Glycoprotein Non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Inherited Lysosomal Storage Disorders
2. Gaucher Cell Biomarkers: Lipids
3. Gaucher Cell Biomarkers: Proteins
4. Emerging Marker: Glycoprotein Non-metastatic Protein B (GPNMB)
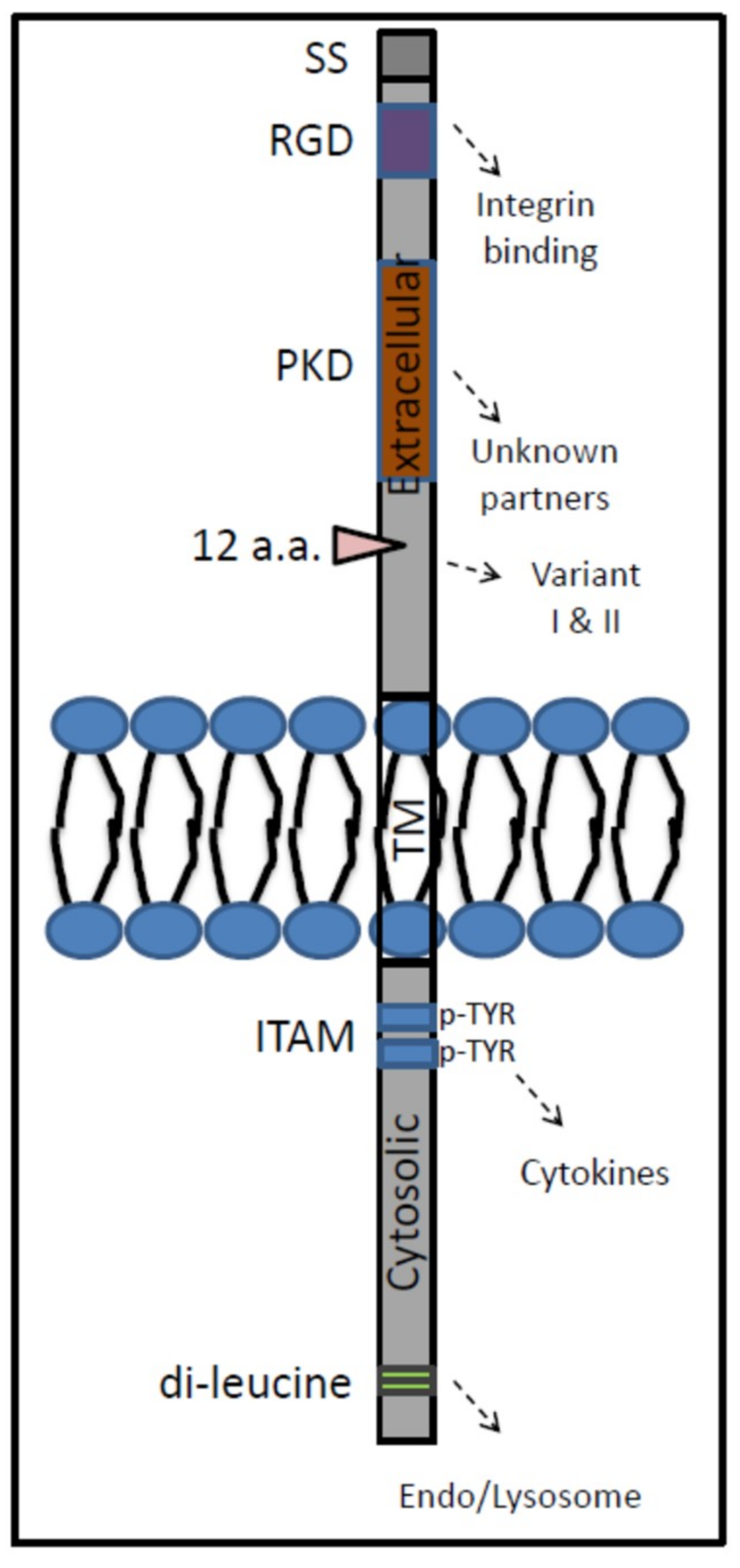
5. GPNMB: Properties
6. Function of GPNMB in Myeloid Cells
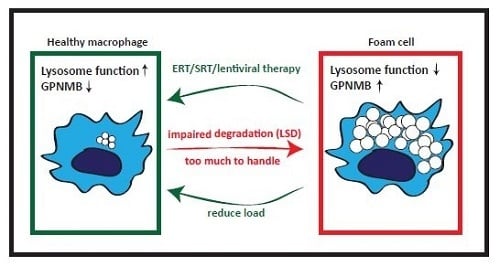
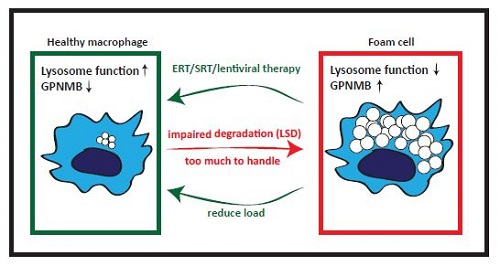
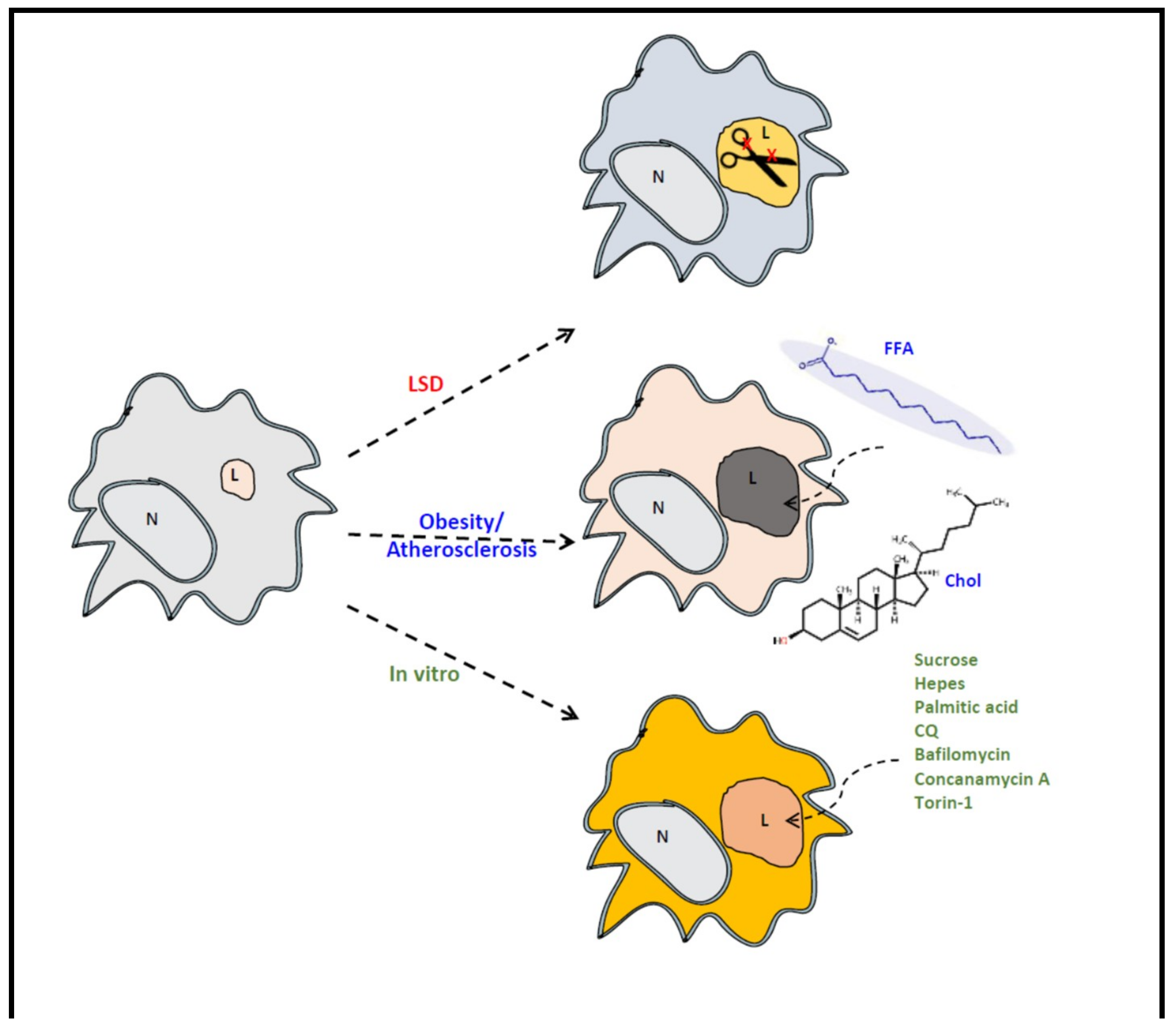
7. GPNMB and Foam Cells in Acquired ‘Metabolic’ Disorders
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neufeld, E.F. Lysosomal Storage Diseases. Annu. Rev. Biochem. 1991, 60, 257–280. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Grabowski, G. Glucosylceramide Lipidosis-Gaucher Disease. In The Metabolic and Molecular Bases of Inherited Diseases, Scriver, C.R., Beaudet , A.L., Sly, W.S., Valle, D., Eds.; 8th ed.; McGraw-Hill: New York, NY, USA, 2001; Volume 8, pp. 3635–3668. [Google Scholar]
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Invest. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, T.; van den Berg, R.J.B.H.N.; Boot, R.G.; van der Marel, G.A.; Overkleeft, H.S.; Aerts, J.M.F.G. Glycosphingolipids—Nature, function, and pharmacological modulation. Angew. Chem. Int. Ed. Engl. 2009, 48, 8848–8869. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; van Breemen, M.J.; Wegdam, W.; Sprenger, R.R.; de Jong, S.; Speijer, D.; Hollak, C.E.; Van Dussen, L.; Hoefsloot, H.C.; Smilde, A.K.; et al. Gaucher disease: A model disorder for biomarker discovery. Expert Rev. Proteomics 2009, 6, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Kallemeijn, W.W.; Mirzaian, M.; Herrera Moro, D.; Marques, A.; Wisse, P.; Boot, R.G.; Willems, L.I.; Overkleeft, H.S.; Aerts, J.M. Gaucher disease and Fabry disease: New markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Hollak, C.; Boot, R.; Groener, A. Biochemistry of glycosphingolipid storage disorders: Implications for therapeutic intervention. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2003, 358, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; Hollak, C.E.M.; Verhoek, M.; Sloof, P.; Poorthuis, B.J.H.M.; Kleijer, W.J.; Wevers, R.A.; van Oers, M.H.J.; Mannens, M.M.A.M.; Aerts, J.M.F.G.; et al. Glucocerebrosidase genotype of Gaucher patients in The Netherlands: Limitations in prognostic value. Hum. Mutat. 1997, 10, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, R.H.; Grant, I.R.; Halsall, D.; Cox, T.M. Twin pairs showing discordance of phenotype in adult Gaucher’s disease. QJM 2004, 97, 199–204. [Google Scholar] [CrossRef]
- Biegstraaten, M.; Van Schaik, I.N.; Aerts, J.M.F.G.; Langeveld, M.; Mannens, M.M.A.M.; Bour, L.J.; Sidransky, E.; Tayebi, N.; Fitzgibbon, E.; Hollak, C.E.M. A monozygotic twin pair with highly discordant Gaucher phenotypes. Blood Cells Mol. Dis. 2011, 46, 39–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastores, G.M.; Weinreb, N.J.; Aerts, H.; Andria, G.; Cox, T.M.; Giralt, M.; Grabowski, G.A.; Mistry, P.K.; Tylki-Szymańska, A. Therapeutic goals in the treatment of Gaucher disease. Semin. Hematol. 2004, 41, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Aggio, M.C.; Andersson, H.C.; Andria, G.; Charrow, J.; Clarke, J.T.R.; Erikson, A.; Giraldo, P.; Goldblatt, J.; Hollak, C.; et al. Gaucher disease type 1: Revised recommendations on evaluations and monitoring for adult patients. Semin. Hematol. 2004, 41, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.M.; Aerts, J.M.F.G.; Belmatoug, N.; Cappellini, M.D.; vom Dahl, S.; Goldblatt, J.; Grabowski, G.A.; Hollak, C.E.M.; Hwu, P.; Maas, M.; et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J. Inherit. Metab. Dis. 2008, 31, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.F.G.; Kallemeijn, W.W.; Wegdam, W.; Joao Ferraz, M.; van Breemen, M.J.; Dekker, N.; Kramer, G.; Poorthuis, B.J.; Groener, J.E.M.; Cox-Brinkman, J.; et al. Biomarkers in the diagnosis of lysosomal storage disorders: Proteins, lipids, and inhibodies. J. Inherit. Metab. Dis. 2011, 34, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Groener, J.E.M.; Poorthuis, B.J.H.M.; Kuiper, S.; Hollak, C.E.M.; Aerts, J.M.F.G. Plasma glucosylceramide and ceramide in type 1 Gaucher disease patients: Correlations with disease severity and response to therapeutic intervention. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2008, 1781, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ghauharali-van der Vlugt, K.; Langeveld, M.; Poppema, A.; Kuiper, S.; Hollak, C.E.M.; Aerts, J.M.; Groener, J.E.M. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin. Chim. Acta 2008, 389, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.M.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.M.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef]
- Murugesan, V.; Chuang, W.-L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef]
