
Transposable Prophages in Leptospira: An Ancient, Now Diverse, Group Predominant in Causative Agents of Weil’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Searching for Transposable Prophages in Leptospira weilii
2.2. Predicting Transposable Prophages in Leptospiraceae
2.3. The Predicted Prophages Predominate in Leptospira Pathogenic Strains
2.4. Sequence Comparison between Predicted Prophages
2.4.1. Identifying (Almost) Identical Prophages
2.4.2. Defining Genera and Species Using Genome Comparison
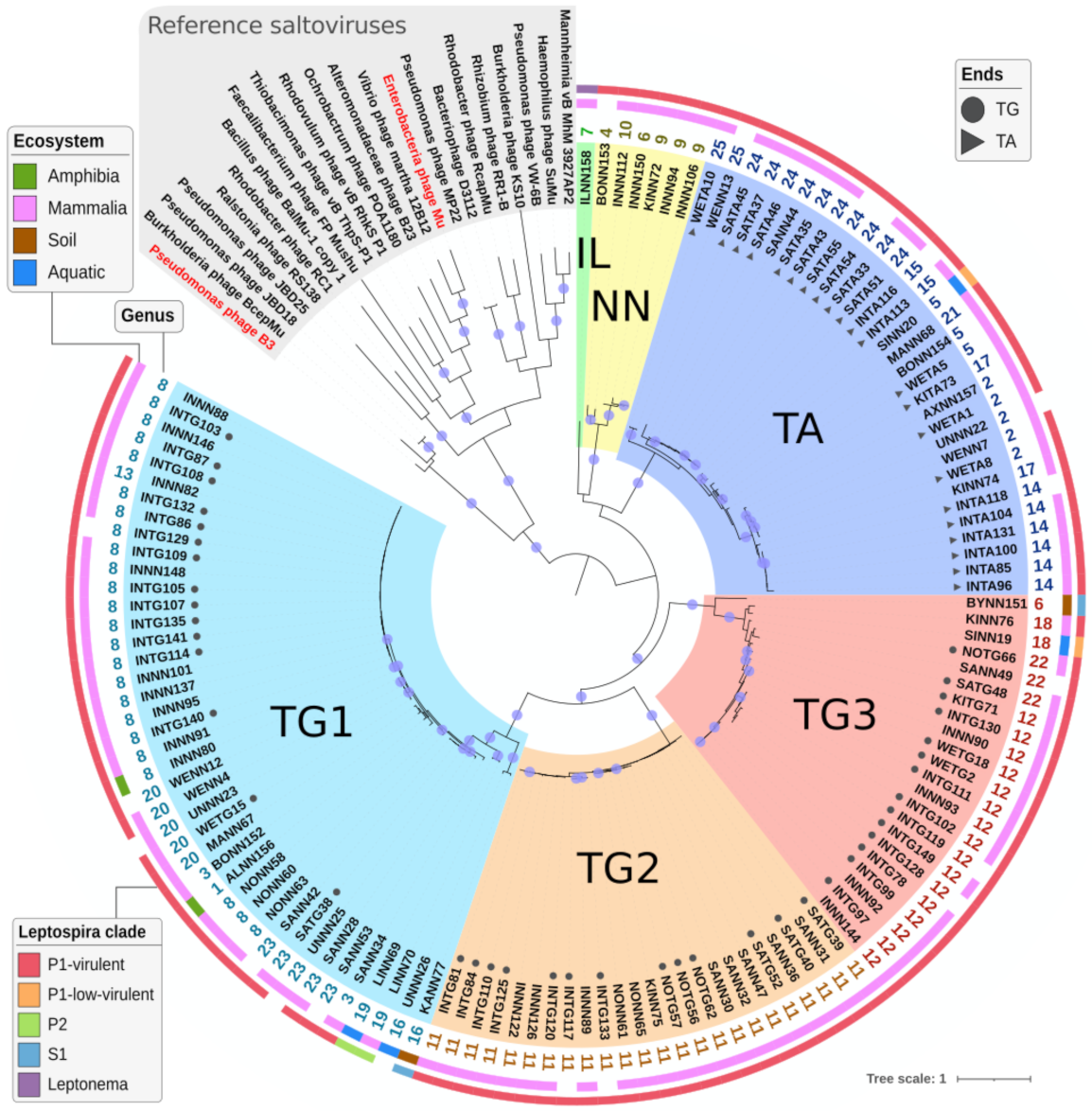
2.4.3. Defining Sub-Families Using the Species Phylogeny
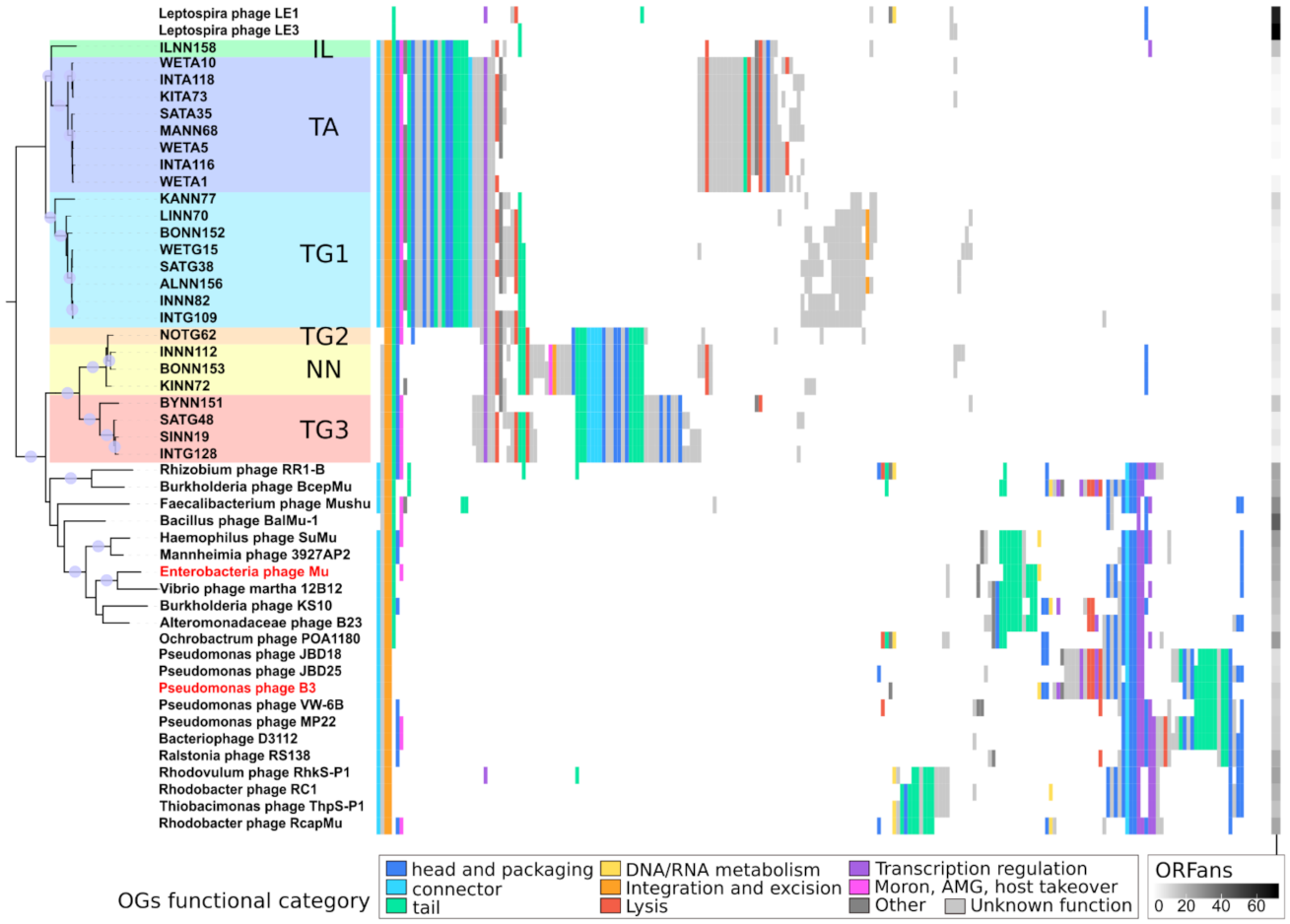
2.4.4. Analyzing Gene Content of Transposable (Pro)Phage
2.4.5. Determining Families Using Phylogeny and Gene Content
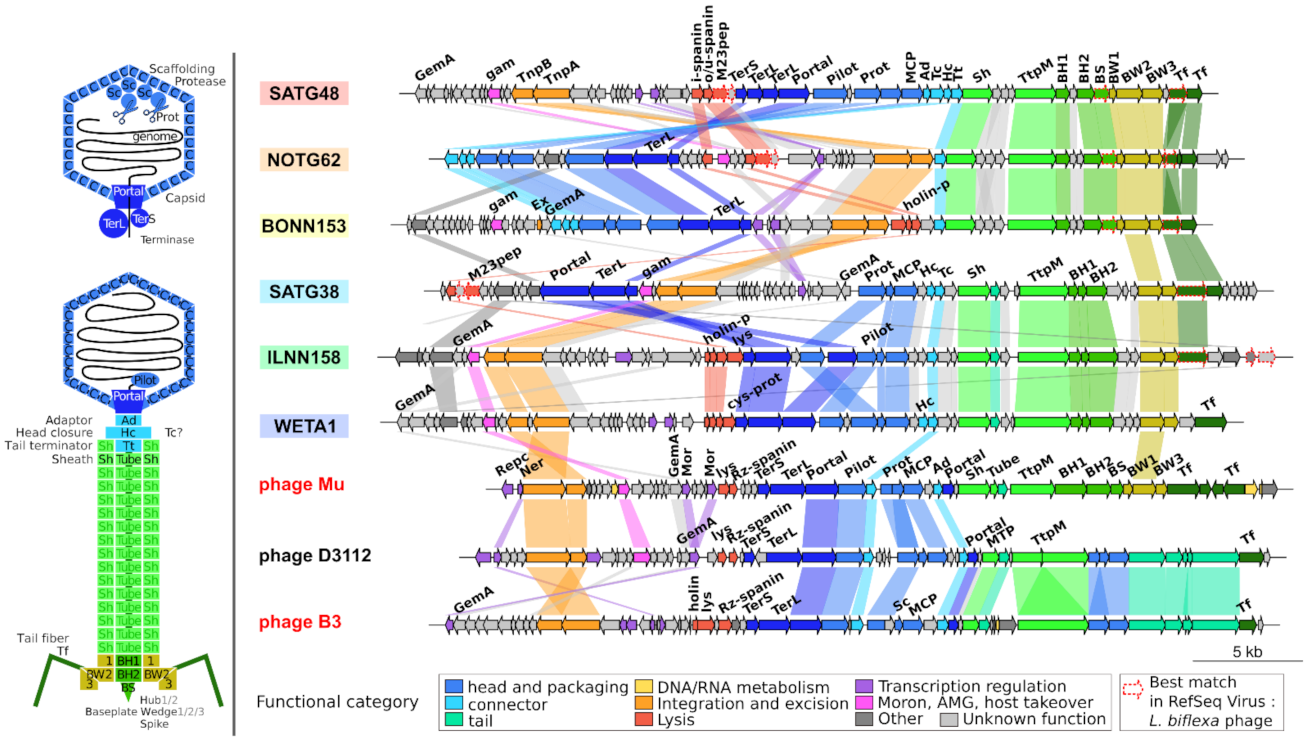
2.5. Genome Organization of Leptospiraceae Prophages
3. Discussion
3.1. A Role in the Virulent Lifestyle Acquisition?
3.2. Complex Relationships: Different Prophages in the Same Host, Different Hosts with the Same Prophages
3.3. Prophages with Unusual Genome Ends
3.4. Transposition and Structural Modules with Similar Yet Different Evolutionary History
3.5. Homologs Are So Divergent That Most Are Not Directly Identifiable
3.6. The Core Genome of Transposable (Pro)Phages Is Limited to … Transposition Genes
3.7. The Evolutionary History of Leptospiraceae Transposable Prophages Remains Elusive
3.8. High Amino Acid Divergence but High Conservation of Genome Length and Organization
4. Material and Methods
4.1. Prophage Search in the RefSeq Database
4.2. Genomic Comparisons
4.3. Two-Step Protein Clustering
4.4. Phylogenies and Gene Content Matrix
4.5. Genomic Maps
4.6. Number of Sequenced Leptospiraceae Species
4.7. Transposase 3D Structure Prediction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weil, A. “Über eine eigenthümliche, mit Milztumor, Icterus und Nephritis einhergehende, acute Infektionskrankheit” [On a strange, acute infectious disease, accompanied by swelling of the spleen, icterus, and nephritis]. In Deutsches Archiv für Klinische Medizin; Ziemssen, H., Ed.; Bergmann: Munich, Germany, 1886; Volume 39, pp. 209–232. (In German) [Google Scholar]
