Dysregulation of Lipid Metabolism in Mkp-1 Deficient Mice during Gram-Negative Sepsis

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
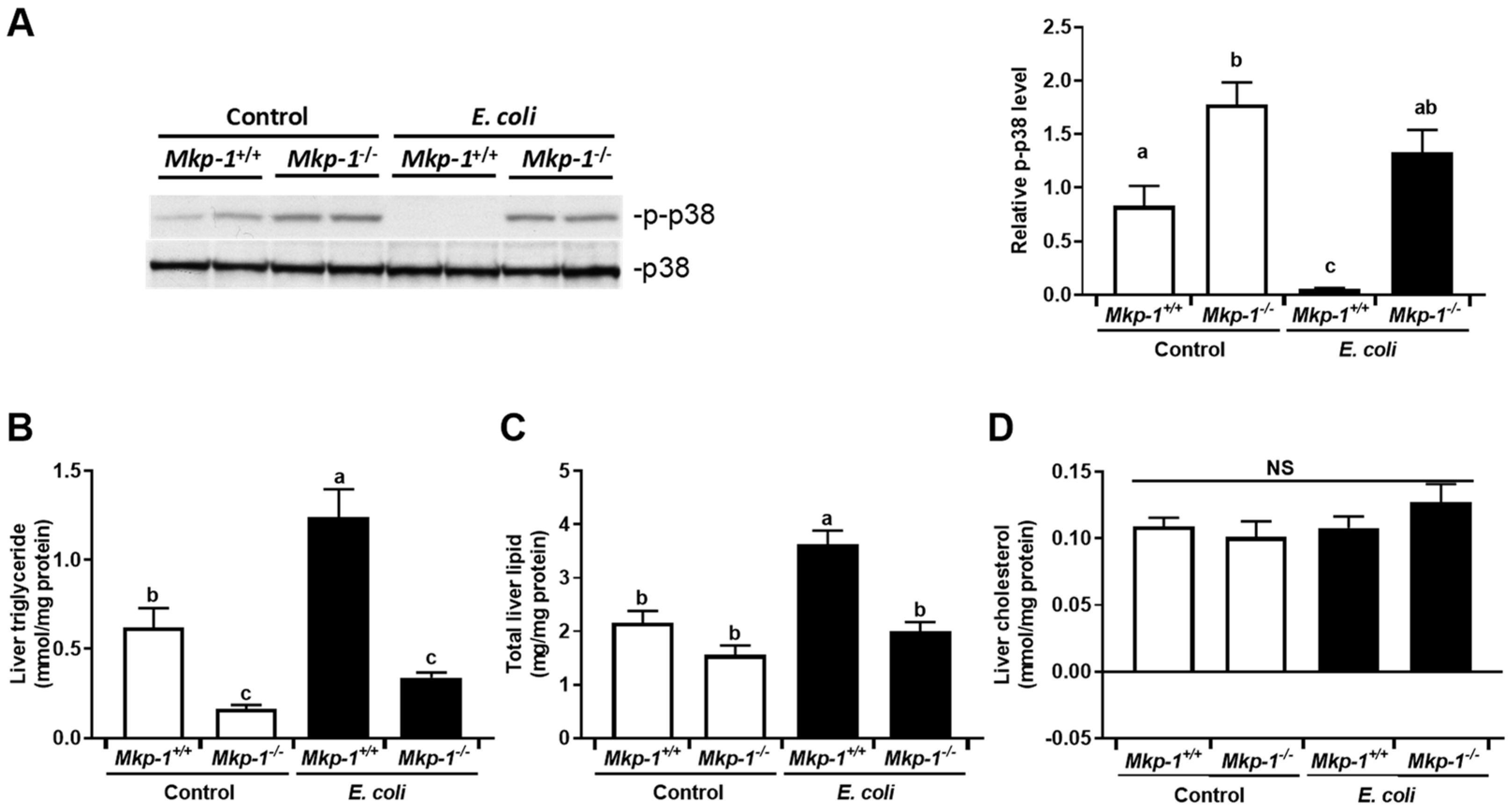
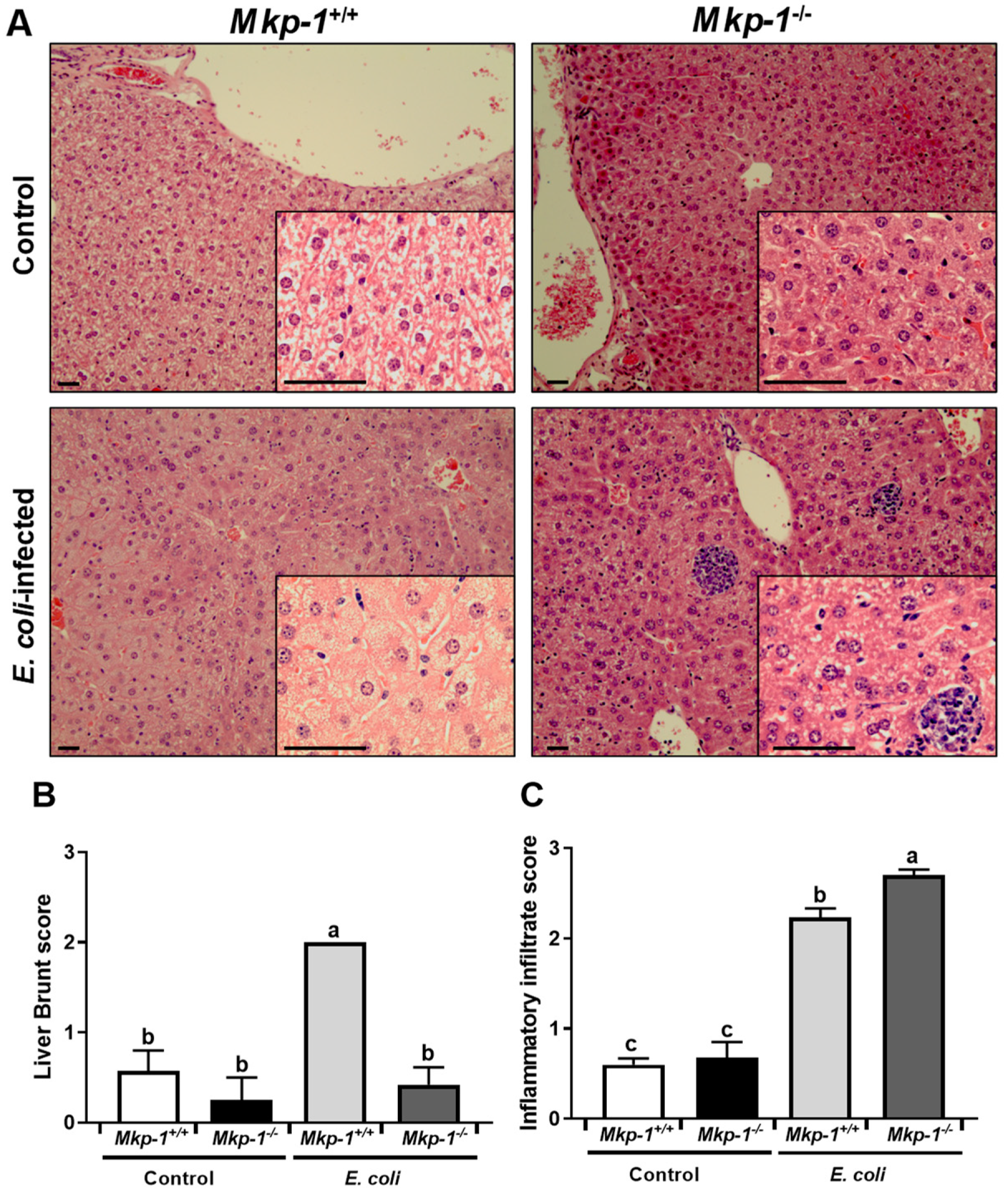
2.1. Changes in p38 Activity and Dysregulation of Lipid Metabolism Caused by Mkp-1 Deficiency and E. coli Infection
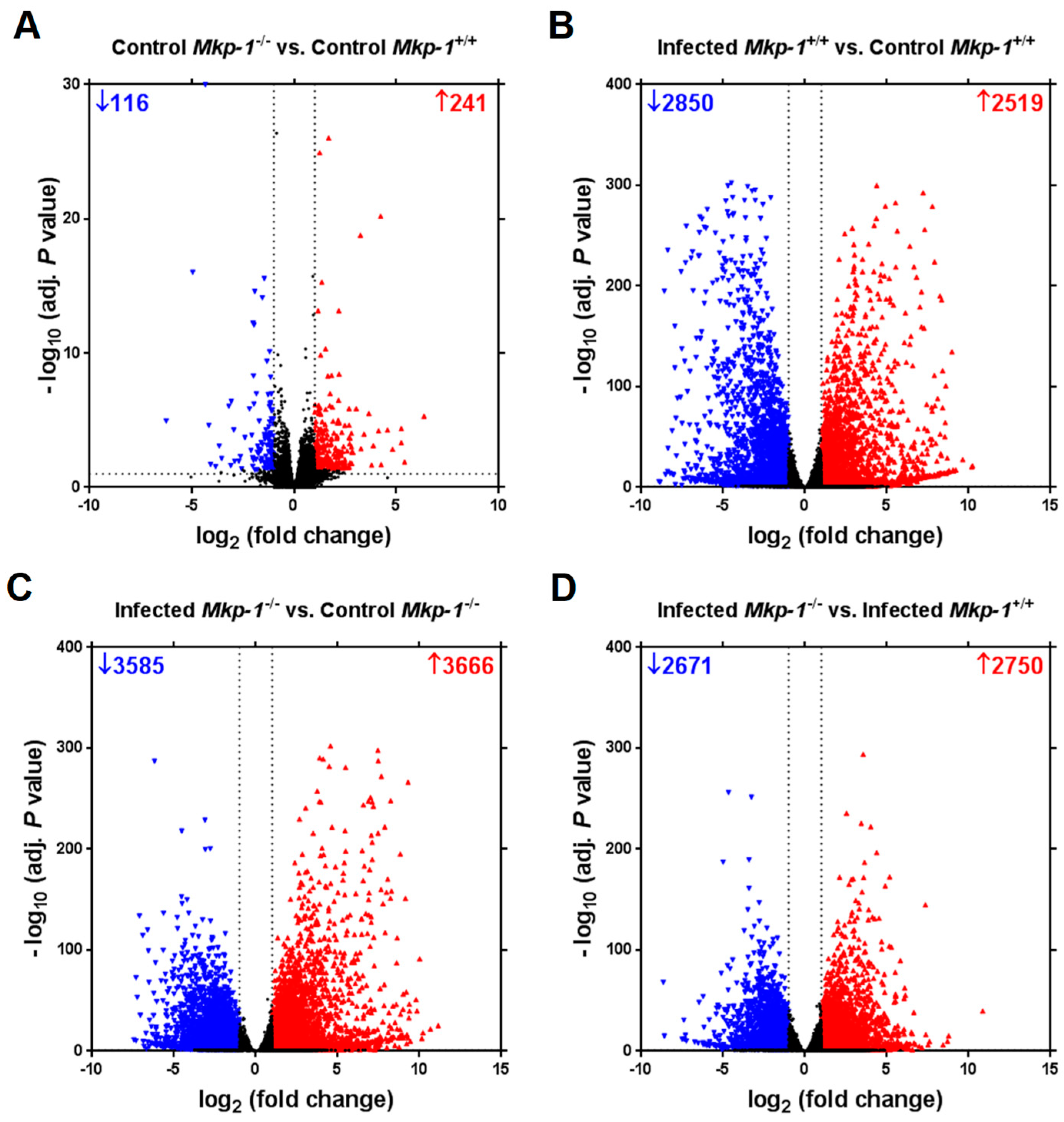
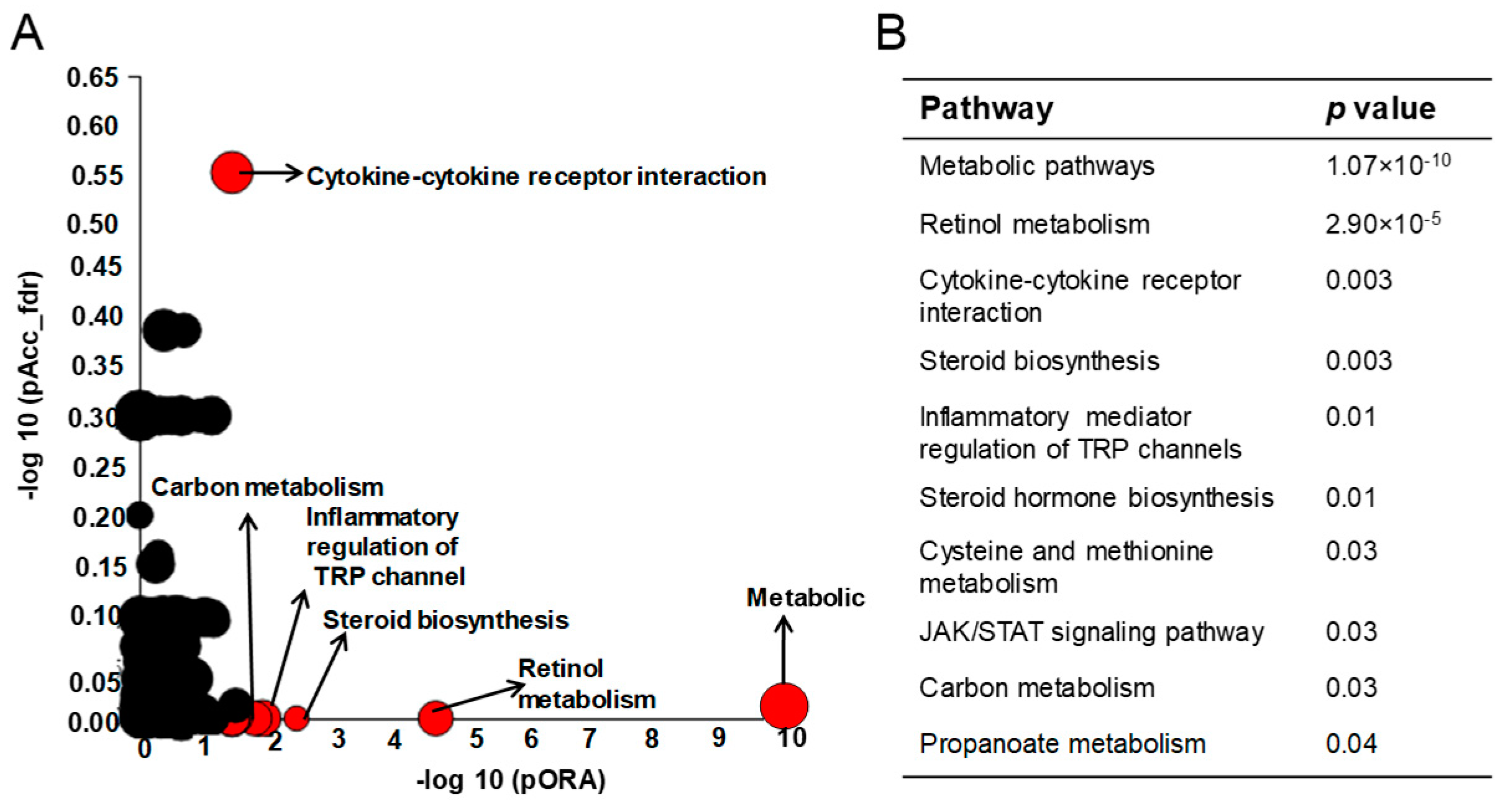
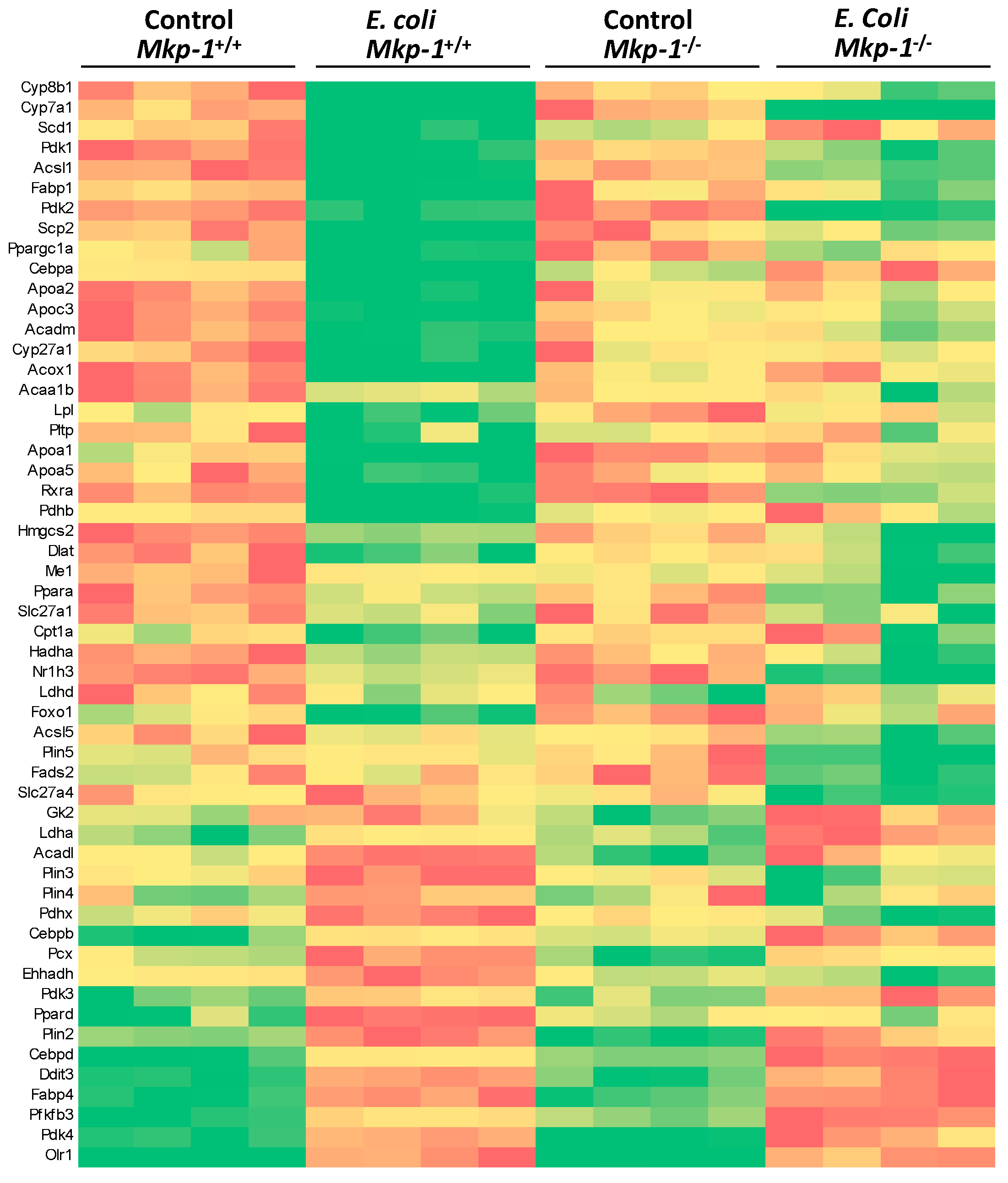
2.2. The Impact of Mkp-1 Deficiency and E. coli Infection on Global Liver Gene Expression Profile
2.3. E. coli Infection Caused a Major Shift in Gene Expression of the Fatty Acid and Glucose Metabolic Programs and Mkp-1 Deficiency Disrupts This Shift
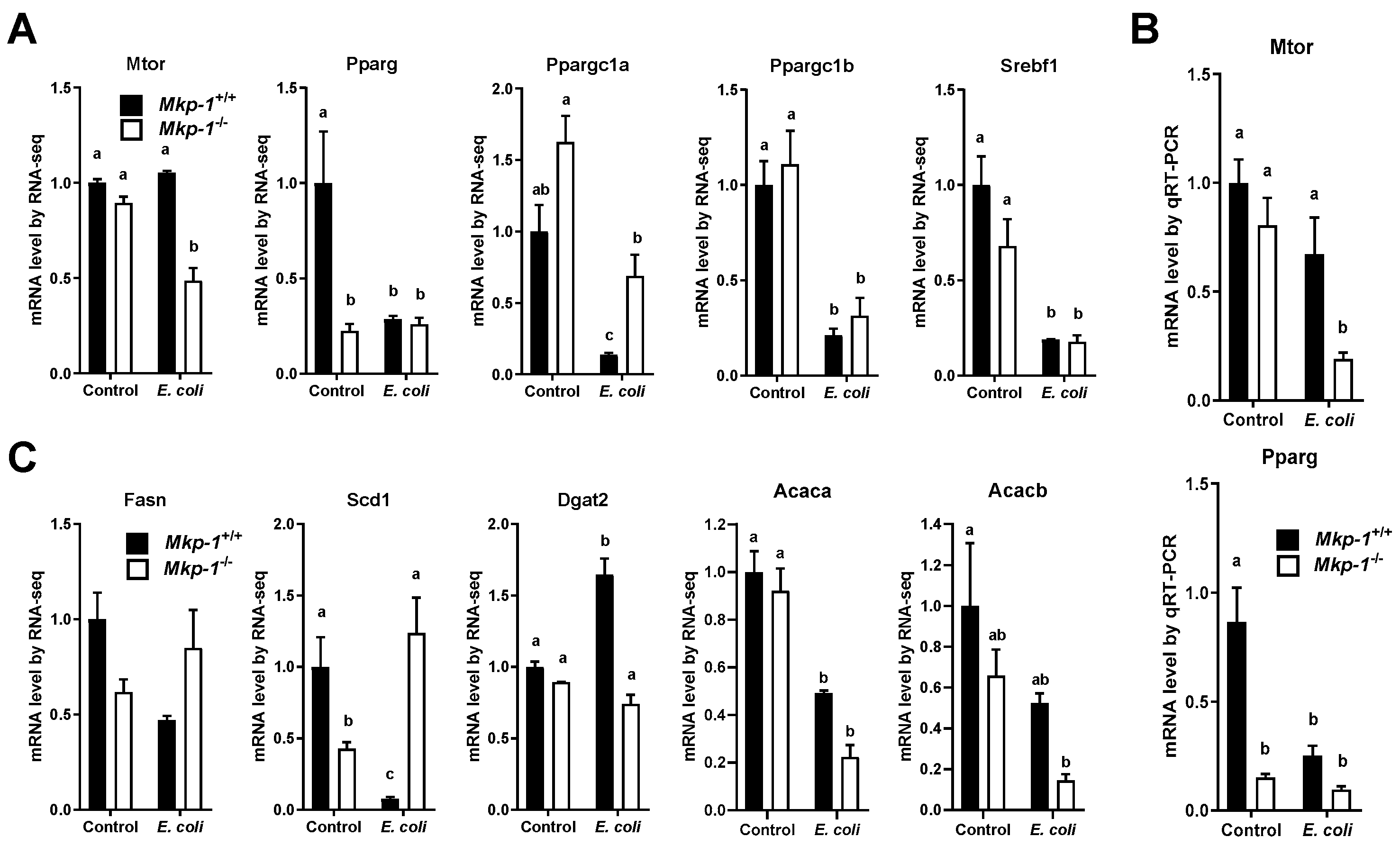
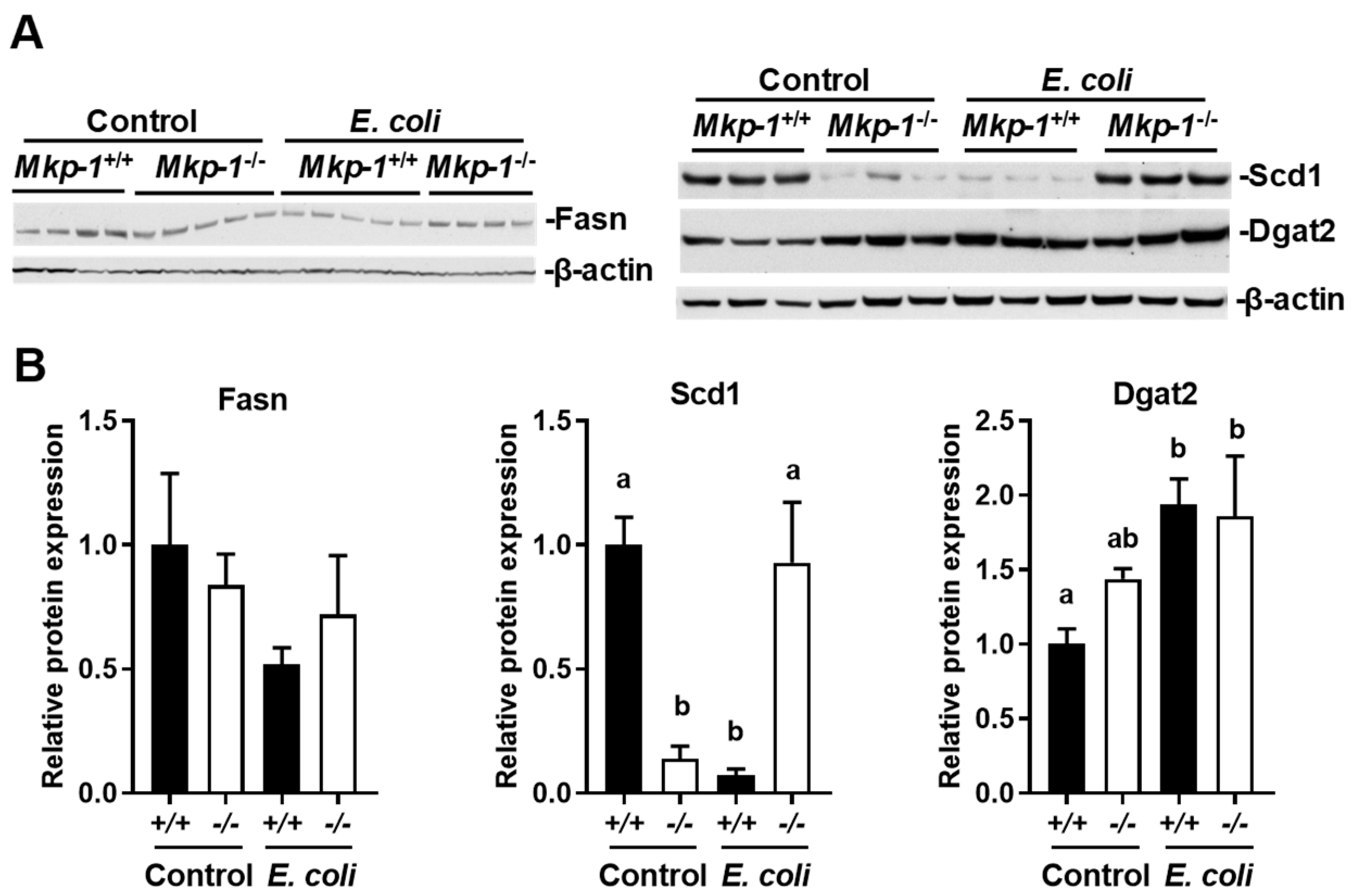
2.4. E. coli Infection Lowers the Expression of Mammalian Target of Rapamycin (mTOR) and Lipogenic Genes
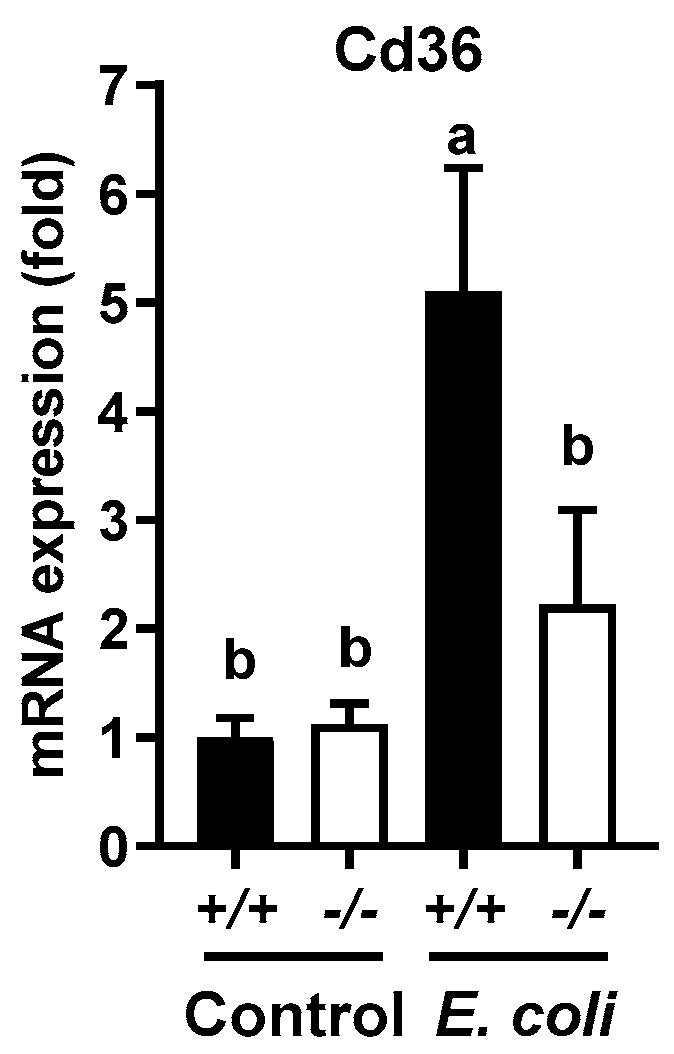
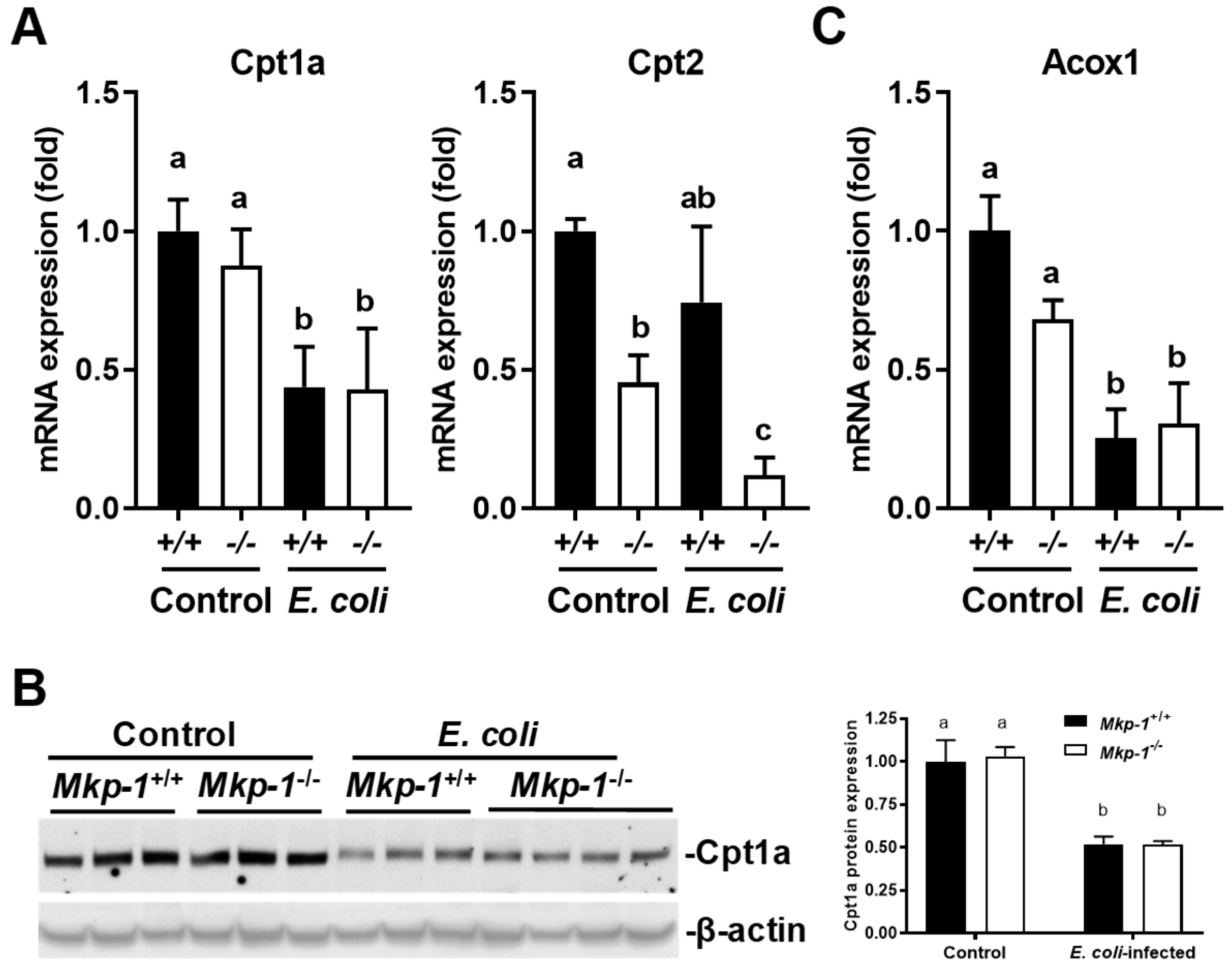
2.5. E. coli Infection Increased the Expression of Genes Involved in Liver Fatty Acid Uptake, and Lowered the Expression of Genes Involved in Mitochondrial and Peroxisomal Fatty Acid Oxidation
2.6. Effects of E. coli Infection and Mkp-1 Deficiency on the Expression of Phosphoenolpyruvate Carboxykinase 1 (Pck1) Protein
3. Discussion
3.1. The Critical Role of Mkp-1 as a p38 Regulator in the Liver
3.2. Mkp-1 and Hyperlipidemia of Sepsis
4. Materials and Methods
4.1. Experimental Animals and E. coli Infection
4.2. Biochemical Assessment of Liver Lipid
4.3. Histological Assessment of Liver Lipid Content
4.4. Liver RNA Extraction
4.5. RNA-Seq
4.6. qRT-PCR
4.7. Western Blotting
4.8. Statistical Analysis
4.9. Data Availability
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Acaa1 | Acetyl-CoA acyltransferase 1 |
| Acaca | Acetyl-CoA carboxylase α |
| Acacb | Acetyl-CoA carboxylase β |
| Acsl1 | Long chain fatty acid-CoA ligase 1 |
| Acot | Acyl-CoA thioesterase |
| Acox1 | Acyl-CoA oxidase 1 |
| ANOVA | Analysis of variance |
| Apo | Apolipoprotein |
| Cd36 CFU | Cluster of differentiation 36 Colony forming units |
| Cebp | CCAAT/enhancer-binding protein |
| Cpt | Carnitine palmitoyltransferase |
| Dgat2 | Diglyceride acyltransferase 2 |
| Fabp1 | Fatty acid-binding protein 1 |
| Fasn | Fatty acid synthase |
| Foxo1 JNK | Forkhead box protein O1 c-Jun N-terminal kinase |
| LPS | Lipopolysaccharide |