- Gaspar, P.; Kallemeijn, W.W.; Strijland, A.; Scheij, S.; Van Eijk, M.; Aten, J.; Overkleeft, H.S.; Balreira, A.; Zunke, F.; Schwake, M.; et al. Action myoclonus-renal failure syndrome: Diagnostic applications of activity-based probes and lipid analysis. J. Lipid Res. 2014, 55, 138–145. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balreira, A.; Gaspar, P.; Caiola, D.; Chaves, J.; Beirao, I.; Lima, J.L.; Azevedo, J.E.; Miranda, M.C.S. A nonsense mutation in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum. Mol. Genet. 2008, 17, 2238–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Gold, H.; Donker-Koopman, W.E.; Verhoek, M.; Overkleeft, H.S.; Boot, R.G.; Kramer, G.; Dekker, N.; et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol. Dis. 2015, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Mellgard, B.; Dinh, Q.; Lan, L.; Qiu, Y.; Cozma, C.; Eichler, S.; Böttcher, T.; Zimran, A. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naïve and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 2017, 122, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, M.J.; Marques, A.R.A.A.; Gaspar, P.; Mirzaian, M.; van Roomen, C.; Ottenhoff, R.; Alfonso, P.; Irún, P.; Giraldo, P.; Wisse, P.; et al. Lyso-glycosphingolipid abnormalities in different murine models of lysosomal storage disorders. Mol. Genet. Metab. 2016, 117, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K. Twenty Five Years of the “Psychosine Hypothesis”: A Personal Perspective of its History and Present Status. Neurochem. Res. 1998, 23, 251–259. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [Green Version]
- Kuchar, L.; Sikora, J.; Gulinello, M.E.; Poupetova, H.; Lugowska, A.; Malinova, V.; Jahnova, H.; Asfaw, B.; Ledvinova, J. Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases. Anal. Biochem. 2017, 525, 73–77. [Google Scholar] [CrossRef]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Marques, A.R.A.; Gaspar, P.; Oussoren, S.V.; Kytidou, K.; Codée, J.D.C.; van der Marel, G.; Overkleeft, H.S.; et al. Simultaneous quantitation of sphingoid bases by UPLC-ESI-MS/MS with identical 13C-encoded internal standards. Clin. Chim. Acta 2017, 466, 178–184. [Google Scholar] [CrossRef]
- Pettazzoni, M.; Froissart, R.; Pagan, C.; Vanier, M.T.; Ruet, S.; Latour, P.; Guffon, N.; Fouilhoux, A.; Germain, D.P.; Levade, T.; et al. LC-MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: A novel tool for the screening of sphingolipidoses and Niemann-Pick type C disease. PLoS ONE 2017, 12, e0181700. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.P.A.B.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.P.A.B. Diagnosis of sphingolipidoses: A new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. 2017, 55, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, E.; Wang, S.; Archer, J.; Dekker, N.; Aerts, J.; Karlsson, S.; Cox, T. B cell lymphoma and myeloma in murine Gaucher’s disease. J. Pathol. 2013, 231, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Cozma, C.; Yang, F.; Kramp, G.; Meyer, A.; Neßlauer, A.-M.; Eichler, S.; Böttcher, T.; Witt, M.; Bräuer, A.U.; et al. Glucosylsphingosine Causes Hematological and Visceral Changes in Mice-Evidence for a Pathophysiological Role in Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 2192. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Choi, L.; Vernon, J.; Kopach, O.; Minett, M.S.; Mills, K.; Clayton, P.T.; Meert, T.; Wood, J.N. The Fabry disease-associated lipid Lyso-Gb3 enhances voltage-gated calcium currents in sensory neurons and causes pain. Neurosci. Lett. 2015, 594, 163–168. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.; Ferraz, M.J.; Mirzaian, M.; Gaspar, P.; Oussoren, S.V.; Wisse, P.; Kuo, C.-L.; Lelieveld, L.T.; Kytidou, K.; Hazeu, M.D.; et al. Lysosomal Storage Diseases. For Better or Worse: Adapting to Defective Lysosomal Glycosphingolipid Breakdown. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2017; pp. 1–13. [Google Scholar]
- Marques, A.R.A.; Mirzaian, M.; Akiyama, H.; Wisse, P.; Ferraz, M.J.; Gaspar, P.; Ghauharali-van der Vlugt, K.; Meijer, R.; Giraldo, P.; Alfonso, P.; et al. Glucosylated cholesterol in mammalian cells and tissues: Formation and degradation by multiple cellular β-glucosidases. J. Lipid Res. 2016, 57, 451–463. [Google Scholar] [CrossRef]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of β-glucosidase 1. Biochem. Biophys. Res. Commun. 2013, 441, 838–843. [Google Scholar] [CrossRef] [PubMed]
- NP-C Guidelines Working Group, J.E.; Wraith, J.E.; Baumgartner, M.R.; Bembi, B.; Covanis, A.; Levade, T.; Mengel, E.; Pineda, M.; Sedel, F.; Topçu, M.; et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol. Genet. Metab. 2009, 98, 152–165. [Google Scholar]
- Porter, F.D.; Scherrer, D.E.; Lanier, M.H.; Langmade, S.J.; Molugu, V.; Gale, S.E.; Olzeski, D.; Sidhu, R.; Dietzen, D.J.; Fu, R.; et al. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2010, 2, 56ra81. [Google Scholar] [CrossRef] [PubMed]