- Costa, F.; Hagan, J.E.; Calcagno, J.; Kane, M.; Torgerson, P.; Martinez-Silveira, M.S.; Stein, C.; Abela-Ridder, B.; Ko, A.I. Global Morbidity and Mortality of Leptospirosis: A Systematic Review. PLoS Negl. Trop. Dis. 2015, 9, e0003898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, B. History of Leptospirosis and Leptospira. In Leptospirosis and Leptospira; Adler, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–9. [Google Scholar]
- Stimson, A.M. Note on an Organism Found in Yellow-Fever Tissue. Public Heal. Reports 1907, 22, 541. [Google Scholar] [CrossRef]
- Inada, R.; Ido, Y.; Hoki, R.; Kaneko, R.; Ito, H. THE ETIOLOGY, MODE OF INFECTION, AND SPECIFIC THERAPY OF WEIL’S DISEASE (SPIROCHÆTOSIS ICTEROHÆMORRHAGICA). J. Exp. Med. 1916, 23, 377–402. [Google Scholar] [CrossRef]
- Hovind-Hougen, K. Leptospiraceae, a New Family to Include Leptospira Noguchi 1917 and Leptonema gen. nov. Int. J. Syst. Bacteriol. 1979, 29, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Levett, P.N. Systematics of Leptospiraceae. In Leptospira and Leptospirosis; Adler, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 11–20. [Google Scholar]
- Xu, Y.; Zhu, Y.; Wang, Y.; Chang, Y.-F.; Zhang, Y.; Jiang, X.; Zhuang, X.; Zhu, Y.; Zhang, J.; Zeng, L.; et al. Whole genome sequencing revealed host adaptation-focused genomic plasticity of pathogenic Leptospira. Sci. Rep. 2016, 6, 20020. [Google Scholar] [CrossRef]
- Fouts, D.E.; Matthias, M.A.; Adhikarla, H.; Adler, B.; Amorim-Santos, L.; Berg, D.E.; Bulach, D.; Buschiazzo, A.; Chang, Y.-F.; Galloway, R.L.; et al. What Makes a Bacterial Species Pathogenic?:Comparative Genomic Analysis of the Genus Leptospira. PLoS Negl. Trop. Dis. 2016, 10, e0004403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.T.; Schiettekatte, O.; Goarant, C.; Neela, V.K.; Bernet, E.; Thibeaux, R.; Ismail, N.; Mohd Khalid, M.K.N.; Amran, F.; Masuzawa, T.; et al. Revisiting the taxonomy and evolution of pathogenicity of the genus Leptospira through the prism of genomics. PLoS Negl. Trop. Dis. 2019, 13, e0007270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibeaux, R.; Iraola, G.; Ferrés, I.; Bierque, E.; Girault, D.; Soupé-Gilbert, M.