Roles of Wnt Target Genes in the Journey of Cancer Stem Cells
by
 ,
,
Jee-Heun Kim
1,†, 
So-Yeon Park
1,2,†,
Youngsoo Jun
1,2,3,
Ji-Young Kim
4 and
Jeong-Seok Nam
1,2,3,* 1
School of Life Sciences, Gwangju Institute of Science and Technology, Gwangju 61005, Korea
2
Cell Logistics Research Center, Gwangju Institute of Science and Technology, Gwangju 61005, Korea
3
Silver Health Bio Research Center, Gwangju Institute of Science and Technology, Gwangju 61005, Korea
4
Laboratory Animal Resource Center, Gwangju Institute of Science and Technology, Gwnagju 61005, Korea
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2017, 18(8), 1604; https://doi.org/10.3390/ijms18081604
Submission received: 28 June 2017
/
Revised: 19 July 2017
/
Accepted: 20 July 2017
/
Published: 25 July 2017
(This article belongs to the Special Issue Cancer Stem Cells)
Abstract
:The importance of Wnt/β-catenin signaling in cancer stem cells (CSCs) has been acknowledged; however, the mechanism through which it regulates the biological function of CSCs and promotes cancer progression remains elusive. Hence, to understand the intricate mechanism by which Wnt controls stemness, the specific downstream target genes of Wnt were established by analyzing the genetic signatures of multiple types of metastatic cancers based on gene set enrichment. By focusing on the molecular function of Wnt target genes, the biological roles of Wnt were interpreted in terms of CSC dynamics from initiation to metastasis. Wnt signaling participates in cancer initiation by generating CSCs from normal stem cells or non-CSCs and augmenting persistent growth at the primary region, which is resistant to anti-cancer therapy. Moreover, it assists CSCs in invading nearby tissues and in entering the blood stream, during which the negative feedback of the Wnt signaling pathway maintains CSCs in a dormant state that is suitable for survival. When CSCs arrive at distant organs, another burst of Wnt signaling induces CSCs to succeed in re-initiation and colonization. This comprehensive understanding of Wnt target genes provides a plausible explanation for how Wnt allows CSCs variation during cancer progression.

1. Introduction
Wnt signaling is a highly complex and evolutionarily conserved pathway that maintains pluripotency during embryonic development and regulates homeostasis in somatic stem cells from various tissues [1]. In recent decades, aberrant activation of Wnt signaling in various types of cancer has been documented and its roles in healthy tissues have been recognized. Genetic mutations that activate Wnt signaling reportedly contribute to cancer initiation [2], and nuclear accumulation of the Wnt signaling molecules β-catenin and lymphoid enhancer-binding factor 1 (LEF1) have been shown to be positively correlated with poor clinical outcomes, such as cancer progression, invasion, metastasis, and recurrence, resulting in low survival rates [2,3,4]. Accordingly, multiple studies on Wnt signaling have reported specific mechanisms that promote cancer initiation and progression and can therefore be investigated as therapeutic targets. In these studies, cancer stem cells (CSCs) have emerged as key players in Wnt-mediated carcinogenesis of various types. CSCs are a subpopulation of cancer cells with properties, such as self-renewal, slow cell cycle, persistent proliferation, homing, and mobilization, similar to those of normal stem cells and are central mediators of radio- and chemo-resistance in cancers as well as recurrence and metastasis [5,6]. Growing evidence has indicated increased Wnt signaling in CSCs compared with that in non-CSCs in multiple solid cancers and leukemia. Similarly, CSCs have elevated expression of Wnt downstream molecules compared with that in non-CSCs, as indicated by the high expression of frizzled receptors (FZD4/5) and increased sensitivity to Wnt3a-induced canonical Wnt signaling [7]. Moreover, Wnt signaling inhibition using genetic modifications or small molecule inhibitors has been shown to limit cancer stemness [8]. Specifically, deletion of the β-catenin gene results in complete regression of CD34+ CSCs in skin tumors. Conversely, expression of a non-degradable β-catenin expands the CSC population [9]. In the context of Wnt ligand secretion, inhibition of porcupine, which palmitoylates Wnt ligands for secretion, effectively decreases colony formation by limiting long-term self-renewal [10]. Similarly, the specific antibody OMP-18R5 blocks the binding of Wnt ligands to FZD [11] and the small molecule inhibitor CWP23228 prevents the formation of β-catenin/T-cell factor (TCF)/LEF complexes, leading to significant suppression of cancer growth, metastasis, and chemo-resistance through CSC inhibition in breast [12] and liver cancers [8]. Although the effects of Wnt on CSC stemness have been investigated in numerous studies, recent studies have suggested that Wnt signaling also plays roles in the generation of CSCs from normal stem cells and cancer cells that lack stemness. Accordingly, loss of adenomatous polyposis coli (APC) elevates the nuclear accumulation of β-catenin in leucine-rich repeat-containing G-protein-coupled receptor 5 LGR5+ normal stem cells and triggers neoplasia by transforming these cells into CSCs [13]. In addition, sustained high level of Wnt signaling leads to the transformation of differentiated gastrointestinal cells, which expressing high levels of doublecortin-like kinase (DCLK1), into CSCs [14]. Hence, Wnt signaling likely plays important roles in the initiation and maintenance of CSCs. However, although phenotypes and consequences of altered Wnt signaling have been reported, details of the associated regulatory mechanisms in CSCs remain unknown. Contributions of Wnt signaling to CSC initiation, persistence, resistance, invasion, and metastasis have been characterized in multiple studies, and upon CSC initiation, persistent growth in primary regions follows enhanced survival, reduced apoptosis, and altered metabolic activities in CSCs and in bulk tumor cells [2,3,4]. Subsequently, the epithelial-to-mesenchymal transition (EMT) allows entry into the process of invasion comprising detachment, intravasation, migration, and extravasation stages [2]. CSCs then progress to metastasis through dormancy, re-initiation, escape of immune surveillance, and establishment of microenvironments. In addition, defense mechanisms against chemo- and radiotherapy are highly activated in CSCs, which desensitize cells to DNA strand breaks, cell cycle arrest, and cytoskeleton or microtubule arrest, allowing continued survival and metastasis [5,6]. In this review, we investigated Wnt-mediated mechanisms that control stemness by examining downstream target genes involved in the characteristic features of CSCs from initiation to metastasis.