- Tint, G.S.; Pentchev, P.; Xu, G.; Batta, A.K.; Shefer, S.; Salen, G.; Honda, A. Cholesterol and oxygenated cholesterol concentrations are markedly elevated in peripheral tissue but not in brain from mice with the Niemann–Pick type C phenotype. J. Inherit. Metab. Dis. 1998, 21, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Sidhu, R.; Porter, F.D.; Yanjanin, N.M.; Speak, A.O.; te Vruchte, D.T.; Platt, F.M.; Fujiwara, H.; Scherrer, D.E.; Zhang, J.; et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res. 2011, 52, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, T.G.; de Oliveira Schmitt Ribas, G.; Saraiva-Pereira, M.L.; Bonatto, M.P.; Kessler, R.G.; Souza, F.T.S.; Trapp, F.; Michelin-Tirelli, K.; Burin, M.G.; Giugliani, R.; et al. Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann-Pick type C patients. Int. J. Dev. Neurosci. 2018, 66, 18–23. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.; Furlan, F.; Kolamunnage, T.; Cananzi, M.; Giordano, L.; Zaninotto, M.; Plebani, M.; Burlina, A. High level of oxysterols in neonatal cholestasis: A pitfall in analysis of biochemical markers for Niemann-Pick type C disease. Clin. Chem. Lab. Med. 2016, 54, 1221–1229. [Google Scholar] [CrossRef]
- Prunet, C.; Petit, J.M.; Ecarnot-Laubriet, A.; Athias, A.; Miguet-Alfonsi, C.; Rohmer, J.F.; Steinmetz, E.; Néel, D.; Gambert, P.; Lizard, G. High circulating levels of 7β- and 7α-hydroxycholesterol and presence of apoptotic and oxidative markers in arterial lesions of normocholesterolemic atherosclerotic patients undergoing endarterectomy. Pathol. Biol. 2006, 54, 22–32. [Google Scholar] [CrossRef]
- Ferderbar, S.; Pereira, E.C.; Apolinário, E.; Bertolami, M.C.; Faludi, A.; Monte, O.; Calliari, L.E.; Sales, J.E.; Gagliardi, A.R.; Xavier, H.T.; et al. Cholesterol oxides as biomarkers of oxidative stress in type 1 and type 2 diabetes mellitus . Diabetes Metab. Res. Rev. 2007, 23, 35–42. [Google Scholar] [CrossRef]
- Alkazemi, D.; Egeland, G.; Vaya, J.; Meltzer, S.; Kubow, S. Oxysterol as a Marker of Atherogenic Dyslipidemia in Adolescence. J. Clin. Endocrinol. Metab. 2008, 93, 4282–4289. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.R.A.; Aten, J.; Ottenhoff, R.; van Roomen, C.P.A.A.; Herrera Moro, D.; Claessen, N.; Vinueza Veloz, M.F.; Zhou, K.; Lin, Z.; Mirzaian, M.; et al. Reducing GBA2 Activity Ameliorates Neuropathology in Niemann-Pick Type C Mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.; Lachmann, R.; Hollak, C.; Aerts, J.; van Weely, S.; Hrebícek, M.; Platt, F.; Butters, T.; Dwek, R.; Moyses, C.; et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 2000, 355, 1481–1485. [Google Scholar] [CrossRef]
- Platt, F.M.; Jeyakumar, M.; Andersson, U.; Priestman, D.A.; Dwek, R.A.; Butters, T.D.; Cox, T.M.; Lachmann, R.H.; Hollak, C.; Aerts, J.M.F.G.; et al. Inhibition of substrate synthesis as a strategy for glycolipid lysosomal storage disease therapy. J. Inherit. Metab. Dis. 2001, 24, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.F.G.; Hollak, C.E.M.; Boot, R.G.; Groener, J.E.M.; Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 2006, 29, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.C.; Vecchio, D.; Prady, H.; Abel, L.; Wraith, J.E. Miglustat for treatment of Niemann-Pick C disease: A randomised controlled study. Lancet Neurol. 2007, 6, 765–772. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Hollak, C.E.M.; Breemen, M.; Maas, M.; Groener, J.E.M.; Boot, R. Identification and use of biomarkers in Gaucher disease and other lysosomal storage diseases. Acta Paediatr. 2007, 94, 43–46. [Google Scholar] [CrossRef]
- Brady, R.O. Enzyme replacement therapy: Conception, chaos and culmination. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2003, 358, 915–919. [Google Scholar] [CrossRef]
- Barton, N.W.; Furbish, F.S.; Murray, G.J.; Garfield, M.; Brady, R.O. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc. Natl. Acad. Sci. 1990, 87. [Google Scholar] [CrossRef]
- McCabe, E.R.B.; Fine, B.A.; Golbus, M.S.; Greenhouse, J.B.; McGrath, G.L.; New, M.; O’Brien, W.E.; Rowley, P.T.; Sly, W.S.; Spence, M.A.; et al. Gaucher Disease. JAMA 1996, 275, 548. [Google Scholar] [CrossRef]
- Hollak, C.; Aerts, J.; van Oers, H. Treatment of Gaucher’s Disease. N. Engl. J. Med. 1993, 328, 1564–1568. [Google Scholar]
- Aerts, J.M.F.G.; Hollak, C.E.M. 4 Plasma and metabolic abnormalities in Gaucher’s disease. Baillieres. Clin. Haematol. 1997, 10, 691–709. [Google Scholar] [CrossRef]
- Hayman, A.R.; Cox, T.M. Tartrate-resistant acid phosphatase: A potential target for therapeutic gold. Cell Biochem. Funct. 2004, 22, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Invest. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Boot, R.G.; Muijsers, A.O.; Donker-Koopman, W.E.; Aerts, J.M. Purification and characterization of human chitotriosidase, a novel member of the chitinase family of proteins. J. Biol. Chem. 1995, 270, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Boot, R.G.; Renkema, G.H.; Strijland, A.; van Zonneveld, A.J.; Aerts, J.M. Cloning of a cDNA encoding chitotriosidase, a human chitinase produced by macrophages. J. Biol. Chem. 1995, 270, 26252–26256. [Google Scholar] [CrossRef] [PubMed]
- Renkema, G.H.; Boot, R.G.; Strijland, A.; Donker-Koopman, W.E.; Berg, M.; Muijsers, A.O.; Aerts, J.M.F.G. Synthesis, Sorting, and Processing into Distinct Isoforms of Human Macrophage Chitotriosidase. Eur. J. Biochem. 1997, 244, 279–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boot, R.G.; Renkema, G.H.; Verhoek, M.; Strijland, A.; Bliek, J.; de Meulemeester, T.M.; Mannens, M.M.; Aerts, J.M. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998, 273, 25680–25685. [Google Scholar] [CrossRef]