| Mkp-1 | MAP kinase phosphatase-1 |
| Mtor | Mammalian target of rapamycin |
| NF-κB | Nuclear factor-κB |
| PBS | Phosphate-buffered saline |
| Pck1/PEPCK-c | Phosphoenolpyruvate carboxykinase, cytosolic isoform |
| Plin | Perilipin |
| Pparg/PPARγ | Peroxisome proliferator-activated receptor γ |
| Ppargc1/PGC-1 | PPARγ coactivator 1 |
| Scd1 | Stearoyl-CoA desaturase 1 |
| qRT-PCR | Quantitative reverse transcription PCR |
| Srebf1/SREBP-1 | Sterol regulatory element-binding transcription factor 1 |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
| VLDL | Very low-density lipoprotein |
References
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of Severe Sepsis in the United States: Analysis of Incidence, Outcome, and Associated Costs of Care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of Severe Sepsis. Virulence 2014, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Trager, K.; DeBacker, D.; Radermacher, P. Metabolic Alterations in Sepsis and Vasoactive Drug-Related Metabolic Effects. Curr. Opin. Crit Care. 2003, 9, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.I.; Wallace, R.J., Jr.; Musher, D.M.; Septimus, E.J.; Kohl, S.; Baughn, R.E. Hypoglycemia As a Manifestation of Sepsis. Am. J. Med. 1980, 68, 649–654. [Google Scholar] [CrossRef]
- Chiolero, R.; Revelly, J.P.; Tappy, L. Energy Metabolism in Sepsis and Injury. Nutrition 1997, 13, 45S–51S. [Google Scholar] [CrossRef]
- Cetinkaya, A.; Erden, A.; Avci, D.; Karagoz, H.; Karahan, S.; Basak, M.; Bulut, K.; Gencer, V.; Mutlu, H. Is Hypertriglyceridemia a Prognostic Factor in Sepsis? Ther. Clin. Risk Manag. 2014, 10, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.M.; Kronfeld, D.S. In Vivo Studies of Hypoglycemia and Lactic Acidosis in Endotoxic Shock. Am. J. Physiol. 1985, 248, E309–E316. [Google Scholar] [CrossRef]
- Berk, J.L.; Hagen, J.F.; Beyer, W.H.; Gerber, M.J. Hypoglycemia of Shock. Ann. Surg. 1970, 171, 400–408. [Google Scholar] [CrossRef]
- Woodske, M.E.; Yokoe, T.; Zou, B.; Romano, L.C.; Rosa, T.C.; Garcia-Ocana, A.; Alonso, L.C.; O’Donnell, C.P.; McVerry, B.J. Hyperinsulinemia Predicts Survival in a Hyperglycemic Mouse Model of Critical Illness. Crit Care Med. 2009, 37, 2596–2603. [Google Scholar] [CrossRef]
