Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors


Abstract
:
1. Introduction
2. Neurodevelopmental Defects
2.1. Pumilio
2.2. Staufen
2.3. Insulin-Like Growth Factor 2 mRNA-Binding Protein (IGF2BP)
2.4. Fragile-X Mental Retardation Protein (FMRP)
2.5. Src-Associated Substrate in Mitosis of 68 kDa (Sam68)
2.6. Cytoplasmic Polyadenylation Element Binding (CPEB)
3. Paraneoplastic Syndromes
3.1. Neuro-Oncological Ventral Antigen ( NOVA)
3.2. Embryonic Lethal/Abnormal Vision-Like (Hu/ELAVL)
4. Early-Onset Neurodegeneration
Survival Motor Neuron (SMN)
5. Late-Onset Neurodegeneration
5.1. TAR DNA-Binding Protein 43 (TDP43)
5.2. Fused in Sarcoma/Translocated in Liposarcoma (FUS/TLS)
5.3. TATA-Box Binding Protein Associated Factor (TAF) 15
5.4. T-Cell Intracellular Antigen (TIA) 1/TIA1-Related/Like Protein (TIAR)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| ALS | Familial Amyotrophic Lateral Sclerosis |
| APA | Alternative Polyadenylation |
| ASD | Autism Spectrum Disorder |
| BDNF | Brain-Derived Neurotrophic Factor |
| BioID | Biotin Identification |
| CAMK2A | Calcium/Calmodulin-Dependent Protein Kinase Type II alpha |
| CPE | Cytoplasmic Polyadenylation Element |
| CPEB | Cytoplasmic Polyadenylation Element Binding |
| DLB | Dementia with Lewy Bodies |
| dsRBD | Double-stranded RNA-Binding Domain |
| eIF | Eukaryotic Initiation Factor |
| ELAV | Embryonic Lethal Abnormal Vision |
| FMRP | Fragile-X Mental Retardation Protein |
| FTD | Frontotemporal Dementia |
| FTLD | Frontotemporallobar Degeneration |
| FUS | Fused in Sarcoma |
| FXS | Fragile-X Syndrome |
| FXTAS | Fragile-X-Associated Tremor/Ataxia Syndrome |
| GMC | Ganglion Mother Cell |
| GWAS | Genome Wide Association Study |
| HD | Huntington’s Disease |
| HITS-CLIP | Cross-Linking Immunoprecipitation High-Throughput Sequencing |
| HNRNP | Heterogeneous Nuclear Ribonucleoprotein |
| IGF2BP | Insulin-Like Growth Factor-2 mRNA-Binding Protein |
| KH | (HNRNP) K-Homology Domain |
| KHDRBS1 | KH Domain-Containing, RNA Binding, Signal Transduction Associated 1 |
| KO | Knockout |
| LTD | Long-Term Depression |
| LTP | Long-Term Potentiation |
| miRNA | MicroRNA |
| MN | Motor Neuron |
| NMJ | Neuromuscular Junction |
| NOVA | Neuro-Oncological Ventral Antigen |
| NPCs | Neural Progenitor Cells |
| P-body | Processing Body |
| PD | Parkinson’s Disease |
| PEM/PSN | Paraneoplastic Encephalomyelopathy/Paraneoplastic Sensory Neuropathy |
| PND | Paraneoplastic Neurodegeneration |
| POMA | Paraneoplastic Opsoclonus Myoclonus Ataxia |
| PSD | Post-Synaptic Density |
| PUM | Pumilio |
| RBD | RNA-Binding Domain |
| RBP | RNA-Binding Protein |
| RNAi | RNA interference |
| RNP | Ribonucleoprotein |
| ROS | Reactive Oxygen Species |
| RRM | RNA Recognition Motif |
| Sam68 | Src-Associated Substrate in Mitosis of 68 kDa |
| SCA1 | Spinocerebellar Ataxia type 1 |
| SCI | Spinal Cord Injury |
| SG | Stress Granule |
| SMA | Spinal Muscular Atrophy |
| SMD | Staufen-Mediated Decay |
| SMN | Survival Motor Neuron |
| snRNPs | Small Nuclear Ribonucleoproteins |
| STAR | Signal Transduction Activator of RNA |
| Stau | Staufen |
| TAF | TBP-Associated Factor |
| TDP43 | Transactive Response DNA Binding Protein 43 kDa |
| TIA1 | T-cell Intracellular Antigen 1 |
| TIAR | TIA1-Related/Like Protein |
| TLE | Temporal Lobe Epilepsy |
| TLS | Translocated in Liposarcoma |
| UTR | Untranslated Region |
References
- Azevedo, F.A.; Carvalho, L.R.; Grinberg, L.T.; Farfel, J.M.; Ferretti, R.E.; Leite, R.E.; Jacob Filho, W.; Lent, R.; Herculano-Houzel, S. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 2009, 513, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- De la Grange, P.; Gratadou, L.; Delord, M.; Dutertre, M.; Auboeuf, D. Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 2010, 38, 2825–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.C.; Zhang, C.; Shah, J.K.; Kulkarni, A.V.; Kalsotra, A.; Cooper, T.A.; Johnson, J.M. Expression of 24,426 human alternative splicing events and predicted cis regulation in 48 tissues and cell lines. Nat. Genet. 2008, 40, 1416–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, T.A.; Schweitzer, A.C.; Chen, T.X.; Staples, M.K.; Lu, G.; Wang, H.; Williams, A.; Blume, J.E. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biol. 2007, 8, R64. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. RNA binding proteins: A common denominator of neuronal function and dysfunction. Neurosci. Bull. 2014, 30, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Daubner, G.M.; Clery, A.; Allain, F.H. Rrm-RNA recognition: Nmr or crystallography...and new findings. Curr. Opin. Struct. Biol. 2013, 23, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, G.; Taylor, I.A.; Ramos, A. Kh-RNA interactions: Back in the groove. Curr. Opin. Struct. Biol. 2015, 30, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.D.; Mansfield, R.E.; Leung, W.; Vaz, P.M.; Loughlin, F.E.; Grant, R.P.; Mackay, J.P. Characterization of a family of ranbp2-type zinc fingers that can recognize single-stranded RNA. J. Mol. Biol. 2011, 407, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Gleghorn, M.L.; Maquat, L.E. ‘Black sheep’ that don’t leave the double-stranded RNA-binding domain fold. Trends Biochem. Sci. 2014, 39, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvelin, A.I.; Noerenberg, M.; Davis, I.; Castello, A. The new (dis)order in RNA regulation. Cell Commun. Signal 2016, 14, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protter, D.S.; Parker, R. Principles and properties of stress granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Cajigas, I.J.; Tushev, G.; Will, T.J.; tom Dieck, S.; Fuerst, N.; Schuman, E.M. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 2012, 74, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Zappulo, A.; van den Bruck, D.; Ciolli Mattioli, C.; Franke, V.; Imami, K.; McShane, E.; Moreno-Estelles, M.; Calviello, L.; Filipchyk, A.; Peguero-Sanchez, E.; et al. Rna localization is a key determinant of neurite-enriched proteome. Nat. Commun. 2017, 8, 583. [Google Scholar] [CrossRef] [PubMed]
- Ainsley, J.A.; Drane, L.; Jacobs, J.; Kittelberger, K.A.; Reijmers, L.G. Functionally diverse dendritic mrnas rapidly associate with ribosomes following a novel experience. Nat. Commun. 2014, 5, 4510. [Google Scholar] [CrossRef] [PubMed]
- Shigeoka, T.; Jung, H.; Jung, J.; Turner-Bridger, B.; Ohk, J.; Lin, J.Q.; Amieux, P.S.; Holt, C.E. Dynamic axonal translation in developing and mature visual circuits. Cell 2016, 166, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Batish, M.; van den Bogaard, P.; Kramer, F.R.; Tyagi, S. Neuronal mrnas travel singly into dendrites. Proc. Natl. Acad. Sci. USA 2012, 109, 4645–4650. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, A.R.; Wu, B.; Singer, R.H. Single beta-actin mRNA detection in neurons reveals a mechanism for regulating its translatability. Science 2014, 343, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zheng, Y.; Wang, Y.; Katz, Z.; Liu, X.; Chen, S.; Singer, R.H.; Gu, W. Specific interaction of kif11 with zbp1 regulates the transport of beta-actin mRNA and cell motility. J. Cell Sci. 2015, 128, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Shan, Z.; Yang, Y.; Liu, C.; Li, J.; Luo, Z.G.; Zhang, M.; Cai, Y.; Wen, W.; Wang, W. The structural basis of miranda-mediated staufen localization during drosophila neuroblast asymmetric division. Nat. Commun. 2015, 6, 8381. [Google Scholar] [CrossRef] [PubMed]
- Schuldt, A.J.; Adams, J.H.; Davidson, C.M.; Micklem, D.R.; Haseloff, J.; St Johnston, D.; Brand, A.H. Miranda mediates asymmetric protein and RNA localization in the developing nervous system. Genes Dev. 1998, 12, 1847–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, Y.; Dohmae, N.; Hirokawa, N. Kinesin transports RNA: Isolation and characterization of an RNA-transporting granule. Neuron 2004, 43, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Schenck, A.; Bardoni, B.; Moro, A.; Bagni, C.; Mandel, J.L. A highly conserved protein family interacting with the fragile × mental retardation protein (fmrp) and displaying selective interactions with fmrp-related proteins fxr1p and fxr2p. Proc. Natl. Acad. Sci. USA 2001, 98, 8844–8849. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.Y.; Lorenz, L.; Richter, J.D. Translational control by neuroguidin, a eukaryotic initiation factor 4e and cpeb binding protein. Mol. Cell. Biol. 2006, 26, 4277–4287. [Google Scholar] [CrossRef] [PubMed]
- Zahr, S.K.; Yang, G.; Kazan, H.; Borrett, M.J.; Yuzwa, S.A.; Voronova, A.; Kaplan, D.R.; Miller, F.D. A translational repression complex in developing mammalian neural stem cells that regulates neuronal specification. Neuron 2018, 97, 520–537. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. Fmrp stalls ribosomal translocation on mrnas linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Richter, J.D. Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Mol. Cell 2006, 24, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, A.S.; Janusz-Kaminska, A.; Switon, K.; Hawthorne, A.L.; Perycz, M.; Urbanska, M.; Bassell, G.J.; Jaworski, J. Zbp1 phosphorylation at serine 181 regulates its dendritic transport and the development of dendritic trees of hippocampal neurons. Sci. Rep. 2017, 7, 1876. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, U.; Nalavadi, V.; Nakamoto, M.; Pallas, D.C.; Ceman, S.; Bassell, G.J.; Warren, S.T. Fmrp phosphorylation reveals an immediate-early signaling pathway triggered by group i mglur and mediated by pp2a. J. Neurosci. 2007, 27, 14349–14357. [Google Scholar] [CrossRef] [PubMed]
- Coffee, R.L., Jr.; Williamson, A.J.; Adkins, C.M.; Gray, M.C.; Page, T.L.; Broadie, K. In vivo neuronal function of the fragile × mental retardation protein is regulated by phosphorylation. Hum. Mol. Genet. 2012, 21, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Jobert, L.; Argentini, M.; Tora, L. Prmt1 mediated methylation of taf15 is required for its positive gene regulatory function. Exp. Cell Res. 2009, 315, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Spassov, D.S.; Jurecic, R. Cloning and comparative sequence analysis of pum1 and pum2 genes, human members of the pumilio family of RNA-binding proteins. Gene 2002, 299, 195–204. [Google Scholar] [CrossRef]
- Gerber, A.P.; Luschnig, S.; Krasnow, M.A.; Brown, P.O.; Herschlag, D. Genome-wide identification of mrnas associated with the translational regulator pumilio in drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2006, 103, 4487–4492. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D.; Bartel, D.P.; Lehmann, R.; Williamson, J.R. The pumilio-rna interaction: A single rna-binding domain monomer recognizes a bipartite target sequence. Biochemistry 1999, 38, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, C.A.; Qiu, C.; Arvola, R.M.; Lou, T.F.; Killingsworth, J.; Campbell, Z.T.; Tanaka Hall, T.M.; Goldstrohm, A.C. Drosophila nanos acts as a molecular clamp that modulates the RNA-binding and repression activities of pumilio. Elife 2016, 5, e17096. [Google Scholar] [CrossRef] [PubMed]
- White, E.K.; Moore-Jarrett, T.; Ruley, H.E. Pum2, a novel murine puf protein, and its consensus RNA-binding site. RNA 2001, 7, 1855–1866. [Google Scholar] [PubMed]
- Cao, Q.; Padmanabhan, K.; Richter, J.D. Pumilio 2 controls translation by competing with eif4e for 7-methyl guanosine cap recognition. RNA 2010, 16, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Vaccani, A.; Xie, Y.; Dahm, R.; Karra, D.; Kiebler, M.A.; Macchi, P. Dendritic localization of the translational repressor pumilio 2 and its contribution to dendritic stress granules. J. Neurosci. 2006, 26, 6496–6508. [Google Scholar] [CrossRef] [PubMed]
- Menon, K.P.; Sanyal, S.; Habara, Y.; Sanchez, R.; Wharton, R.P.; Ramaswami, M.; Zinn, K. The translational repressor pumilio regulates presynaptic morphology and controls postsynaptic accumulation of translation factor eif-4e. Neuron 2004, 44, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Arvola, R.M.; Weidmann, C.A.; Tanaka Hall, T.M.; Goldstrohm, A.C. Combinatorial control of messenger rnas by pumilio, nanos and brain tumor proteins. RNA Biol. 2017, 14, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Dubnau, J.; Chiang, A.S.; Grady, L.; Barditch, J.; Gossweiler, S.; McNeil, J.; Smith, P.; Buldoc, F.; Scott, R.; Certa, U.; et al. The staufen/pumilio pathway is involved in drosophila long-term memory. Curr. Biol. 2003, 13, 286–296. [Google Scholar] [CrossRef]
- Ye, B.; Petritsch, C.; Clark, I.E.; Gavis, E.R.; Jan, L.Y.; Jan, Y.N. Nanos and pumilio are essential for dendrite morphogenesis in drosophila peripheral neurons. Curr. Biol. 2004, 14, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Muraro, N.I.; Weston, A.J.; Gerber, A.P.; Luschnig, S.; Moffat, K.G.; Baines, R.A. Pumilio binds para mRNA and requires nanos and brat to regulate sodium current in drosophila motoneurons. J. Neurosci. 2008, 28, 2099–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhu, T.; Chen, Y.; Xu, E.Y. Loss of preimplantation embryo resulting from a pum1 gene trap mutation. Biochem. Biophys. Res. Commun. 2015, 462, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gennarino, V.A.; Singh, R.K.; White, J.J.; De Maio, A.; Han, K.; Kim, J.Y.; Jafar-Nejad, P.; di Ronza, A.; Kang, H.; Sayegh, L.S.; et al. Pumilio1 haploinsufficiency leads to sca1-like neurodegeneration by increasing wild-type ataxin1 levels. Cell 2015, 160, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Gennarino, V.A.; Palmer, E.E.; McDonell, L.M.; Wang, L.; Adamski, C.J.; Koire, A.; See, L.; Chen, C.A.; Schaaf, C.P.; Rosenfeld, J.A.; et al. A mild pum1 mutation is associated with adult-onset ataxia, whereas haploinsufficiency causes developmental delay and seizures. Cell 2018, 172, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, D.; Xia, J.; Han, W.; Cui, X.; Neuenkirchen, N.; Hermes, G.; Sestan, N.; Lin, H. Post-transcriptional regulation of mouse neurogenesis by pumilio proteins. Genes Dev. 2017, 31, 1354–1369. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Schoderboeck, L.; Gingl, E.; Luzi, E.; Riefler, J.; Di Leva, F.; Karra, D.; Thomas, S.; Kiebler, M.A.; Macchi, P. Mammalian pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc. Natl. Acad. Sci. USA 2010, 107, 3222–3227. [Google Scholar] [CrossRef] [PubMed]