- Fusetti, F.; von Moeller, H.; Houston, D.; Rozeboom, H.J.; Dijkstra, B.W.; Boot, R.G.; Aerts, J.M.F.G.; van Aalten, D.M.F. Structure of human chitotriosidase. Implications for specific inhibitor design and function of mammalian chitinase-like lectins. J. Biol. Chem. 2002, 277, 25537–25544. [Google Scholar] [CrossRef]
- Boven, L.A.; van Meurs, M.; Boot, R.G.; Mehta, A.; Boon, L.; Aerts, J.M.; Laman, J.D. Gaucher Cells Demonstrate a Distinct Macrophage Phenotype and Resemble Alternatively Activated Macrophages. Am. J. Clin. Pathol. 2004, 122, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, M.; van Roomen, C.P.A.A.; Renkema, G.H.; Bussink, A.P.; Andrews, L.; Blommaart, E.F.C.; Sugar, A.; Verhoeven, A.J.; Boot, R.G.; Aerts, J.M.F.G. Characterization of human phagocyte-derived chitotriosidase, a component of innate immunity. Int. Immunol. 2005, 17, 1505–1512. [Google Scholar] [CrossRef] [Green Version]
- Bussink, A.P.; van Eijk, M.; Renkema, G.H.; Aerts, J.M.; Boot, R.G. The Biology of the Gaucher Cell: The Cradle of Human Chitinases. Int. Rev. Cytol. 2006, 252, 71–128. [Google Scholar] [PubMed]
- Bussink, A.P.; Speijer, D.; Aerts, J.M.F.G.; Boot, R.G. Evolution of mammalian chitinase(-like) members of family 18 glycosyl hydrolases. Genetics 2007, 177, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, B.; Ghauharali-van der Vlugt, K.; Helmond, M.T.J.; Out, J.M.M.; Donker-Koopman, W.E.; Groener, J.E.M.; Boot, R.G.; Renkema, G.H.; van der Marel, G.A.; van Boom, J.H.; et al. Transglycosidase activity of chitotriosidase: Improved enzymatic assay for the human macrophage chitinase. J. Biol. Chem. 2003, 278, 40911–40916. [Google Scholar] [CrossRef] [PubMed]
- Schoonhoven, A.; Rudensky, B.; Elstein, D.; Zimran, A.; Hollak, C.E.M.; Groener, J.E.; Aerts, J.M.F.G. Monitoring of Gaucher patients with a novel chitotriosidase assay. Clin. Chim. Acta 2007, 381, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Van Dussen, L.; Hendriks, E.J.; Groener, J.E.M.; Boot, R.G.; Hollak, C.E.M.; Aerts, J.M.F.G. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014, 37, 991–1001. [Google Scholar] [CrossRef]
- Boot, R.G.; van Achterberg, T.A.E.; van Aken, B.E.; Renkema, G.H.; Jacobs, M.J.H.M.; Aerts, J.M.F.G.; de Vries, C.J.M. Strong Induction of Members of the Chitinase Family of Proteins in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; van Eijk, M.; Silva, E.; Hatta, M.; Faber, W.; Aerts, J.M.F.G.; Das, P.K. Increased chitotriosidase activity in serum of leprosy patients: Association with bacillary leprosy. Clin. Immunol. 2009, 131, 501–509. [Google Scholar] [CrossRef]
- Boot, R.G.; Hollak, C.E.M.; Verhoek, M.; Alberts, C.; Jonkers, R.E.; Aerts, J.M. Plasma chitotriosidase and CCL18 as surrogate markers for granulomatous macrophages in sarcoidosis. Clin. Chim. Acta 2010, 411, 31–36. [Google Scholar] [CrossRef]
- Guo, Y.; He, W.; Boer, A.M.; Wevers, R.A.; de Bruijn, A.M.; Groener, J.E.M.; Hollak, C.E.M.; Aerts, J.M.F.G.; Galjaard, H.; van Diggelen, O.P. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J. Inherit. Metab. Dis. 1995, 18, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vedder, A.C.; Cox-Brinkman, J.; Hollak, C.E.M.; Linthorst, G.E.; Groener, J.E.M.; Helmond, M.T.J.; Scheij, S.; Aerts, J.M.F.G. Plasma chitotriosidase in male Fabry patients: A marker for monitoring lipid-laden macrophages and their correction by enzyme replacement therapy. Mol. Genet. Metab. 2006, 89, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Schaefer, E.; Lührs, T.; Mani, L.; Kuhn, J.; Vanier, M.T.; Krummenauer, F.; Gal, A.; Beck, M.; Mengel, E. Critical assessment of chitotriosidase analysis in the rational laboratory diagnosis of children with Gaucher disease and Niemann–Pick disease type A/B and C. J. Inherit. Metab. Dis. 2006, 29, 647–652. [Google Scholar] [CrossRef]
- Moran, M.T.; Schofield, J.P.; Hayman, A.R.; Shi, G.P.; Young, E.; Cox, T.M. Pathologic gene expression in Gaucher disease: Up-regulation of cysteine proteinases including osteoclastic cathepsin K. Blood 2000, 96, 1969–1978. [Google Scholar]
- Boot, R.G.; Verhoek, M.; de Fost, M.; Hollak, C.E.M.; Maas, M.; Bleijlevens, B.; van Breemen, M.J.; van Meurs, M.; Boven, L.A.; Laman, J.D.; et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: A novel surrogate marker for assessing therapeutic intervention. Blood 2004, 103, 33–39. [Google Scholar] [CrossRef]
- Deegan, P.B.; Moran, M.T.; McFarlane, I.; Schofield, J.P.; Boot, R.G.; Aerts, J.M.F.G.; Cox, T.M. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol. Dis. 2005, 35, 259–267. [Google Scholar] [CrossRef]
- Chang, K.-L.; Hwu, W.-L.; Yeh, H.-Y.; Lee, N.-C.; Chien, Y.-H. CCL18 as an alternative marker in Gaucher and Niemann-Pick disease with chitotriosidase deficiency. Blood Cells Mol. Dis. 2010, 44, 38–40. [Google Scholar] [CrossRef]