- Frazier, W.J.; Wang, X.; Wancket, L.M.; Li, X.A.; Meng, X.; Nelin, L.D.; Cato, A.C.; Liu, Y. Increased Inflammation, Impaired Bacterial Clearance, and Metabolic Disruption after Gram-Negative Sepsis in Mkp-1-Deficient Mice. J. Immunol. 2009, 183, 7411–7419. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, R.L.; Matson, C.F.; Rowberg, A.H.; Beisel, W.R. Defective Lipid Disposal Mechanisms During Bacterial Infection in Rhesus Monkeys. Metabolism 1976, 25, 615–624. [Google Scholar] [CrossRef]
- Griffiths, J.; Groves, A.C.; Leung, F.Y. Hypertriglyceridemia and Hypoglycemia in Gram-Negative Sepsis in the Dog. Surg. Gynecol. Obstet. 1973, 136, 897–903. [Google Scholar] [PubMed]
- Gallin, J.I.; Kaye, D.; O’Leary, W.M. Serum Lipids in Infection. N. Engl. J. Med. 1969, 281, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.W.; Gosnell, J.E.; Kumwenda, Z.L. The Lipemia of Sepsis: Triglyceride-Rich Lipoproteins As Agents of Innate Immunity. J. Endotoxin. Res. 2000, 6, 421–430. [Google Scholar] [PubMed]
- Wendel, M.; Paul, R.; Heller, A.R. Lipoproteins in Inflammation and Sepsis. II. Clinical Aspects. Intensive Care Med. 2007, 33, 25–35. [Google Scholar] [CrossRef]
- Lefer, A.M. Significance of Lipid Mediators in Shock States. Circ. Shock 1989, 27, 3–12. [Google Scholar] [PubMed]
- Kawakami, M.; Cerami, A. Studies of Endotoxin-Induced Decrease in Lipoprotein Lipase Activity. J. Exp. Med. 1981, 154, 631–639. [Google Scholar] [CrossRef]
- Lanza-Jacoby, S.; Lansey, S.C.; Cleary, M.P.; Rosato, F.E. Alterations in Lipogenic Enzymes and Lipoprotein Lipase Activity During Gram-Negative Sepsis in the Rat. Arch. Surg. 1982, 117, 144–147. [Google Scholar] [CrossRef]
- Beutler, B.; Greenwald, D.; Hulmes, J.D.; Chang, M.; Pan, Y.C.; Mathison, J.; Ulevitch, R.; Cerami, A. Identity of Tumour Necrosis Factor and the Macrophage-Secreted Factor Cachectin. Nature 1985, 316, 552–554. [Google Scholar] [CrossRef]
- Beutler, B.A.; Cerami, A. Recombinant Interleukin 1 Suppresses Lipoprotein Lipase Activity in 3T3-L1 Cells. J. Immunol. 1985, 135, 3969–3971. [Google Scholar]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, Inflammation and Innate Immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Aspichueta, P.; Perez-Agote, B.; Perez, S.; Ochoa, B.; Fresnedo, O. Impaired Response of VLDL Lipid and ApoB Secretion to Endotoxin in the Fasted Rat Liver. J. Endotoxin. Res. 2006, 12, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Masaki, T.; Seike, M.; Yoshimatsu, H. TNF-Alpha Induces Hepatic Steatosis in Mice by Enhancing Gene Expression of Sterol Regulatory Element Binding Protein-1c (SREBP-1c). Exp. Biol. Med. 2007, 232, 614–621. [Google Scholar]
