Causative Links between Protein Aggregation and Oxidative Stress: A Review

 , and
, and
Abstract
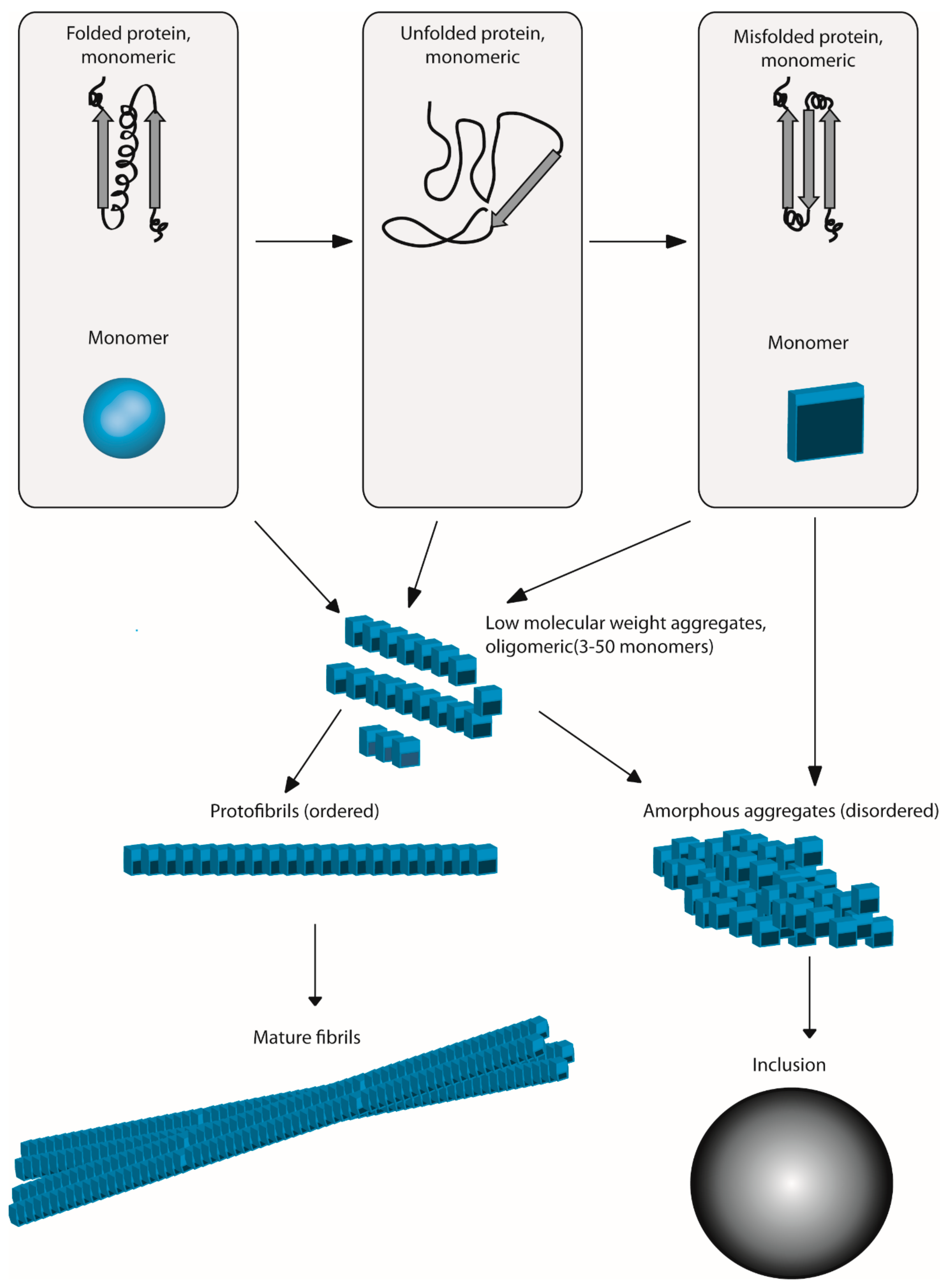
:1. Introduction
2. Oxidative Stress Can Produce Aggregation: A Mechanistic View
2.1. Oxidation of Critical Amino Acids Induces Structural Changes within Proteins, Leading to Aggregation
2.1.1. Cysteine Oxidation
2.1.2. Other Aminoacids Involved
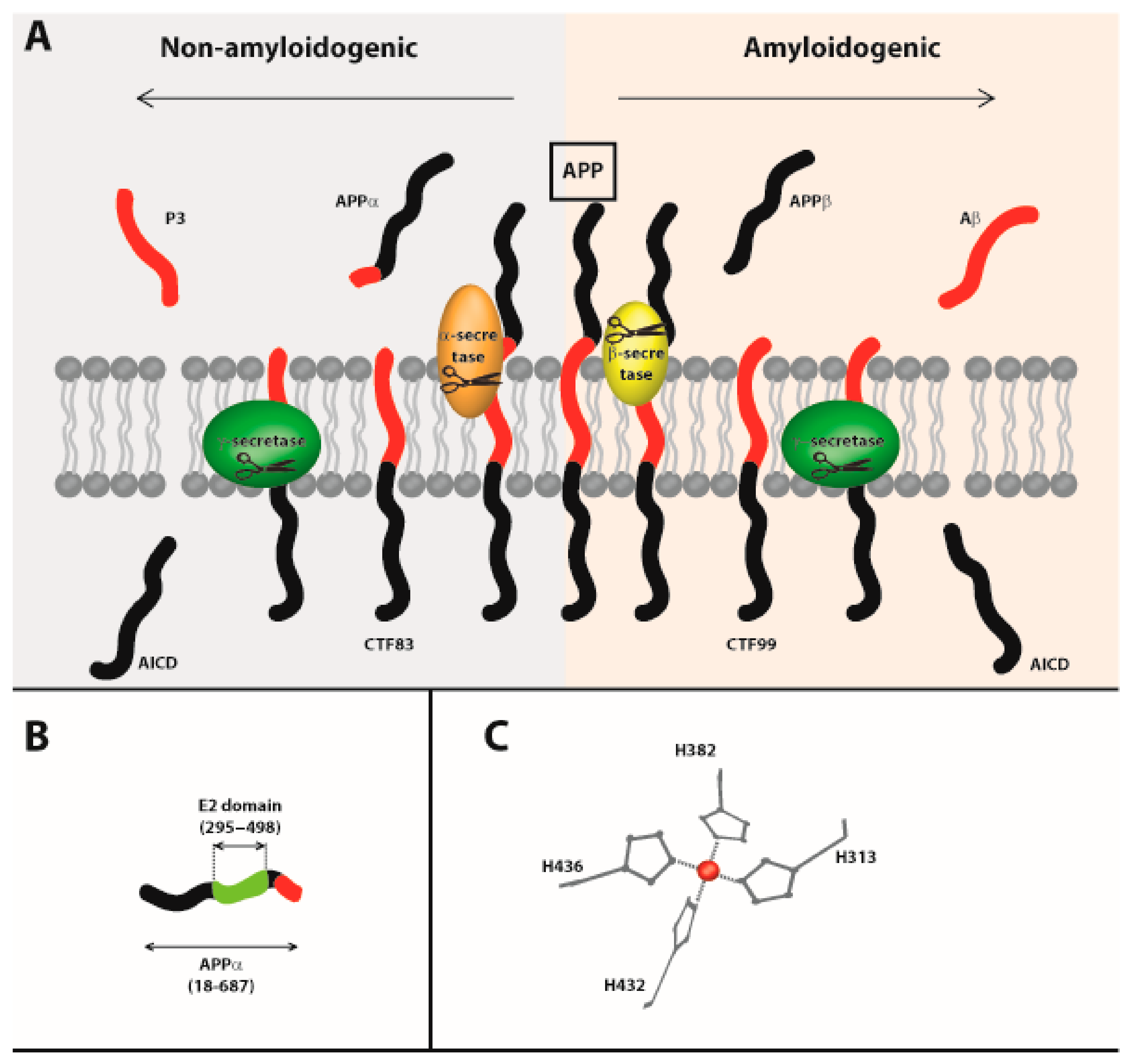
2.2. Role of Metals
2.3. Carbonylation Leads to Aggregation
2.4. Protein Oxidation Influences Aggregation by Modulating Chaperone Protein Activity
2.5. Protein Oxidation Influences Aggregation by Perturbing the Translational Process
2.6. Oxidative Stress Contributes to Aggregation by Modulating the Proteasome and Autophagy Capacity
3. Aggregation Can, in Turn, Produce Oxidative Stress, or Protect Against Oxidative Insults
3.1. Pro-Oxidative Effects
3.2. Anti-Oxidative Effects
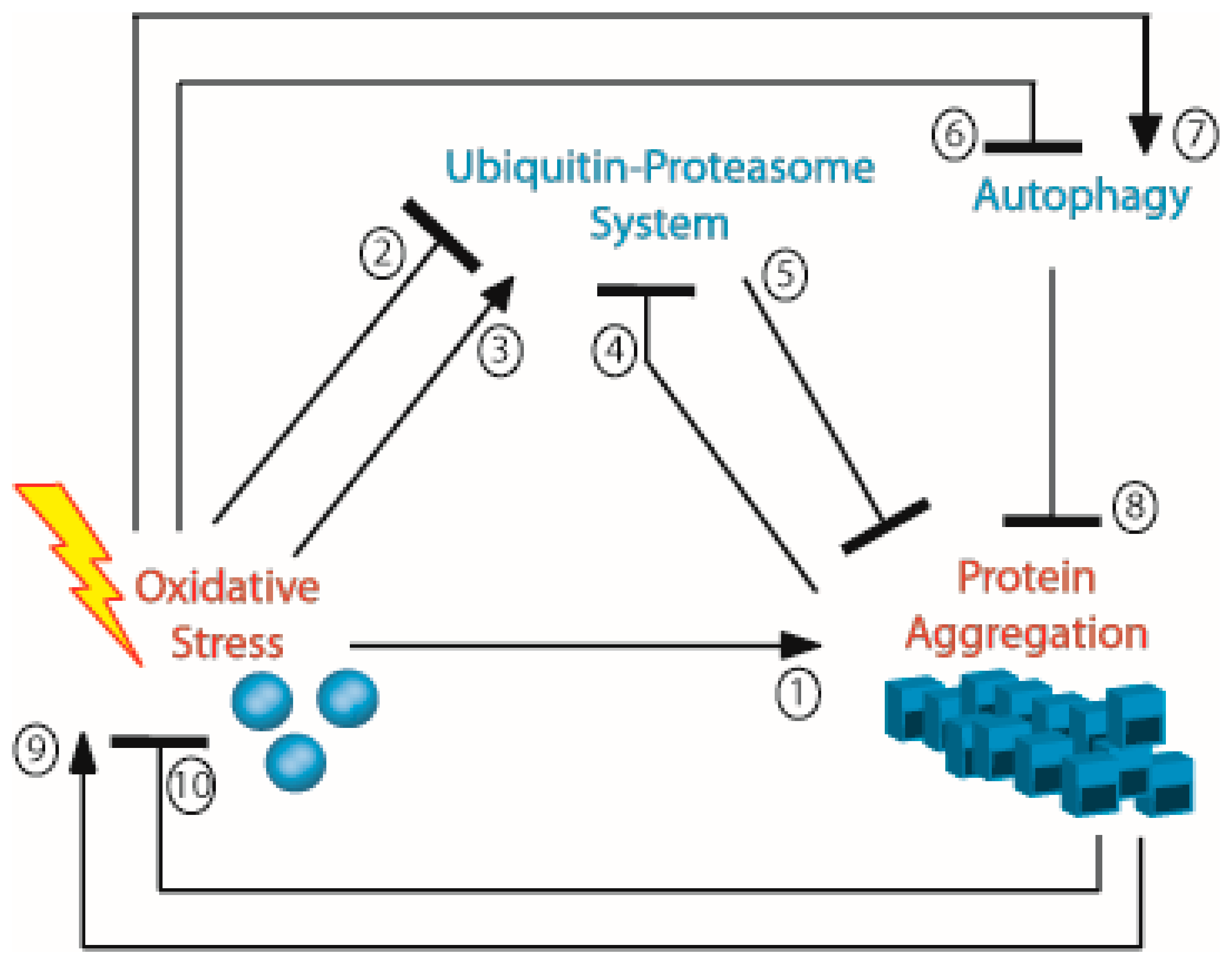
4. Aggregation and Oxidation can be Parts of a Vicious Circle
Author Contributions
Funding
Conflicts of Interest
References
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Labbadia, J.; Morimoto, R.I. Rethinking HSF1 in Stress, Development, and Organismal Health. Trends Cell Biol. 2017, 27, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Scherzinger, E.; Sittler, A.; Schweiger, K.; Heiser, V.; Lurz, R.; Hasenbank, R.; Bates, G.P.; Lehrach, H.; Wanker, E.E. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington’s disease pathology. Proc. Natl. Acad. Sci. USA 1999, 96, 4604–4609. [Google Scholar] [CrossRef] [PubMed]
- Beerten, J.; Schymkowitz, J.; Rousseau, F. Aggregation prone regions and gatekeeping residues in protein sequences. Curr. Top. Med. Chem. 2012, 12, 2470–2478. [Google Scholar] [CrossRef] [PubMed]
- Trotter, E.W.; Berenfeld, L.; Krause, S.A.; Petsko, G.A.; Gray, J.V. Protein misfolding and temperature up-shift cause G1 arrest via a common mechanism dependent on heat shock factor in Saccharomycescerevisiae. Proc. Natl. Acad. Sci. USA 2001, 98, 7313–7318. [Google Scholar] [CrossRef] [PubMed]
- Weids, A.J.; Ibstedt, S.; Tamas, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef] [Green Version]
- Ciric, D.; Richard, C.A.; Moudjou, M.; Chapuis, J.; Sibille, P.; Daude, N.; Westaway, D.; Adrover, M.; Beringue, V.; Martin, D.; et al. Interaction between Shadoo and PrP Affects the PrP-Folding Pathway. J. Virol. 2015, 89, 6287–6293. [Google Scholar] [CrossRef] [Green Version]
- Pepe, A.; Avolio, R.; Matassa, D.S.; Esposito, F.; Nitsch, L.; Zurzolo, C.; Paladino, S.; Sarnataro, D. Regulation of sub-compartmental targeting and folding properties of the Prion-like protein Shadoo. Sci. Rep. 2017, 7, 3731. [Google Scholar] [CrossRef]
- Sarnataro, D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3081. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Debulpaep, M.; Kuemmerer, N.; Lopez de la Paz, M.; Martins, I.C.; Reumers, J.; Morris, K.L.; Copland, A.; Serpell, L.; Serrano, L.; et al. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 2010, 7, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Sabate, R.; Rousseau, F.; Schymkowitz, J.; Ventura, S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015, 11, e1004013. [Google Scholar] [CrossRef] [PubMed]