2. Target Genes of Wnt/β-Catenin Signaling
Wnt signaling regulates the expression of various genes through multiple pathways. In the canonical pathway, low β-catenin expression is maintained through phosphorylation of Ser/Thr residues and ubiquitination by protein degradation complexes. Following the stimulation of Wnt signaling by canonical ligands, degradation complexes are disrupted and de-phosphorylated β-catenin is translocated into the nucleus to bind LEF1/TCF4 family transcription factors, thereby transforming them from transcriptional repressors into transcriptional activators. In contrast, binding of non-canonical ligands to Wnt/FZD receptors transduces signals through intracellular calcium ions, c-Jun N-terminal kinases (JNK), receptor tyrosine kinase (RYK) or receptor tyrosine kinase-like orphan receptor (ROR), but not through β-catenin [15]. The canonical pathway has been studied more comprehensively than the non-canonical pathway and has been found to be activated by multiple mechanisms, including the destruction of degradation complexes, such as APC, and the consequent nuclear accumulation of β-catenin, in various types of cancer. Furthermore, the notion that CSCs possess higher activity of Wnt signaling covers canonical pathway as the LEF/TCF binding element showed higher transcriptional activity in CSCs than non-CSCs [16,17,18,19]. Therefore, to investigate the contributions of Wnt signaling to CSC regulation, we considered genes that are regulated by the canonical pathway and discuss their specific molecular mechanisms. Initially, we performed gene set enrichment analysis (GSEA) and identified a group of genes that are induced by β-catenin/LEF1/TCF4 complexes and are significantly upregulated in metastatic liver, colon, gastric and ovarian cancers compared with non-metastatic ones (Figure 1). In addition, we compared gene expression profiles in metastatic primary cancer (duke stage D) and early-stage non-metastatic primary cancers (duke stage A) using microarray data of colon cancers (GSE14333) and ovarian cancers (GSE2109) from the Gene Expression Omnibus. Subsequently, we identified genetic profiles that drive metastasis and applied gene expression data to GSEA to determine whether expression levels of the a priori defined set of genes differ significantly between the biological states. In these computations, genes that are upregulated by LEF1 were significantly enriched in metastatic cancers. Moreover, metastatic liver and gastric cancers showed increased expression of LEF1 target genes (TCGA). Thus, to further investigate the ensuing molecular mechanisms in CSC regulation, we generated target gene list from various source e.g., LEF1_UP.v1_UP gene set browsed from MSigDB, β-catenin target gene [20], and β-catenin chip assay result [21], and considered the biological functions of these genes and accordingly categorized them using Ingenuity Pathway Analysis (Ingenuity® System, Available online: http://www.ingenuity.com, Redwood City, CA, USA). Subsequently, we interpreted these biological functions in terms of initiation, persistence, maintenance, resistance to anticancer therapy, invasion of neighboring tissues, and metastasis of CSCs (Table 1). The full list of gene (Table S1) and the detailed analytical method can be found in Supplementary Materials.