- Ohhira, M.; Motomura, W.; Fukuda, M.; Yoshizaki, T.; Takahashi, N.; Tanno, S.; Wakamiya, N.; Kohgo, Y.; Kumei, S.; Okumura, T. Lipopolysaccharide Induces Adipose Differentiation-Related Protein Expression and Lipid Accumulation in the Liver Through Inhibition of Fatty Acid Oxidation in Mice. J. Gastroenterol. 2007, 42, 969–978. [Google Scholar] [CrossRef]
- Liu, Y.; Shepherd, E.G.; Nelin, L.D. MAPK Phosphatases—Regulating the Immune Response. Nat. Rev. Immunol. 2007, 7, 202–212. [Google Scholar] [CrossRef]
- Wang, X.; Nelin, L.D.; Kuhlman, J.R.; Meng, X.; Welty, S.E.; Liu, Y. The Role of MAP Kinase Phosphatase-1 in the Protective Mechanism of Dexamethasone Against Endotoxemia. Life Sci. 2008, 83, 671–680. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Q.; Matta, R.; Meng, X.; Liu, X.; Liu, C.G.; Nelin, L.D.; Liu, Y. Inducible Nitric-Oxide Synthase Expression Is Regulated by Mitogen-Activated Protein Kinase Phosphatase-1. J. Biol. Chem. 2009, 284, 27123–27134. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Shepherd, E.G.; Manson, M.E.; Nelin, L.D.; Sorokin, A.; Liu, Y. The Role of Mitogen-Activated Protein Kinase Phosphatase-1 in the Response of Alveolar Macrophages to Lipopolysaccharide: Attenuation of Proinflammatory Cytokine Biosynthesis Via Feedback Control of P38. J. Biol. Chem. 2005, 280, 8101–8108. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, X.; Nelin, L.D.; Yao, Y.; Matta, R.; Manson, M.E.; Baliga, R.S.; Meng, X.; Smith, C.V.; Bauer, J.A.; et al. MAP Kinase Phosphatase 1 Controls Innate Immune Responses and Suppresses Endotoxic Shock. J. Exp. Med. 2006, 203, 131–140. [Google Scholar] [CrossRef]
- Lang, R.; Hammer, M.; Mages, J. DUSP Meet Immunology: Dual Specificity MAPK Phosphatases in Control of the Inflammatory Response. J. Immunol. 2006, 177, 7497–7504. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.C.; Kraft, A.S. Conditional Expression of the Mitogen-Activated Protein Kinase (MAPK) Phosphatase MKP-1 Preferentially Inhibits P38 MAPK and Stress-Activated Protein Kinase in U937 Cells. J. Biol. Chem. 1997, 272, 16917–16923. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Barry, S.P.; Roth, R.J.; Wu, J.J.; Jones, E.A.; Bennett, A.M.; Flavell, R.A. Dynamic Regulation of Pro- and Anti-Inflammatory Cytokines by MAPK Phosphatase 1 (MKP-1) in Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 2274–2279. [Google Scholar] [CrossRef] [PubMed]