- Soldi, G.; Bemporad, F.; Torrassa, S.; Relini, A.; Ramazzotti, M.; Taddei, N.; Chiti, F. Amyloid formation of a protein in the absence of initial unfolding and destabilization of the native state. Biophys. J. 2005, 89, 4234–4244. [Google Scholar] [CrossRef] [PubMed]
- Dumoulin, M.; Kumita, J.R.; Dobson, C.M. Normal and aberrant biological self-assembly: Insights from studies of human lysozyme and its amyloidogenic variants. Acc. Chem. Res. 2006, 39, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Vannocci, T.; Varela, L.; Azuaga, A.I.; Chiti, F. A model for the aggregation of the acylphosphatase from Sulfolobus solfataricus in its native-like state. Biochim. Biophys. Acta 2008, 1784, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pardo, J.; Grana-Montes, R.; Fernandez-Mendez, M.; Ruyra, A.; Roher, N.; Aviles, F.X.; Lorenzo, J.; Ventura, S. Amyloid formation by human carboxypeptidase D transthyretin-like domain under physiological conditions. J. Biol. Chem. 2014, 289, 33783–33796. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, P.I.; Reddy, G.; Straub, J.E.; Thirumalai, D. Propensity to form amyloid fibrils is encoded as excitations in the free energy landscape of monomeric proteins. J. Mol. Biol. 2014, 426, 2653–2666. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, H.; Shahab, U.; Rehman, S.; Rafi, Z.; Khan, M.Y.; Ansari, A.; Siddiqui, Z.; Ashraf, J.M.; Abdullah, S.M.; et al. Protein oxidation: An overview of metabolism of sulphur containing amino acid, cysteine. Front. Biosci. 2017, 9, 71–87. [Google Scholar] [CrossRef]
- Serebryany, E.; Woodard, J.C.; Adkar, B.V.; Shabab, M.; King, J.A.; Shakhnovich, E.I. An Internal Disulfide Locks a Misfolded Aggregation-prone Intermediate in Cataract-linked Mutants of Human gammaD-Crystallin. J. Biol. Chem. 2016, 291, 19172–19183. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Sykes, C.E.; Geddes, T.J.; Jaunarajs, K.L.; Bishop, C. Tryptophan hydroxylase 2 aggregates through disulfide cross-linking upon oxidation: Possible link to serotonin deficits and non-motor symptoms in Parkinson’s disease. J. Neurochem. 2011, 116, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Fraga, H.; Grana-Montes, R.; Illa, R.; Covaleda, G.; Ventura, S. Association between foldability and aggregation propensity in small disulfide-rich proteins. Antioxid. Redox Signal. 2014, 21, 368–383. [Google Scholar] [CrossRef] [PubMed]
- Jenness, R. Comparative aspects of milk proteins. J. Dairy Res. 1979, 46, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Lemus, E.; Silva, E.; Leinisch, F.; Dorta, E.; Lorentzen, L.G.; Davies, M.J.; Lopez-Alarcon, C. Alpha- and beta-casein aggregation induced by riboflavin-sensitized photo-oxidation occurs via di-tyrosine cross-links and is oxygen concentration dependent. Food Chem. 2018, 256, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Amano, W.; Kubo, T.; Fukuhara, A.; Ihara, H.; Azuma, Y.T.; Tajima, H.; Inui, T.; Sawa, A.; Takeuchi, T. Glyceraldehyde-3-phosphate dehydrogenase aggregate formation participates in oxidative stress-induced cell death. J. Biol. Chem. 2009, 284, 34331–34341. [Google Scholar] [CrossRef] [PubMed]