3. Initiation
Cancer initiation is associated with various intrinsic and extrinsic factors, including direct DNA damage by genotoxic compounds, viral infection, and inherent and acquired genetic mutations. Tumorigenesis is strongly associated with the presence of CSCs, which cause heterogeneity of cancer cells according to proposed hierarchical models [36]. Although the exact mechanisms behind the production of CSCs and acquisition of pluripotency remain unknown, transformation of normal stem cells into CSCs or re-acquisition of stemness in subpopulations of cancer cells have been related to Wnt signaling and the expression of its target molecules [37]. For example, the intestinal stem cell marker LGR5 is expressed in 5–10% of adenoma cells and is a direct Wnt target gene that is induced by direct β-catenin binding [21]. In addition, β-catenin accumulation upon APC loss increases RAC1 expression, leading to increased generation of reactive oxygen species (ROS) and nuclear transcription factor-κB (NF-κB) signaling, which are known to stimulate the expansion of LGR5+ cell populations [38]. The positive correlation between LGR5+ cells and Wnt activation was also confirmed in localization analyses of LGR5+ cells [13], which were transformed into neoplasia following nuclear accumulation of β-catenin due to APC deletion [13]. A recent study has also shown that LGR5 regulates the tumor-initiating ability of CSCs and that selective apoptosis of LGR5+ cells decreases tumor formation in human organoids [39]. Moreover, LGR5 is known to bind the Wnt receptor component R-spondin [37], creating a positive feedback loop between aberrant Wnt signaling and expansion of LGR5+ cell populations and contributing to tumor initiation [40].
DCLK1, the transcription factor, is regulated by Wnt signaling through the LEF-binding site at its promoter region [22] and has been shown to be enriched in metastatic liver and colon cancers [41,42,43]. DCLK1 is generally known as a marker for gastrointestinal tuft cells but has recently been considered a CSC marker because it is expressed in quiescent cancer cells and because high fractions of DCLK1+ cells exhibit the stem-like characteristics and the ability to overcome quiescence with sufficient niche signals [44,45]. Although DCLK1 has been associated with various signaling pathways, such as those mediated by Notch, Hedgehog, and NF-kB, the resulting control of stemness has not yet been clearly elucidated [46,47,48]. DCLK1 is positively correlated with nuclear translocation of β-catenin upon APC deletion [49], and β-catenin knockdown in APCmin/+ mice leads to significant reduction in DCLK1 protein levels, which consequently attenuates crypt hyperplasia and tumorigenesis in APCmin/+ mice without affecting downstream NF-kB signaling [50]. In addition, DCLK1 knockdown in APCmin/+ mice attenuates intestinal adenoma and adenocarcinoma, whereas DCLK1 overexpression facilitates intestinal tumorigenesis in this model [51]. Wang et al. have investigated DCLK1 expression under normal and pathological conditions and have demonstrated their roles in colon cancer initiation [45]. In their study, DCLK1 expression was specifically increased in long-lived tuft cells, which originated from LGR5+ stem cells. Long-lived DCLK1+ tuft cells were also involved in regeneration upon chemical or microbial-induced injury and likely contributed to colon cancer as a tumor-initiating population with persistent Wnt activation [45]. Taken together, these studies have suggested that DCLK1 is a downstream molecule of the Wnt signaling pathway and is associated with direct LEF binding and cancer initiation.
Other CSC biomarkers include cluster of differentiation 44 (CD44), cluster of differentiation 133 (CD133), and aldehyde dehydrogenase (ALDH), which are positively regulated by Wnt/β-catenin signaling. Accordingly, compared with non-CSCs, ALDH+ or CD44+/CD24− breast CSCs exhibit higher TCF/LEF-dependent transcriptional activity and treatment with Wnt3a further increases relative numbers of CSCs, whereas Wnt ligand knockdown decreases them [52]. Similarly, relative numbers of ALDH1+/CD133+ liver CSCs decrease following the blockade of β-catenin-dependent transcription [8]. However, further studies are needed to determine whether these CSC biomarkers are involved in Wnt-mediated generation of CSCs.
4. Persistence
Both CSCs and non-CSCs can grow persistently, whereas only CSCs adapt to the surrounding environment to avoid cell death. Accordingly, small subpopulations of CSCs of patients have been maintained for years in differentiated culture systems, and non-CSCs can be de-differentiated to CSCs using cytokine supplements under appropriate culture conditions [53,54] and non-CSCs are reportedly derived from persistent CSCs [55] which proliferate more rapidly, express anti-apoptotic genes, and have consequent higher survival rates. In a recent study, genetic disruption of proliferation was found to deplete the maintenance of CSC populations remaining non-CSCs population intact among human epithelial breast and colon cancer cells [56]. CSCs have also been shown to resist the effects of radio- and chemotherapy conditions, which are extremely deleterious for tumor and surrounding cell populations [5].
Kruppel-like factor (KLF5) is a zinc-finger transcription factor that is critical for maintaining stem cell integrity and regulating the cell cycle [57]. The KLF5 gene is upregulated in various cancer types, including hepatocellular carcinoma and breast and intestinal cancers, and KLF5 deletion in the presence of induced mutant β-catenin suppresses the transcription of histone deacetylase 1 (HDAC1), which promotes colorectal cancer by promoting both stem and transit-amplifying cell proliferation [57]. KLF5 is constantly upregulated in CD44high/CD133high CSC populations, and KLF5 overexpression enhances colony-forming ability and resistance to anticancer drugs, such as cisplatin and 5-fluorouracil, reflecting increased anchorage-independent growth [58]. Regulatory roles of KLF5 on cancer stemness have also been indicated in siRNA-mediated KLF5-knockdown experiments that have shown reduced numbers of CD44high/CD133high cells [58]. In mice, inducible deletion of KLF5 in LGR5+ stem cells suppresses their proliferation and survival in association with nuclear localization of β-catenin and the generation of abnormal apoptotic cells in intestinal crypts [57].