- Salojin, K.V.; Owusu, I.B.; Millerchip, K.A.; Potter, M.; Platt, K.A.; Oravecz, T. Essential Role of MAPK Phosphatase-1 in the Negative Control of Innate Immune Responses. J. Immunol. 2006, 176, 1899–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.J.; Roth, R.J.; Anderson, E.J.; Hong, E.G.; Lee, M.K.; Choi, C.S.; Neufer, P.D.; Shulman, G.I.; Kim, J.K.; Bennett, A.M. Mice Lacking MAP Kinase Phosphatase-1 Have Enhanced MAP Kinase Activity and Resistance to Diet-Induced Obesity. Cell Metab. 2006, 4, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. An Emerging Role of MTOR in Lipid Biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cases, S.; Stone, S.J.; Zhou, P.; Yen, E.; Tow, B.; Lardizabal, K.D.; Voelker, T.; Farese, R.V., Jr. Cloning of DGAT2, a Second Mammalian Diacylglycerol Acyltransferase, and Related Family Members. J. Biol. Chem. 2001, 276, 38870–38876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lardizabal, K.D.; Mai, J.T.; Wagner, N.W.; Wyrick, A.; Voelker, T.; Hawkins, D.J. DGAT2 Is a New Diacylglycerol Acyltransferase Gene Family: Purification, Cloning, and Expression in Insect Cells of Two Polypeptides From Mortierella Ramanniana With Diacylglycerol Acyltransferase Activity. J. Biol. Chem. 2001, 276, 38862–38869. [Google Scholar] [CrossRef]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic Fatty Acid Transporter Cd36 Is a Common Target of LXR, PXR, and PPARgamma in Promoting Steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of Energy Metabolism by Long-Chain Fatty Acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Bonnefont, J.P.; Djouadi, F.; Prip-Buus, C.; Gobin, S.; Munnich, A.; Bastin, J. Carnitine Palmitoyltransferases 1 and 2: Biochemical, Molecular and Medical Aspects. Mol. Asp. Med. 2004, 25, 495–520. [Google Scholar] [CrossRef]
- Moreno-Fernandez, M.E.; Giles, D.A.; Stankiewicz, T.E.; Sheridan, R.; Karns, R.; Cappelletti, M.; Lampe, K.; Mukherjee, R.; Sina, C.; Sallese, A.; et al. Peroxisomal Beta-Oxidation Regulates Whole Body Metabolism, Inflammatory Vigor, and Pathogenesis of Nonalcoholic Fatty Liver Disease. JCI Insight 2018, 3, 93626. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, P.; Johnson, M.T.; Yang, J.; Lepage, D.F.; Conlon, R.A.; Kalhan, S.C.; Reshef, L.; Tilghman, S.M.; Hanson, R.W. Phosphoenolpyruvate Carboxykinase and the Critical Role of Cataplerosis in the Control of Hepatic Metabolism. Nutr. Metab. 2005, 2, 33. [Google Scholar] [CrossRef] [PubMed]