- Cumming, R.C.; Schubert, D. Amyloid-beta induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. FASEB J. 2005, 19, 2060–2062. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Tajima, H.; Kuwae, T.; Takeshima, T.; Nakano, T.; Tanaka, M.; Sunaga, K.; Fukuhara, Y.; Nakashima, K.; Ohama, E.; et al. Pro-apoptotic protein glyceraldehyde-3-phosphate dehydrogenase promotes the formation of Lewy body-like inclusions. Eur. J. Neurosci. 2005, 21, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Knaupp, A.S.; Kass, I.; Kleifeld, O.; Marijanovic, E.M.; Hughes, V.A.; Lupton, C.J.; Buckle, A.M.; Bottomley, S.P.; Medcalf, R.L. Oxidation of an exposed methionine instigates the aggregation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2014, 289, 26922–26936. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.T.; Hata, F.; Inui, T.; Takeuchi, T. The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J. Biol. Chem. 2007, 282, 26562–26574. [Google Scholar] [CrossRef]
- Sayre, L.M.; Perry, G.; Atwood, C.S.; Smith, M.A. The role of metals in neurodegenerative diseases. Cell. Mol. Biol. 2000, 46, 731–741. [Google Scholar]
- Abeyawardhane, D.L.; Fernandez, R.D.; Murgas, C.J.; Heitger, D.R.; Forney, A.K.; Crozier, M.K.; Lucas, H.R. Iron Redox Chemistry Promotes Antiparallel Oligomerization of alpha-Synuclein. J. Am. Chem. Soc. 2018, 140, 5028–5032. [Google Scholar] [CrossRef] [PubMed]
- Del Giudice, R.; Pesce, A.; Cozzolino, F.; Monti, M.; Relini, A.; Piccoli, R.; Arciello, A.; Monti, D.M. Effects of iron on the aggregation propensity of the N-terminal fibrillogenic polypeptide of human apolipoprotein AI. BioMetals 2018, 31, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Jones, M.; Atrian-Blasco, E.; Kieffer, I.; Faller, P.; Collin, F.; Hureau, C. Identification of key structural features of the elusive Cu-Abeta complex that generates ROS in Alzheimer’s disease. Chem. Sci. 2017, 8, 5107–5118. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Young, T.R.; Pukala, T.L.; Cappai, R.; Wedd, A.G.; Xiao, Z. The Human Amyloid Precursor Protein Binds Copper Ions Dominated by a Picomolar-Affinity Site in the Helix-Rich E2 Domain. Biochemistry 2018, 57, 4165–4176. [Google Scholar] [CrossRef]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed]
- Dienemann, C.; Coburger, I.; Mehmedbasic, A.; Andersen, O.M.; Than, M.E. Mutants of metal binding site M1 in APP E2 show metal specific differences in binding of heparin but not of sorLA. Biochemistry 2015, 54, 2490–2499. [Google Scholar] [CrossRef]
- Weng, S.L.; Huang, K.Y.; Kaunang, F.J.; Huang, C.H.; Kao, H.J.; Chang, T.H.; Wang, H.Y.; Lu, J.J.; Lee, T.Y. Investigation and identification of protein carbonylation sites based on position-specific amino acid composition and physicochemical features. MC Bioinform. 2017, 18, 1105. [Google Scholar] [CrossRef]
- Arena, S.; Salzano, A.M.; Renzone, G.; D’Ambrosio, C.; Scaloni, A. Non-enzymatic glycation and glycoxidation protein products in foods and diseases: An interconnected, complex scenario fully open to innovative proteomic studies. Mass Spectrom. Rev. 2014, 33, 49–77. [Google Scholar] [CrossRef]