- Follwaczny, P.; Schieweck, R.; Riedemann, T.; Demleitner, A.; Straub, T.; Klemm, A.H.; Bilban, M.; Sutor, B.; Popper, B.; Kiebler, M.A. Pumilio2-deficient mice show a predisposition for epilepsy. Dis. Model. Mech. 2017, 10, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Huang, H.; Huang, Y.Y.; Yuan, J.X.; Zhou, X.; Chen, Y.M. Reduced pumilio-2 expression in patients with temporal lobe epilepsy and in the lithium-pilocarpine induced epilepsy rat model. Epilepsy Behav. 2015, 50, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Siemen, H.; Colas, D.; Heller, H.C.; Brustle, O.; Pera, R.A. Pumilio-2 function in the mouse nervous system. PLoS ONE 2011, 6, e25932. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Fortes, P.; Beloso, A.; Dotti, C.; Ortin, J. A human sequence homologue of staufen is an rna-binding protein that is associated with polysomes and localizes to the rough endoplasmic reticulum. Mol. Cell. Biol. 1999, 19, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Wickham, L.; Duchaine, T.; Luo, M.; Nabi, I.R.; DesGroseillers, L. Mammalian staufen is a double-stranded-RNA- and tubulin-binding protein which localizes to the rough endoplasmic reticulum. Mol. Cell. Biol. 1999, 19, 2220–2230. [Google Scholar] [CrossRef] [PubMed]
- Buchner, G.; Bassi, M.T.; Andolfi, G.; Ballabio, A.; Franco, B. Identification of a novel homolog of the drosophila staufen protein in the chromosome 8q13-q21.1 region. Genomics 1999, 62, 113–118. [Google Scholar] [CrossRef] [PubMed]
- St Johnston, D.; Beuchle, D.; Nusslein-Volhard, C. Staufen, a gene required to localize maternal rNAS in the drosophila egg. Cell 1991, 66, 51–63. [Google Scholar] [CrossRef]
- Duchaine, T.F.; Hemraj, I.; Furic, L.; Deitinghoff, A.; Kiebler, M.A.; DesGroseillers, L. Staufen2 isoforms localize to the somatodendritic domain of neurons and interact with different organelles. J. Cell Sci. 2002, 115, 3285–3295. [Google Scholar] [PubMed]
- Schwarz, M.S.; Wang, T.J.; Miller, H.C.; Esselman, W.J. Detection of thy-1.2 membrane complexes shed from thymocytes and lymphoma cells by an immunoradiometric assay. Mol. Immunol. 1980, 17, 1381–1388. [Google Scholar] [CrossRef]
- Li, P.; Yang, X.; Wasser, M.; Cai, Y.; Chia, W. Inscuteable and staufen mediate asymmetric localization and segregation of prospero RNA during drosophila neuroblast cell divisions. Cell 1997, 90, 437–447. [Google Scholar] [CrossRef]
- Vessey, J.P.; Amadei, G.; Burns, S.E.; Kiebler, M.A.; Kaplan, D.R.; Miller, F.D. An asymmetrically localized staufen2-dependent RNA complex regulates maintenance of mammalian neural stem cells. Cell Stem Cell 2012, 11, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Kusek, G.; Campbell, M.; Doyle, F.; Tenenbaum, S.A.; Kiebler, M.; Temple, S. Asymmetric segregation of the double-stranded RNA binding protein staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell 2012, 11, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; Maher-Laporte, M.; Topolnik, L.; Laurent, C.E.; Sossin, W.; Desgroseillers, L.; Lacaille, J.C. Staufen1 regulation of protein synthesis-dependent long-term potentiation and synaptic function in hippocampal pyramidal cells. Mol. Cell. Biol. 2008, 28, 2896–2907. [Google Scholar] [CrossRef] [PubMed]
- Vessey, J.P.; Macchi, P.; Stein, J.M.; Mikl, M.; Hawker, K.N.; Vogelsang, P.; Wieczorek, K.; Vendra, G.; Riefler, J.; Tubing, F.; et al. A loss of function allele for murine staufen1 leads to impairment of dendritic staufen1-rnp delivery and dendritic spine morphogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 16374–16379. [Google Scholar] [CrossRef] [PubMed]
- Goetze, B.; Tuebing, F.; Xie, Y.; Dorostkar, M.M.; Thomas, S.; Pehl, U.; Boehm, S.; Macchi, P.; Kiebler, M.A. The brain-specific double-stranded RNA-binding protein staufen2 is required for dendritic spine morphogenesis. J. Cell Biol. 2006, 172, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Furic, L.; Maher-Laporte, M.; DesGroseillers, L. A genome-wide approach identifies distinct but overlapping subsets of cellular mrnas associated with staufen1- and staufen2-containing ribonucleoprotein complexes. RNA 2008, 14, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Maher-Laporte, M.; DesGroseillers, L. Genome wide identification of staufen2-bound mrnas in embryonic rat brains. BMB Rep. 2010, 43, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Heraud-Farlow, J.E.; Sharangdhar, T.; Li, X.; Pfeifer, P.; Tauber, S.; Orozco, D.; Hormann, A.; Thomas, S.; Bakosova, A.; Farlow, A.R.; et al. Staufen2 regulates neuronal target rnas. Cell Rep. 2013, 5, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Furic, L.; Desgroseillers, L.; Maquat, L.E. Mammalian staufen1 recruits upf1 to specific mRNA 3’utrs so as to elicit mRNA decay. Cell 2005, 120, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Dugre-Brisson, S.; Elvira, G.; Boulay, K.; Chatel-Chaix, L.; Mouland, A.J.; DesGroseillers, L. Interaction of staufen1 with the 5′ end of mRNA facilitates translation of these rnas. Nucleic Acids Res. 2005, 33, 4797–4812. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Gleghorn, M.L.; Maquat, L.E. Staufen2 functions in staufen1-mediated mRNA decay by binding to itself and its paralog and promoting upf1 helicase but not atpase activity. Proc. Natl. Acad. Sci. USA 2013, 110, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Kiebler, M.A.; Hemraj, I.; Verkade, P.; Kohrmann, M.; Fortes, P.; Marion, R.M.; Ortin, J.; Dotti, C.G. The mammalian staufen protein localizes to the somatodendritic domain of cultured hippocampal neurons: Implications for its involvement in mRNA transport. J. Neurosci. 1999, 19, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Kohrmann, M.; Luo, M.; Kaether, C.; DesGroseillers, L.; Dotti, C.G.; Kiebler, M.A. Microtubule-dependent recruitment of staufen-green fluorescent protein into large rna-containing granules and subsequent dendritic transport in living hippocampal neurons. Mol. Biol. Cell 1999, 10, 2945–2953. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; Miller, L.C.; Tartas, M.; McAdam, R.; Laplante, I.; Badeaux, F.; DesGroseillers, L.; Sossin, W.S.; Lacaille, J.C. Staufen 2 regulates mglur long-term depression and map1b mRNA distribution in hippocampal neurons. Learn. Mem. 2011, 18, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.G.; Martinez Tosar, L.J.; Loschi, M.; Pasquini, J.M.; Correale, J.; Kindler, S.; Boccaccio, G.L. Staufen recruitment into stress granules does not affect early mRNA transport in oligodendrocytes. Mol. Biol. Cell 2005, 16, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.H.; Nam, Y.J.; Kim, S.Y.; Kim, E.G.; Jeong, J.; Kim, H.K. The transport of staufen2-containing ribonucleoprotein complexes involves kinesin motor protein and is modulated by mitogen-activated protein kinase pathway. J. Neurochem. 2007, 102, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Kim, H.K. Role of staufen in dendritic mRNA transport and its modulation. Neurosci. Lett. 2006, 397, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.J.; Meulemans, D.; Vazquez, L.; Colaco, N.; Schuman, E. A role for a rat homolog of staufen in the transport of RNA to neuronal dendrites. Neuron 2001, 32, 463–475. [Google Scholar] [CrossRef]
- Thomas, M.G.; Martinez Tosar, L.J.; Desbats, M.A.; Leishman, C.C.; Boccaccio, G.L. Mammalian staufen 1 is recruited to stress granules and impairs their assembly. J. Cell Sci. 2009, 122, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.L.; Wachter, K.; Muhleck, B.; Pazaitis, N.; Kohn, M.; Lederer, M.; Huttelmaier, S. Insulin-like growth factor 2 mRNA-binding proteins (igf2bps): Post-transcriptional drivers of cancer progression? Cell Mol. Life Sci. 2013, 70, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; Nielsen, J.; Kristensen, M.A.; Koch, G.; Christiansen, J. Cytoplasmic trafficking of igf-ii mrna-binding protein by conserved kh domains. J. Cell Sci. 2002, 115, 2087–2097. [Google Scholar] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. Par-clip—A method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 2010, 41, 2034. [Google Scholar] [CrossRef] [PubMed]
- Conway, A.E.; Van Nostrand, E.L.; Pratt, G.A.; Aigner, S.; Wilbert, M.L.; Sundararaman, B.; Freese, P.; Lambert, N.J.; Sathe, S.; Liang, T.Y.; et al. Enhanced clip uncovers imp protein-RNA targets in human pluripotent stem cells important for cell adhesion and survival. Cell Rep. 2016, 15, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Hammer, N.A.; Nielsen, J.; Madsen, M.; Dalbaeck, C.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. Dwarfism and impaired gut development in insulin-like growth factor ii mRNA-binding protein 1-deficient mice. Mol. Cell. Biol. 2004, 24, 4448–4464. [Google Scholar] [CrossRef] [PubMed]
- Dai, N.; Zhao, L.; Wrighting, D.; Kramer, D.; Majithia, A.; Wang, Y.; Cracan, V.; Borges-Rivera, D.; Mootha, V.K.; Nahrendorf, M.; et al. Igf2bp2/imp2-deficient mice resist obesity through enhanced translation of ucp1 mRNA and other mrnas encoding mitochondrial proteins. Cell Metab. 2015, 21, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Boylan, K.L.; Mische, S.; Li, M.; Marques, G.; Morin, X.; Chia, W.; Hays, T.S. Motility screen identifies drosophila igf-ii mRNA-binding protein-zipcode-binding protein acting in oogenesis and synaptogenesis. PLoS Genet. 2008, 4, e36. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, K.; Fainsod, A.; Kalcheim, C.; Yisraeli, J.K. The RNA-binding protein vg1 rbp is required for cell migration during early neural development. Development 2003, 130, 5649–5661. [Google Scholar] [CrossRef] [PubMed]
- Nishino, J.; Kim, S.; Zhu, Y.; Zhu, H.; Morrison, S.J. A network of heterochronic genes including imp1 regulates temporal changes in stem cell properties. Elife 2013, 2, e00924. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Kishi, Y.; Gotoh, Y. Imp2 regulates differentiation potentials of mouse neocortical neural precursor cells. Genes Cells 2013, 18, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Tiruchinapalli, D.M.; Oleynikov, Y.; Kelic, S.; Shenoy, S.M.; Hartley, A.; Stanton, P.K.; Singer, R.H.; Bassell, G.J. Activity-dependent trafficking and dynamic localization of zipcode binding protein 1 and beta-actin mRNA in dendrites and spines of hippocampal neurons. J. Neurosci. 2003, 23, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Eom, T.; Oleynikov, Y.; Shenoy, S.M.; Liebelt, D.A.; Dictenberg, J.B.; Singer, R.H.; Bassell, G.J. Neurotrophin-induced transport of a beta-actin mrnp complex increases beta-actin levels and stimulates growth cone motility. Neuron 2001, 31, 261–275. [Google Scholar] [CrossRef]
- Jonson, L.; Vikesaa, J.; Krogh, A.; Nielsen, L.K.; Hansen, T.; Borup, R.; Johnsen, A.H.; Christiansen, J.; Nielsen, F.C. Molecular composition of imp1 ribonucleoprotein granules. Mol. Cell. Proteom. 2007, 6, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Weidensdorfer, D.; Stohr, N.; Baude, A.; Lederer, M.; Kohn, M.; Schierhorn, A.; Buchmeier, S.; Wahle, E.; Huttelmaier, S. Control of c-myc mRNA stability by igf2bp1-associated cytoplasmic rnps. RNA 2009, 15, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Rouanet, J.P.; Donlin-Asp, P.G.; Guo, P.; Zhang, H.; Singer, R.H.; Rossoll, W.; Bassell, G.J. Dynamics of survival of motor neuron (smn) protein interaction with the mrna-binding protein imp1 facilitates its trafficking into motor neuron axons. Dev. Neurobiol. 2014, 74, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Hüttelmaier, S.; Zenklusen, D.; Lederer, M.; Dictenberg, J.; Lorenz, M.; Meng, X.; Bassell, G.J.; Condeelis, J.; Singer, R.H. Spatial regulation of beta-actin translation by src-dependent phosphorylation of zbp1. Nature 2005, 438, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.J.; Wu, B.; Buxbaum, A.R.; Das, S.; Tsai, A.; English, B.P.; Grimm, J.B.; Lavis, L.D.; Singer, R.H. Glutamate-induced RNA localization and translation in neurons. Proc. Natl. Acad. Sci. USA 2016, 113, E6877–E6886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, T.; Antar, L.N.; Singer, R.H.; Bassell, G.J. Localization of a beta-actin messenger ribonucleoprotein complex with zipcode-binding protein modulates the density of dendritic filopodia and filopodial synapses. J. Neurosci. 2003, 23, 10433–10444. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Sasaki, Y.; Wen, Z.; Bassell, G.J.; Zheng, J.Q. An essential role for beta-actin mRNA localization and translation in Ca2+-dependent growth cone guidance. Nat. Neurosci. 2006, 9, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.M.; van Horck, F.P.; Lin, A.C.; Allison, R.; Standart, N.; Holt, C.E. Asymmetrical beta-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat. Neurosci. 2006, 9, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Welshhans, K.; Bassell, G.J. Netrin-1-induced local beta-actin synthesis and growth cone guidance requires zipcode binding protein 1. J. Neurosci. 2011, 31, 9800–9813. [Google Scholar] [CrossRef] [PubMed]