-E.; Picardeau, M.; Goarant, C. Deciphering the unexplored Leptospira diversity from soils uncovers genomic evolution to virulence. Microb. Genomics 2018, 4. [Google Scholar] [CrossRef]
- Picardeau, M. Toolbox of Molecular Techniques for Studying Leptospira Spp. In Spirochete Biology: The Post Genomic Era; Adler, B., Ed.; Springer: Cham, Switzerland, 2017; pp. 141–162. [Google Scholar]
- Ricaldi, J.N.; Fouts, D.E.; Selengut, J.D.; Harkins, D.M.; Patra, K.P.; Moreno, A.; Lehmann, J.S.; Purushe, J.; Sanka, R.; Torres, M.; et al. Whole Genome Analysis of Leptospira licerasiae Provides Insight into Leptospiral Evolution and Pathogenicity. PLoS Negl. Trop. Dis. 2012, 6, e1853. [Google Scholar] [CrossRef] [Green Version]
- Haapa-Paananen, S.; Savilahti, H. Applications of the Bacteriophage Mu In Vitro Transposition Reaction and Genome Manipulation via Electroporation of DNA Transposition Complexes. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Humana Press: New York, NY, USA, 2018; pp. 279–286. [Google Scholar]
- Patrick Higgins, N. Muprints and Whole Genome Insertion Scans: Methods for Investigating Chromosome Accessibility and DNA Dynamics using Bacteriophage Mu. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Humana Press: New York, NY, USA, 2018; pp. 303–314. [Google Scholar]
- Toussaint, A. Transposable Bacteriophages as Genetic Tools. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Humana Press: New York, NY, USA, 2018; pp. 263–278. [Google Scholar]
- Van Gijsegem, F. Use of RP4: Mini-Mu for Gene Transfer. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Humana Press: New York, NY, USA, 2018; pp. 287–302. [Google Scholar]
- Toussaint, A.; Rice, P.A. Transposable phages, DNA reorganization and transfer. Curr. Opin. Microbiol. 2017, 38, 88–94. [Google Scholar] [CrossRef]