- Pineda, M.; Perez-Poyato, M.S.; O’Callaghan, M.; Vilaseca, M.A.; Pocovi, M.; Domingo, R.; Portal, L.R.; Pérez, A.V.; Temudo, T.; Gaspar, A.; et al. Clinical experience with miglustat therapy in pediatric patients with Niemann-Pick disease type C: A case series. Mol. Genet. Metab. 2010, 99, 358–366. [Google Scholar] [CrossRef]
- De Castro-Orós, I.; Irún, P.; Cebolla, J.J.; Rodriguez-Sureda, V.; Mallén, M.; Pueyo, M.J.; Mozas, P.; Dominguez, C.; Pocoví, M. Assessment of plasma chitotriosidase activity, CCL18/PARC concentration and NP-C suspicion index in the diagnosis of Niemann-Pick disease type C: A prospective observational study. J. Transl. Med. 2017, 15, 43. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Yasothan, U.; Kirkpatrick, P. Velaglucerase alfa. Nat. Rev. Drug Discov. 2010, 9, 837–838. [Google Scholar] [CrossRef] [Green Version]
- Zimran, A.; Brill-Almon, E.; Chertkoff, R.; Petakov, M.; Blanco-Favela, F.; Muñoz, E.T.; Solorio-Meza, S.E.; Amato, D.; Duran, G.; Giona, F.; et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011, 118, 5767–5773. [Google Scholar] [CrossRef] [Green Version]
- Elstein, D.; Hollak, C.; Aerts, J.M.F.G.; van Weely, S.; Maas, M.; Cox, T.M.; Lachmann, R.H.; Hrebicek, M.; Platt, F.M.; Butters, T.D.; et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 2004, 27, 757–766. [Google Scholar] [CrossRef]
- Cox, T.M.; Drelichman, G.; Cravo, R.; Balwani, M.; Burrow, T.A.; Martins, A.M.; Lukina, E.; Rosenbloom, B.; Ross, L.; Angell, J.; et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: A phase 3, randomised, open-label, non-inferiority trial. Lancet 2015, 385, 2355–2362. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lukina, E.; Ben Turkia, H.; Amato, D.; Baris, H.; Dasouki, M.; Ghosn, M.; Mehta, A.; Packman, S.; Pastores, G.; et al. Effect of Oral Eliglustat on Splenomegaly in Patients With Gaucher Disease Type 1. JAMA 2015, 313, 695. [Google Scholar] [CrossRef] [Green Version]
- Smid, B.E.; Ferraz, M.J.; Verhoek, M.; Mirzaian, M.; Wisse, P.; Overkleeft, H.S.; Hollak, C.E.; Aerts, J.M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 2016, 11, 28. [Google Scholar] [CrossRef]
- Hashimoto, S.; Yamada, M.; Motoyoshi, K.; Akagawa, K.S.; Matsushima, K. Enhancement of macrophage colony-stimulating factor-induced growth and differentiation of human monocytes by interleukin-10. Blood 1997, 89, 315–321. [Google Scholar]
- Kramer, G.; Wegdam, W.; Donker-Koopman, W.; Ottenhoff, R.; Gaspar, P.; Verhoek, M.; Nelson, J.; Gabriel, T.; Kallemeijn, W.; Boot, R.G.; et al. Elevation of glycoprotein nonmetastatic melanoma protein B in type 1 Gaucher disease patients and mouse models. FEBS Open Bio. 2016, 6, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Dahl, M.; Doyle, A.; Olsson, K.; Månsson, J.-E.; Marques, A.R.A.; Mirzaian, M.; Aerts, J.M.; Ehinger, M.; Rothe, M.; Modlich, U.; et al. Lentiviral gene therapy using cellular promoters cures type 1 Gaucher disease in mice. Mol. Ther. 2015, 23, 835–844. [Google Scholar] [CrossRef]
- Pavlova, E.V.; Archer, J.; Z Wang, S.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J. Pathol. 2015, 235, 113–124. [Google Scholar] [CrossRef]
- Xu, Y.-H.; Jia, L.; Quinn, B.; Zamzow, M.; Stringer, K.; Aronow, B.; Sun, Y.; Zhang, W.; Setchell, K.D.; Grabowski, G.A. Global gene expression profile progression in Gaucher disease mouse models. BMC Genomics 2011, 12, 20. [Google Scholar] [CrossRef]
- Zigdon, H.; Savidor, A.; Levin, Y.; Meshcheriakova, A.; Schiffmann, R.; Futerman, A.H. Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS ONE 2015, 10, e0120194. [Google Scholar] [CrossRef]
- Murugesan, V.; Liu, J.; Yang, R.; Lin, H.; Lischuk, A.; Pastores, G.; Zhang, X.; Chuang, W.-L.; Mistry, P.K. Validating glycoprotein non-metastatic melanoma B (gpNMB, osteoactivin), a new biomarker of Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 47–53. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Gabriel, T.L.; Aten, J.; van Roomen, C.P.A.A.; Ottenhoff, R.; Claessen, N.; Alfonso, P.; Irún, P.; Giraldo, P.; Aerts, J.M.F.G.; et al. Gpnmb Is a Potential Marker for the Visceral Pathology in Niemann-Pick Type C Disease. PLoS ONE 2016, 11, e0147208. [Google Scholar] [CrossRef]
- Alam, M.S.; Getz, M.; Safeukui, I.; Yi, S.; Tamez, P.; Shin, J.; Velázquez, P.; Haldar, K. Genomic Expression Analyses Reveal Lysosomal, Innate Immunity Proteins, as Disease Correlates in Murine Models of a Lysosomal Storage Disorder. PLoS ONE 2012, 7, e48273. [Google Scholar] [CrossRef]
- Cluzeau, C.V.M.; Watkins-Chow, D.E.; Fu, R.; Borate, B.; Yanjanin, N.; Dail, M.K.; Davidson, C.D.; Walkley, S.U.; Ory, D.S.; Wassif, C.A.; et al. Microarray expression analysis and identification of serum biomarkers for Niemann–Pick disease, type C1. Hum. Mol. Genet. 2012, 21, 3632–3646. [Google Scholar] [CrossRef]
- UniProtKB-Q14956. Available online: https://www.uniprot.org/uniprot/Q14956 (accessed on 24 November 2018).
- Strausberg, R.L.; Feingold, E.A.; Grouse, L.H.; Derge, J.G.; Klausner, R.D.; Collins, F.S.; Wagner, L.; Shenmen, C.M.; Schuler, G.D.; Altschul, S.F.; et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [Green Version]
- UniProtKB-Q99P91. Available online: https://www.uniprot.org/uniprot/Q99P91 (accessed on 24 November 2018).