- Haigis, M.C.; Yankner, B.A. The aging stress response. Mol. Cell 2010, 40, 333–344. [Google Scholar] [CrossRef]
- Castro, J.P.; Ott, C.; Jung, T.; Grune, T.; Almeida, H. Carbonylation of the cytoskeletal protein actin leads to aggregate formation. Free Radic. Biol. Med. 2012, 53, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Tanase, M.; Urbanska, A.M.; Zolla, V.; Clement, C.C.; Huang, L.; Morozova, K.; Follo, C.; Goldberg, M.; Roda, B.; Reschiglian, P.; et al. Role of Carbonyl Modifications on Aging-Associated Protein Aggregation. Sci. Rep. 2016, 6, 19311. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Bhat, W.F.; Afsar, M.; Khan, M.S.; Al-Bagmi, M.S.; Bano, B. Modification of chickpea cystatin by reactive dicarbonyl species: Glycation, oxidation and aggregation. Arch. Biochem. Biophys. 2018, 650, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Karri, S.; Singh, S.; Paripati, A.K.; Marada, A.; Krishnamoorthy, T.; Guruprasad, L.; Balasubramanian, D.; Sepuri, N.B.V. Adaptation of Mge1 to oxidative stress by local unfolding and altered Interaction with mitochondrial Hsp70 and Mxr2. Mitochondrion 2018, 46, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Rauch, J.N.; Jinwal, U.K.; Thompson, A.D.; Srinivasan, S.; Dickey, C.A.; Gestwicki, J.E. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem. Biol. 2012, 19, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Rand, J.D.; Grant, C.M. The thioredoxin system protects ribosomes against stress-induced aggregation. Mol. Biol. Cell 2006, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Weids, A.J.; Grant, C.M. The yeast peroxiredoxin Tsa1 protects against protein-aggregate-induced oxidative stress. J. Cell Sci. 2014, 127, 1327–1335. [Google Scholar] [CrossRef]
- Wickner, R.B. [URE3] as an altered URE2 protein: Evidence for a prion analog in Saccharomyces cerevisiae. Science 1994, 264, 566–569. [Google Scholar] [CrossRef]
- Doronina, V.A.; Staniforth, G.L.; Speldewinde, S.H.; Tuite, M.F.; Grant, C.M. Oxidative stress conditions increase the frequency of de novo formation of the yeast [PSI+] prion. Mol. Microbiol. 2015, 96, 163–174. [Google Scholar] [CrossRef]
- Jamar, N.H.; Kritsiligkou, P.; Grant, C.M. The non-stop decay mRNA surveillance pathway is required for oxidative stress tolerance. Nucleic Acids Res. 2017, 45, 6881–6893. [Google Scholar] [CrossRef] [Green Version]
- Njomen, E.; Osmulski, P.A.; Jones, C.L.; Gaczynska, M.; Tepe, J.J. Small Molecule Modulation of Proteasome Assembly. Biochemistry 2018, 57, 4214–4224. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Ito, H.; Taniguchi, Y.; Inoue, H.; Takeda, S.; Takahashi, R. Proteasome inhibition in medaka brain induces the features of Parkinson’s disease. J. Neurochem. 2010, 115, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M.; et al. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Zhao, J.; Zhang, J.; Liu, W.; Zhao, M.; Li, J.; Lv, J.; Li, Y. Effect of lysosomal and ubiquitin-proteasome system dysfunction on the abnormal aggregation of alpha-synuclein in PC12 cells. Exp. Ther. Med. 2015, 9, 2088–2094. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Singh, D.; Singh, B.K.; Singh, S.; Mittra, N.; Jha, R.R.; Patel, D.K.; Singh, C. Alpha-synuclein aggregation, Ubiquitin proteasome system impairment, and L-Dopa response in zinc-induced Parkinsonism: Resemblance to sporadic Parkinson’s disease. Mol. Cell. Biochem. 2018, 444, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.M.; Netto, L.E.S.; Simões, V.; Santos, L.F.A.; Gozzo, F.C.; Demasi, M.A.A.; Oliveira, C.L.P.; Bicev, R.N.; Klitzke, C.F.; Sogayar, M.C.; et al. Redox Control of 20S Proteasome Gating. Antioxid. Redox Signal. 2012, 16, 1183–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korovila, I.; Hugo, M.; Castro, J.P.; Weber, D.; Hohn, A.; Grune, T.; Jung, T. Proteostasis, oxidative stress and aging. Redox Biol. 2017, 13, 550–567. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Abraham, E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J. Biol. Chem. 2009, 284, 22213–22221. [Google Scholar] [CrossRef] [PubMed]
- Demasi, M.; Shringarpure, R.; Davies, K.J. Glutathiolation of the proteasome is enhanced by proteolytic inhibitors. Arch. Biochem. Biophys. 2001, 389, 254–263. [Google Scholar] [CrossRef]
- Silva, G.M.; Netto, L.E.; Discola, K.F.; Piassa-Filho, G.M.; Pimenta, D.C.; Barcena, J.A.; Demasi, M. Role of glutaredoxin 2 and cytosolic thioredoxins in cysteinyl-based redox modification of the 20S proteasome. FEBS J. 2008, 275, 2942–2955. [Google Scholar] [CrossRef]
- Bridgford, J.L.; Xie, S.C.; Cobbold, S.A.; Pasaje, C.F.A.; Herrmann, S.; Yang, T.; Gillett, D.L.; Dick, L.R.; Ralph, S.A.; Dogovski, C.; et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat. Commun. 2018, 9, 3801. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.-H.; Cho, K.; Kang, H.-J.; Jeon, E.-Y.; Kim, H.-S.; Kwon, H.-J.; Kim, H.-M.; Kim, D.-H.; Yoon, S.-Y. Autophagy in microglia degrades extracellular β-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy 2014, 10, 1761–1775. [Google Scholar] [CrossRef]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 2016, 22, 865. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant α-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292. [Google Scholar] [CrossRef]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [Green Version]
- Yung, C.; Sha, D.; Li, L.; Chin, L.-S. Parkin Protects Against Misfolded SOD1 Toxicity by Promoting Its Aggresome Formation and Autophagic Clearance. Mol. Neurobiol. 2016, 53, 6270–6287. [Google Scholar] [CrossRef]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.-S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, H.; Guan, Y.; Wang, Q.; Zhou, F.; Jie, L.; Ju, J.; Pu, L.; Du, H.; Wang, X. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am. J. Transl. Res. 2015, 7, 1574–1587. [Google Scholar] [PubMed]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox regulation of antioxidants, autophagy, and the response to stress: Implications for electrophile therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Cheung, E.C.; Vousden, K.H. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009, 28, 3015–3026. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040. [Google Scholar] [CrossRef]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular metabolic and autophagic pathways: Traffic control by redox signaling. Free Radic. Biol. Med. 2013, 63, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef]