Endothelin1 (EDN1) is primarily expressed in vascular epithelial cells, where it maintains vascular tone. EDN1 is reportedly secreted by many solid tumors, and it induces persistent growth and survival by promoting cell proliferation and suppressing apoptosis. Although the mechanisms that lead to pathological EDN1 levels are unclear, β-catenin suppression has shown to be closely associated with reduced EDN1 mRNA expression [23]. Moreover, END1 peptide secretions from colorectal cancer cells are significantly reduced after the introduction of dominant-negative TCF4 or β-catenin mutants, whereas overexpression of wild-type β-catenin or TCF results in increased END1 peptide production. In addition, treatments of APC-induced growth arrested cells with exogenous EDN1 restore proliferation by suppressing cell death. Furthermore, END1 overexpression enhances cell proliferation in vitro and in xenotransplantation assays and is accompanied with increases in the expression of several cell cycle and proliferation molecules [59]. Chip assays and sequencing analyses have also indicated direct regulation of EDN1 by β-catenin, and four potential TCF-binding elements have been identified in the EDN1 promoter region. Subsequent luciferase reporter assays have confirmed that β-catenin directly regulates EDN1 expression through these TCF-binding elements [23], and Rosano et al. have shown that EDN1 and its receptor are upregulated in cisplatinum-resistant and taxo-resistant cell lines, reflecting decreased sensitivity to cytotoxic drugs and increased survival and proliferation due to enhanced MAPK and Akt signaling [60].
Achaete-scute homolog 2 (ASCL2) is an intestinal stem cells transcription factor that contains a basic helix-loop-helix domain and a downstream Wnt signaling target [61]. ASCL2 overexpression is observed in colorectal cancers [62], and ASCL2 overexpression shifts the hierarchy of stem/progenitor cells in liver metastases and affects clinical outcomes [63]. Hence, dysregulated ASCL2 expression is thought to facilitate colorectal cancer cell proliferation [64,65]. Elevated ASCL2 expression in patients with osteosarcoma is also associated with osteosarcoma metastasis and poor prognosis [66]. Hence, with β-catenin and TCF, ASCL2 activates genes that are fundamental for maintaining the stem cell state, suggesting that ASCL2 forms a transcriptional switch that is Wnt-responsive and Wnt-dependent and defines stem cell identity. Moreover, ASCL2 is regulated in a direct autocrine loop that leads to distinct on/off expression patterns, and Wnt/R-spondin reportedly activates this regulatory loop [24]. Lastly, compared with control cells, ASCL2-overexpressing cells exhibit 5-FU resistance due to greater overall survival and fewer apoptotic cells [67].
As a member of the activator protein-1 superfamily, Fos-related antigen-1 (FRA1) positively regulates transcription and post-transcriptional processes. Accordingly, elevated FRA1 mRNA levels are associated with aberrant β-catenin accumulation in lung [68], brain [69], breast [70], and bladder [71] cancers. Furthermore, β-catenin accumulation and FRA-1 are positively correlated with WHO disease grades [69], and Chip assays have shown direct biding of β-catenin and FRA1 [21]. In experimental systems, FRA1 overexpression is sufficient to increase cell motility and anchorage-independent growth, and these are representative features of stem cells. In addition, compared with FRA1 expression, FRA1 knockdown prevents cell cycle progression, cell growth, and colony formation under anchorage-independent conditions and increases sensitivity to cisplatin [72]. At the protein level, FRA1 overexpression reduces p53 and increases MDM2 expression levels, consistent with apoptosis inhibition [68].