- Perycz, M.; Urbanska, A.S.; Krawczyk, P.S.; Parobczak, K.; Jaworski, J. Zipcode binding protein 1 regulates the development of dendritic arbors in hippocampal neurons. J. Neurosci. 2011, 31, 5271–5285. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.J.; Willis, D.E.; Xu, M.; Tep, C.; Jiang, C.; Yoo, S.; Schanen, N.C.; Kirn-Safran, C.B.; van Minnen, J.; English, A.; et al. Limited availability of zbp1 restricts axonal mRNA localization and nerve regeneration capacity. EMBO J. 2011, 30, 4665–4677. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.T.; Wilkinson, K.D.; Reines, D.; Warren, S.T. Fmr1 protein: Conserved rnp family domains and selective RNA binding. Science 1993, 262, 563–566. [Google Scholar] [CrossRef]
- Hinds, H.L.; Ashley, C.T.; Sutcliffe, J.S.; Nelson, D.L.; Warren, S.T.; Housman, D.E.; Schalling, M. Tissue specific expression of fmr-1 provides evidence for a functional role in fragile × syndrome. Nat. Genet. 1993, 3, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. The protein product of the fragile × gene, fmr1, has characteristics of an RNA-binding protein. Cell 1993, 74, 291–298. [Google Scholar] [CrossRef]
- Feng, Y.; Gutekunst, C.A.; Eberhart, D.E.; Yi, H.; Warren, S.T.; Hersch, S.M. Fragile × mental retardation protein: Nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 1997, 17, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, D.E.; Malter, H.E.; Feng, Y.; Warren, S.T. The fragile × mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 1996, 5, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Hollingworth, D.; Pastore, A. G-quartet-dependent recognition between the fmrp rgg box and RNA. RNA 2003, 9, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Choi, M.; Siomi, M.C.; Nussbaum, R.L.; Dreyfuss, G. Essential role for kh domains in RNA binding: Impaired RNA binding by a mutation in the kh domain of fmr1 that causes fragile × syndrome. Cell 1994, 77, 33–39. [Google Scholar] [CrossRef]
- De Boulle, K.; Verkerk, A.J.; Reyniers, E.; Vits, L.; Hendrickx, J.; Van Roy, B.; Van den Bos, F.; de Graaff, E.; Oostra, B.A.; Willems, P.J. A point mutation in the fmr-1 gene associated with fragile × mental retardation. Nat. Genet. 1993, 3, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Zang, J.B.; Nosyreva, E.D.; Spencer, C.M.; Volk, L.J.; Musunuru, K.; Zhong, R.; Stone, E.F.; Yuva-Paylor, L.A.; Huber, K.M.; Paylor, R.; et al. A mouse model of the human fragile × syndrome i304n mutation. PLoS Genet. 2009, 5, e1000758. [Google Scholar] [CrossRef] [PubMed]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. Fmrp targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Chopra, P.; Suhl, J.A.; Warren, S.T.; Bassell, G.J. Identification of consensus binding sites clarifies fmrp binding determinants. Nucleic Acids Res. 2016, 44, 6649–6659. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Maihara, T.; Hattori, H.; Furusho, K.; Okazawa, H.; Ishizu, K.; Yonekura, Y. [18f]-fluorodeoxyglucose-positron emission tomography findings in protein infants with severe periventricular leukomalacia and hypsarrhythmia. Eur. J. Pediatr. 1997, 156, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G.; Warren, S.T. Variation of the cgg repeat at the fragile × site results in genetic instability: Resolution of the sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Pieretti, M.; Zhang, F.P.; Fu, Y.H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the fmr-1 gene in fragile × syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P. Identification of a gene (fmr-1) containing a cgg repeat coincident with a breakpoint cluster region exhibiting length variation in fragile × syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Sutcliffe, J.S.; Nelson, D.L.; Zhang, F.; Pieretti, M.; Caskey, C.T.; Saxe, D.; Warren, S.T. DNA methylation represses fmr-1 transcription in fragile × syndrome. Hum. Mol. Genet. 1992, 1, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Colak, D.; Zaninovic, N.; Cohen, M.S.; Rosenwaks, Z.; Yang, W.-Y.; Gerhardt, J.; Disney, M.D.; Jaffrey, S.R. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile × syndrome. Science 2014, 343, 1002–1005. [Google Scholar] [CrossRef] [PubMed]
- Drozd, M.; Bardoni, B.; Capovilla, M. Modeling fragile × syndrome in drosophila. Front. Mol. Neurosci. 2018, 11, 124. [Google Scholar] [CrossRef] [PubMed]
- Dahlhaus, R. Of men and mice: Modeling the fragile × syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, C.; Shang, S.; Cai, Y.; Deng, X.; Zhang, J.; Shao, F.; Zhu, D.; Liu, Y.; Chen, G.; et al. Loss of fmrp impaired hippocampal long-term plasticity and spatial learning in rats. Front. Mol. Neurosci. 2017, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.M.; Green, J.R.; Veeraragavan, S.; Yuva, L.; McCoy, A.; Wu, Y.; Warren, J.; Little, L.; Ji, D.; Cui, X.; et al. Fmr1 and nlgn3 knockout rats: Novel tools for investigating autism spectrum disorders. Behav. Neurosci. 2014, 128, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Fmr1 knockout mice: A model to study fragile × mental retardation. The dutch-belgian fragile × consortium. Cell 1994, 78, 23–33. [Google Scholar]
- Mientjes, E.J.; Nieuwenhuizen, I.; Kirkpatrick, L.; Zu, T.; Hoogeveen-Westerveld, M.; Severijnen, L.; Rife, M.; Willemsen, R.; Nelson, D.L.; Oostra, B.A. The generation of a conditional fmr1 knock out mouse model to study fmrp function in vivo. Neurobiol. Dis. 2006, 21, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Bailey, A.M.; Matthies, H.J.; Renden, R.B.; Smith, M.A.; Speese, S.D.; Rubin, G.M.; Broadie, K. Drosophila fragile x-related gene regulates the map1b homolog futsch to control synaptic structure and function. Cell 2001, 107, 591–603. [Google Scholar] [CrossRef]
- Dockendorff, T.C.; Su, H.S.; McBride, S.M.; Yang, Z.; Choi, C.H.; Siwicki, K.K.; Sehgal, A.; Jongens, T.A. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 2002, 34, 973–984. [Google Scholar] [CrossRef]
- Sabaratnam, M. Pathological and neuropathological findings in two males with fragile-x syndrome. J. Intellect. Disabil. Res. JIDR 2000, 44 Pt 1, 81–85. [Google Scholar] [CrossRef]
- Cheng, Y.; Corbin, J.G.; Levy, R.J. Programmed cell death is impaired in the developing brain of fmr1 mutants. Dev. Neurosci. 2013, 35, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Gatto, C.L.; Broadie, K. Fragile × mental retardation protein is required for programmed cell death and clearance of developmentally-transient peptidergic neurons. Dev. Biol. 2011, 356, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Irwin, S.A.; Patel, B.; Idupulapati, M.; Harris, J.B.; Crisostomo, R.A.; Larsen, B.P.; Kooy, F.; Willems, P.J.; Cras, P.; Kozlowski, P.B.; et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-x syndrome: A quantitative examination. Am. J. Med. Genet. 2001, 98, 161–167. [Google Scholar] [CrossRef]
- Grossman, A.W.; Aldridge, G.M.; Weiler, I.J.; Greenough, W.T. Local protein synthesis and spine morphogenesis: Fragile × syndrome and beyond. J. Neurosci. 2006, 26, 7151–7155. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Zhang, Y.Q.; Woodruff, E.; Broadie, K. The drosophila fragile × gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 2004, 14, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Martin, A.; Crespo, M.; Portera-Cailliau, C. Delayed stabilization of dendritic spines in fragile × mice. J. Neurosci. 2010, 30, 7793–7803. [Google Scholar] [CrossRef] [PubMed]
- Suresh, A.; Dunaevsky, A. Relationship between synaptic ampar and spine dynamics: Impairments in the fxs mouse. Cereb Cortex 2017, 27, 4244–4256. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Huot, M.E.; Tremblay, S.; Filion, C.; Labelle, Y.; Khandjian, E.W. Trapping of messenger RNA by fragile × mental retardation protein into cytoplasmic granules induces translation repression. Hum. Mol. Genet. 2002, 11, 3007–3017. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Sharma, M.R.; Shi, X.; Agrawal, R.K.; Joseph, S. Fragile × mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 2014, 54, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Fraser, C.E.; Mostovetsky, O.; Stefani, G.; Jones, T.A.; Eddy, S.R.; Darnell, R.B. Kissing complex rNAS mediate interaction between the fragile-x mental retardation protein kh2 domain and brain polyribosomes. Genes Dev. 2005, 19, 903–918. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Absher, D.; Eberhart, D.E.; Brown, V.; Malter, H.E.; Warren, S.T. Fmrp associates with polyribosomes as an mrnp, and the i304n mutation of severe fragile × syndrome abolishes this association. Mol. Cell 1997, 1, 109–118. [Google Scholar] [CrossRef]
- Napoli, I.; Mercaldo, V.; Boyl, P.P.; Eleuteri, B.; Zalfa, F.; De Rubeis, S.; Di Marino, D.; Mohr, E.; Massimi, M.; Falconi, M.; et al. The fragile × syndrome protein represses activity-dependent translation through cyfip1, a new 4e-bp. Cell 2008, 134, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Plante, I.; Davidovic, L.; Ouellet, D.L.; Gobeil, L.-A.; Tremblay, S.; Khandjian, E.W.; Provost, P. Dicer-derived micrornas are utilized by the fragile × mental retardation protein for assembly on target rNAS. J. Biomed. Biotechnol. 2006, 2006, 64347. [Google Scholar] [CrossRef] [PubMed]
- Muddashetty, R.S.; Nalavadi, V.C.; Gross, C.; Yao, X.; Xing, L.; Laur, O.; Warren, S.T.; Bassell, G.J. Reversible inhibition of psd-95 mRNA translation by mir-125a, fmrp phosphorylation, and mglur signaling. Mol. Cell 2011, 42, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, W.; Zhang, L.R.; Zhang, C.Y. Fmrp regulates mir196a-mediated repression of hoxb8 via interaction with the ago2 mid domain. Mol. Biosyst. 2014, 10, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by fmrp-associated micrornas mir-125b and mir-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Khayachi, A.; Gwizdek, C.; Poupon, G.; Alcor, D.; Chafai, M.; Cassé, F.; Maurin, T.; Prieto, M.; Folci, A.; De Graeve, F.; et al. Sumoylation regulates fmrp-mediated dendritic spine elimination and maturation. Nat. Commun. 2018, 9, 757. [Google Scholar] [CrossRef] [PubMed]
- Dolzhanskaya, N.; Merz, G.; Aletta, J.M.; Denman, R.B. Methylation regulates the intracellular protein-protein and protein-RNA interactions of fmrp. J. Cell Sci. 2006, 119, 1933–1946. [Google Scholar] [CrossRef] [PubMed]
- Siomi, M.C.; Higashijima, K.; Ishizuka, A.; Siomi, H. Casein kinase ii phosphorylates the fragile × mental retardation protein and modulates its biological properties. Mol. Cell. Biol. 2002, 22, 8438–8447. [Google Scholar] [CrossRef] [PubMed]
- Bartley, C.M.; O’Keefe, R.A.; Bordey, A. Fmrp s499 is phosphorylated independent of mtorc1-s6k1 activity. PLoS ONE 2014, 9, e96956. [Google Scholar] [CrossRef] [PubMed]
- Bartley, C.M.; O’Keefe, R.A.; Blice-Baum, A.; Mihailescu, M.R.; Gong, X.; Miyares, L.; Karaca, E.; Bordey, A. Mammalian fmrp s499 is phosphorylated by ck2 and promotes secondary phosphorylation of fmrp. eNeuro 2016, 3, ENEURO-0092. [Google Scholar] [CrossRef] [PubMed]
- Ceman, S.; O’Donnell, W.T.; Reed, M.; Patton, S.; Pohl, J.; Warren, S.T. Phosphorylation influences the translation state of fmrp-associated polyribosomes. Hum. Mol. Genet. 2003, 12, 3295–3305. [Google Scholar] [CrossRef] [PubMed]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Gresack, J.; Allis, C.D.; Darnell, R.B. Excess translation of epigenetic regulators contributes to fragile × syndrome and is alleviated by brd4 inhibition. Cell 2017, 170, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.T.; Ye, S.H.; Yang, H.X.; Zhou, Y.T.; Zhao, Q.H.; Sun, W.W.; Gao, M.M.; Yi, Y.H.; Long, Y.S. A novel role of fragile × mental retardation protein in pre-mRNA alternative splicing through RNA-binding protein 14. Neuroscience 2017, 349, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Alpatov, R.; Lesch, B.J.; Nakamoto-Kinoshita, M.; Blanco, A.; Chen, S.; Stutzer, A.; Armache, K.J.; Simon, M.D.; Xu, C.; Ali, M.; et al. A chromatin-dependent role of the fragile × mental retardation protein fmrp in the DNA damage response. Cell 2014, 157, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, C.; Barbon, A.; Barlati, S. Activity regulation of adenosine deaminases acting on RNA (adars). Mol. Neurobiol. 2012, 45, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, G.; Lundin, D.; Navon, R.; Ohman, M. Deregulation of the a-to-i RNA editing mechanism in psychiatric disorders. Hum. Mol. Genet. 2012, 21, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Bonini, D.; Lacoux, C.; Pacini, L.; Zingariello, M.; Sancillo, L.; Bosisio, D.; Salvi, V.; Mingardi, J.; La Via, L.; et al. Absence of the fragile × mental retardation protein results in defects of RNA editing of neuronal mrnas in mouse. RNA Biol. 2017, 14, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Jepson, J.E.; Savva, Y.A.; Pepper, A.S.; Reenan, R.A.; Jongens, T.A. Modulation of dadar-dependent RNA editing by the drosophila fragile × mental retardation protein. Nat. Neurosci. 2011, 14, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Shamay-Ramot, A.; Khermesh, K.; Porath, H.T.; Barak, M.; Pinto, Y.; Wachtel, C.; Zilberberg, A.; Lerer-Goldshtein, T.; Efroni, S.; Levanon, E.Y.; et al. Fmrp interacts with adar and regulates RNA editing, synaptic density and locomotor activity in zebrafish. PLoS Genet. 2015, 11, e1005702. [Google Scholar] [CrossRef] [PubMed]
- Strumbos, J.G.; Brown, M.R.; Kronengold, J.; Polley, D.B.; Kaczmarek, L.K. Fragile × mental retardation protein is required for rapid experience-dependent regulation of the potassium channel kv3.1b. J. Neurosci. 2010, 30, 10263–10271. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.Y.; Rotman, Z.; Blundon, J.A.; Cho, Y.; Cui, J.; Cavalli, V.; Zakharenko, S.S.; Klyachko, V.A. Fmrp regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via bk channels. Neuron 2013, 77, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Antar, L.N.; Li, C.; Zhang, H.; Carroll, R.C.; Bassell, G.J. Local functions for fmrp in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell Neurosci. 2006, 32, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Miyashiro, K.Y.; Beckel-Mitchener, A.; Purk, T.P.; Becker, K.G.; Barret, T.; Liu, L.; Carbonetto, S.; Weiler, I.J.; Greenough, W.T.; Eberwine, J. Rna cargoes associating with fmrp reveal deficits in cellular functioning in fmr1 null mice. Neuron 2003, 37, 417–431. [Google Scholar] [CrossRef]