- Guerrero-Gomez, D.; Mora-Lorca, J.A.; Saenz-Narciso, B.; Naranjo-Galindo, F.J.; Munoz-Lobato, F.; Parrado-Fernandez, C.; Goikolea, J.; Cedazo-Minguez, Á.; Link, C.D.; Neri, C.; et al. Loss of glutathione redox homeostasis impairs proteostasis by inhibiting autophagy-dependent protein degradation. Cell Death Differ. 2019. Available online: https://www.nature.com/articles/s41418-018-0270-9 (accessed on 15 February 2019). [CrossRef]
- Civiletto, G.; Dogan, S.A.; Cerutti, R.; Fagiolari, G.; Moggio, M.; Lamperti, C.; Beninca, C.; Viscomi, C.; Zeviani, M. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol. Med. 2018, 10, e8799. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.; Galvan, V.; Lin, A.-L.; Oddo, S. How longevity research can lead to therapies for Alzheimer’s disease: The rapamycin story. Exp. Gerontol. 2015, 68, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Wang, R.; Gao, Y.; Che, H.; Pan, Y.; Fu, P. Evaluating the Effectiveness of GTM-1, Rapamycin, and Carbamazepine on Autophagy and Alzheimer Disease. Med. Sci. Monit. 2017, 23, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thellung, S.; Scoti, B.; Corsaro, A.; Villa, V.; Nizzari, M.; Gagliani, M.C.; Porcile, C.; Russo, C.; Pagano, A.; Tacchetti, C.; et al. Pharmacological activation of autophagy favors the clearing of intracellular aggregates of misfolded prion protein peptide to prevent neuronal death. Cell Death Dis. 2018, 9, 166. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kim, D.H.; Park, Y.G.; Chun, B.G.; Choi, S.H. Protective effects of rilmenidine and AGN 192403 on oxidative cytotoxicity and mitochondrial inhibitor-induced cytotoxicity in astrocytes. Free Radic. Biol. Med. 2002, 33, 1321–1333. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2017, 14, 534–551. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.K.; Seo, J.H.; Ryu, H.S.; Lim, K.S.; Jeong, M.H.; Kang, D.H.; Kang, S.W. A rapamycin derivative, biolimus, preferentially activates autophagy in vascular smooth muscle cells. Sci. Rep. 2018, 8, 16551. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.; Hornburg, D.; Antonin, M.; Krahmer, N.; Collado, J.; Schaffer, M.; Zubaite, G.; Luchtenborg, C.; Sachsenheimer, T.; Brugger, B.; et al. Molecular and structural architecture of polyQ aggregates in yeast. Proc. Natl. Acad. Sci. USA 2018, 115, E3446–E3453. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Rub, C.; Bruderek, M.; Voos, W. Amyloid beta-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 2016, 27, 3257–3272. [Google Scholar] [CrossRef]
- Wang, X.; Becker, K.; Levine, N.; Zhang, M.; Lieberman, A.P.; Moore, D.J.; Ma, J. Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 2019, 7, 41. [Google Scholar] [CrossRef]
- Chaudhary, R.K.; Patel, K.A.; Patel, M.K.; Joshi, R.H.; Roy, I. Inhibition of Aggregation of Mutant Huntingtin by Nucleic Acid Aptamers In vitro and in a Yeast Model of Huntington’s Disease. Mol. Ther. 2015, 23, 1912–1926. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.A.; Kolluri, T.; Jain, S.; Roy, I. Designing Aptamers which Respond to Intracellular Oxidative Stress and Inhibit Aggregation of Mutant Huntingtin. Free Radic. Biol. Med. 2018, 120, 311–316. [Google Scholar] [CrossRef]
