
MET Exon 14 Skipping: A Case Study for the Detection of Genetic Variants in Cancer Driver Genes by Deep Learning

 ,
, 
Abstract
:1. Introduction
2. Results
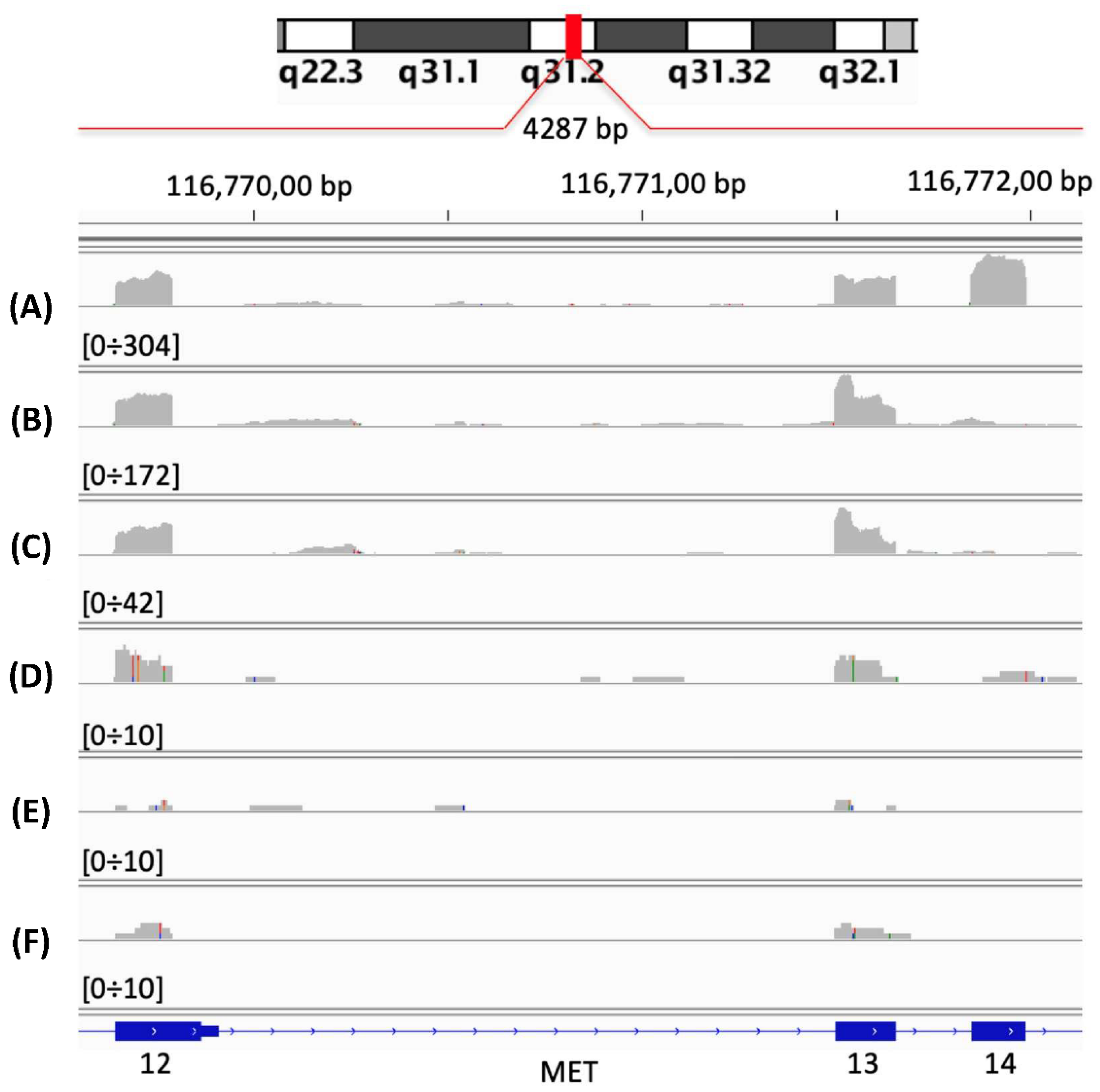
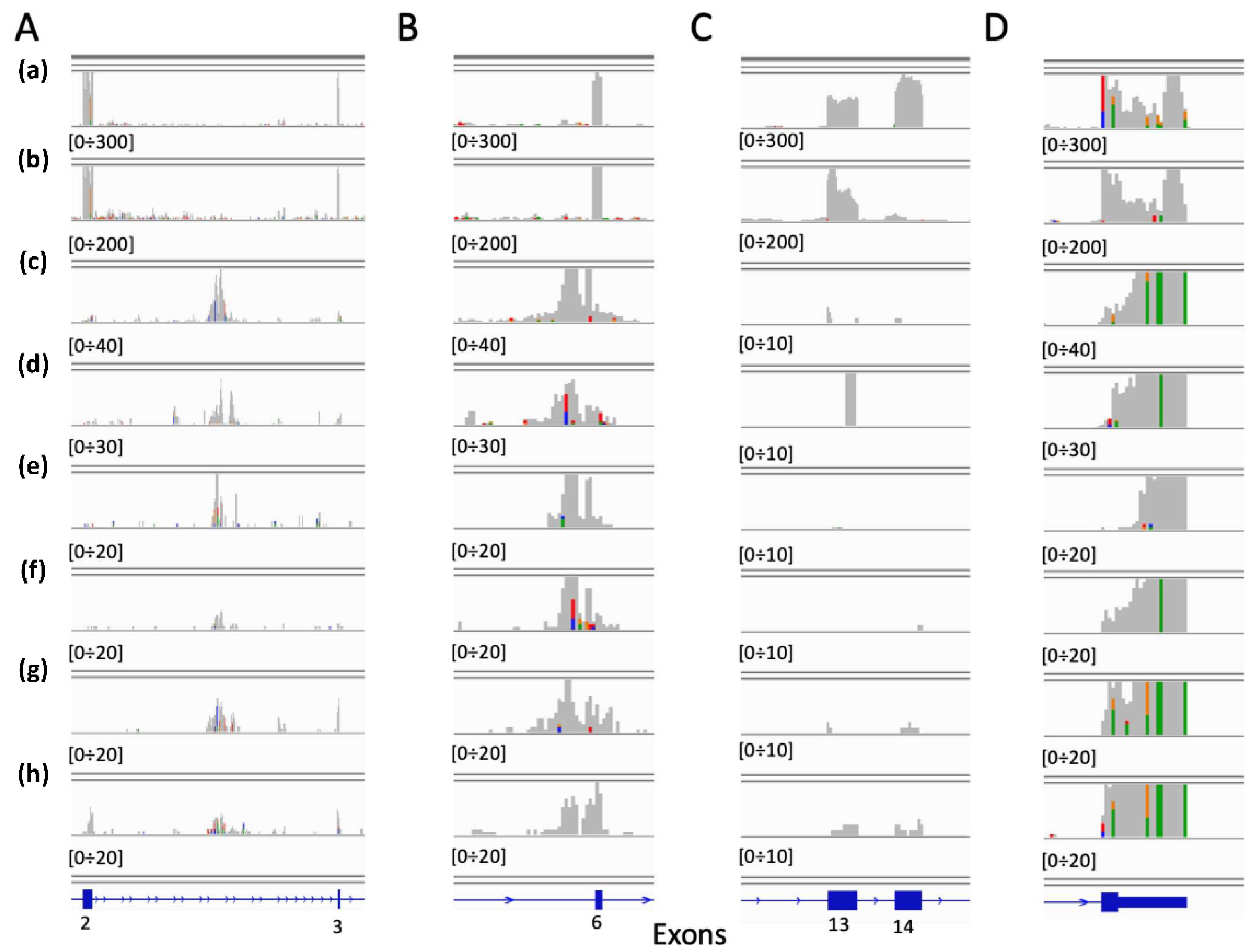
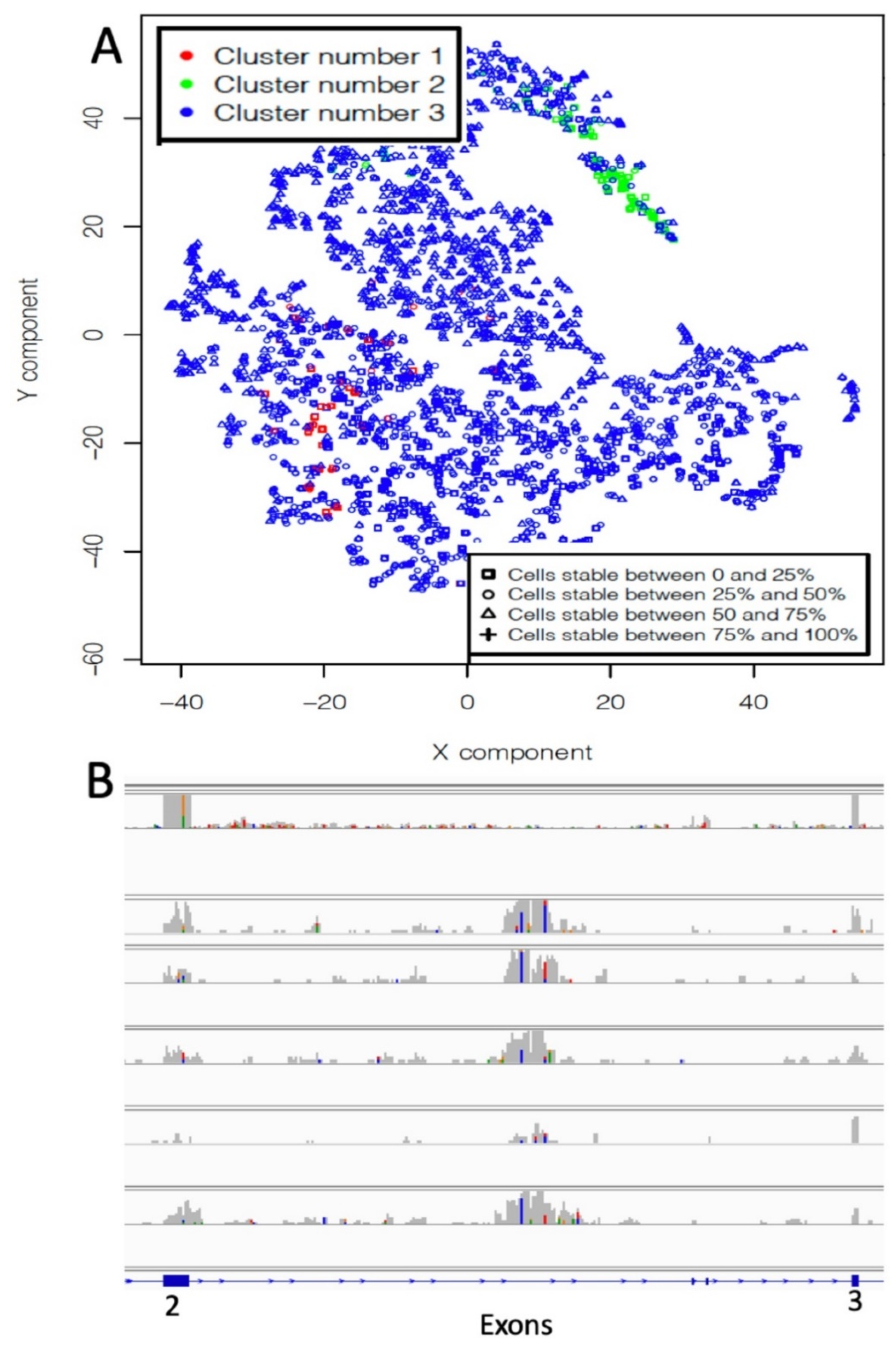
2.1. Neural Network for the Detection of MET Exon 14 Skipping (METΔ14).
Neural Network Validation and Discovery on TCGA Samples
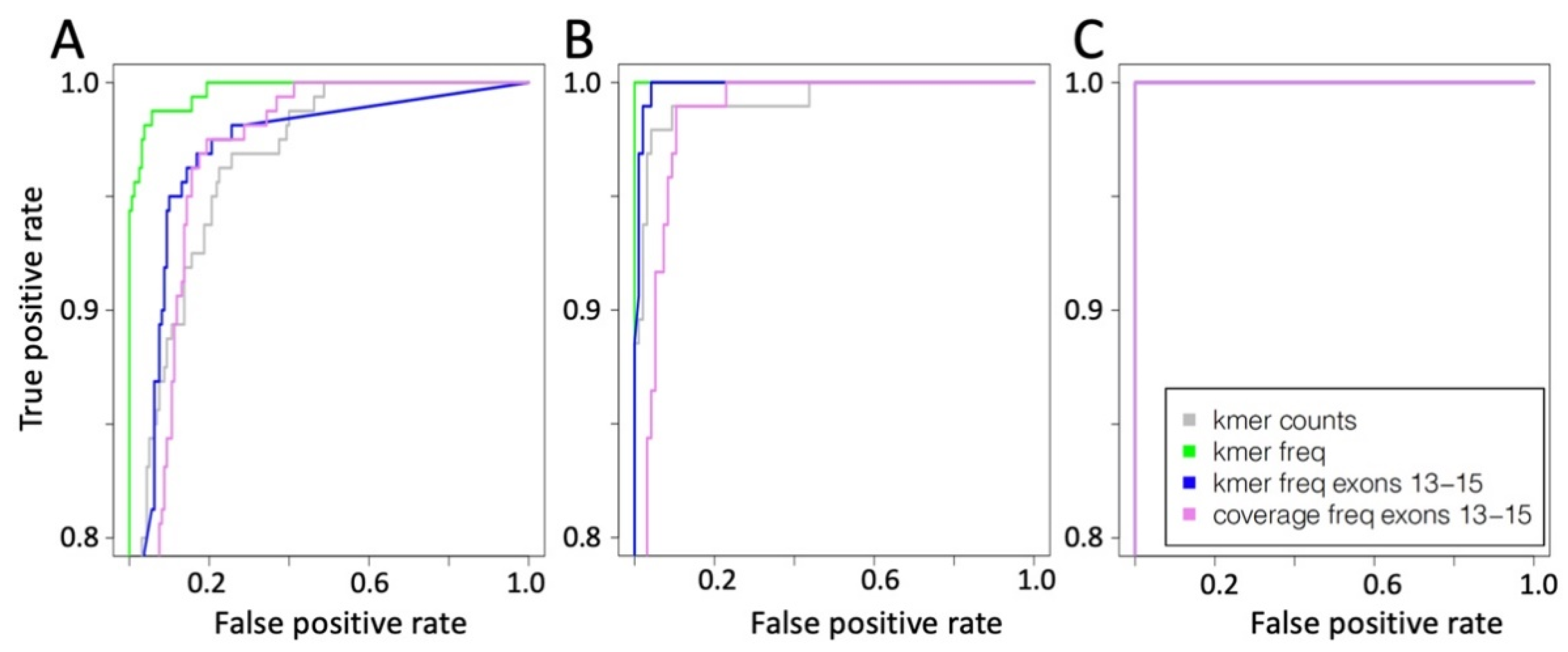
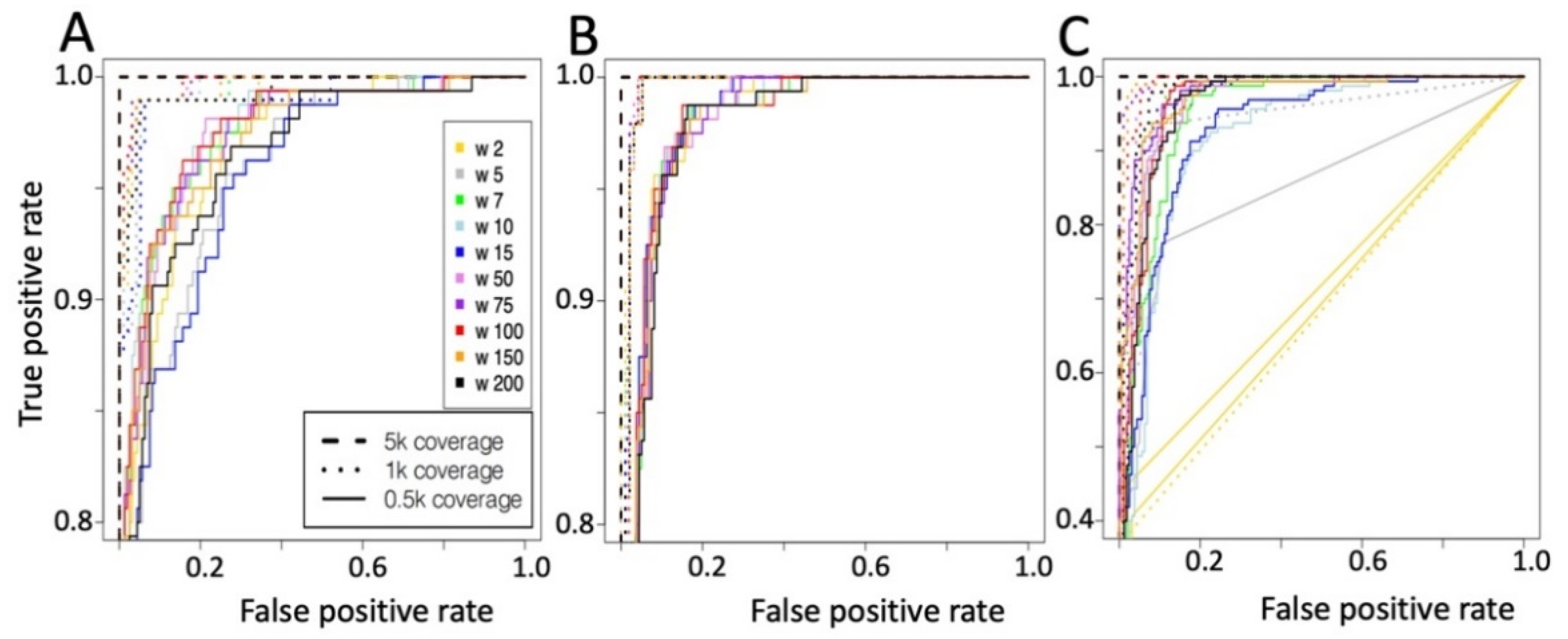
2.2. Convolutional Neural Network (CNN) for the Detection of METΔ14
Convolutional Neural Network Validation on Bronchus and Lung Samples
2.3. Sparsely Connected Autoencoders (SCA) to Detect MET Non-Canonical Isoforms
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Generating the Data for the Neural Network Training and Test Set
4.2.1. 16/31. k-mer Training Set
4.2.2. 16/31. k-mer Test Set
4.2.3. Coverage Training and Test Set
4.3. TCGA RNAseq Datasets
4.4. Model Coding and Hyperparameter Selection for NN
4.5. Model Coding and Hyperparameter Selection for CNN
4.6. Model Coding and Hyperparameter Selection for Sparsely Connected Autoencoders (SCA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, L. Alternative splicing: Human disease and quantitative analysis from high-throughput sequencing. Comput. Struct. Biotechnol. J. 2021, 19, 183–195. [Google Scholar] [CrossRef]