- Barbee, S.A.; Estes, P.S.; Cziko, A.-M.; Hillebrand, J.; Luedeman, R.A.; Coller, J.M.; Johnson, N.; Howlett, I.C.; Geng, C.; Ueda, R.; et al. Staufen- and fmrp-containing neuronal rnps are structurally and functionally related to somatic p bodies. Neuron 2006, 52, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Flores, C.M.; Cervero, F.; Hargreaves, K.M. The RNA binding and transport proteins staufen and fragile × mental retardation protein are expressed by rat primary afferent neurons and localize to peripheral and central axons. Neuroscience 2006, 141, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Dictenberg, J.B.; Swanger, S.A.; Antar, L.N.; Singer, R.H.; Bassell, G.J. A direct role for fmrp in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile × syndrome. Dev. Cell 2008, 14, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dictenberg, J.B.; Ku, L.; Li, W.; Bassell, G.J.; Feng, Y. Dynamic association of the fragile × mental retardation protein as a messenger ribonucleoprotein between microtubules and polyribosomes. Mol. Biol. Cell 2008, 19, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Akins, M.R.; Berk-Rauch, H.E.; Kwan, K.Y.; Mitchell, M.E.; Shepard, K.A.; Korsak, L.I.; Stackpole, E.E.; Warner-Schmidt, J.L.; Sestan, N.; Cameron, H.A.; et al. Axonal ribosomes and mrnas associate with fragile × granules in adult rodent and human brains. Hum. Mol. Genet. 2017, 26, 192–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Taylor, S.J.; Shalloway, D. Specificity and determinants of sam68 RNA binding. Implications for the biological function of k homology domains. J. Biol. Chem. 1997, 272, 27274–27280. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Haga, I.; Li, Q.H.; Fujisawa, J. Identification of cellular mRNA targets for RNA-binding protein sam68. Nucleic Acids Res. 2002, 30, 5452–5464. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.J.; Anafi, M.; Pawson, T.; Shalloway, D. Functional interaction between c-src and its mitotic target, sam 68. J. Biol. Chem. 1995, 270, 10120–10124. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Messina, V.; Bianchi, E.; Barchi, M.; Vogel, G.; Moretti, C.; Palombi, F.; Stefanini, M.; Geremia, R.; Richard, S.; et al. Sam68 regulates translation of target mrnas in male germ cells, necessary for mouse spermatogenesis. J. Cell Biol. 2009, 185, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Younts, T.J.; Castillo, P.E.; Jordan, B.A. Rna-binding protein sam68 controls synapse number and local beta-actin mRNA metabolism in dendrites. Proc. Natl. Acad. Sci. USA 2013, 110, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Richard, S. Sam68, the kh domain-containing superstar. Biochim. Biophys. Acta 2003, 1653, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, L.; Qian, R.; Liu, J.; Yao, Y.; Jiang, Z.; Song, X.; Ren, J.; Zhang, F. Expression of sam68 associates with neuronal apoptosis and reactive astrocytes after spinal cord injury. Cell. Mol. Neurobiol. 2017, 37, 487–498. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Bielli, P.; Compagnucci, C.; Cesari, E.; Volpe, E.; Farioli Vecchioli, S.; Sette, C. Sam68 promotes self-renewal and glycolytic metabolism in mouse neural progenitor cells by modulating aldh1a3 pre-mrna 3′-end processing. Elife 2016, 5, e20750. [Google Scholar] [CrossRef] [PubMed]
- Ben Fredj, N.; Grange, J.; Sadoul, R.; Richard, S.; Goldberg, Y.; Boyer, V. Depolarization-induced translocation of the rna-binding protein sam68 to the dendrites of hippocampal neurons. J. Cell Sci. 2004, 117, 1079–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, T.; Wu, K.; Witte, H.; Hanno-Iijima, Y.; Glatter, T.; Richard, S.; Scheiffele, P. Sam68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell 2011, 147, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Achsel, T.; Massiello, A.; Chalfant, C.E.; Sette, C. The RNA-binding protein sam68 modulates the alternative splicing of bcl-x. J. Cell Biol. 2007, 176, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Baltimore, D. Sam68 is required for both nf-kappab activation and apoptosis signaling by the tnf receptor. Mol. Cell 2011, 43, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Grange, J.; Boyer, V.; Fabian-Fine, R.; Fredj, N.B.; Sadoul, R.; Goldberg, Y. Somatodendritic localization and mRNA association of the splicing regulatory protein sam68 in the hippocampus and cortex. J. Neurosci. Res. 2004, 75, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; He, J.J. Sam68 relocalization into stress granules in response to oxidative stress through complexing with tia-1. Exp. Cell Res. 2009, 315, 3381–3395. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in fxtas patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Pedrotti, S.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Stamm, S.; Manley, J.L.; Sette, C. The splicing regulator sam68 binds to a novel exonic splicing silencer and functions in smn2 alternative splicing in spinal muscular atrophy. EMBO J. 2010, 29, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Rochette, C.F.; Gilbert, N.; Simard, L.R. Smn gene duplication and the emergence of the smn2 gene occurred in distinct hominids: Smn2 is unique to homo sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, V.; Pelosi, L.; Bustamante, M.B.; Nobili, A.; Berardinelli, M.G.; D’Amelio, M.; Musaro, A.; Sette, C. Sam68 is a physiological regulator of smn2 splicing in spinal muscular atrophy. J. Cell Biol. 2015, 211, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Narcis, J.O.; Tapia, O.; Tarabal, O.; Piedrafita, L.; Caldero, J.; Berciano, M.T.; Lafarga, M. Accumulation of poly(a) RNA in nuclear granules enriched in sam68 in motor neurons from the smndelta7 mouse model of sma. Sci. Rep. 2018, 8, 9646. [Google Scholar] [CrossRef] [PubMed]
- Welk, J.F.; Charlesworth, A.; Smith, G.D.; MacNicol, A.M. Identification and characterization of the gene encoding human cytoplasmic polyadenylation element binding protein. Gene 2001, 263, 113–120. [Google Scholar] [CrossRef]
- Kurihara, Y.; Tokuriki, M.; Myojin, R.; Hori, T.; Kuroiwa, A.; Matsuda, Y.; Sakurai, T.; Kimura, M.; Hecht, N.B.; Uesugi, S. Cpeb2, a novel putative translational regulator in mouse haploid germ cells. Biol. Reprod. 2003, 69, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Theis, M.; Si, K.; Kandel, E.R. Two previously undescribed members of the mouse cpeb family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc. Natl. Acad. Sci. USA 2003, 100, 9602–9607. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Klann, E. Making synaptic plasticity and memory last: Mechanisms of translational regulation. Genes Dev. 2009, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Kan, M.C.; Lin, C.L.; Richter, J.D. Cpeb3 and cpeb4 in neurons: Analysis of RNA-binding specificity and translational control of ampa receptor glur2 mRNA. EMBO J. 2006, 25, 4865–4876. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D. Cpeb: A life in translation. Trends Biochem. Sci. 2007, 32, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, T.; Swanger, S.A.; Takeuchi, K.; Kim, J.H.; Nalavadi, V.; Shin, J.; Lorenz, L.J.; Zukin, R.S.; Bassell, G.J.; Richter, J.D. Bidirectional control of mRNA translation and synaptic plasticity by the cytoplasmic polyadenylation complex. Mol. Cell 2012, 47, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.C.; Ryan, K.; Manley, J.L.; Richter, J.D. Symplekin and xgld-2 are required for cpeb-mediated cytoplasmic polyadenylation. Cell 2004, 119, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Richter, J.D. Dissolution of the maskin-eif4e complex by cytoplasmic polyadenylation and poly(a)-binding protein controls cyclin b1 mrna translation and oocyte maturation. EMBO J. 2002, 21, 3852–3862. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eif4e inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.M.; Davare, M.A.; Oh, M.C.; Derkach, V.; Soderling, T.R. Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase ii and protein phosphatase 1 during hippocampal long-term potentiation. J. Neurosci. 2005, 25, 5604–5610. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.M.; Nozaki, N.; Shigeri, Y.; Soderling, T.R. Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase ii. J. Neurosci. 2004, 24, 5193–5201. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Jung, M.Y.; Sarkissian, M.; Richter, J.D. N-methyl-d-aspartate receptor signaling results in aurora kinase-catalyzed cpeb phosphorylation and alpha camkii mRNA polyadenylation at synapses. EMBO J. 2002, 21, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Sarkissian, M.; Mendez, R.; Richter, J.D. Progesterone and insulin stimulation of cpeb-dependent polyadenylation is regulated by aurora a and glycogen synthase kinase-3. Genes Dev. 2004, 18, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Bava, F.A.; Eliscovich, C.; Ferreira, P.G.; Minana, B.; Ben-Dov, C.; Guigo, R.; Valcarcel, J.; Mendez, R. Cpeb1 coordinates alternative 3′-utr formation with translational regulation. Nature 2013, 495, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wells, D.; Tay, J.; Mendis, D.; Abbott, M.A.; Barnitt, A.; Quinlan, E.; Heynen, A.; Fallon, J.R.; Richter, J.D. Cpeb-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-camkii mRNA at synapses. Neuron 1998, 21, 1129–1139. [Google Scholar] [CrossRef]
- Alarcon, J.M.; Hodgman, R.; Theis, M.; Huang, Y.-S.; Kandel, E.R.; Richter, J.D. Selective modulation of some forms of schaffer collateral-ca1 synaptic plasticity in mice with a disruption of the cpeb-1 gene. Learn. Mem. 2004, 11, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Zearfoss, N.R.; Alarcon, J.M.; Trifilieff, P.; Kandel, E.; Richter, J.D. A molecular circuit composed of cpeb-1 and c-jun controls growth hormone-mediated synaptic plasticity in the mouse hippocampus. J. Neurosci. 2008, 28, 8502–8509. [Google Scholar] [CrossRef] [PubMed]
- Berger-Sweeney, J.; Zearfoss, N.R.; Richter, J.D. Reduced extinction of hippocampal-dependent memories in cpeb knockout mice. Learn. Mem. 2006, 13, 4–7. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, M.; Cao, G.; Montero Llopis, P.; Kundel, M.; Jones, K.; Hofler, C.; Shin, C.; Wells, D.G. Cytoplasmic polyadenylation element binding protein 1-mediated mRNA translation in purkinje neurons is required for cerebellar long-term depression and motor coordination. J. Neurosci. 2007, 27, 6400–6411. [Google Scholar] [CrossRef] [PubMed]
- Pavlopoulos, E.; Trifilieff, P.; Chevaleyre, V.; Fioriti, L.; Zairis, S.; Pagano, A.; Malleret, G.; Kandel, E.R. Neuralized1 activates cpeb3: A function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell 2011, 147, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Richter, J.D. Activity-dependent polyadenylation in neurons. RNA 2005, 11, 1340–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanger, S.A.; He, Y.A.; Richter, J.D.; Bassell, G.J. Dendritic glun2a synthesis mediates activity-induced nmda receptor insertion. J. Neurosci. 2013, 33, 8898–8908. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.Y.; Kundel, M.; Wells, D.G. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J. Neurosci. 2004, 24, 9425–9433. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Richter, J.D. Analysis of mRNA translation in cultured hippocampal neurons. Methods Enzymol. 2007, 431, 143–162. [Google Scholar] [PubMed]
- Silva, A.J.; Stevens, C.F.; Tonegawa, S.; Wang, Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase ii mutant mice. Science 1992, 257, 201–206. [Google Scholar] [CrossRef]
- Oruganty-Das, A.; Ng, T.; Udagawa, T.; Goh, E.L.; Richter, J.D. Translational control of mitochondrial energy production mediates neuron morphogenesis. Cell Metab. 2012, 16, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, B.B.; Gioio, A.E.; Hillefors, M.; Aschrafi, A. Axonal protein synthesis and the regulation of local mitochondrial function. Results Probl. Cell Differ. 2009, 48, 225–242. [Google Scholar] [PubMed]
- Bestman, J.E.; Cline, H.T. The RNA binding protein cpeb regulates dendrite morphogenesis and neuronal circuit assembly in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 20494–20499. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Carson, J.H.; Barbarese, E.; Richter, J.D. Facilitation of dendritic mRNA transport by cpeb. Genes Dev. 2003, 17, 638–653. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Wilczynska, A.; Aigueperse, C.; Kress, M.; Dautry, F.; Weil, D. The translational regulator cpeb1 provides a link between dcp1 bodies and stress granules. J. Cell Sci. 2005, 118, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Posner, J.B.; Darnell, R.B. Nova, the paraneoplastic ri antigen, is homologous to an rna-binding protein and is specifically expressed in the developing motor system. Neuron 1993, 11, 657–672. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Yin, G.L.; Darnell, R.B. The neuronal rna-binding protein nova-2 is implicated as the autoantigen targeted in poma patients with dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 13254–13259. [Google Scholar] [CrossRef] [PubMed]