- Hulo, C.; Masson, P.; Le Mercier, P.; Toussaint, A. A structured annotation frame for the transposable phages: A new proposed family “Saltoviridae” within the Caudovirales. Virology 2015, 477, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.P.; Lou, Z.; Meng, L.; Harshey, R.M. Transposable Prophage Mu Is Organized as a Stable Chromosomal Domain of E. coli. PLoS Genet. 2013, 9, e1003902. [Google Scholar] [CrossRef] [Green Version]
- Toussaint, A. Transposable Mu-like phages in Firmicutes: New instances of divergence generating retroelements. Res. Microbiol. 2013, 164, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, A.; Van Gijsegem, F. Extension of the transposable bacterial virus family: Two genomic organisations among phages and prophages with a Tn552-related transposase. Res. Microbiol. 2018, 169, 495–499. [Google Scholar] [CrossRef]
- Cazares, A.; Mendoza-Hernández, G.; Guarneros, G. Core and accessory genome architecture in a group of Pseudomonas aeruginosa Mu-like phages. BMC Genomics 2014, 15, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold-Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, S.; Bu, D.; Xu, J. Protein threading using residue co-variation and deep learning. Bioinformatics 2018, 34, i263–i273. [Google Scholar] [CrossRef] [Green Version]
- Xu, J. Distance-based protein folding powered by deep learning. Proc. Natl. Acad. Sci. USA 2019, 116, 16856–16865. [Google Scholar] [CrossRef] [Green Version]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Pérez Bucio, R.E.; Mom, R.; Toussaint, A.; Petit, M.-A.; Enault, F. PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR Genomics Bioinforma. 2021, 3. [Google Scholar] [CrossRef] [PubMed]
- Akroyd, J.; Symonds, N. Localization of the gam gene of bacteriophage Mu and characterisation of the gene product. Gene 1986, 49, 273–282. [Google Scholar] [CrossRef]