- Kim, P.; Yang, M.; Yiya, K.; Zhao, W.; Zhou, X. ExonSkipDB: Functional annotation of exon skipping event in human. Nucleic Acids Res. 2019, 48, D896–D907. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via Diverse Exon 14 Splicing Alterations Occurs in Multiple Tumor Types and Confers Clinical Sensitivity to MET Inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.C. MET Receptor Juxtamembrane Exon 14 Alternative Spliced Variant: Novel Cancer Genomic Predictive Biomarker. Cancer Discov. 2015, 5, 802–805. [Google Scholar] [CrossRef] [Green Version]
- Champagnac, A.; Bringuier, P.-P.; Barritault, M.; Isaac, S.; Watkin, E.; Forest, F.; Maury, J.-M.; Girard, N.; Brevet, M. Frequency of MET exon 14 skipping mutations in non-small cell lung cancer according to technical approach in routine diagnosis: Results from a real-life cohort of 2369 patients. J. Thorac. Dis. 2020, 12, 2172–2178. [Google Scholar] [CrossRef]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Awad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non–Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Van Der Steen, N.; Giovannetti, E.; Pauwels, P.; Peters, G.J.; Hong, D.S.; Cappuzzo, F.; Hirsch, F.R.; Rolfo, C. cMET Exon 14 Skipping: From the Structure to the Clinic. J. Thorac. Oncol. 