- Horike, N.; Sakoda, H.; Kushiyama, A.; Ono, H.; Fujishiro, M.; Kamata, H.; Nishiyama, K.; Uchijima, Y.; Kurihara, Y.; Kurihara, H.; et al. AMP-Activated Protein Kinase Activation Increases Phosphorylation of Glycogen Synthase Kinase 3beta and Thereby Reduces CAMP-Responsive Element Transcriptional Activity and Phosphoenolpyruvate Carboxykinase C Gene Expression in the Liver. J. Biol. Chem. 2008, 283, 33902–33910. [Google Scholar] [CrossRef]
- Gomez-Valades, A.G.; Mendez-Lucas, A.; Vidal-Alabro, A.; Blasco, F.X.; Chillon, M.; Bartrons, R.; Bermudez, J.; Perales, J.C. Pck1 Gene Silencing in the Liver Improves Glycemia Control, Insulin Sensitivity, and Dyslipidemia in Db/Db Mice. Diabetes 2008, 57, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lei, Q.Y.; Zhao, S.; Guan, K.L. Regulation of Glycolysis and Gluconeogenesis by Acetylation of PKM and PEPCK. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawan, A.; Zhang, L.; Gatzke, F.; Min, K.; Jurczak, M.J.; Al-Mutairi, M.; Richter, P.; Camporez, J.P.; Couvillon, A.; Pesta, D.; et al. Hepatic Mitogen-Activated Protein Kinase Phosphatase 1 Selectively Regulates Glucose Metabolism and Energy Homeostasis. Mol. Cell Biol. 2015, 35, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.E.; Firestein, G.S. Mitogen Activated Protein Kinase Inhibitors: Where are we now and where are we going? Ann. Rheum. Dis. 2006, 65 (Suppl. 3), iii83–iii88. [Google Scholar] [CrossRef]
- Chen, P.; Li, J.; Barnes, J.; Kokkonen, G.C.; Lee, J.C.; Liu, Y. Restraint of Proinflammatory Cytokine Biosynthesis by Mitogen-Activated Protein Kinase Phosphatase-1 in Lipopolysaccharide-Stimulated Macrophages. J. Immunol. 2002, 169, 6408–6416. [Google Scholar] [CrossRef] [Green Version]
- Hammer, M.; Mages, J.; Dietrich, H.; Servatius, A.; Howells, N.; Cato, A.C.; Lang, R. Dual Specificity Phosphatase 1 (DUSP1) Regulates a Subset of LPS-Induced Genes and Protects Mice From Lethal Endotoxin Shock. J. Exp. Med. 2006, 203, 15–20. [Google Scholar] [CrossRef]
- Lee, J.C.; Kassis, S.; Kumar, S.; Badger, A.; Adams, J.L. P38 Mitogen-Activated Protein Kinase Inhibitors—Mechanisms and Therapeutic Potentials. Pharmacol. Ther. 1999, 82, 389–397. [Google Scholar] [CrossRef]
