; Heyn, J.; Beiras-Fernandez, A.; Kreth, S. MicroRNA-665 is involved in the regulation of the expression of the cardioprotective cannabinoid receptor CB2 in patients with severe heart failure. Biochem. Biophys. Res. Commun. 2014, 451, 516–521. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Lv, P.; Zhao, X.; Wang, X.; Ma, X.; Meng, W.; Meng, X.; Dong, S. Predictive value of circulating miR-328 and miR-134 for acute myocardial infarction. Mol. Cell. Biochem. 2014, 394, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Sakata, Y.; Nakatani, D.; Suna, S.; Mizuno, H.; Shimizu, M.; Usami, M.; Sasaki, T.; Sato, H.; Kawahara, Y.; et al. A subset of circulating microRNAs are predictive for cardiac death after discharge for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2012, 427, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Hsiao, Y.-W.; Chang, S.-L. Circulating microRNAs in arrhythmogenic right ventricular cardiomyopathy with ventricular arrhythmia. EP Eur. 2017, 20, f37–f45. [Google Scholar] [CrossRef] [PubMed]
- Yang, V.K.; Loughran, K.A.; Meola, D.M.; Juhr, C.; Thane, K.; Davis, A.; Hoffman, A. Circulating exosome microRNA associated with heart failure secondary to myxomatous mitral valve disease in a naturally occurring canine model. J. Extracell. Vesicles 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Coffey, S.; Williams, M.J.A.; Phillips, L.V.; Jones, G.T. Circulating microRNA Profiling Needs Further Refinement Before Clinical Use in Patients with Aortic Stenosis. J. Am. Heart Assoc. 2015, 4, e002150. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hou, S.; Huang, D.; Luo, X.; Zhang, J.; Chen, J.; Xu, W. Expression profile analysis of circulating microRNAs and their effects on ion channels in chinese atrial fibrillation patients. Int. J. Clin. Exp. Med. 2015, 8, 845–853. [Google Scholar] [PubMed]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Qiao, Y.; Wang, Q.; Liu, B.; Hou, J.; Li, R.; Tang, C. Down-regulation of lncRNA MEG3 promotes hypoxia-induced human pulmonary artery smooth muscle cell proliferation and migration via repressing PTEN by sponging miR-21. Biochem. Biophys. Res. Commun. 2018, 495, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Nie, X.; Sun, S.; Dong, S.; Yuan, C.; Li, Y.; Xiao, B.; Jie, D.; Liu, Y. Long Non-Coding RNA MEG3 Downregulation Triggers Human Pulmonary Artery Smooth Muscle Cell Proliferation and Migration via the p53 Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2569–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Xu, H.; Chang, H.; Tong, Y.; Zhang, T.; Guo, G. Knockdown of long non-coding RNA MEG3 protects H9c2 cells from hypoxia-induced injury by targeting microRNA-183. J. Cell. Biochem. 2018, 119, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, M.T.; Gupta, S.K.; Viereck, J.; Foinquinos, A.; Samolovac, S.; Kramer, F.; Garg, A.; Remke, J.; Zimmer, K.; Batkai, S.; et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ. Res. 2017, 121, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Jaé, N.; Holdt, L.; Dimmeler, S. Long Noncoding RNAs from Clinical Genetics to Therapeutic Targets? J. Am. Coll. Cardiol. 2016, 67, 1214–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhong, Y.; Wang, Y.; Zhang, X.; Batista, D.; Gejman, R.; Ansell, P.; Zhao, J.; Weng, C.; Klibanski, A. Activation of p53 by MEG3 non-coding RNA. J. Biol. Chem. 2007, 282, 24731–24742. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.; Sarma, K.; Song, J.; Kingston, R.; Borrowsky, M.; Lee, J. Genome-wide Identification of Polycomb-Associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Das, P.P.; Hendrix, D.A.; Apostolou, E.; Buchner, A.; Canver, M.; Beyaz, S.; Ljuboja, D.; Kuintzle, R.; Kim, W.; Kamik, R.; et al. PRC2 Is Required to Maintain Expression of the Maternal Gtl2-Rian-Mirg Locus by Preventing De Novo DNA Methylation in Mouse Embryonic Stem Cells. Cell Rep. 2015, 12, 1456–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic of Dlk1-Dio3 ncRNA locus features. The mouse Dlk1-Dio3 ncRNA locus (top panel) harbors sixty-one miRNAs and three lncRNA genes. Several miRNAs (shaded in light purple) reside within Gtl2, anti-Rtl1, Rian, and Mirg coding regions (shaded in light purple), whereas many miRNAs (shaded in light blue) exist between the aforementioned coding regions. The human DLK1-DIO3 ncRNA locus (bottom panel) harbors fifty-three miRNAs, which are largely present in the same regions as the mouse locus. Both loci are regulated by a 5′ intergenic differentially methylated region (IG-DMR), which is upstream of the Gtl2-DMR. While most lncRNAs are homologous between mouse and human gene annotations, it is important to note the mouse Mirg lncRNA harbors miRNAs, whereas the human Meg9 lncRNA does not. Instead, Meg9 resides downstream of all annotated human locus miRNAs. NcRNAs are expressed from the non-methylated maternal allele, whereas methylation of the IG-DMR in the paternal chromosome (not depicted) prevents expression of ncRNAs. Protein-coding genes (shaded in black) are expressed predominantly from the paternal allele [33].