2016, 11, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Marczyk, M.; Fu, C.; Lau, R.; Du, L.; Trevarton, A.J.; Sinn, B.; Gould, R.E.; Pusztai, L.; Hatzis, C.; Symmans, W.F. The impact of RNA extraction method on accurate RNA sequencing from formalin-fixed paraf-fin-embedded tissues. BMC Cancer 2019, 19, 1189. [Google Scholar] [CrossRef] [PubMed]
- Pruis, M.; Geurts-Giele, W.; Von Der, T.J.; Meijssen, I.; Dinjens, W.; Aerts, J.; Dingemans, A.; Lolkema, M.; Paats, M.; Dubbink, H. Highly accurate DNA-based detection and treatment results of MET exon 14 skipping mutations in lung cancer. Lung Cancer 2020, 140, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J. Thorac. Oncol. 2019, 14, 737–741. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, X.; MacLeod, J.; Liu, J. Discerning novel splice junctions derived from RNA-seq alignment: A deep learning approach. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Du, X.; Hu, C.; Yao, Y.; Sun, S.; Zhang, Y. Analysis and Prediction of Exon Skipping Events from RNA-Seq with Sequence Information Using Rotation Forest. Int. J. Mol. Sci. 2017, 18, 2691. [Google Scholar] [CrossRef] [Green Version]
- Zuallaert, J.; Godin, F.; Kim, M.; Soete, A.; Saeys, Y.; De Neve, W. SpliceRover: Interpretable convolutional neural networks for improved splice site prediction. Bioinformatics 2018, 34, 4180–4188. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [Green Version]