The transcription factor Myc proto-oncogene protein (MYC) favors cell growth and proliferation and suppresses cell cycle arrest [73,74]. These roles of MYC have been well documented in lymphocytes using conditional knockout systems [75,76]. In addition, MYC overexpression in B lymphocytes is sufficient to increase cell proliferation [77], and ectopic expression of MYC in hepatocytes using an adenoviral gene transfer system in vivo results in significant cell growth [78]. MYC is reportedly essential for Wnt-mediated growth, and its absence reduces Wnt-induced cell growth and proliferation in colonic crypts [78]. MYC can also switch metabolic phenotypes between aerobic glycolysis and oxidative phosphorylation by directly upregulating the transcription of glucose transporter-1, hexokinase-2, enolase, and lactate dehydrogenase [79]. In general, highly proliferative cancer cells depend on aerobic glycolysis rather than oxidative phosphorylation, and the ensuing rapid energy and lactic acid production is known as the Warburg effect [80]. Compared with non-CSCs, CSCs prefer Warburg-type energy metabolism, thereby allowing constitutive proliferation and resistance to cell damage by radiation-induced ROS [80,81]; this potentially reflects scavenging of ROS by abundant lactate in these cells [82]. In addition to metabolic and proliferative contributions to stemness, CSCs have to escape immune surveillance. MYC has been shown to directly regulate the expression of the tumor cell surface immune checkpoint protein cluster of differentiation 47 (CD47) and programmed death-ligand 1 (PD-L1), which mediate “don’t eat me” and “don’t find me” signals, respectively, following MYC binding to their promoters. MYC inactivation in mouse tumors also leads to decreased CD47 and PD-L1 expression and enhanced antitumor immune responses. However, MYC inactivation in tumors due to enforced CD47 or PD-L1 expression leads to suppressed immune responses and continued tumor growth [83,84]. Thus, MYC likely initiates and maintains tumorigenesis, in part, by modulating immune regulatory molecules.
Cyclin D1 (CCND1) is a traditional Wnt/β-catenin target gene, and time-lapse photography and quantitative image analyses have shown that CCND1 facilitates continued cell cycle progression [85]. This study has also revealed a highly ordered mechanism underlying continued proliferation, in which the decision to continue the cell cycle occurs at the G2 phase under conditions of elevated CCND1 expression. These conditions are maintained through the G1 phase and are required for initiation of the S phase, during which CCND1 levels are automatically reduced to allow DNA synthesis. High CCND1 expression is again required upon entry into the G2 phase. Consistent with this fact, CCND1 expression was found to be higher in metastatic lymph nodes than in primary regions of papillary thyroid carcinomas that were co-localized with β-catenin [86]. Another study demonstrated that CCND1 expression is upregulated in lung CSCs and promotes cell proliferation and clonogenic formation [87] in association with resistance against progestin [88] and cisplatin [89]. Collectively, these studies have indicated that Wnt-mediated re-colonization at distant orgnas is partly dependent on CCND1.
Chemo-resistance has also been associated with the ATP-binding cassette subfamily B member 1 (ABCB1), which is an ATP-dependent drug efflux pump that transports molecules across cell membranes. ABCB1 overexpression and its product P-glycoprotein are responsible for multiple drug resistance (MDR). The basal promoter of ABCB1 has several β-catenin/TCF4/LEF1-binding sites, suggesting that the canonical Wnt/β-catenin pathway regulates ABCB1, as shown in early colorectal [25], neuroblastoma [90] and breast [91] cancers. Furthermore, ABCB1 is highly overexpressed in doxorubicin-resistant cell lines [90], and β-catenin depletion in a chronic myeloid leukemia cell line leads to reduced ABCB1 mRNA expression, whereas Wnt signaling increases ABCB1 mRNA expression [92]. Taken together, these studies have indicated that resistance in CSCs can be achieved indirectly by mechanisms that lead to constitutive proliferation and resistance to apoptosis or directly by defense mechanisms that desensitize CSCs to anticancer agents and facilitate persistent growth.
5. Invasion and Migration
Although the origin of CSCs remains controversial, re-acquisition of CSC phenotypes in differentiated non-CSCs has been reported in multiple studies, and EMT is considered a major driver. EMT is a crucial process for morphogenesis during embryonic development and was initially recognized by developmental biologists. However, studies conducted in the last decade have shown that EMT can be aberrantly rebooted in adult tissues under pathological conditions, such as fibrosis and cancer in terms of poor wound healing and cancer invasiveness, respectively. EMT also contributes to the acquisition of invasion potential during breast carcinogenesis and chemotherapy. Specifically, during EMT, morphologic and phenotypic changes increase cancer cell motility, dissemination, invasiveness, and dedifferentiation [93,94]. Multiple signaling pathways are involved in the EMT process, and their downstream transcription factors, such as Snail, Twist, and Zeb, function as master controllers of the EMT process [95]. Moreover, among various signaling pathways, Wnt/β-catenin signaling is known to strongly promote EMT through LEF/TCF-dependent transcriptional activation [96]. Wnt/β-catenin activation is also required for generating CSCs from de-differentiated non-CSCs. In particular, inhibition of glycogen synthase kinase-3β (GSK-3β) expression promotes nuclear translocation of β-catenin and restores stem cell phenotypes in differentiated colorectal cells [97]. Moreover, activation of the E-twenty-six-related gene–FZD4 axis causes nuclear accumulation of β-catenin and transforms prostate cancer cells through EMT [98]. More recently, it has been confirmed that β-catenin forms a complex with Twist1/TCF4 and enhances its transactional activity, and consequently promotes tumorigenesis and re-acquisition of the CSC phenotype [99]. The invasion potential of CSCs is reportedly higher than that of non-CSCs, and this has been demonstrated in established cancer cell lines and patient-derived cancer cells. These experiments have indicated that glioma CSCs exhibit higher invasion activity than non-CSCs in the frontal region of tumors [100]. Similarly, ECM matrix penetration and cellular motility are enhanced in various types of CSCs, including those from breast [101] and liver [102] cancers, and some Wnt/β-catenin target genes have been shown to promote CSC invasion and migration.