- Shikano, S.; Bonkobara, M.; Zukas, P.K.; Ariizumi, K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 8125–8134. [Google Scholar] [CrossRef]
- Hoashi, T.; Sato, S.; Yamaguchi, Y.; Passeron, T.; Tamaki, K.; Hearing, V.J. Glycoprotein nonmetastatic melanoma protein b, a melanocytic cell marker, is a melanosome-specific and proteolytically released protein. FASEB J. 2010, 24, 1616–1629. [Google Scholar] [CrossRef] [Green Version]
- Furochi, H.; Tamura, S.; Mameoka, M.; Yamada, C.; Ogawa, T.; Hirasaka, K.; Okumura, Y.; Imagawa, T.; Oguri, S.; Ishidoh, K.; et al. Osteoactivin fragments produced by ectodomain shedding induce MMP-3 expression via ERK pathway in mouse NIH-3T3 fibroblasts. FEBS Lett. 2007, 581, 5743–5750. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.A.N.; Annis, M.G.; Dong, Z.; Pepin, F.; Hallett, M.; Park, M.; Siegel, P.M. ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS ONE 2010, 5, e12093. [Google Scholar] [CrossRef]
- Li, B.; Castano, A.P.; Hudson, T.E.; Nowlin, B.T.; Lin, S.-L.; Bonventre, J.V.; Swanson, K.D.; Duffield, J.S. The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 4767–4781. [Google Scholar] [CrossRef] [Green Version]
- Theos, A.C.; Watt, B.; Harper, D.C.; Janczura, K.J.; Theos, S.C.; Herman, K.E.; Marks, M.S. The PKD domain distinguishes the trafficking and amyloidogenic properties of the pigment cell protein PMEL and its homologue GPNMB. Pigment Cell Melanoma Res. 2013, 26, 470–486. [Google Scholar] [CrossRef] [Green Version]
- Weterman, M.A.J.; Ajubi, N.; van Dinter, I.M.R.; Degen, W.G.J.; van Muijen, G.N.P.; Ruiter, D.J.; Bloemers, H.P.J. nmb, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int. J. Cancer 1995, 60, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Tissue expression of GPNMB-Summary-The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000136235-GPNMB/tissue (accessed on 3 December 2018).
- Ripoll, V.M.; Irvine, K.M.; Ravasi, T.; Sweet, M.J.; Hume, D.A. Gpnmb is induced in macrophages by IFN-gamma and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J. Immunol. 2007, 178, 6557–6566. [Google Scholar] [CrossRef]
- Sheng, M.H.-C.; Wergedal, J.E.; Mohan, S.; Lau, K.-H.W. Osteoactivin is a novel osteoclastic protein and plays a key role in osteoclast differentiation and activity. FEBS Lett. 2008, 582, 1451–1458. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, V.M.; Meadows, N.A.; Raggatt, L.-J.; Chang, M.K.; Pettit, A.R.; Cassady, A.I.; Hume, D.A. Microphthalmia transcription factor regulates the expression of the novel osteoclast factor GPNMB. Gene 2008, 413, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Maric, G.; Rose, A.A.; Annis, M.G.; Siegel, P.M. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. Onco. Targets. Ther. 2013, 6, 839–852. [Google Scholar]
- Le Borgne, R.; Planque, N.; Martin, P.; Dewitte, F.; Saule, S.; Hoflack, B. The AP-3-dependent targeting of the melanosomal glycoprotein QNR-71 requires a di-leucine-based sorting signal. J. Cell Sci. 2001, 114, 2831–2841. [Google Scholar]
- Zhang, P.; Liu, W.; Zhu, C.; Yuan, X.; Li, D.; Gu, W.; Ma, H.; Xie, X.; Gao, T. Silencing of GPNMB by siRNA Inhibits the Formation of Melanosomes in Melanocytes in a MITF-Independent Fashion. PLoS ONE 2012, 7, e42955. [Google Scholar] [CrossRef]
- Anderson, M.G.; Libby, R.T.; Mao, M.; Cosma, I.M.; Wilson, L.A.; Smith, R.S.; John, S.W. Genetic context determines susceptibility to intraocular pressure elevation in a mouse pigmentary glaucoma. BMC Biol. 2006, 4, 20. [Google Scholar] [CrossRef]
- Anderson, M.G.; Smith, R.S.; Hawes, N.L.; Zabaleta, A.; Chang, B.; Wiggs, J.L.; John, S.W.M. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat. Genet. 2001, 30, 81–85. [Google Scholar] [CrossRef]
- Yang, C.-F.; Lin, S.-P.; Chiang, C.-P.; Wu, Y.-H.; H’ng, W.S.; Chang, C.-P.; Chen, Y.-T.; Wu, J.-Y. Loss of GPNMB Causes Autosomal-Recessive Amyloidosis Cutis Dyschromica in Humans. Am. J. Hum. Genet. 2018, 102, 219–232. [Google Scholar] [CrossRef]
- Bandari, P.S.; Qian, J.; Yehia, G.; Joshi, D.D.; Maloof, P.B.; Potian, J.; Oh, H.S.; Gascon, P.; Harrison, J.S.; Rameshwar, P. Hematopoietic growth factor inducible neurokinin-1 type: A transmembrane protein that is similar to neurokinin 1 interacts with substance P. Regul. Pept. 2003, 111, 169–178. [Google Scholar] [CrossRef]
- Safadi, F.F.; Xu, J.; Smock, S.L.; Rico, M.C.; Owen, T.A.; Popoff, S.N. Cloning and Characterization of Osteoactivin, A Novel cDNA Expressed in Osteoblasts. J. Cell. Biochem. 2002, 84, 12–26. [Google Scholar] [CrossRef]
- Turque, N.; Denhez, F.; Martin, P.; Planque, N.; Bailly, M.; Bègue, A.; Stéhelin, D.; Saule, S. Characterization of a new melanocyte-specific gene (QNR-71) expressed in v-myc-transformed quail neuroretina. EMBO J. 1996, 15, 3338–3350. [Google Scholar] [CrossRef]
- Aksan, I.; Goding, C.R. Targeting the microphthalmia basic helix-loop-helix-leucine zipper transcription factor to a subset of E-box elements in vitro and in vivo. Mol. Cell. Biol. 1998, 18, 6930–6938. [Google Scholar] [CrossRef]
- Du, J.; Miller, A.J.; Widlund, H.R.; Horstmann, M.A.; Ramaswamy, S.; Fisher, D.E. MLANA/MART1 and SILV/PMEL17/GP100 are transcriptionally regulated by MITF in melanocytes and melanoma. Am. J. Pathol. 2003, 163, 333–343. [Google Scholar] [CrossRef]
- Gutknecht, M.; Geiger, J.; Joas, S.; Dörfel, D.; Salih, H.R.; Müller, M.R.; Grünebach, F.; Rittig, S.M. The transcription factor MITF is a critical regulator of GPNMB expression in dendritic cells. Cell Commun. Signal. 2015, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, T.L.; Tol, M.J.; Ottenhof, R.; van Roomen, C.; Aten, J.; Claessen, N.; Hooibrink, B.; de Weijer, B.; Serlie, M.J.; Argmann, C.; et al. Lysosomal stress in obese adipose tissue macrophages contributes to MITF-dependent Gpnmb induction. Diabetes 2014, 63, 3310–3323. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Li, H.; Puertollano, R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. life Sci. C. 2014, 71, 2483–2497. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J. Insight into the microphthalmia gene. Trends Genet. 1995, 11, 442–448. [Google Scholar] [CrossRef]
- Hemesath, T.J.; Steingrímsson, E.; McGill, G.; Hansen, M.J.; Vaught, J.; Hodgkinson, C.A.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A.; Fisher, D.E. microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 1994, 8, 2770–2780. [Google Scholar] [CrossRef]