- Bagby, G.J.; Spitzer, J.A. Lipoprotein Lipase Activity in Rat Heart and Adipose Tissue During Endotoxic Shock. Am. J. Physiol. 1980, 238, H325–H330. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.W.; Grunfeld, C.; Feingold, K.R.; Rapp, J.H. Human Very Low Density Lipoproteins and Chylomicrons Can Protect Against Endotoxin-Induced Death in Mice. J. Clin. Investig. 1990, 86, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The Interaction of Hepatic Lipid and Glucose Metabolism in Liver Diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Mahoney, J.; Le, T.N.; Pekala, P.; Cerami, A. Purification of Cachectin, a Lipoprotein Lipase-Suppressing Hormone Secreted by Endotoxin-Induced RAW 264.7 Cells. J. Exp. Med. 1985, 161, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.A.; Milsark, I.W.; Cerami, A. Cachectin/Tumor Necrosis Factor: Production, Distribution, and Metabolic Fate in Vivo. J. Immunol. 1985, 135, 3972–3977. [Google Scholar] [PubMed]
- Chen, H.C.; Farese, R.V., Jr. DGAT and Triglyceride Synthesis: A New Target for Obesity Treatment? Trends Cardiovasc. Med. 2000, 10, 188–192. [Google Scholar] [CrossRef]
- Vluggens, A.; Andreoletti, P.; Viswakarma, N.; Jia, Y.; Matsumoto, K.; Kulik, W.; Khan, M.; Huang, J.; Guo, D.; Yu, S.; et al. Reversal of Mouse Acyl-CoA Oxidase 1 (ACOX1) Null Phenotype by Human ACOX1b Isoform. Lab Investig. 2010, 90, 696–708. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, J.; Yan, W.; Zhou, K.; Cao, Y.; Cai, W. P38alpha MAPK Antagonizing JNK to Control the Hepatic Fat Accumulation in Pediatric Patients Onset Intestinal Failure. Cell Death Dis. 2017, 8, e3110. [Google Scholar] [CrossRef]
- Lawan, A.; Shi, H.; Gatzke, F.; Bennett, A.M. Diversity and Specificity of the Mitogen-Activated Protein Kinase Phosphatase-1 Functions. Cell Mol. Life Sci. 2013, 70, 223–237. [Google Scholar] [CrossRef]
- Lawan, A.; Bennett, A.M. Mitogen-Activated Protein Kinase Regulation in Hepatic Metabolism. Trends Endocrinol. Metab. 2017, 28, 868–878. [Google Scholar] [CrossRef]
- Dorfman, K.; Carrasco, D.; Gruda, M.; Ryan, C.; Lira, S.A.; Bravo, R. Disruption of the Erp/Mkp-1 Gene Does Not Affect Mouse Development: Normal MAP Kinase Activity in ERP/MKP-1-Deficient Fibroblasts. Oncogene 1996, 13, 925–931. [Google Scholar] [PubMed]