- Luque, F.A.; Furneaux, H.M.; Ferziger, R.; Rosenblum, M.K.; Wray, S.H.; Schold, S.C.; Glantz, M.J.; Jaeckle, K.A.; Biran, H.; Lesser, M. Anti-ri: An antibody associated with paraneoplastic opsoclonus and breast cancer. Ann. Neurol. 1991, 29, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Hormigo, A.; Dalmau, J.; Rosenblum, M.K.; River, M.E.; Posner, J.B. Immunological and pathological study of anti-ri-associated encephalopathy. Ann. Neurol. 1994, 36, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Hayakawa-Yano, Y.; Mele, A.; Darnell, R.B. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron 2010, 66, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Racca, C.; Gardiol, A.; Eom, T.; Ule, J.; Triller, A.; Darnell, R.B. The neuronal splicing factor nova co-localizes with target rNAS in the dendrite. Front. Neural Circuits 2010, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Musunuru, K.; Lewis, H.A.; Burley, S.K.; Darnell, R.B. The tetranucleotide ucay directs the specific recognition of RNA by the nova k-homology 3 domain. Proc. Natl. Acad. Sci. USA 2000, 97, 5740–5745. [Google Scholar] [CrossRef] [PubMed]
- Lewis, H.A.; Chen, H.; Edo, C.; Buckanovich, R.J.; Yang, Y.Y.; Musunuru, K.; Zhong, R.; Darnell, R.B.; Burley, S.K. Crystal structures of nova-1 and nova-2 k-homology RNA-binding domains. Structure 1999, 7, 191–203. [Google Scholar] [CrossRef]
- Jensen, K.B.; Dredge, B.K.; Stefani, G.; Zhong, R.; Buckanovich, R.J.; Okano, H.J.; Yang, Y.Y.; Darnell, R.B. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 2000, 25, 359–371. [Google Scholar] [CrossRef]
- Ule, J.; Darnell, R.B. RNA binding proteins and the regulation of neuronal synaptic plasticity. Curr. Opin. Neurobiol. 2006, 16, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ruggiu, M.; Herbst, R.; Kim, N.; Jevsek, M.; Fak, J.J.; Mann, M.A.; Fischbach, G.; Burden, S.J.; Darnell, R.B. Rescuing z+ agrin splicing in nova null mice restores synapse formation and unmasks a physiologic defect in motor neuron firing. Proc. Natl. Acad. Sci. USA 2009, 106, 3513–3518. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Shi, S.-H.; Ule, J.; Ruggiu, M.; Barker, L.A.; Darnell, R.B.; Jan, Y.N.; Jan, L.Y. Common molecular pathways mediate long-term potentiation of synaptic excitation and slow synaptic inhibition. Cell 2005, 123, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Cline, H. Synaptogenesis: A balancing act between excitation and inhibition. Curr. Biol. 2005, 15, R203–R205. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Stefani, G.; Mele, A.; Ruggiu, M.; Wang, X.; Taneri, B.; Gaasterland, T.; Blencowe, B.J.; Darnell, R.B. An RNA map predicting nova-dependent splicing regulation. Nature 2006, 444, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Miranda-Rottmann, S.; Ruggiu, M.; Park, C.Y.; Fak, J.J.; Zhong, R.; Duncan, J.S.; Fabella, B.A.; Junge, H.J.; Chen, Z.; et al. Nova2-mediated RNA regulation is required for axonal pathfinding during development. Elife 2016, 5, e14371. [Google Scholar] [CrossRef] [PubMed]
- Darnell, R.B. Hits-clip: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Darnell, R.B. RNA processing and its regulation: Global insights into biological networks. Nat. Rev. Genet. 2010, 11, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. Hits-clip yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Eom, T.; Zhang, C.; Wang, H.; Lay, K.; Fak, J.; Noebels, J.L.; Darnell, R.B. Nova-dependent regulation of cryptic nmd exons controls synaptic protein levels after seizure. eLife 2013, 2, e00178. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Ule, A.; Spencer, J.; Williams, A.; Hu, J.S.; Cline, M.; Wang, H.; Clark, T.; Fraser, C.; Ruggiu, M.; et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005, 37, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Leggere, J.C.; Saito, Y.; Darnell, R.B.; Tessier-Lavigne, M.; Junge, H.J.; Chen, Z. Nova regulates dcc alternative splicing during neuronal migration and axon guidance in the spinal cord. Elife 2016, 5, e14264. [Google Scholar] [CrossRef] [PubMed]
- Polydorides, A.D.; Okano, H.J.; Yang, Y.Y.; Stefani, G.; Darnell, R.B. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of nova to regulate neuron-specific alternative splicing. Proc. Natl. Acad. Sci. USA 2000, 97, 6350–6355. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.R.; Grossman, D.; White, K. Mutant alleles at the locus elav in drosophila melanogaster lead to nervous system defects. A developmental-genetic analysis. J. Neurogenet. 1985, 2, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Okano, H.J.; Darnell, R.B. A hierarchy of hu RNA binding proteins in developing and adult neurons. J. Neurosci. 1997, 17, 3024–3037. [Google Scholar] [CrossRef] [PubMed]
- Ince-Dunn, G.; Okano, H.J.; Jensen, K.B.; Park, W.-Y.; Zhong, R.; Ule, J.; Mele, A.; Fak, J.J.; Yang, C.; Zhang, C.; et al. Neuronal elav-like (hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron 2012, 75, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhausser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide analysis of regulatory interactions of the rna-binding protein hur. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Good, P.J. A conserved family of elav-like genes in vertebrates. Proc. Natl. Acad. Sci. USA 1995, 92, 4557–4561. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. Rna stabilization by the au-rich element binding protein, hur, an elav protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.C.; Steitz, J.A. Hns, a nuclear-cytoplasmic shuttling sequence in hur. Proc. Natl. Acad. Sci. USA 1998, 95, 15293–15298. [Google Scholar] [CrossRef] [PubMed]
- Hinman, M.N.; Zhou, H.L.; Sharma, A.; Lou, H. All three RNA recognition motifs and the hinge region of huc play distinct roles in the regulation of alternative splicing. Nucleic Acids Res. 2013, 41, 5049–5061. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.D.; Sengupta, J.; Morin, M.; Neve, R.L.; Valenzuela, C.F.; Perrone-Bizzozero, N.I. Overexpression of hud accelerates neurite outgrowth and increases gap-43 mRNA expression in cortical neurons and retinoic acid-induced embryonic stem cells in vitro. Exp. Neurol. 2001, 168, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Bolognani, F.; Tanner, D.C.; Merhege, M.; Deschenes-Furry, J.; Jasmin, B.; Perrone-Bizzozero, N.I. In vivo post-transcriptional regulation of gap-43 mRNA by overexpression of the rna-binding protein hud. J. Neurochem. 2006, 96, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Terashima, K.; Yamamoto, K.; Sakashita, E.; Sakamoto, H. Cytoplasmic localization is required for the mammalian elav-like protein hud to induce neuronal differentiation. Genes Cells Devoted Mol. Cell. Mech. 1999, 4, 667–683. [Google Scholar] [CrossRef]
- Sun, K.; Li, X.; Chen, X.; Bai, Y.; Zhou, G.; Kokiko-Cochran, O.N.; Lamb, B.; Hamilton, T.A.; Lin, C.Y.; Lee, Y.S.; et al. Neuron-specific hur-deficient mice spontaneously develop motor neuron disease. J. Immunol. 2018, 201, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Quattrone, A.; Pascale, A.; Nogues, X.; Zhao, W.; Gusev, P.; Pacini, A.; Alkon, D.L. Posttranscriptional regulation of gene expression in learning by the neuronal elav-like mrna-stabilizing proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 11668–11673. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Kakumoto, K.; Yoshida, T.; Kuwako, K.I.; Miyazaki, T.; Yamaguchi, J.; Konno, A.; Hata, J.; Uchiyama, Y.; Hirai, H.; et al. Elavl3 is essential for the maintenance of purkinje neuron axons. Sci. Rep. 2018, 8, 2722. [Google Scholar] [CrossRef] [PubMed]
- DeBoer, E.M.; Azevedo, R.; Vega, T.A.; Brodkin, J.; Akamatsu, W.; Okano, H.; Wagner, G.C.; Rasin, M.R. Prenatal deletion of the RNA-binding protein hud disrupts postnatal cortical circuit maturation and behavior. J. Neurosci. 2014, 34, 3674–3686. [Google Scholar] [CrossRef] [PubMed]
- Bolognani, F.; Qiu, S.; Tanner, D.C.; Paik, J.; Perrone-Bizzozero, N.I.; Weeber, E.J. Associative and spatial learning and memory deficits in transgenic mice overexpressing the RNA-binding protein hud. Neurobiol. Learn. Mem. 2007, 87, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, W.; Fujihara, H.; Mitsuhashi, T.; Yano, M.; Shibata, S.; Hayakawa, Y.; Okano, H.J.; Sakakibara, S.; Takano, H.; Takano, T.; et al. The rna-binding protein hud regulates neuronal cell identity and maturation. Proc. Natl. Acad. Sci. USA 2005, 102, 4625–4630. [Google Scholar] [CrossRef] [PubMed]
- Hao le, T.; Duy, P.Q.; An, M.; Talbot, J.; Iyer, C.C.; Wolman, M.; Beattie, C.E. Hud and the survival motor neuron protein interact in motoneurons and are essential for motoneuron development, function, and mRNA regulation. J. Neurosci. 2017, 37, 11559–11571. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Hutchison, E.R.; Lee, E.K.; Kuwano, Y.; Kim, M.M.; Masuda, K.; Srikantan, S.; Subaran, S.S.; Marasa, B.S.; Mattson, M.P.; et al. Mir-375 inhibits differentiation of neurites by lowering hud levels. Mol. Cell. Biol. 2010, 30, 4197–4210. [Google Scholar] [CrossRef] [PubMed]
- Pignolet, B.S.; Gebauer, C.M.; Liblau, R.S. Immunopathogenesis of paraneoplastic neurological syndromes associated with anti-hu antibodies: A beneficial antitumor immune response going awry. Oncoimmunology 2013, 2, e27384. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Iwayama, Y.; Hattori, E.; Iwamoto, K.; Toyota, T.; Ohnishi, T.; Ohba, H.; Maekawa, M.; Kato, T.; Yoshikawa, T. Genome-wide association study of schizophrenia in japanese population. PLoS ONE 2011, 6, e20468. [Google Scholar] [CrossRef] [PubMed]
- Noureddine, M.A.; Qin, X.J.; Oliveira, S.A.; Skelly, T.J.; van der Walt, J.; Hauser, M.A.; Pericak-Vance, M.A.; Vance, J.M.; Li, Y.J. Association between the neuron-specific rna-binding protein elavl4 and parkinson disease. Hum. Genet. 2005, 117, 27–33. [Google Scholar] [CrossRef] [PubMed]
- DeStefano, A.L.; Latourelle, J.; Lew, M.F.; Suchowersky, O.; Klein, C.; Golbe, L.I.; Mark, M.H.; Growdon, J.H.; Wooten, G.F.; Watts, R.; et al. Replication of association between elavl4 and parkinson disease: The genepd study. Hum. Genet. 2008, 124, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Pascale, A.; Wang, J.; Ho, L.; Quattrone, A.; Gandy, S.; Haroutunian, V.; Racchi, M.; Pasinetti, G.M. Nelav proteins alteration in alzheimer’s disease brain: A novel putative target for amyloid-beta reverberating on abetapp processing. J. Alzheimers Dis. 2009, 16, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Abdelmohsen, K.; Hutchison, E.R.; Mitchell, S.J.; Grammatikakis, I.; Guo, R.; Noh, J.H.; Martindale, J.L.; Yang, X.; Lee, E.K.; et al. Hud regulates coding and noncoding RNA to induce app->abeta processing. Cell Rep. 2014, 7, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Fukao, A.; Sasano, Y.; Imataka, H.; Inoue, K.; Sakamoto, H.; Sonenberg, N.; Thoma, C.; Fujiwara, T. The elav protein hud stimulates cap-dependent translation in a poly(a)- and eif4a-dependent manner. Mol. Cell 2009, 36, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M., Jr.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative regulatory mapping indicates that the rna-binding protein hur couples pre-mrna processing and mRNA stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Hinman, M.N.; Barron, V.A.; Geng, C.; Zhou, G.; Luo, G.; Siegel, R.E.; Lou, H. Hu proteins regulate alternative splicing by inducing localized histone hyperacetylation in an RNA-dependent manner. Proc. Natl. Acad. Sci. USA 2011, 108, E627–E635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragkouli, A.; Koukouraki, P.; Vlachos, I.S.; Paraskevopoulou, M.D.; Hatzigeorgiou, A.G.; Doxakis, E. Neuronal elavl proteins utilize auf-1 as a co-partner to induce neuron-specific alternative splicing of app. Sci. Rep. 2017, 7, 44507. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Fallini, C.; Colombrita, C.; Pascale, A.; Laforenza, U.; Quattrone, A.; Silani, V. Post-transcriptional regulation of neuro-oncological ventral antigen 1 by the neuronal RNA-binding proteins elav. J. Biol. Chem. 2008, 283, 7531–7541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, H.L.; Hasman, R.A.; Lou, H. Hu proteins regulate polyadenylation by blocking sites containing u-rich sequences. J. Biol. Chem. 2007, 282, 2203–2210. [Google Scholar] [CrossRef] [PubMed]
- Antic, D.; Lu, N.; Keene, J.D. Elav tumor antigen, hel-n1, increases translation of neurofilament m mRNA and induces formation of neurites in human teratocarcinoma cells. Genes Dev. 1999, 13, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Bolognani, F.; Perrone-Bizzozero, N.I. Rna-protein interactions and control of mRNA stability in neurons. J. Neurosci. Res. 2008, 86, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.L.; Wait, R.; Mahtani, K.R.; Sully, G.; Clark, A.R.; Saklatvala, J. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor hur. Mol. Cell. Biol. 2001, 21, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.C.; Steitz, J.A. Overexpression of hur, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of are-containing mrnas. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.C.; Myer, V.E.; Steitz, J.A. Au-rich elements target small nuclear rNAS as well as mrnas for rapid degradation. Genes Dev. 1997, 11, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Fabian, M.R.; Sonenberg, N.; Bhattacharyya, S.N.; Filipowicz, W. Hur protein attenuates mirna-mediated repression by promoting mirisc dissociation from the target rna. Nucleic Acids Res. 2012, 40, 5088–5100. [Google Scholar] [CrossRef] [PubMed]
- Meisner, N.-C.; Filipowicz, W. Properties of the regulatory rna-binding protein hur and its role in controlling mirna repression. Adv. Exp. Med. Biol. 2011, 700, 106–123. [Google Scholar] [PubMed]