- Büttner, C.R.; Wu, Y.; Maxwell, K.L.; Davidson, A.R. Baseplate assembly of phage Mu: Defining the conserved core components of contractile-tailed phages and related bacterial systems. Proc. Natl. Acad. Sci. USA 2016, 113, 10174–10179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, J.; Young, R. Phage Lysis: Multiple Genes for Multiple Barriers. In Advances in Virus Research; Kielian, M., Mettenleiter, T.C., Rossinck, M.J., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 33–70. [Google Scholar]
- Wang, F.; Xiong, Y.; Xiao, Y.; Han, J.; Deng, X.; Lin, L. MMPphg from the thermophilic Meiothermus bacteriophage MMP17 as a potential antimicrobial agent against both Gram-negative and Gram-positive bacteria. Virol. J. 2020, 17, 130. [Google Scholar] [CrossRef] [PubMed]
- Schuch, R.; Fischetti, V.A. The Secret Life of the Anthrax Agent Bacillus anthracis: Bacteriophage-Mediated Ecological Adaptations. PLoS ONE 2009, 4, e6532. [Google Scholar] [CrossRef] [Green Version]
- Ristow, P.; Bourhy, P.; da Cruz McBride, F.W.; Figueira, C.P.; Huerre, M.; Ave, P.; Girons, I.; Saint Girons, I.; Ko, A.I.; Picardeau, M. The OmpA-Like Protein Loa22 Is Essential for Leptospiral Virulence. PLoS Pathog. 2007, 3, e97. [Google Scholar] [CrossRef]
- Davies, E.V.; James, C.E.; Williams, D.; O’Brien, S.; Fothergill, J.L.; Haldenby, S.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. Temperate phages both mediate and drive adaptive evolution in pathogen biofilms. Proc. Natl. Acad. Sci. USA 2016, 113, 8266–8271. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Kümmerli, R.; Paterson, S.; Winstanley, C.; Brockhurst, M.A. Transposable temperate phages promote the evolution of divergent social strategies in Pseudomonas aeruginosa populations. Proc. Biol. Sci. 2019, 286, 20191794. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Imbroglios of Viral Taxonomy: Genetic Exchange and Failings of Phenetic Approaches. J. Bacteriol. 2002, 184, 4891–4905. [Google Scholar] [CrossRef] [Green Version]
- Pato, M.L.; Howe, M.M.; Higgins, N.P. A DNA gyrase-binding site at the center of the bacteriophage Mu genome is required for efficient replicative transposition. Proc. Natl. Acad. Sci. USA 1990, 87, 8716–8720. [Google Scholar] [CrossRef] [Green Version]