- Le, N.Q.K.; Yapp, E.K.Y.; Nagasundaram, N.; Yeh, H.-Y. Classifying Promoters by Interpreting the Hidden Information of DNA Sequences via Deep Learning and Combination of Continuous FastText N-Grams. Front. Bioeng. Biotechnol. 2019, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Le, N.Q.K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.-T.; Nguyen, N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef]
- Wen, J.; Liu, Y.; Shi, Y.; Huang, H.; Deng, B.; Xiao, X. A classification model for lncRNA and mRNA based on k-mers and a convolutional neural network. BMC Bioinform. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Miglio, U.; Berrino, E.; Panero, M.; Ferrero, G.; Tarrero, L.C.; Miano, V.; Dell’Aglio, C.; Sarotto, I.; Annaratone, L.; Marchiò, C.; et al. The expression of LINE1-MET chimeric transcript identifies a subgroup of aggressive breast cancers. Int. J. Cancer 2018, 143, 2838–2848. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Alessandri, L.; Cordero, F.; Beccuti, M.; Licheri, N.; Arigoni, M.; Olivero, M.; Di Renzo, M.F.; Sapino, A.; Calogero, R. Sparsely-connected autoencoder (SCA) for single cell RNAseq data mining. NPJ Syst. Biol. Appl. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Ye, X.; Ho, J.W.K. Ultrafast clustering of single-cell flow cytometry data using FlowGrid. BMC Syst. Biol. 2019, 13, 35. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Wang, J.; Torre, E.; Dueck, H.; Shaffer, S.; Bonasio, R.; Murray, J.I.; Raj, A.; Li, M.; Zhang, N.R. SAVER: Gene expression recovery for single-cell RNA sequencing. Nat. Methods 2018, 15, 539–542. [Google Scholar] [CrossRef]