Matrix metalloproteinase-7 (MMP7) is a well-known β-catenin target gene. Accordingly, ectopic LEF1 expression increases β-catenin binding to TCF-binding sequences and activates the MMP7 promoter [26]. Moreover, the conserved LEF1 recognition site is present on the promoter region of MMP-7 and is bound directly by LEF1 in oral squamous cell carcinoma cell lines and human oral squamous cell carcinoma tissues [103]. Members of the MMP family contribute to extracellular matrix (ECM) degradation and are classified according to their substrates as gelatinases (MMP2 and MMP9), collagenases (MMP1 and MMP13), metalloelastase (MMP12), and matrilysin (MMP7) [104]. MMP7 is synthesized and secreted into the extracellular matrix to increase invasion potential of cells via proteolytic degradation of ECM proteins. Although most MMPs degrade their ECM substrates, MMP7 is expressed in many epithelial cell types [105] and is reportedly expressed at high levels in multiple cancer types, including pancreatic [106], colon [107], and gastric [108] cancers. In particular, MMP7 is associated with highly invasive phenotypes that lead to poor prognoses in cancer patients. In addition to ECM degradation, MMP7 promotes cancer invasion by converting inactive forms of other MMPs, such as MMP2 and MMP9, into active forms [104]. MMP7 is involved in Wnt-mediated invasion in cancer cells. Accordingly, Wnt signaling activation by transfection with a mutant β-catenin that lacks the GSK-3β-specific phosphorylation site leads to morphological changes in oral squamous cancer cells from polygonal shape to spindle shape and decreases cell–cell interactions of cells with high invasion and migration capacity by inducing MMP7 [103]. These studies have implicated MMP7 as a mediator of increased CSC invasion potential in response to Wnt/β-catenin signaling. Similarly, membrane type 1-matrix metalloproteinase 1 (MT1-MMP) is directly regulated by the β-catenin/TCF complex through direct binding to its TCF promoter-binding site [109]. MT1-MMP increases invasion capacity through actin-rich cell protrusions that are known as invadopodia and are responsible for matrix degradation [110]; ectopic MT1-MMP overexpression promotes EMT [111]. Furthermore, CD133+ ovarian CSCs exhibit high MT1-MMP expression, and MT1-MMP knockdown specifically inhibits CSC invasiveness without affecting non-CSCs [112]. These experiments have indicated that MT1-MMP is a target of Wnt/β-catenin signaling that facilitates CSC infiltration into surrounding tissues.
In a previous study, inhibition of hyaluronan (HA) synthase-2 (HAS2), a biosynthetic enzyme of hyaluoran which is known as one of MMP7 regulators, was shown to decrease MMP7 expression under the condition of HAS2 inhibition and consequently inhibit the invasion ability of colon cancer cells [113]. HAS2 expression is also higher in bone-metastatic sub-clone breast cancer cells than in parental cells, and it increases the perforation of basement membranes by enhancing MMP activities without affecting their expression, reflecting inhibited expression of tissue metalloproteinase inhibitor 1 [114]. HAS2 expression is regulated by Wnt/β-catenin signaling, and LEF1 overexpression increases HAS2 mRNA expression in colon cancer cell lines by through its multiple binding sites on the promoter region of HAS2. Furthermore, HAS2 protein levels are indirectly increased by LEF1 via enhanced expression of HAS2 antisense RNA, which stabilizes HAS2 mRNA [27]. Consistent with this finding, breast CSCs with invasive phenotypes have been shown to express HAS2 at high levels, and subsequent treatment with the HAS2 inhibitor 4-methylumbelliferone has been shown to decrease the ability of CSC migration and invasion [115].
HA biosynthesis is activated as a consequence of increased HAS2 expression following Wnt signaling activation. Thereupon, the extracellular matrix components enter into HA-rich condition. As CD44 has HA binding site in its extracellular domain, HA binds to CD44 and transfers signals that mediate cellular responses to microenvironments, thereby increasing its motility [116]. This HA-induced invasive phenotype is dependent on C-X-C chemokine receptor type 4 (CXCR4) at least in part, due to its nearby location of HA binding site on CD44. The binding of HA on CD44 facilitates CXCR4 activation with its ligand, C-X-C motif chemokine ligand 12 (CXCL12), which consequently enhances MMP expression and CSC invasion [117]. Singh et al. revealed that the induction of CXCL12 enhanced the expression of MMP family members such as MMP1, MMP13, MMP9, MMP3, MMP10, MMP11 and MMP14 which are able to destruct the extraceullar matrix to increase invasion potentials [117]. For several decades, CD44 has been considered as one of the traditional Wnt target gene because CD44+ populations tend to expand when Wnt signaling is activated by APC mutation. In contrast, CD44+ populations are depleted by abrogation of the TCF4/β-catenin complex following genetic disruption of the TCF4/β-catenin-binding site [28]. Similarly, under HA-rich conditions, CD44+ populations frequently bound to CXCR4/CXCL12 [118], suggesting that indirect CXCR4 regulation by Wnt and HAS2 may increase the invasiveness of CD44+ CSCs with TCF/β-catenin-dependent expression.