- Li, J.; Sapper, T.N.; Mah, E.; Rudraiah, S.; Schill, K.E.; Chitchumroonchokchai, C.; Moller, M.V.; McDonald, J.D.; Rohrer, P.R.; Manautou, J.E.; et al. Green Tea Extract Provides Extensive Nrf2-Independent Protection Against Lipid Accumulation and NFkappaB Pro-Inflammatory Responses During Nonalcoholic Steatohepatitis in Mice Fed a High-Fat Diet. Mol. Nutr. Food Res. 2016, 60, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. Nonalcoholic Fatty Liver Disease (NAFLD) Activity Score and the Histopathologic Diagnosis in NAFLD: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 Kills Breast Cancer Cells in Vitro by Targeting Mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef] [PubMed]
- Benhalevy, D.; Gupta, S.K.; Danan, C.H.; Ghosal, S.; Sun, H.W.; Kazemier, H.G.; Paeschke, K.; Hafner, M.; Juranek, S.A. The Human CCHC-Type Zinc Finger Nucleic Acid-Binding Protein Binds G-Rich Elements in Target MRNA Coding Sequences and Promotes Translation. Cell Rep. 2017, 18, 2979–2990. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−∆∆Ct Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| 18s | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Acox1 | CAGGAAGAGCAAGGAAGTGG | CCTTTCTGGCTGATCCCATA |
| Cd36 | ATGGGCTGTGATCGGAACTG | TTTGCCACGTCATCTGGGTTT |
| Cpt1a | CAGAGGATGGACACTGTAAAGG | CGGCACTTCTTGATCAAGCC |
| Cpt2 | GGATAAACAGAATAAGCACACCA | GAAGGAACAAAGCGGATGAG |
| Mtor | ATTCAATCCATAGCCCCGTC | TGCATCACTCGTTCATCCTG |
| Pck1 | TCTCTGATCCAGACCTTCCAA | GAAGTCCAGACCGTTATGCAG |
| Pparg | CCAGAGTCTGCTGATCTGCG | GCCACCTCTTTGCTCTGATC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Wang, X.; Ackerman, W.E., IV; Batty, A.J.; Kirk, S.G.; White, W.M.; Wang, X.; Anastasakis, D.; Samavati, L.; Buhimschi, I.; et al. Dysregulation of Lipid Metabolism in Mkp-1 Deficient Mice during Gram-Negative Sepsis. Int. J. Mol. Sci. 2018, 19, 3904. https://doi.org/10.3390/ijms19123904
Li J, Wang X, Ackerman WE IV, Batty AJ, Kirk SG, White WM, Wang X, Anastasakis D, Samavati L, Buhimschi I, et al. Dysregulation of Lipid Metabolism in Mkp-1 Deficient Mice during Gram-Negative Sepsis. International Journal of Molecular Sciences. 2018; 19(12):3904. https://doi.org/10.3390/ijms19123904
Chicago/Turabian StyleLi, Jinhui, Xiantao Wang, William E. Ackerman, IV, Abel J. Batty, Sean G. Kirk, William M. White, Xianxi Wang, Dimitrios Anastasakis, Lobelia Samavati, Irina Buhimschi, and et al. 2018. "Dysregulation of Lipid Metabolism in Mkp-1 Deficient Mice during Gram-Negative Sepsis" International Journal of Molecular Sciences 19, no. 12: 3904. https://doi.org/10.3390/ijms19123904