- Huang, C.; Zou, Q.; Liu, H.; Qiu, B.; Li, Q.; Lin, Y.; Liang, Y. Management of Non-small Cell Lung Cancer Patients with MET Exon 14 Skipping Mutations. Curr. Treat. Options Oncol. 2020, 21, 1–15. [Google Scholar] [CrossRef]
- Apicella, M.; Migliore, C.; Capelôa, T.; Menegon, S.; Cargnelutti, M.; Degiuli, M.; Sapino, A.; Sottile, A.; Sarotto, I.; Casorzo, L.; et al. Dual MET/EGFR therapy leads to complete response and resistance prevention in a MET-amplified gas-troesophageal xenopatient cohort. Oncogene 2017, 36, 1200–1210. [Google Scholar] [CrossRef]
- Lee, J.; Tran, P.; Klempner, S. Targeting the MET Pathway in Gastric and Oesophageal Cancers: Refining the Optimal Approach. Clin. Oncol. 2016, 28, e35–e44. [Google Scholar] [CrossRef]
- Lee, J.; Ou, S.H.I.; Lee, J.M.; Kim, H.C.; Hong, M.; Kim, S.Y.; Jang, J.; Ahn, S.; Kang, S.Y.; Lee, S.; et al. Gastrointestinal malignancies harbor actionable MET exon 14 deletions. Oncotarget 2015, 6, 28211–28222. [Google Scholar] [CrossRef] [Green Version]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutterbach, B.; Zeng, Q.; Davis, L.J.; Hatch, H.; Hang, G.; Kohl, N.E.; Gibbs, J.B.; Pan, B.-S. Lung Cancer Cell Lines Harboring MET Gene Amplification Are Dependent on Met for Growth and Survival. Cancer Res. 2007, 67, 2081–2088. [Google Scholar] [CrossRef] [Green Version]