- Fukao, A.; Mishima, Y.; Takizawa, N.; Oka, S.; Imataka, H.; Pelletier, J.; Sonenberg, N.; Thoma, C.; Fujiwara, T. Micrornas trigger dissociation of eif4ai and eif4aii from target mrnas in humans. Mol. Cell 2014, 56, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Atlas, R.; Behar, L.; Elliott, E.; Ginzburg, I. The insulin-like growth factor mRNA binding-protein imp-1 and the ras-regulatory protein g3bp associate with tau mRNA and hud protein in differentiated p19 neuronal cells. J. Neurochem. 2004, 89, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Akten, B.; Kye, M.J.; Hao le, T.; Wertz, M.H.; Singh, S.; Nie, D.; Huang, J.; Merianda, T.T.; Twiss, J.L.; Beattie, C.E.; et al. Interaction of survival of motor neuron (smn) and hud proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc. Natl. Acad. Sci. USA 2011, 108, 10337–10342. [Google Scholar] [CrossRef] [PubMed]
- Sosanya, N.M.; Huang, P.P.; Cacheaux, L.P.; Chen, C.J.; Nguyen, K.; Perrone-Bizzozero, N.I.; Raab-Graham, K.F. Degradation of high affinity hud targets releases kv1.1 mRNA from mir-129 repression by mtorc1. J. Cell Biol. 2013, 202, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Sosanya, N.M.; Cacheaux, L.P.; Workman, E.R.; Niere, F.; Perrone-Bizzozero, N.I.; Raab-Graham, K.F. Mammalian target of rapamycin (mtor) tagging promotes dendritic branch variability through the capture of Ca2+/calmodulin-dependent protein kinase ii alpha (camkiialpha) mrnas by the rna-binding protein hud. J. Biol. Chem. 2015, 290, 16357–16371. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Kim, H.H.; Kim, P.; Donnelly, C.J.; Kalinski, A.L.; Vuppalanchi, D.; Park, M.; Lee, S.J.; Merianda, T.T.; Perrone-Bizzozero, N.I.; et al. A hud-zbp1 ribonucleoprotein complex localizes gap-43 mRNA into axons through its 3′ untranslated region au-rich regulatory element. J. Neurochem. 2013, 126, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Vanevski, F.; Xu, B. Hud interacts with bdnf mRNA and is essential for activity-induced bdnf synthesis in dendrites. PLoS ONE 2015, 10, e0117264. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, K.; Sakashita, E.; Saito, K.; Sakamoto, H. Complex formation of the neuron-specific elav-like hu rna-binding proteins. Nucleic Acids Res. 2002, 30, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Aronov, S.; Aranda, G.; Behar, L.; Ginzburg, I. Visualization of translated tau protein in the axons of neuronal p19 cells and characterization of tau rnp granules. J. Cell Sci. 2002, 115, 3817–3827. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Kasashima, K.; Saito, K.; Fukuda, M.; Fukao, A.; Sasano, Y.; Inoue, K.; Fujiwara, T.; Sakamoto, H. Microtubule association of a neuronal rna-binding protein hud through its binding to the light chain of map1b. Biochimie 2011, 93, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Tiruchinapalli, D.M.; Caron, M.G.; Keene, J.D. Activity-dependent expression of elav/hu rbps and neuronal mrnas in seizure and cocaine brain. J. Neurochem. 2008, 107, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Zhang, H.; Su, Y.; Silani, V.; Singer, R.H.; Rossoll, W.; Bassell, G.J. The survival of motor neuron (smn) protein interacts with the mrna-binding protein hud and regulates localization of poly(a) mRNA in primary motor neuron axons. J. Neurosci. 2011, 31, 3914–3925. [Google Scholar] [CrossRef] [PubMed]
- La Bella, V.; Cisterni, C.; Salaun, D.; Pettmann, B. Survival motor neuron (smn) protein in rat is expressed as different molecular forms and is developmentally regulated. Eur. J. Neurosci. 1998, 10, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the smn gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Raimer, A.C.; Gray, K.M.; Matera, A.G. Smn—A chaperone for nuclear rnp social occasions? RNA Biol. 2017, 14, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Jangi, M.; Fleet, C.; Cullen, P.; Gupta, S.V.; Mekhoubad, S.; Chiao, E.; Allaire, N.; Bennett, C.F.; Rigo, F.; Krainer, A.R.; et al. Smn deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc. Natl. Acad. Sci. USA 2017, 114, E2347–E2356. [Google Scholar] [CrossRef] [PubMed]
- Doktor, T.K.; Hua, Y.; Andersen, H.S.; Broner, S.; Liu, Y.H.; Wieckowska, A.; Dembic, M.; Bruun, G.H.; Krainer, A.R.; Andresen, B.S. Rna-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of u12-dependent introns. Nucleic Acids Res. 2017, 45, 395–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Custer, S.K.; Gilson, T.D.; Li, H.; Todd, A.G.; Astroski, J.W.; Lin, H.; Liu, Y.; Androphy, E.J. Altered mRNA splicing in smn-depleted motor neuron-like cells. PLoS ONE 2016, 11, e0163954. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Gavrilov, D.K.; Shi, X.; Das, K.; Gilliam, T.C.; Wang, C.H. Differential smn2 expression associated with sma severity. Nat. Genet. 1998, 20, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burlet, P.; Liu, Q.; Bertrandy, S.; Clermont, O.; Munnich, A.; Dreyfuss, G.; Melki, J. Correlation between severity and smn protein level in spinal muscular atrophy. Nat. Genet. 1997, 16, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.N.; Androphy, E.J.; Singh, R.N. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Vickers, T.A.; Baker, B.F.; Bennett, C.F.; Krainer, A.R. Enhancement of smn2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007, 5, e73. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of smn2 splicing in the cns rescues necrosis in a type iii sma mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral smn restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Garcera, A.; Mincheva, S.; Gou-Fabregas, M.; Caraballo-Miralles, V.; Llado, J.; Comella, J.X.; Soler, R.M. A new model to study spinal muscular atrophy: Neurite degeneration and cell death is counteracted by bcl-x(l) overexpression in motoneurons. Neurobiol. Dis. 2011, 42, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Schrank, B.; Gotz, R.; Gunnersen, J.M.; Ure, J.M.; Toyka, K.V.; Smith, A.G.; Sendtner, M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 1997, 94, 9920–9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler-Botija, C.; Ferrer, I.; Gich, I.; Baiget, M.; Tizzano, E.F. Neuronal death is enhanced and begins during foetal development in type i spinal muscular atrophy spinal cord. Brain 2002, 125, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Lotti, F.; Imlach, W.L.; Saieva, L.; Beck, E.S.; Hao le, T.; Li, D.K.; Jiao, W.; Mentis, G.Z.; Beattie, C.E.; McCabe, B.D.; et al. An smn-dependent u12 splicing event essential for motor circuit function. Cell 2012, 151, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Kariya, S.; Park, G.H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced smn protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes-Diaz, C.; Nicole, S.; Velasco, M.E.; Borra-Cebrian, C.; Panozzo, C.; Frugier, T.; Millet, G.; Roblot, N.; Joshi, V.; Melki, J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 2002, 11, 1439–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowerman, M.; Shafey, D.; Kothary, R. Smn depletion alters profilin ii expression and leads to upregulation of the rhoa/rock pathway and defects in neuronal integrity. J. Mol. Neurosci. 2007, 32, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kroning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Nolle, A.; Zeug, A.; van Bergeijk, J.; Tonges, L.; Gerhard, R.; Brinkmann, H.; Al Rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The spinal muscular atrophy disease protein smn is linked to the rho-kinase pathway via profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Pan, F.; Hong, D.; Shenoy, S.M.; Singer, R.H.; Bassell, G.J. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 2003, 23, 6627–6637. [Google Scholar] [CrossRef] [PubMed]
- Saal, L.; Briese, M.; Kneitz, S.; Glinka, M.; Sendtner, M. Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA 2014, 20, 1789–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallini, C.; Donlin-Asp, P.G.; Rouanet, J.P.; Bassell, G.J.; Rossoll, W. Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J. Neurosci. 2016, 36, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Rossoll, W.; Kroning, A.K.; Ohndorf, U.M.; Steegborn, C.; Jablonka, S.; Sendtner, M. Specific interaction of smn, the spinal muscular atrophy determining gene product, with hnrnp-r and gry-rbp/hnrnp-q: A role for smn in RNA processing in motor axons? Hum. Mol. Genet. 2002, 11, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Donlin-Asp, P.G.; Fallini, C.; Campos, J.; Chou, C.C.; Merritt, M.E.; Phan, H.C.; Bassell, G.J.; Rossoll, W. The survival of motor neuron protein acts as a molecular chaperone for mrnp assembly. Cell Rep. 2017, 18, 1660–1673. [Google Scholar] [CrossRef] [PubMed]
- Glinka, M.; Herrmann, T.; Funk, N.; Havlicek, S.; Rossoll, W.; Winkler, C.; Sendtner, M. The heterogeneous nuclear ribonucleoprotein-r is necessary for axonal beta-actin mRNA translocation in spinal motor neurons. Hum. Mol. Genet. 2010, 19, 1951–1966. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Yang, X.; Pan, D.; Huang, J.; Sahin, M.; Zhou, J. Smn deficiency reduces cellular ability to form stress granules, sensitizing cells to stress. Cell Mol. Neurobiol. 2011, 31, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, tdp-43, that binds to human immunodeficiency virus type 1 tar DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [PubMed]
- Tsuiji, H.; Iguchi, Y.; Furuya, A.; Kataoka, A.; Hatsuta, H.; Atsuta, N.; Tanaka, F.; Hashizume, Y.; Akatsu, H.; Murayama, S.; et al. Spliceosome integrity is defective in the motor neuron diseases als and sma. EMBO Mol. Med. 2013, 5, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Casafont, I.; Bengoechea, R.; Tapia, O.; Berciano, M.T.; Lafarga, M. Tdp-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 2009, 167, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. Tdp-43 and fus rna-binding proteins bind distinct sets of cytoplasmic messenger rnas and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012, 287, 15635–15647. [Google Scholar] [CrossRef] [PubMed]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of tdp-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mrna depletion and RNA missplicing contribute to neuronal vulnerability from loss of tdp-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by tdp-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Sanelli, T.; Dib, S.; Sheps, D.; Findlater, J.; Bilbao, J.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. Rna targets of tdp-43 identified by uv-clip are deregulated in als. Mol. Cell Neurosci. 2011, 47, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. Tdp-43 repression of nonconserved cryptic exons is compromised in als-ftd. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Yalamanchili, H.K.; Park, J.; De Maio, A.; Lu, H.C.; Wan, Y.W.; White, J.J.; Bondar, V.V.; Sayegh, L.S.; Liu, X.; et al. Extensive cryptic splicing upon loss of rbm17 and tdp43 in neurodegeneration models. Hum. Mol. Genet. 2016, 25, 5083–5093. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Dork, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor tdp-43 and sr proteins promote in vitro and in vivo cftr exon 9 skipping. EMBO J. 2001, 20, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Passoni, M.; De Conti, L.; Baralle, M.; Buratti, E. Ug repeats/tdp-43 interactions near 5’ splice sites exert unpredictable effects on splicing modulation. J. Mol. Biol. 2012, 415, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Fiesel, F.C.; Weber, S.S.; Supper, J.; Zell, A.; Kahle, P.J. Tdp-43 regulates global translational yield by splicing of exon junction complex component skar. Nucleic Acids Res. 2012, 40, 2668–2682. [Google Scholar] [CrossRef] [PubMed]
- Mercado, P.A.; Ayala, Y.M.; Romano, M.; Buratti, E.; Baralle, F.E. Depletion of tdp 43 overrides the need for exonic and intronic splicing enhancers in the human apoa-ii gene. Nucleic Acids Res. 2005, 33, 6000–6010. [Google Scholar] [CrossRef] [PubMed]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic mutations in rna-binding proteins and their roles in als. Hum. Genet. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded c9orf72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-atg translation in c9ftd/als. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding als: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [PubMed]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of tdp-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. Tdp-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered ribostasis: Rna-protein granules in degenerative disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. Tar DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.J.; Yang, C.H.; Fang, Y.H.; Cho, K.H.; Chien, W.L.; Wang, W.T.; Wu, T.W.; Lin, C.P.; Fu, W.M.; Shen, C.K. Elevated expression of tdp-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking ftld-u. J. Exp. Med. 2010, 207, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Swarup, V.; Phaneuf, D.; Bareil, C.; Robertson, J.; Rouleau, G.A.; Kriz, J.; Julien, J.P. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with tdp-43 genomic fragments. Brain 2011, 134, 2610–2626. [Google Scholar] [CrossRef] [PubMed]
- Handley, E.E.; Pitman, K.A.; Dawkins, E.; Young, K.M.; Clark, R.M.; Jiang, T.C.; Turner, B.J.; Dickson, T.C.; Blizzard, C.A. Synapse dysfunction of layer v pyramidal neurons precedes neurodegeneration in a mouse model of tdp-43 proteinopathies. Cereb Cortex 2017, 27, 3630–3647. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic tdp-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968. [Google Scholar] [CrossRef] [PubMed]