Moreover, the direct regulation of Wnt on CXCL12 has been confirmed in previous studies [29]. Specifically, under conditions of Wnt activation in stem-like basal cancer cells, β-catenin binds to the promoter region of CXCL12 via a LEF/TCF-binding site and increases its mRNA expression. Moreover, this signaling mechanism is inhibited following β-catenin disruption by treatment with ICG-001 [29]. In another study, dense intra-tumoral microvessels have been observed near CD133+ glioma CSCs that co-express the CXCR4 [119], and one study has shown that CXCR4+ subpopulations of CD133+ pancreatic CSCs can evade primary tumors and disseminate into the blood stream [120]. Various experiments using CXCR4 inhibitors have suggested that CSCs exploit the CXCR4/CXCL12 axis to induce the secretion of vascular endothelial growth factor and further promote invasion into vessels [121,122]. Collectively, these studies have indicated that Wnt/β-catenin in CSCs promote invasive phenotypes via CXCR4/CXCL12 axis-mediated angiogenesis and lymphangiogenesis. Subsequently, invasive CSCs penetrate into surrounding tissues and into proximal blood and lymphatic vessels, leading to systemic distribution in the circulation system and arrival at metastatic sites.
6. Metastasis
Both cancer cell and CSC populations are heterogeneous, and CSC subpopulations with higher metastatic potency are likely to be disseminated into nearby tissues. In addition, migration to distant sites through the blood stream leads to the colonization of secondary organs and outgrowth. Circulating disseminated CD44+/CD24−/low or ALDH1high/CD24−/low expressing CSCs have been discovered in patients with metastatic breast cancer [123]. Similarly, disseminated CD133-expressing CSCs have been identified in patients with metastatic prostate [124] and recurrent colon [125] cancer. During circulation in the blood stream or at pre-metastatic niches, CSCs maintain dormancy and increase survival potential. Although the regulatory roles of Wnt in dormancy remain controversial, a recent study has suggested that Wnt-related mechanisms are involved in the regulation of latent competency, which is a dormant state with metastatic potential. Specifically, following Wnt signaling activation, the CXCR4/CXCL12 axis contributes to survival and dormancy status. In addition, analyses of patients with breast cancer have indicated that the expression of proto-oncogene tyrosine-protein kinase (Src) is positively correlated with late relapse in bone, but not in other tissues, and further mechanistic studies have shown that CXCR4/CXCL12-activated Src supports the survival of indolent breast cancer cells in bone marrow by activating Akt [126].
Dickkopf-related protein (DKK1) is a Wnt downstream molecule and a negative feedback component that likely plays essential roles in the regulation of late competency. Upon activation, Wnt reportedly binds TCF in the promoter region of DKK1 and induces its expression [31]. Li et al. have shown that instead of directly binding to the Wnt ligand, DKK1 antagonizes Wnt signaling by forming a complex with LDL receptor related protein 6 (LRP6) and Kremen and then removing LRP6 from the cell surface, thereby promoting its degradation and inhibiting Wnt binding and LRP6 turnover [127]. As an inhibitor of this Wnt autocrine loop, DKK1 decelerates the cell cycle and reduces the expression of innate immune sensors, allowing evasion of NK-cell-mediated immune surveillance [128]. Taken together, these data have indicated that Wnt activation may be essential for CSC transformation into dormancy and that it likely acts by upregulating DKK1 expression.
Moreover, one study has indicated that Wnt/β-catenin signaling controls metastatic colonization of target organs and that human lung adenocarcinomas enhance the competence of lung adenocarcinoma cells to colonize bone and brain tissues using distinct Wnt signaling pathways through LEF1 and HOXB9 [129]. During re-colonization at distant organs, CSCs escape from dormancy and re-initiate proliferation. This can be achieved by direct transcriptional activation in CSCs or by stimulation from cells, such as fibroblasts and immune cells, in the microenvironment. Wnt/β-catenin signaling can re-initiate cell cycle progression in dormant CSCs by upregulating c-MYC (MYC) expression through direct promoter binding [130,21]. Accordingly, MYC is central to the core interactome in metastatic breast cancer patients, and stable MYC knockdown decreases colony formation at secondary organs [131]. These studies have suggested that MYC mediates Wnt-induced metastatic colonization by re-initiating the cell cycle and cell growth.