- Pogenberg, V.; Ögmundsdóttir, M.H.; Bergsteinsdóttir, K.; Schepsky, A.; Phung, B.; Deineko, V.; Milewski, M.; Steingrímsson, E.; Wilmanns, M.; Ogmundsdóttir, M.H.; et al. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012, 26, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013, 200, 475–491. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Takemoto, C.; Kobayashi, I.; Watanabe, A.; Nobukuni, Y.; Fisher, D.E.; Tachibana, M. Ser298 of MITF, a mutation site in Waardenburg syndrome type 2, is a phosphorylation site with functional significance. Hum. Mol. Genet. 2000, 9, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.-S.; Sato, K.; Dougherty, I.I.; Cruz, P.D.; Ariizumi, K. DC-HIL is a negative regulator of T lymphocyte activation. Blood 2007, 109, 4320–4327. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.-S.; Dougherty, I.; Cruz, P.D.; Ariizumi, K. Syndecan-4 mediates the coinhibitory function of DC-HIL on T cell activation. J. Immunol. 2007, 179, 5778–5784. [Google Scholar] [CrossRef]
- Chung, J.-S.; Bonkobara, M.; Tomihari, M.; Cruz, P.D.; Ariizumi, K. The DC-HIL/syndecan-4 pathway inhibits human allogeneic T-cell responses. Eur. J. Immunol. 2009, 39, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.-S.; Tamura, K.; Akiyoshi, H.; Cruz, P.D.; Ariizumi, K. The DC-HIL/syndecan-4 pathway regulates autoimmune responses through myeloid-derived suppressor cells. J. Immunol. 2014, 192, 2576–2584. [Google Scholar] [CrossRef]
- Smith, L.L.; Giachelli, C.M. Structural Requirements for α9β1-Mediated Adhesion and Migration to Thrombin-Cleaved Osteopontin. Exp. Cell Res. 1998, 242, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Redick, S.D.; Settles, D.L.; Briscoe, G.; Erickson, H.P. Defining fibronectin’s cell adhesion synergy site by site-directed mutagenesis. J. Cell Biol. 2000, 149, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Tomihari, M.; Hwang, S.-H.; Chung, J.-S.; Cruz, P.D.; Ariizumi, K.; Ariizumi, K. Gpnmb is a melanosome-associated glycoprotein that contributes to melanocyte/keratinocyte adhesion in a RGD-dependent fashion. Exp. Dermatol. 2009, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Sondag, G.R.; Malcuit, C.; Kim, M.-H.; Safadi, F.F. Macrophage-Associated Osteoactivin/GPNMB Mediates Mesenchymal Stem Cell Survival, Proliferation, and Migration Via a CD44-Dependent Mechanism. J. Cell. Biochem. 2016, 117, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Alboslemy, T.; Safadi, F.; Kim, M.-H. Glycoprotein Nonmelanoma Clone B Regulates the Crosstalk between Macrophages and Mesenchymal Stem Cells toward Wound Repair. J. Invest. Dermatol. 2018, 138, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, P.; Xu, Z.; Chen, H.; Xie, X. GPNMB enhances bone regeneration by promoting angiogenesis and osteogenesis: Potential role for tissue engineering bone. J. Cell. Biochem. 2013, 114, 2729–2737. [Google Scholar] [CrossRef] [PubMed]
- Abdelmagid, S.M.; Barbe, M.F.; Rico, M.C.; Salihoglu, S.; Arango-Hisijara, I.; Selim, A.H.; Anderson, M.G.; Owen, T.A.; Popoff, S.N.; Safadi, F.F. Osteoactivin, an anabolic factor that regulates osteoblast differentiation and function. Exp. Cell Res. 2008, 314, 2334–2351. [Google Scholar] [CrossRef] [PubMed]
- Katayama, A.; Nakatsuka, A.; Eguchi, J.; Murakami, K.; Teshigawara, S.; Kanzaki, M.; Nunoue, T.; Hida, K.; Wada, N.; Yasunaka, T.; et al. Beneficial impact of Gpnmb and its significance as a biomarker in nonalcoholic steatohepatitis. Sci. Rep. 2015, 5, 16920. [Google Scholar] [CrossRef] [Green Version]
- Onaga, M.; Ido, A.; Hasuike, S.; Uto, H.; Moriuchi, A.; Nagata, K.; Hori, T.; Hayash, K.; Tsubouchi, H. Osteoactivin expressed during cirrhosis development in rats fed a choline-deficient, l-amino acid-defined diet, accelerates motility of hepatoma cells. J. Hepatol. 2003, 39, 779–785. [Google Scholar] [CrossRef]
- Haralanova-Ilieva, B.; Ramadori, G.; Armbrust, T. Expression of osteoactivin in rat and human liver and isolated rat liver cells. J. Hepatol. 2005, 42, 565–572. [Google Scholar] [CrossRef]
- Sasaki, T.; Ishikawa, T.; Abe, R.; Nakayama, R.; Asada, A.; Matsuki, N.; Ikegaya, Y. Astrocyte calcium signaling orchestrates neuronal synchronization in organotypic hippocampal slices. J. Physiol. 2014, 592, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Patel-Chamberlin, M.; Wang, Y.; Satirapoj, B.; Phillips, L.M.; Nast, C.C.; Dai, T.; Watkins, R.A.; Wu, X.; Natarajan, R.; Leng, A.; et al. Hematopoietic growth factor inducible neurokinin-1 (Gpnmb/Osteoactivin) is a biomarker of progressive renal injury across species. Kidney Int. 2011, 79, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Uto, H.; Takami, Y.; Takahama, Y.; Hasuike, S.; Kodama, M.; Nagata, K.; Moriuchi, A.; Numata, M.; Ido, A.; et al. Transgenic expression of osteoactivin in the liver attenuates hepatic fibrosis in rats. Biochem. Biophys. Res. Commun. 2007, 356, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Tabu, K.; Sasaki, F.; Takami, Y.; Morinaga, Y.; Mawatari, S.; Hashimoto, S.; Tanoue, S.; Kanmura, S.; Tamai, T.; et al. Glycoprotein Nonmetastatic Melanoma B (Gpnmb)-Positive Macrophages Contribute to the Balance between Fibrosis and Fibrolysis during the Repair of Acute Liver Injury in Mice. PLoS ONE 2015, 10, e0143413. [Google Scholar] [CrossRef] [PubMed]
- Pahl, M.V.; Vaziri, N.D.; Yuan, J.; Adler, S.G. Upregulation of monocyte/macrophage HGFIN (Gpnmb/Osteoactivin) expression in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, F.; Kumagai, K.; Uto, H.; Takami, Y.; Kure, T.; Tabu, K.; Nasu, Y.; Hashimoto, S.; Kanmura, S.; Numata, M.; et al. Expression of glycoprotein nonmetastatic melanoma protein B in macrophages infiltrating injured mucosa is associated with the severity of experimental colitis in mice. Mol. Med. Rep. 2015, 12, 7503–7511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Feferman, L.; Sharma, G.; Tobacman, J.K. Increased GPNMB, phospho-ERK1/2, and MMP-9 in cystic fibrosis in association with reduced arylsulfatase B. Mol. Genet. Metab. 2018, 124, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Takahashi, K.; Hojo, H. An ultrastructural and experimental study on the development of tubular structures in the lysosomes of Gaucher cells. Lab. Invest. 1988, 58, 590–598. [Google Scholar] [PubMed]