- Mattson, M.P.; Goodman, Y. Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res. 1995, 676, 219–224. [Google Scholar] [CrossRef]
- Lim, Y.A.; Rhein, V.; Baysang, G.; Meier, F.; Poljak, A.; Raftery, M.J.; Guilhaus, M.; Ittner, L.M.; Eckert, A.; Gotz, J. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics 2010, 10, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Bhowmick, D.C.; Pany, S.; Joe, M.; Zaghlula, N.; Jeremic, A.M. Apoptosis signal regulating kinase-1 and NADPH oxidase mediate human amylin evoked redox stress and apoptosis in pancreatic beta-cells. Biochim. Biophys. Acta 2018, 1860, 1721–1733. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R. Soc. B: Biol. Sci. 2005, 360, 2309–2314. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Abramov, A.Y.; Duchen, M.R. Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 2011, 134, 1658–1672. [Google Scholar] [CrossRef]
- Palazzi, L.; Bruzzone, E.; Bisello, G.; Leri, M.; Stefani, M.; Bucciantini, M.; Polverino de Laureto, P. Oleuropein aglycone stabilizes the monomeric alpha-synuclein and favours the growth of non-toxic aggregates. Sci. Rep. 2018, 8, 8337. [Google Scholar] [CrossRef]
- Baruch-Suchodolsky, R.; Fischer, B. Soluble amyloid beta1-28-copper(I)/copper(II)/Iron(II) complexes are potent antioxidants in cell-free systems. Biochemistry 2008, 47, 7796–7806. [Google Scholar] [CrossRef]
- Baruch-Suchodolsky, R.; Fischer, B. Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry 2009, 48, 4354–4370. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Rodriguez, W.; Yatsimirsky, A.K.; Glabe, C.G. Binding of Zn(II), Cu(II), and Fe(II) ions to Alzheimer’s A beta peptide studied by fluorescence. Bioorganic Med. Chem. Lett. 1999, 9, 2243–2248. [Google Scholar] [CrossRef]
- Khan, A.; Dobson, J.P.; Exley, C. Redox cycling of iron by Abeta42. Free Radic. Biol. Med. 2006, 40, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.Y.; Guo-Ross, S.X.; Bondy, S.C. The stabilization of ferrous iron by a toxic beta-amyloid fragment and by an aluminum salt. Brain Res. 1999, 839, 221–226. [Google Scholar] [CrossRef]
- Carija, A.; Navarro, S.; de Groot, N.S.; Ventura, S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 2017, 12, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Sanchez de Groot, N.; Gomes, R.A.; Villar-Pique, A.; Babu, M.M.; Coelho, A.V.; Ventura, S. Proteome response at the edge of protein aggregation. Open Biol. 2015, 5, 140221. [Google Scholar] [CrossRef] [PubMed]
- Sideri, T.C.; Stojanovski, K.; Tuite, M.F.; Grant, C.M. Ribosome-associated peroxiredoxins suppress oxidative stress-induced de novo formation of the [PSI+] prion in yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 6394–6399. [Google Scholar] [CrossRef] [PubMed]
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. Alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marín-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. αSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef] [PubMed]