Figure 1.
Schematic of Dlk1-Dio3 ncRNA locus features. The mouse Dlk1-Dio3 ncRNA locus (top panel) harbors sixty-one miRNAs and three lncRNA genes. Several miRNAs (shaded in light purple) reside within Gtl2, anti-Rtl1, Rian, and Mirg coding regions (shaded in light purple), whereas many miRNAs (shaded in light blue) exist between the aforementioned coding regions. The human DLK1-DIO3 ncRNA locus (bottom panel) harbors fifty-three miRNAs, which are largely present in the same regions as the mouse locus. Both loci are regulated by a 5′ intergenic differentially methylated region (IG-DMR), which is upstream of the Gtl2-DMR. While most lncRNAs are homologous between mouse and human gene annotations, it is important to note the mouse Mirg lncRNA harbors miRNAs, whereas the human Meg9 lncRNA does not. Instead, Meg9 resides downstream of all annotated human locus miRNAs. NcRNAs are expressed from the non-methylated maternal allele, whereas methylation of the IG-DMR in the paternal chromosome (not depicted) prevents expression of ncRNAs. Protein-coding genes (shaded in black) are expressed predominantly from the paternal allele [33].

Figure 2.
Dlk1-Dio3 ncRNA Expression in the Heart and Cardiomyocyte Differentiation. (a) General trends of locus ncRNA expression over time: Many ncRNAs are expressed at their highest levels during fetal and early-postnatal cardiac development, and are gradually downregulated to low basal levels during adulthood. However, disease or stress triggers marked upregulation of numerous locus ncRNAs. (b) Locus ncRNAs are dynamically expressed throughout phases of cardiomyocyte lineage commitment and differentiation. This plot was generated using supplementary data from Wamstad et al. [62], which analyzed the transcriptome during cardiomyocyte-directed differentiation. To generate the plot, raw values of individual ncRNAs were normalized to their maximal expression, and subsequently grouped by the differentiation phase at which maximal ncRNA expression was reached. The relative levels conveyed represent the average of all ncRNAs within a given group, and are comprised as follows: Group 1 = miR-1906, -770, -493, -337, -540, -665, -432, -1188, -882, -299, -380-5p, -323-5p, -758, -679, -666, -654, -544, -485, -453, -412, and -369-5p; Group 2 = miR-673-5p, -341, -370, -494, -667+, -376b+, -300+, -and 541; Group 3 = miR-431+, -127+, -434-5p+, -1197, -323-3p++, -1193++, -543++, -495++, -539, -134+, -668, -496, -409-3p, and -410+; Group 4 = Gtl2, miR-379+, -411+, -376c++, -376a+, -381+, and -382+; Group 5 = miR-433+, -136++, -380-3p, -487b+, -154+, -377+, -409-5p+, and -369-3p++. To account for levels, ncRNAs annotated with “+” indicate measurements over 100 reads, and “++” for over 1000 reads. Although groups 2, 3, and 4 all peak in cardiac precursors, these ncRNAs were subdivided into groups based on striking differences in expression levels during the mesoderm (early) and cardiomyocyte (later) phases. These ncRNA patterns illustrate the dynamic regulation of the locus. Consistent with the high levels reported in the fetal and postnatal heart, locus ncRNAs are enriched in later phases of cardiac differentiation.
Figure 2.