- Asaoka, Y.; Tada, M.; Ikenoue, T.; Seto, M.; Imai, M.; Miyabayashi, K.; Yamamoto, K.; Yamamoto, S.; Kudo, Y.; Mohri, D.; et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem. Biophys. Res. Commun. 2010, 394, 1042–1046. [Google Scholar] [CrossRef]
- Li, B.; Torossian, A.; Sun, Y.; Du, R.; Dicker, A.P.; Lu, B. Higher Levels of c-Met Expression and Phosphorylation Identify Cell Lines With Increased Sensitivity to AMG-458, a Novel Selective c-Met Inhibitor With Radiosensitizing Effects. Int. J. Radiat. Oncol. 2012, 84, e525–e531. [Google Scholar] [CrossRef]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic Mutations Lead to an Oncogenic Deletion of Met in Lung Cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Melsted, P.; Pritchard, J.K. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinform. 2011, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Status |
RNAseq (Million Reads) |
MET (Thousand Reads) |
|---|---|---|---|
| EBC-1 | Amplified MET | 113 | 1447 |
| Hs746T | Amplified METΔ14 | 95 | 846 |
| A549 | MET | 115 | 109 |
| NCI-H596 | METΔ14 | 118 | 114 |
| TCGA Tissue | # Inspected Tissue | # Detected METΔ14 | # Detected False METΔ14 |
|---|---|---|---|
| Adrenal gland | 10 | 0 | 0 |
| Bladder | 280 | 1 | 0 |
| Brain | 28 | 0 | 0 |
| Breast | 162 | 0 | 0 |