- Tabner, B.J.; El-Agnaf, O.M.; German, M.J.; Fullwood, N.J.; Allsop, D. Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem. Soc. Trans. 2005, 33, 1082–1086. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Beck, M.V.; Griffith, J.D.; Deshmukh, M.; Dokholyan, N.V. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. 2018, 115, 4661–4665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrawell, N.E.; Lambert-Smith, I.; Mitchell, K.; McKenna, J.; McAlary, L.; Ciryam, P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. SOD1(A4V) aggregation alters ubiquitin homeostasis in a cell model of ALS. J. Cell Sci. 2018, 131, jcs209122. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedeoglu, A.; Cormier, K.; Payton, S.; Tseitlin, K.A.; Kremsky, J.N.; Lai, L.; Li, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; et al. Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp. Gerontol. 2004, 39, 1641–1649. [Google Scholar] [CrossRef]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42 Peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Mehta, K.; Govindaraju, T. Hybrid Multifunctional Modulators Inhibit Multifaceted Abeta Toxicity and Prevent Mitochondrial Damage. ACS Chem. Neurosci. 2018, 9, 1432–1440. [Google Scholar] [CrossRef]
- Hilt, S.; Altman, R.; Kalai, T.; Maezawa, I.; Gong, Q.; Wachsmann-Hogiu, S.; Jin, L.W.; Voss, J.C. A Bifunctional Anti-Amyloid Blocks Oxidative Stress and the Accumulation of Intraneuronal Amyloid-Beta. Molecules 2018, 23, 2010. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Protein(s) Involved | Mechanism | Residue Involved | Related Disease/Pathway | Reference |
|---|---|---|---|---|
| γD-crystallin | Cys32–Cys41 internal disulfide bond formation leads to the stabilization of a partially unfolded domain, which is prone to further intermolecular interactions. Internal disulfide bond formation provokes aggregation under physiological conditions. | Cysteine | Cataracts in older people | [20] |
| Tryptophan hydroxylase 2 (TPH2) | Intra- and inter-molecular disulfide bonds responsible for high molecular weight aggregates. | Cysteine | Parkinson’s disease | [21] |
| Milk caseins | Oxidation of Tryptophan, Tyrosine, Methionine, and Histidine residues decreases di-Tyr and di-Trp formation but allows increased protein aggregation. | Tryptophan, Tyrosine, Methionine, Histidine | None | [24] |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Oxidation of Methionine allows subsequent misfolding and further aggregation of GAPDH. | Methionine | None | [28] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. https://doi.org/10.3390/ijms20163896
Lévy E, El Banna N, Baïlle D, Heneman-Masurel A, Truchet S, Rezaei H, Huang M-E, Béringue V, Martin D, Vernis L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. International Journal of Molecular Sciences. 2019; 20(16):3896. https://doi.org/10.3390/ijms20163896
Chicago/Turabian StyleLévy, Elise, Nadine El Banna, Dorothée Baïlle, Amélie Heneman-Masurel, Sandrine Truchet, Human Rezaei, Meng-Er Huang, Vincent Béringue, Davy Martin, and Laurence Vernis. 2019. "Causative Links between Protein Aggregation and Oxidative Stress: A Review" International Journal of Molecular Sciences 20, no. 16: 3896. https://doi.org/10.3390/ijms20163896