Dlk1-Dio3 ncRNA Expression in the Heart and Cardiomyocyte Differentiation. (a) General trends of locus ncRNA expression over time: Many ncRNAs are expressed at their highest levels during fetal and early-postnatal cardiac development, and are gradually downregulated to low basal levels during adulthood. However, disease or stress triggers marked upregulation of numerous locus ncRNAs. (b) Locus ncRNAs are dynamically expressed throughout phases of cardiomyocyte lineage commitment and differentiation. This plot was generated using supplementary data from Wamstad et al. [62], which analyzed the transcriptome during cardiomyocyte-directed differentiation. To generate the plot, raw values of individual ncRNAs were normalized to their maximal expression, and subsequently grouped by the differentiation phase at which maximal ncRNA expression was reached. The relative levels conveyed represent the average of all ncRNAs within a given group, and are comprised as follows: Group 1 = miR-1906, -770, -493, -337, -540, -665, -432, -1188, -882, -299, -380-5p, -323-5p, -758, -679, -666, -654, -544, -485, -453, -412, and -369-5p; Group 2 = miR-673-5p, -341, -370, -494, -667+, -376b+, -300+, -and 541; Group 3 = miR-431+, -127+, -434-5p+, -1197, -323-3p++, -1193++, -543++, -495++, -539, -134+, -668, -496, -409-3p, and -410+; Group 4 = Gtl2, miR-379+, -411+, -376c++, -376a+, -381+, and -382+; Group 5 = miR-433+, -136++, -380-3p, -487b+, -154+, -377+, -409-5p+, and -369-3p++. To account for levels, ncRNAs annotated with “+” indicate measurements over 100 reads, and “++” for over 1000 reads. Although groups 2, 3, and 4 all peak in cardiac precursors, these ncRNAs were subdivided into groups based on striking differences in expression levels during the mesoderm (early) and cardiomyocyte (later) phases. These ncRNA patterns illustrate the dynamic regulation of the locus. Consistent with the high levels reported in the fetal and postnatal heart, locus ncRNAs are enriched in later phases of cardiac differentiation.


{kind=link}
{kind=link}
Table 1.
Summary of Dlk1-Dio3 noncoding RNAs with known functions or differential expression in heart development and disease.
Table 1.
Summary of Dlk1-Dio3 noncoding RNAs with known functions or differential expression in heart development and disease.
| ncRNA | Development | Species | Disease | Species |
|---|---|---|---|---|
| miRNAs | ||||
| All * | dynamically expressed during mESC cardiomyocyte-directed differentiation [62] | m [62] | ||
| miR-493 | Maturation [51] | s [51] | — | |
| miR-337 | — | MI [77], HF [86], angiogenesis [86], fibrosis [86], remodeling [86]. Targets STK35 [86] | h [86], m [77,86] | |
| miR-665 | — | CHF [92], HF [93] | h [92,93] | |
| miR-431 | Proliferation [15] | m [15], r [15] | — | |
| miR-433 | VSD [68] | h [68] | MI [77,79], fibrosis [79], ventricular remodeling [79]. Targets AZIN1 and JNK1 [79] | m [77,79] |
| miR-127 | Valve morphogenesis [66], maturation [51] | r [66], d [66], mk [66], s [51] | MI [77] | m [77] |
| miR-434 | — | MI [77] | m [77] | |
| miR-432 | Maturation [51] | s [51] | Atrial fibrillation [99] | h [99] |
| miR-136 | — | MI [77] | m [77] | |
| miR-370 | — | MI77, CAD [81], coronary microembolism [80] | m [77], h [81], p [80] | |
| miR-379 | VSD68, maturation [51] | h [68], s [51] | MI [77] | m [77] |
| miR-411 | Proliferation [15], maturation [51] | m [15], r [15], s [51] | MI [77] | m [77] |
| miR-299a | — | Congestive HF [91] | h [91] | |
| miR-299b | — | Congestive HF [91] | h [91] | |
| miR-380 | Proliferation [15], maturation [51] | m [15], r [15], s [51] | MI [95] | h [95] |
| miR-323 | — | Coronary microembolism [80] | p [80] | |
| miR-329 | Maturation [51] | s [51] | MI77, IR-induced neovascularization [85] | m [51,85] |