- Pato, M.L. Central location of the Mu strong gyrase binding site is obligatory for optimal rates of replicative transposition. Proc. Natl. Acad. Sci. USA 1994, 91, 7056–7060. [Google Scholar] [CrossRef] [Green Version]
- Pato, M.L.; Banerjee, M. The Mu strong gyrase-binding site promotes efficient synapsis of the prophage termini. Mol. Microbiol. 1996, 22, 283–292. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Enright, A.J. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Söding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics 2014, 15, 1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2; Springer: New York, NY, USA, 2009. [Google Scholar]
- Guy, L.; Roat Kultima, J.; Andersson, S.G.E. genoPlotR: Comparative gene and genome visualization in R. Bioinformatics 2010, 26, 2334–2335. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, J.; Rasche, H.; Maughmer, C.; Criscione, A.; Mijalis, E.; Liu, M.; Hu, J.C.; Young, R.; Gill, J.J. Galaxy and Apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PLOS Comput. Biol. 2020, 16, e1008214. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olo Ndela, E.; Enault, F.; Toussaint, A. Transposable Prophages in Leptospira: An Ancient, Now Diverse, Group Predominant in Causative Agents of Weil’s Disease. Int. J. Mol. Sci. 2021, 22, 13434. https://doi.org/10.3390/ijms222413434
Olo Ndela E, Enault F, Toussaint A. Transposable Prophages in Leptospira: An Ancient, Now Diverse, Group Predominant in Causative Agents of Weil’s Disease. International Journal of Molecular Sciences. 2021; 22(24):13434. https://doi.org/10.3390/ijms222413434
Chicago/Turabian StyleOlo Ndela, Eric, François Enault, and Ariane Toussaint. 2021. "Transposable Prophages in Leptospira: An Ancient, Now Diverse, Group Predominant in Causative Agents of Weil’s Disease" International Journal of Molecular Sciences 22, no. 24: 13434. https://doi.org/10.3390/ijms222413434