- Iguchi, Y.; Katsuno, M.; Niwa, J.; Takagi, S.; Ishigaki, S.; Ikenaka, K.; Kawai, K.; Watanabe, H.; Yamanaka, K.; Takahashi, R.; et al. Loss of tdp-43 causes age-dependent progressive motor neuron degeneration. Brain 2013, 136, 1371–1382. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ferris, J.; Gao, F.B. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein tdp-43 promotes dendritic branching. Mol. Brain 2009, 2, 30. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The als disease protein tdp-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.; Feiger, Z.; Shilton, A.K.; Luo, C.C.; Silverman, E.; Rodal, A.A. Role of bmp receptor traffic in synaptic growth defects in an als model. Mol. Biol. Cell 2016, 27, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R.K.; Mangelsdorf, M.; Panwar, A.; Butler, T.J.; Noakes, P.G.; Wallace, R.H. Identification of RNA bound to the tdp-43 ribonucleoprotein complex in the adult mouse brain. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013, 14, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.-C.; Hazelett, D.J.; Stewart, J.A.; Morton, D.B. Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a drosophila, tdp-43 loss of function model of als. Brain Res. 2014, 1584, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Klima, R.; Buratti, E.; Verstreken, P.; Baralle, F.E.; Feiguin, F. Chronological requirements of tdp-43 function in synaptic organization and locomotive control. Neurobiol. Dis. 2014, 71, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Godena, V.K.; Romano, G.; Romano, M.; Appocher, C.; Klima, R.; Buratti, E.; Baralle, F.E.; Feiguin, F. Tdp-43 regulates drosophila neuromuscular junctions growth by modulating futsch/map1b levels and synaptic microtubules organization. PLoS ONE 2011, 6, e17808. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Siddegowda, B.B.; Estes, P.S.; Johannesmeyer, J.; Kovalik, T.; Daniel, S.G.; Pearson, A.; Bowser, R.; Zarnescu, D.C. Futsch/map1b mRNA is a translational target of tdp-43 and is neuroprotective in a drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 15962–15974. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.; Yao, S.; Zarnescu, D.C. Dynamic duo—FMRP and tdp-43: Regulating common targets, causing different diseases. Brain Res. 2018, 1693, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wu, F.; Xu, W.; Shi, J.; Hu, W.; Jin, N.; Qian, W.; Wang, X.; Iqbal, K.; Gong, C.X.; et al. Tdp-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017, 45, 6177–6193. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Chen, F.; Iqbal, K.; Gong, C.X.; Wang, X.; Liu, F. Transactive response DNA-binding protein 43 (tdp-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J. Biol. Chem. 2017, 292, 10600–10612. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. Tdp-43 gains function due to perturbed autoregulation in a tardbp knock-in mouse model of als-ftd. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of tdp-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. Tdp-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in als/ftd. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. Tdp-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderweyde, T.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; et al. Tar DNA binding protein-43 (tdp-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, S.J.; Meyerowitz, J.; James, J.L.; Liddell, J.R.; Crouch, P.J.; Kanninen, K.M.; White, A.R. Endogenous tdp-43 localized to stress granules can subsequently form protein aggregates. Neurochem. Int. 2012, 60, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of tdp-43 mRNA granules is impaired by als-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, A.; Kimura, N.; Watanabe, Y.; Watanabe, S.; Ishihama, A. Tdp-43 binds and transports g-quadruplex-containing mrnas into neurites for local translation. Genes Cells Devoted Mol. Cell. Mech. 2016, 21, 466–481. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. Tar DNA-binding protein 43 (tdp-43) regulates stress granule dynamics via differential regulation of g3bp and tia-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Lin, A.Y.; Ebata, A.; Boon, J.Y.; Reid, W.; Xu, Y.-F.; Kobrin, K.; Murphy, G.J.; Petrucelli, L.; Wolozin, B. Als-linked mutations enlarge tdp-43-enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 2014, 34, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.F.; Wu, L.-S.; Chang, H.-Y.; Shen, C.K.J. Tdp-43, the signature protein of ftld-u, is a neuronal activity-responsive factor. J. Neurochem. 2008, 105, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like tdp-43 rnp granules in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E2466–E2475. [Google Scholar] [CrossRef] [PubMed]
- Rogelj, B.; Easton, L.E.; Bogu, G.K.; Stanton, L.W.; Rot, G.; Curk, T.; Zupan, B.; Sugimoto, Y.; Modic, M.; Haberman, N.; et al. Widespread binding of fus along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012, 2, 603. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of als-linked proteins fus/tls and tdp-43 intersect in processing long pre-mrnas. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Lerga, A.; Hallier, M.; Delva, L.; Orvain, C.; Gallais, I.; Marie, J.; Moreau-Gachelin, F. Identification of an RNA binding specificity for the potential splicing factor tls. J. Biol. Chem. 2001, 276, 6807–6816. [Google Scholar] [CrossRef] [PubMed]
- Masuda, A.; Takeda, J.; Okuno, T.; Okamoto, T.; Ohkawara, B.; Ito, M.; Ishigaki, S.; Sobue, G.; Ohno, K. Position-specific binding of fus to nascent RNA regulates mRNA length. Genes Dev. 2015, 29, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. Tls/fus (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, S.; Masuda, A.; Fujioka, Y.; Iguchi, Y.; Katsuno, M.; Shibata, A.; Urano, F.; Sobue, G.; Ohno, K. Position-dependent fus-rna interactions regulate alternative splicing events and transcriptions. Sci. Rep. 2012, 2, 529. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Reed, R. Fus functions in coupling transcription to splicing by mediating an interaction between rnap ii and u1 snrnp. Proc. Natl. Acad. Sci. USA 2015, 112, 8608–8613. [Google Scholar] [CrossRef] [PubMed]
- Zinszner, H.; Sok, J.; Immanuel, D.; Yin, Y.; Ron, D. Tls (fus) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell Sci. 1997, 110 Pt 15, 1741–1750. [Google Scholar]
- Kapeli, K.; Pratt, G.A.; Vu, A.Q.; Hutt, K.R.; Martinez, F.J.; Sundararaman, B.; Batra, R.; Freese, P.; Lambert, N.J.; Huelga, S.C.; et al. Distinct and shared functions of als-associated proteins tdp-43, fus and taf15 revealed by multisystem analyses. Nat. Commun. 2016, 7, 12143. [Google Scholar] [CrossRef] [PubMed]
- Morlando, M.; Dini Modigliani, S.; Torrelli, G.; Rosa, A.; Di Carlo, V.; Caffarelli, E.; Bozzoni, I. Fus stimulates microrna biogenesis by facilitating co-transcriptional drosha recruitment. EMBO J. 2012, 31, 4502–4510. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncrnas allosterically modify rna-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of fus and hdac1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in fus, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the fus/tls gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobagyi, T.; et al. Als mutant fus disrupts nuclear localization and sequesters wild-type fus within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of tdp-43 and fus/tls proteins. J. Neurochem. 2016, 138 (Suppl. S1), 95–111. [Google Scholar] [CrossRef]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R. A new subtype of frontotemporal lobar degeneration with fus pathology. Brain 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [PubMed]
- Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Gijselinck, I.; Van den Broeck, M.; Mattheijssens, M.; Peeters, K.; De Deyn, P.P.; et al. Genetic contribution of fus to frontotemporal lobar degeneration. Neurology 2010, 74, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Matsuoka, M. Overexpression of nuclear fus induces neuronal cell death. Neuroscience 2015, 287, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.J.; Wang, D.; Wei, X.; Xia, X.G. Fus transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. Als-associated mutant fus induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Bercier, V.; Lissouba, A.; Liao, M.; Brustein, E.; Rouleau, G.A.; Drapeau, P. Fus and tardbp but not sod1 interact in genetic models of amyotrophic lateral sclerosis. PLoS Genet. 2011, 7, e1002214. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The als-associated proteins fus and tdp-43 function together to affect drosophila locomotion and life span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef] [PubMed]
- Soo, K.Y.; Sultana, J.; King, A.E.; Atkinson, R.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.; Farg, M.A.; Atkin, J.D. Als-associated mutant fus inhibits macroautophagy which is restored by overexpression of rab1. Cell Death Discov. 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ling, S.C.; Qiu, J.; Albuquerque, C.P.; Zhou, Y.; Tokunaga, S.; Li, H.; Qiu, H.; Bui, A.; Yeo, G.W.; et al. Als-causative mutations in fus/tls confer gain and loss of function by altered association with smn and u1-snrnp. Nat. Commun. 2015, 6, 6171. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.; Fumoto, K.; Blokhuis, A.M.; Engelen-Lee, J.; Zhou, Y.; van den Heuvel, D.M.; Koppers, M.; van Diggelen, F.; van Heest, J.; Demmers, J.A.; et al. Als-associated mutations in fus disrupt the axonal distribution and function of smn. Hum. Mol. Genet. 2013, 22, 3690–3704. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J., Jr.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H., Jr.; Hayward, L.J. Mutant fus proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Fujii, R.; Watanabe, Y.; Okabe, S.; Fukui, K.; Takumi, T. Myosin-va facilitates the accumulation of mrna/protein complex in dendritic spines. Curr. Biol. 2006, 16, 2345–2351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Neubert, T.A.; Jordan, B.A. RNA binding proteins accumulate at the postsynaptic density with synaptic activity. J. Neurosci. 2012, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Higashi, S.; Kawakami, I.; Kobayashi, Z.; Hosokawa, M.; Katsuse, O.; Togo, T.; Hirayasu, Y.; Akiyama, H. Localization of fused in sarcoma (fus) protein to the post-synaptic density in the brain. Acta Neuropathol. 2012, 124, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Belly, A.; Moreau-Gachelin, F.; Sadoul, R.; Goldberg, Y. Delocalization of the multifunctional RNA splicing factor tls/fus in hippocampal neurones: Exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neurosci. Lett. 2005, 379, 152–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, R.; Okabe, S.; Urushido, T.; Inoue, K.; Yoshimura, A.; Tachibana, T.; Nishikawa, T.; Hicks, G.G.; Takumi, T. The RNA binding protein tls is translocated to dendritic spines by mglur5 activation and regulates spine morphology. Curr. Biol. 2005, 15, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Fujii, R.; Takumi, T. Tls facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J. Cell Sci. 2005, 118, 5755–5765. [Google Scholar] [CrossRef] [PubMed]
- Schoen, M.; Reichel, J.M.; Demestre, M.; Putz, S.; Deshpande, D.; Proepper, C.; Liebau, S.; Schmeisser, M.J.; Ludolph, A.C.; Michaelis, J.; et al. Super-resolution microscopy reveals presynaptic localization of the als/ftd related protein fus in hippocampal neurons. Front. Cell. Neurosci. 2015, 9, 496. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Zhang, H.; Loiselle, D.; Haystead, T.; Macara, I.G.; Mili, S. The rna-binding protein fus directs translation of localized mrnas in apc-rnp granules. J. Cell Biol. 2013, 203, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, F.; Maragkakis, M.; Alexiou, P.; Maronski, M.A.; Dichter, M.A.; Mourelatos, Z. Identification of in vivo, conserved, taf15 RNA binding sites reveals the impact of taf15 on the neuronal transcriptome. Cell Rep. 2013, 3, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ameur, A.; Zaghlool, A.; Halvardson, J.; Wetterbom, A.; Gyllensten, U.; Cavelier, L.; Feuk, L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol. 2011, 18, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Sjogren, H.; Meis-Kindblom, J.; Kindblom, L.G.; Aman, P.; Stenman, G. Fusion of the ews-related gene taf2n to tec in extraskeletal myxoid chondrosarcoma. Cancer Res. 1999, 59, 5064–5067. [Google Scholar] [PubMed]
- Martini, A.; La Starza, R.; Janssen, H.; Bilhou-Nabera, C.; Corveleyn, A.; Somers, R.; Aventin, A.; Foa, R.; Hagemeijer, A.; Mecucci, C.; et al. Recurrent rearrangement of the ewing’s sarcoma gene, ewsr1, or its homologue, taf15, with the transcription factor ciz/nmp4 in acute leukemia. Cancer Res. 2002, 62, 5408–5412. [Google Scholar] [PubMed]