- van Breemen, M.J.; de Fost, M.; Voerman, J.S.A.; Laman, J.D.; Boot, R.G.; Maas, M.; Hollak, C.E.M.; Aerts, J.M.; Rezaee, F. Increased plasma macrophage inflammatory protein (MIP)-1α and MIP-1β levels in type 1 Gaucher disease. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 788–796. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, E.; van Roomen, C.P.A.A.; van Puijvelde, G.H.; Ottenhoff, R.; van Eijk, M.; Aten, J.; Kuiper, J.; Overkleeft, H.S.; Groen, A.K.; Verhoeven, A.J.; et al. Correction of Liver Steatosis by a Hydrophobic Iminosugar Modulating Glycosphingolipids Metabolism. PLoS ONE 2012, 7, e38520. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, D.A.E.; van Scheppingen, J.; van der Poel, M.; Bossers, K.; Schuurman, K.G.; van Eden, C.G.; Hol, E.M.; Hamann, J.; Huitinga, I. Gene Expression Profiling of Multiple Sclerosis Pathology Identifies Early Patterns of Demyelination Surrounding Chronic Active Lesions. Front. Immunol. 2017, 8, 1810. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jüllig, M.; Middleditch, M.J.; Cooper, G.J.S. Modelling atherosclerosis by proteomics: Molecular changes in the ascending aortas of cholesterol-fed rabbits. Atherosclerosis 2015, 242, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Grijalva, A.; Skowronski, A.; van Eijk, M.; Serlie, M.J.; Ferrante, A.W. Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013, 18, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Tol, M.J.; van der Lienden, M.J.C.; Gabriel, T.L.; Hagen, J.J.; Scheij, S.; Veenendaal, T.; Klumperman, J.; Donker-Koopman, W.E.; Verhoeven, A.J.; Overkleeft, H.; et al. HEPES activates a MiT/TFE-dependent lysosomal-autophagic gene network in cultured cells: A call for caution. Autophagy 2018, 14, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, K.; Hirata, H.; Ohbuchi, K.; Nishi, K.; Maeda, A.; Kuniyasu, A.; Yamada, D.; Maeda, T.; Tsuji, A.; Sawada, M.; et al. The novel monoclonal antibody 9F5 reveals expression of a fragment of GPNMB/osteoactivin processed by furin-like protease(s) in a subpopulation of microglia in neonatal rat brain. Glia 2016, 64, 1938–1961. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-J.; Ma, W.-J.; Yokoyama, S. Expression and immunolocalization of Gpnmb, a glioma-associated glycoprotein, in normal and inflamed central nervous systems of adult rats. Brain Behav. 2012, 2, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Duan, S.; Cui, J.; Yan, X.; Li, H.; Wang, Y.; Chen, F.; Zhang, L.; Liu, J.; Xie, X. Induction of Matrix Metalloproteinase-3 (MMP-3) Expression in the Microglia by Lipopolysaccharide (LPS) via Upregulation of Glycoprotein Nonmetastatic Melanoma B (GPNMB) Expression. J. Mol. Neurosci. 2014, 54, 234–242. [Google Scholar] [CrossRef]
- Tyburczy, M.E.; Kotulska, K.; Pokarowski, P.; Mieczkowski, J.; Kucharska, J.; Grajkowska, W.; Roszkowski, M.; Jozwiak, S.; Kaminska, B. Novel Proteins Regulated by mTOR in Subependymal Giant Cell Astrocytomas of Patients with Tuberous Sclerosis Complex and New Therapeutic Implications. Am. J. Pathol. 2010, 176, 1878–1890. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.L.; Boddeke, H.W.G.M.; et al. Glioma-Associated Microglia/Macrophages Display an Expression Profile Different from M1 and M2 Polarization and Highly Express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [PubMed]
- Hudson, A.L.; Parker, N.R.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Chen, J.; Wheeler, H.R.; Howell, V.M. Glioblastoma Recurrence Correlates With Increased APE1 and Polarization Toward an Immuno-Suppressive Microenvironment. Front. Oncol. 2018, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.-T.; Wakiya, K.; Dowell, J.M.; Herndon, J.E.; Reardon, D.A.; Graner, M.W.; Riggins, G.J.; Wikstrand, C.J.; Bigner, D.D.; Herndon Ii, J.E.; et al. Glycoprotein Nonmetastatic Melanoma Protein B, a Potential Molecular Therapeutic Target in Patients with Glioblastoma Multiforme. Clin. Cancer Res. 2006, 12, 1970–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, H.; Shimazawa, M.; Kimura, M.; Takata, M.; Tsuruma, K.; Yamada, M.; Takahashi, H.; Hozumi, I.; Niwa, J.; Iguchi, Y.; et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci. Rep. 2012, 2, 573. [Google Scholar] [CrossRef]
- Nakano, Y.; Suzuki, Y.; Takagi, T.; Kitashoji, A.; Ono, Y.; Tsuruma, K.; Yoshimura, S.; Shimazawa, M.; Iwama, T.; Hara, H. Glycoprotein nonmetastatic melanoma protein B (GPNMB) as a novel neuroprotective factor in cerebral ischemia–reperfusion injury. Neuroscience 2014, 277, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Budge, K.M.; Neal, M.L.; Richardson, J.R.; Safadi, F.F. Glycoprotein NMB: An Emerging Role in Neurodegenerative Disease. Mol. Neurobiol. 2018, 55, 5167–5176. [Google Scholar] [CrossRef] [PubMed]
- Hüttenrauch, M.; Ogorek, I.; Klafki, H.; Otto, M.; Stadelmann, C.; Weggen, S.; Wiltfang, J.; Wirths, O. Glycoprotein NMB: A novel Alzheimer’s disease associated marker expressed in a subset of activated microglia. Acta Neuropathol. Commun. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.L.; Boyle, A.M.; Budge, K.M.; Safadi, F.F.; Richardson, J.R. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflammation 2018, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet. Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Mosharov, E.; Talloczy, Z.; Zucca, F.A.; Simon, J.D.; Zecca, L. Neuronal pigmented autophagic vacuoles: Lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J. Neurochem. 2008, 106, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Perrett, R.M.; Alexopoulou, Z.; Tofaris, G.K. The endosomal pathway in Parkinson’s disease. Mol. Cell. Neurosci. 2015, 66, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Pericleous, M.; Kelly, C.; Wang, T.; Livingstone, C.; Ala, A. Wolman’s disease and cholesteryl ester storage disorder: The phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2017, 2, 670–679. [Google Scholar] [CrossRef]
- Vom Dahl, S.; Harzer, K.; Rolfs, A.; Albrecht, B.; Niederau, C.; Vogt, C.; van Weely, S.; Aerts, J.; Müller, G.; Häussinger, D. Hepatosplenomegalic lipidosis: What unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase −1 exon 8 splice junction mutation. J. Hepatol. 1999, 31, 741–746. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Lienden, M.J.C.; Gaspar, P.; Boot, R.; Aerts, J.M.F.G.; Van Eijk, M. Glycoprotein Non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages. Int. J. Mol. Sci. 2019, 20, 66. https://doi.org/10.3390/ijms20010066
Van der Lienden MJC, Gaspar P, Boot R, Aerts JMFG, Van Eijk M. Glycoprotein Non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages. International Journal of Molecular Sciences. 2019; 20(1):66. https://doi.org/10.3390/ijms20010066
Chicago/Turabian StyleVan der Lienden, Martijn J.C., Paulo Gaspar, Rolf Boot, Johannes M.F.G. Aerts, and Marco Van Eijk. 2019. "Glycoprotein Non-Metastatic Protein B: An Emerging Biomarker for Lysosomal Dysfunction in Macrophages" International Journal of Molecular Sciences 20, no. 1: 66. https://doi.org/10.3390/ijms20010066