| Bronchus and lung | 690 | 17 | 1 |
| Cervix (uterus) | 236 | 0 | 6 |
| Corpus uteri | 109 | 0 | 4 |
| Esophagus | 165 | 0 | 0 |
| Hearth/mediastinum/pleura | 78 | 0 | 1 |
| Kidney | 435 | 0 | 3 |
| Pancreas | 89 | 0 | 0 |
| Skin | 288 | 0 | 1 |
| Soft tissues | 35 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nosi, V.; Luca, A.; Milan, M.; Arigoni, M.; Benvenuti, S.; Cacchiarelli, D.; Cesana, M.; Riccardo, S.; Di Filippo, L.; Cordero, F.; et al. MET Exon 14 Skipping: A Case Study for the Detection of Genetic Variants in Cancer Driver Genes by Deep Learning. Int. J. Mol. Sci. 2021, 22, 4217. https://doi.org/10.3390/ijms22084217
Nosi V, Luca A, Milan M, Arigoni M, Benvenuti S, Cacchiarelli D, Cesana M, Riccardo S, Di Filippo L, Cordero F, et al. MET Exon 14 Skipping: A Case Study for the Detection of Genetic Variants in Cancer Driver Genes by Deep Learning. International Journal of Molecular Sciences. 2021; 22(8):4217. https://doi.org/10.3390/ijms22084217
Chicago/Turabian StyleNosi, Vladimir, Alessandrì Luca, Melissa Milan, Maddalena Arigoni, Silvia Benvenuti, Davide Cacchiarelli, Marcella Cesana, Sara Riccardo, Lucio Di Filippo, Francesca Cordero, and et al. 2021. "MET Exon 14 Skipping: A Case Study for the Detection of Genetic Variants in Cancer Driver Genes by Deep Learning" International Journal of Molecular Sciences 22, no. 8: 4217. https://doi.org/10.3390/ijms22084217