- Ticozzi, N.; Vance, C.; Leclerc, A.L.; Keagle, P.; Glass, J.D.; McKenna-Yasek, D.; Sapp, P.C.; Silani, V.; Bosco, D.A.; Shaw, C.E.; et al. Mutational analysis reveals the fus homolog taf15 as a candidate gene for familial amyotrophic lateral sclerosis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A yeast functional screen predicts new candidate als disease genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881–20890. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. Fet proteins taf15 and ews are selective markers that distinguish ftld with fus pathology from amyotrophic lateral sclerosis with fus mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.; Vlassis, A.; Guialis, A.; Leichter, M. Domains involved in taf15 subcellular localisation: Dependence on cell type and ongoing transcription. Gene 2012, 506, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Blechingberg, J.; Luo, Y.; Bolund, L.; Damgaard, C.K.; Nielsen, A.L. Gene expression responses to fus, ews, and taf15 reduction and stress granule sequestration analyses identifies fet-protein non-redundant functions. PLoS ONE 2012, 7, e46251. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.K.; Stahlberg, A.; Arvidsson, Y.; Olofsson, A.; Semb, H.; Stenman, G.; Nilsson, O.; Aman, P. The multifunctional fus, ews and taf15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Tian, Q.; Duan, X.; Streuli, M.; Schlossman, S.F.; Anderson, P. Identification and functional characterization of a tia-1-related nucleolysin. Proc. Natl. Acad. Sci. USA 1992, 89, 8681–8685. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.R.; Medley, Q.G.; O’Brien, S.; Anderson, P.; Streuli, M. Structure, tissue distribution and genomic organization of the murine rrm-type RNA binding proteins tia-1 and tiar. Nucleic Acids Res. 1996, 24, 3829–3835. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Yu, J.; Zhang, Z.; Gygi, M.P.; Krainer, A.R.; Gygi, S.P.; Reed, R. Sr proteins function in coupling rnap ii transcription to pre-mrna splicing. Mol. Cell 2007, 26, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Suswam, E.A.; Li, Y.Y.; Mahtani, H.; King, P.H. Novel DNA-binding properties of the rna-binding protein tiar. Nucleic Acids Res. 2005, 33, 4507–4518. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Headey, S.J.; Yoga, Y.M.; Scanlon, M.J.; Gorospe, M.; Wilce, M.C.; Wilce, J.A. Distinct binding properties of tiar rrms and linker region. RNA Biol. 2013, 10, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hasman, R.A.; Young, K.M.; Kedersha, N.L.; Lou, H. U1 snrnp-dependent function of tiar in the regulation of alternative RNA processing of the human calcitonin/cgrp pre-mrna. Mol. Cell. Biol. 2003, 23, 5959–5971. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Hinman, M.N.; Hasman, R.A.; Mehta, P.; Lou, H. Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mrna. Mol. Cell. Biol. 2008, 28, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Kayikci, M.; Briese, M.; Zarnack, K.; Luscombe, N.M.; Rot, G.; Zupan, B.; Curk, T.; Ule, J. Iclip predicts the dual splicing effects of tia-rna interactions. PLoS Biol. 2010, 8, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Aznarez, I.; Barash, Y.; Shai, O.; He, D.; Zielenski, J.; Tsui, L.C.; Parkinson, J.; Frey, B.J.; Rommens, J.M.; Blencowe, B.J. A systematic analysis of intronic sequences downstream of 5′ splice sites reveals a widespread role for u-rich motifs and tia1/tial1 proteins in alternative splicing regulation. Genome Res. 2008, 18, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Garzia, A.; Mazzola, M.; Gerstberger, S.; Molina, H.; Tuschl, T. The tia1 rna-binding protein family regulates eif2ak2-mediated stress response and cell cycle progression. Mol. Cell 2018, 69, 622–635. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of tia-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.; Hennig, J.; Jagtap, P.K.; Sonntag, M.; Valcarcel, J.; Sattler, M. Structure, dynamics and RNA binding of the multi-domain splicing factor tia-1. Nucleic Acids Res. 2014, 42, 5949–5966. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Silanes, I.; Galban, S.; Martindale, J.L.; Yang, X.; Mazan-Mamczarz, K.; Indig, F.E.; Falco, G.; Zhan, M.; Gorospe, M. Identification and functional outcome of mrnas associated with RNA-binding protein tia-1. Mol. Cell. Biol. 2005, 25, 9520–9531. [Google Scholar] [CrossRef] [PubMed]
- Bley, N.; Lederer, M.; Pfalz, B.; Reinke, C.; Fuchs, T.; Glass, M.; Moller, B.; Huttelmaier, S. Stress granules are dispensable for mRNA stabilization during cellular stress. Nucleic Acids Res. 2015, 43, e26. [Google Scholar] [CrossRef] [PubMed]
- Arimoto-Matsuzaki, K.; Saito, H.; Takekawa, M. Tia1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 2016, 7, 10252. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. Tia1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017, 95, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Piecyk, M.; Wax, S.; Beck, A.R.; Kedersha, N.; Gupta, M.; Maritim, B.; Chen, S.; Gueydan, C.; Kruys, V.; Streuli, M.; et al. Tia-1 is a translational silencer that selectively regulates the expression of tnf-alpha. EMBO J. 2000, 19, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.R.; Miller, I.J.; Anderson, P.; Streuli, M. Rna-binding protein tiar is essential for primordial germ cell development. Proc. Natl. Acad. Sci. USA 1998, 95, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Karalok, H.M.; Aydin, E.; Saglam, O.; Torun, A.; Guzeloglu-Kayisli, O.; Lalioti, M.D.; Kristiansson, H.; Duke, C.M.; Choe, G.; Flannery, C.; et al. Mrna-binding protein tia-1 reduces cytokine expression in human endometrial stromal cells and is down-regulated in ectopic endometrium. J. Clin. Endocrinol. Metab. 2014, 99, E2610–E2619. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; Balch, G.C.; Kedersha, N.; Anderson, P.; Zimmerman, G.A.; Beauchamp, R.D.; Prescott, S.M. Regulation of cyclooxygenase-2 expression by the translational silencer tia-1. J. Exp. Med. 2003, 198, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Gueydan, C.; Droogmans, L.; Chalon, P.; Huez, G.; Caput, D.; Kruys, V. Identification of tiar as a protein binding to the translational regulatory au-rich element of tumor necrosis factor alpha mrna. J. Biol. Chem. 1999, 274, 2322–2326. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Cok, S.J.; Zeng, C.; Morrison, A.R. Translational repression of human matrix metalloproteinases-13 by an alternatively spliced form of t-cell-restricted intracellular antigen-related protein (tiar). J. Biol. Chem. 2003, 278, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Heck, M.V.; Azizov, M.; Stehning, T.; Walter, M.; Kedersha, N.; Auburger, G. Dysregulated expression of lipid storage and membrane dynamics factors in tia1 knockout mouse nervous tissue. Neurogenetics 2014, 15, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Kharraz, Y.; Salmand, P.A.; Camus, A.; Auriol, J.; Gueydan, C.; Kruys, V.; Morello, D. Impaired embryonic development in mice overexpressing the rna-binding protein tiar. PLoS ONE 2010, 5, e11352. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Jimenez, C.; Izquierdo, J.M. T-cell intracellular antigen (tia)-proteins deficiency in murine embryonic fibroblasts alters cell cycle progression and induces autophagy. PLoS ONE 2013, 8, e75127. [Google Scholar] [CrossRef] [PubMed]
- Tak, H.; Eun, J.W.; Kim, J.; Park, S.J.; Kim, C.; Ji, E.; Lee, H.; Kang, H.; Cho, D.H.; Lee, K.; et al. T-cell-restricted intracellular antigen 1 facilitates mitochondrial fragmentation by enhancing the expression of mitochondrial fission factor. Cell Death Differ. 2017, 24, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Carrascoso, I.; Alcalde, J.; Sanchez-Jimenez, C.; Gonzalez-Sanchez, P.; Izquierdo, J.M. T-cell intracellular antigens and hu antigen r antagonistically modulate mitochondrial activity and dynamics by regulating optic atrophy 1 gene expression. Mol. Cell. Biol. 2017, 37, e00174-17. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Li, W.; Nagayama, T.; He, X.; Sinor, A.D.; Chang, J.; Mao, X.; Graham, S.H.; Simon, R.P.; Greenberg, D.A. Expression of the rna-binding protein tiar is increased in neurons after ischemic cerebral injury. J. Neurosci. Res. 2000, 59, 767–774. [Google Scholar] [CrossRef]
- Kolarova, M.; Garcia-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and pathology of tau protein in alzheimer disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; Lummertz da Rocha, E.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the rna-binding protein tia1 regulates tau pathophysiology and toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Apicco, D.J.; Ash, P.E.A.; Maziuk, B.; LeBlang, C.; Medalla, M.; Al Abdullatif, A.; Ferragud, A.; Botelho, E.; Ballance, H.I.; Dhawan, U.; et al. Reducing the RNA binding protein tia1 protects against tau-mediated neurodegeneration in vivo. Nat. Neurosci. 2018, 21, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of rna-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]



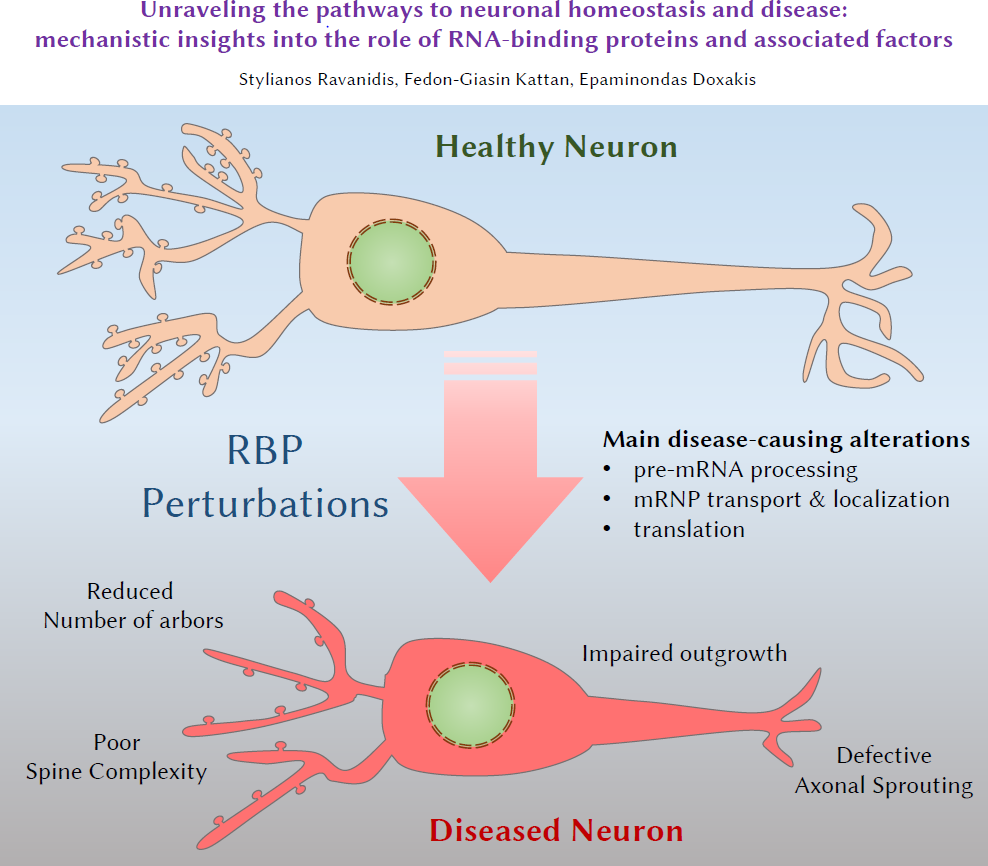{kind=link}
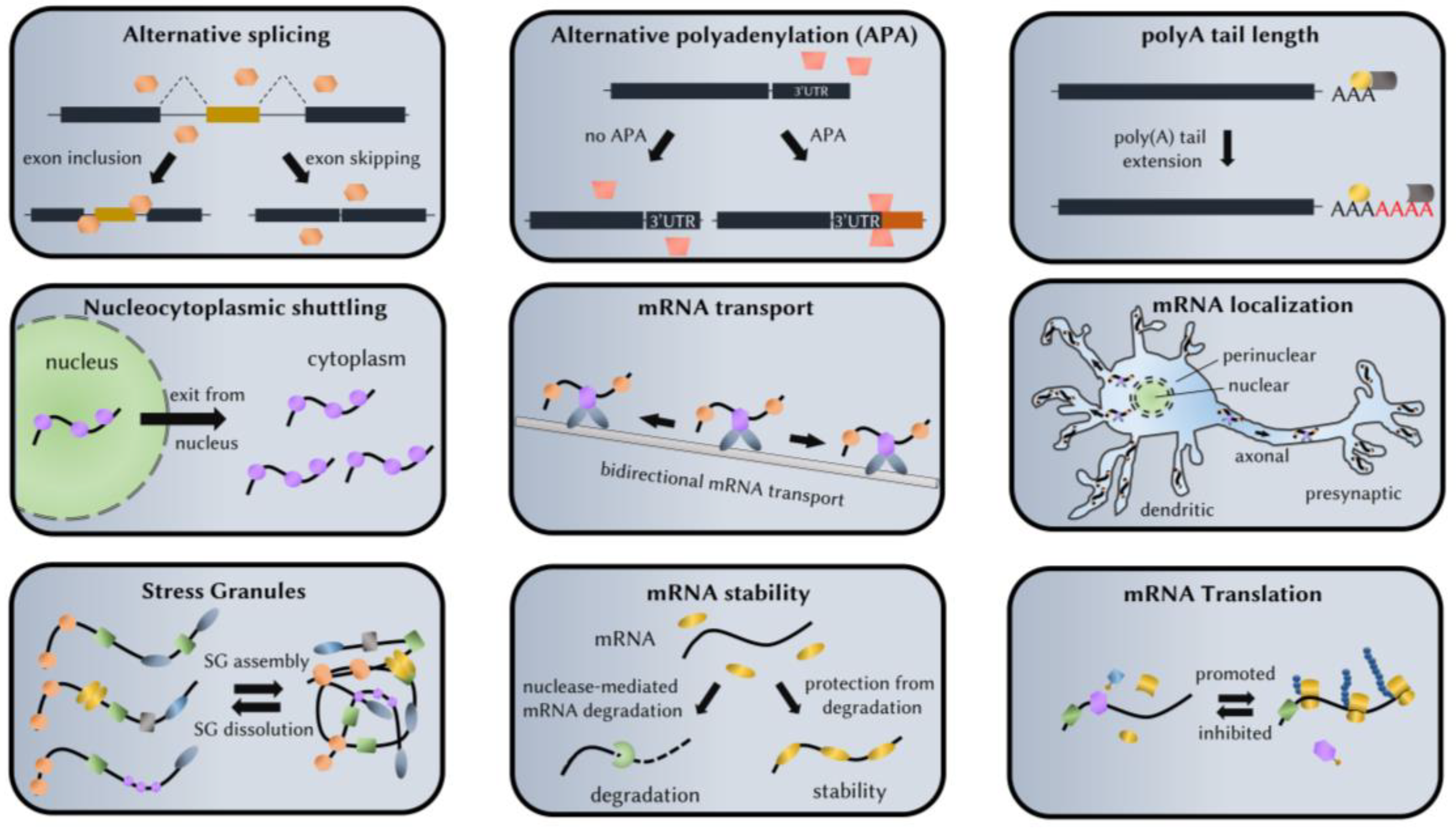{kind=link}
{kind=link}
| Protein | Binding Motif(s) | Main Functions | Human-Associated Pathology | Rodent Knockouts |
|---|---|---|---|---|
| Pumilio | UGUANAUA | Translation repressor Induce SG assembly RNP granule transport | Temporal Lobe Epilepsy (TLE) | Reduced number of neural stem cells Enhanced dendritic outgrowth and arborization SCA1-like neurodegeneration Progressive motor dysfunction Impaired learning and memory |
| Staufen | RNA stem–loops | RNP granule transport Polarized mRNA transport Inhibit SG assembly Translation enhancement Staufen mediated decay (SMD) | - | Reduced number of neural stem cells Reduced dendritic tree arborization Fewer synapses Deficits in locomotor activity |
| Insulin-like Growth Factor 2 mRNA-Binding Protein (IGF2BP) | CAUH (H = A, U, or C) or CA-rich motifs | RNP transport Translation inhibition | - | Perinatal death Smaller animal size Compromised central and peripheral synaptogenesis PSC and NSC depletion and accelerated differentiation Axonal growth deficits after peripheral nerve injury |
| FragileX Mental Retardation Protein (FMRP) | TGGA | Transcriptional activation Alternative splicing Inhibit RNA A-I editing Translation inhibition RNP transport miRISC recruitment on target DNA damage response | Fragile-X Syndrome (FXS) Fragile-X-Associated Tremor/Ataxia Syndrome (FXTAS) | More and shorter dendritic spines and delayed maturation Reduced mobility of growth cones Deficits in learning and memory Hyperactivity |
| Src-Associated substrate in Mitosis of 68 kDa (Sam68) | UAAA or UUAA | Intracellular signaling adaptor Promotes translation Alternative splicing RNP granule transport Proapoptotic roles after injury | Fragile-X-Associated Tremor /Ataxia Syndrome (FXTAS) Spinal Muscular Atrophy (SMA) | Reduced NPC proliferation Fewer dendritic spines Motor coordination deficits |
| Cytoplasmic Polyadenylation Element Binding protein (CPEB) | UUUAU or UUUUAAU | Cytoplasmic polyadenylation Alternative Polyadenylation RNP granule transport Local translation Promote SG assembly | - | Impaired mitochondrial function Reduced dendritic mRNA transport Reduced theta burst-induced LTP Inability to extinguish memories Motor coordination deficits Motor learning delay |
| Neuro-Oncological Ventral Antigen (NOVA) | YCAY (Y is C or U) | Alternative splicing Alternative polyadenylation Inhibit nonsense mediated decay RNP granule transport | Paraneoplastic Opsoclonus Myoclonus Ataxia (POMA) | Nova1 KO: Death within 3 weeks of birth Axonal outgrowth defects Profound motor failure Nova2 KO: Death within 2 weeks of birth Aberrant migration of cortical and Purkinje neurons Axonal outgrowth defects No LTP following external stimulation Double KO: death soon after birth due to lack of lung motor innervation |
| Embryonic Lethal/Abnormal Vision-Like (ELAVL) | U-rich (with G or A intermittently) | Transcription rate Alternative splicing Alternative polyadenylation Compete with miRNA binding to mRNAs mRNA stability Translation enhancement RNP transport | Paraneoplastic Encephalomyelopathy/Paraneoplastic Sensory Neuropathy (PEM/PSN) Schizophrenia Parkinson’s disease (PD) | Neuron targeted HuR KO: motor neuron disease (poor balance, decreased movement and strength) HuC KO: Purkinje cell impaired functionality and morphology; Cerebral Ataxia; Epileptic seizures; Impaired spatial learning HuD KO: Impaired spatial learning; Lower levels of anxiety and activity; Predisposition towards auditory-induced seizures; Fewer differentiated neurons; Reduced axodendritic complexity; Cortical and motor deficits HuC/D doubleKO: perinatal death |
| Survival Motor Neuron (SMN) | not an RBP | snRNP assembly Alternative splicing RNP granule transport Axonal protein synthesis Induce SG assembly | Spinal Muscular Atrophy (SMA) | Embryos die prenatally Reduced association of RNPs with microtubules and actin filaments Shorter neurites, fewer branches and poor terminal arborization Enhanced neuronal death Smaller RNP granules |
| TAR DNA-binding Protein 43 (TDP43) | UG repeats | Alternative splicing mRNA stability RNP granule transport Induce SG assembly miRNA biogenesis Translation regulation Nuclear pore transport | Familial Amyotrophic Lateral Sclerosis (fALS) Frontotemporal dementia (FTD) Alzheimer’s Disease (AD) Dementia with Lewy bodies (DLB) Huntington’s Disease (HD) | Degeneration of large motor axons Skeletal muscle grouped atrophy Denervation of the neuromuscular junction |
| Fused in Sarcoma (FUS) | GUGGU | Alternative splicing Alternative polyadenylation Nucleocytoplasmic shuttling miRNA biogenesis RNP granule transport DNA damage response | Familial Amyotrophic Lateral Sclerosis (fALS) Frontotemporal dementia (FTD) | Perinatal lethality Enhanced neuronal cell death |
| TATA-box binding protein Associated Factor 15 (TAF15) | GGUAAGU | Alternative splicing RNA stability RNP granule transport | Familial Amyotrophic Lateral Sclerosis (fALS) | - |
| T-cell-restricted Intracellular Antigen 1 (TIA1) & TIA1-Related (TIAR) | T-rich motifs (DNA) U-rich motifs (RNA) | Alternative splicing Translation inhibition SG nucleation/assembly Inhibit nonsense mediated decay DNA damage response | Familial Amyotrophic Lateral Sclerosis (fALS) Tauopathies | TIAR: embryonic lethality TIA1: high rate of perinatal death, widespread inflammation TIA1/TIAR doubleKO: embryonic lethality |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravanidis, S.; Kattan, F.-G.; Doxakis, E. Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors. Int. J. Mol. Sci. 2018, 19, 2280. https://doi.org/10.3390/ijms19082280
Ravanidis S, Kattan F-G, Doxakis E. Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors. International Journal of Molecular Sciences. 2018; 19(8):2280. https://doi.org/10.3390/ijms19082280
Chicago/Turabian StyleRavanidis, Stylianos, Fedon-Giasin Kattan, and Epaminondas Doxakis. 2018. "Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors" International Journal of Molecular Sciences 19, no. 8: 2280. https://doi.org/10.3390/ijms19082280