| miR-494 | — | I/R-induced apoptosis [89], IR-induced neovascularization [85], HF [93], arrhythmogenic RV-cardiomyopathy [96]. Targets PTEN, ROCK1, CamKIIδ, FGFR2, and LIF [89] | m [85,89], h [93,96] | |
| miR-1193 | — | MI [77] | m [77] | |
| miR-543 | Maturation [51] | s [51] | MI [77] | m [77] |
| miR-495 | Proliferation [15,69], angiogenic differentiation [65]. Targets VEZF1 [65], Cited2 [69] | h [65], m [15], r [15,69] | MI neovascularization [65], fibrosis [84], MI [72,77], IR neovascularization [85], mitral valve disease [97] | h [65,84], m [72,77,85], d [97] |
| miR-376c | — | MI [77] | m [77] | |
| miR-654 | VSD [68] | h [68] | — | |
| miR-376b | — | MI [77] | m [77] | |
| miR-376a | — | MI [77] | m [77] | |
| miR-300 | Differentiation [63], target of Bmi1 [63] | m [63] | — | |
| miR-381 | — | Hypertrophy [73] | c [73] | |
| miR-487b | VSD [68], maturation [51] | h [68], s [51] | MI [77], IR neovascularization [85] | m [77,85] |
| miR-539 | Proliferation [15] | m [15], r [15] | MI [77,78], MI apoptosis [28]. Targets OGA [78] and PHB2 [28] | m [28,77,78] |
| miR-544 | — | MI [77] | m [77] | |
| miR-382 | Maturation [51] | s [51] | MI77, comorbid aortic stenosis and CAD [98] | m [77], h [98] |
| miR-134 | Proliferation [64]. Directly targets Meis2 [64] | h [64] | MI [38,94] | h [38,94] |
| miR-668 | Proliferation [15], maturation [70] | m [15,70], r [15] | — | |
| miR-485 | Maturation [51] | s [51] | Hypertrophy [75], MI77, remodeling [75]. Targets MAPL [75] | m [75,77] |
| miR-154 | Proliferation [15], maturation [70] | m [15,70], r [15] | Hypertrophy [74], fibrosis [74,82,83], apoptosis [83]. Targets p15 [74] and DKK2 [82] | m [74,82,83] |
| miR-377 | — | I/R neovascularization [86], MI neovascularization [87], fibrosis [87], Cyclosporin-mediated apoptosis [71]. Targets XIAP [71], NRP2 [71], and VEGF [87] | m [86,87], r [71] | |
| miR-541 | — | Hypertrophy [76], MI 77]. Repressed by MITF [76] | m [76,77] | |
| miR-409 | VSD [68], maturation [51] | h [68], s [51] | MI [77] | m [77] |
| miR-369 | — | MI [77] | m [77] | |
| miR-410 | Proliferation [15,69], maturation [51]. Targets Cited2 [69] | r [15,69], m [15], s [51] | MI [72,77], I/R mitophagy [88]. | m [72,77,88] |
| lncRNAs | ||||
| Gtl2 | Angiogenesis [100,101] | h [100,101] | I/R apoptosis [102], fibrosis [103], hypertension [100], neovascularization [104]. Decoys miR-183 [102] | M [100,103,104], r [102] |
Key: HF = Heart Failure, MI = Myocardial Infarction, CHF = Chronic heart failure, VSD = ventricular septal defect, CAD = coronary artery disease, RV = right-ventricular, IR = ischemia-reperfusion. For species column, h = human, m = mouse, r = rat, p = pig, s = sheep, d = dog, c = cat, mk = monkey. * All locus miRNAs, except miR-2932, -3544, -3070a, -3070b, -3071, -329, and -3072. The following miRNAs have not been reported in any publications at this time: miR-2932, miR-1906-1, miR-770, miR-673, miR-3544, miR-540, miR-3070a, miR-3070b, miR-3071, miR-341, miR-1188, miR-882, miR-1197, miR-758, miR-679, miR-666, miR-453, miR-667, miR-496a, miR-412, miR-3077, Rtl1, Rian, and Mirg.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dill, T.L.; Naya, F.J. A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease. J. Cardiovasc. Dev. Dis. 2018, 5, 37. https://doi.org/10.3390/jcdd5030037
AMA Style
Dill TL, Naya FJ. A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease. Journal of Cardiovascular Development and Disease. 2018; 5(3):37. https://doi.org/10.3390/jcdd5030037
Chicago/Turabian StyleDill, Tiffany L., and Francisco J. Naya. 2018. "A Hearty Dose of Noncoding RNAs: The Imprinted DLK1-DIO3 Locus in Cardiac Development and Disease" Journal of Cardiovascular Development and Disease 5, no. 3: 37. https://doi.org/10.3390/jcdd5030037
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.