Claudin1 (CLDN1) is a tight junction protein (TJP) that regulates the permeability of epithelial barriers through which small ions and neutral solutes enter [132]. Recently, the roles of CLDN1 in metastasis have received increasing attention because increased permeability of endothelial cells is required for cancer cells to intravasate and extravasate [133]. Moreover, CLDN1 expression in cancer cells is positively correlated with cancer progression because TJP is essential for cell–cell interactions that promote carcinogenesis and metastasis [134]. However, although CLDN1 overexpression in lung cancer cells increases cell–cell connections and prevents dissemination into proximal tissues [135], CLDN1 induces EMT in hepatocellular carcinomas and promotes invasion and metastasis by activating cellular signaling via Ras and extracellular signal-regulated kinases [136]. Moreover, inhibition of CLDN1 expression in gastric cancer cells results in decreased re-initiation of proliferation and leads to anoikis [137]. Similarly, CLDN1 augments anchorage-independent growth to favor metastatic colonization. However, clinical evidence has indicated that CLDN1 is a poor prognostic marker because higher CLDN1 levels are present in tumor tissues from distant metastatic sites in patients than in matched primary tumors [138,139]. Although further studies are needed to elucidate the precise mechanisms of CLDN1 in CSCs, resistance to anoikis is a typical phenotype of CSCs and is essential for anchorage-independent survival and colonization. Accordingly, CLDN1 expression is increased in CSCs compared with that in non-CSCs [140], and CLDN1 is directly upregulated by Wnt signaling. Specifically, ectopic APC expression decreases CLDN1 expression in Wnt-activated APC-deficient colon cancer cells, and CLDN1 requires its TCF-binding site for transcriptional activation [32]. In addition, the typical intestinal transcription factor caudal homeobox protein binds to the promoter of CLDN1 and increases its expression by forming a complex with TCF4 [141]. Hence, CLDN1 may mediate Wnt-induced re-initiation of CSCs at metastatic sites. Collectively, these studies have indicated that Wnt signaling re-activation at secondary organs can induce CLDN1 overexpression and trigger dormant CSC proliferation by promoting resistance to anoikis.
Metastatic CSCs has recently emerged which refers the specific subpopulations of CSCs that colonize better at the distant organs than other cell populations [142,143]. Therefore, various ongoing studies are directed at the discovery of biomarkers for metastatic CSCs, and the variant isoforms of CD44 (CD44v) have gained attention as one of novel biomarkers. CD44v are generated through alternative splicing from CD44 and gain the additional extracellular domains providing more glycosylation sites which enhance the ability of capturing various microenvironmental ligands including hepatocellular growth factor (HGF), osteopontin (OPN), vascular endothelial growth factor (VEGF), fibroblast growth factors (FGF), and CXCL12. Hence, these ligands are concentrated nearby CD44v and activate their downstream signalings such as PI3K-Akt, Smad, Src, and β-catenin pathway promoting metastasis in CD44v+ CSC [116,118,144]. For these reasons, CD44v makes CSCs respond to microenvironmental cytokines facilitating metastatic signalings via diverse pathways. Among existing numerous variants, CD44v6+ CSCs are recently documented as a subpopulation that forming outgrowths at distant metastatic sites by exhibiting constitutive reprogramming [33]. The specific inhibition of CD44v6 using antibodies have proved that metastasis of head and neck cancer is dependent of CD44v6+ CSCs [118]. Interestingly, in colon cancer, CD44v6+ CSCs are more frequently observed from the secondary organ outgrowth site with elevated β-catenin accumulation level, while CD44v6+ CSCs are hardly observed from the primary tumor site [33]. In addition, the presence of CD44v6+ populations was significantly decreased under the conditions of TCF4 loss [28]. Accordingly, the genetic profiles present that Wnt-signaling molecules are up-regulated in CD44v6+ CSCs compared to that of CD44v6− counterpart, and the LEF/TCF-dependent transcriptional activity is activated in CD44v6+ CSCs as well [33]. Moreover, Wnt3a ligand-mediated activation of Wnt signaling increases the existing population of CD44v6+ CSCs [33]. From these together, Wnt signaling enhances metastatic potential of CSCs through CD44v6 which activates metastatic signaling exhibiting more sensitive response to microenvironment.
In pre-metastatic niches, various components of secondary microenvironments can stimulate CSC colonization by activating Wnt signaling. For example, the extracellular matrix protein tenascin C is commonly found in stem cell niches and supports Wnt-mediated outgrowth of breast cancer micrometastases by increasing the expression of Syndecan 4, which has been implicated as a co-receptor of the Wnt receptor FZD7 [145,146,147]. Periostin is another matrix protein in stem cell niches that promotes the outgrowth of micrometastatic colonies by facilitating Wnt ligand secretion from tumor cells [148]. Tumor-associated macrophages also secrete interleukin-1β, which activates Wnt signaling in colon cancers by phosphorylating GSK-3β, stabilizing β-catenin, and enhancing TCF target gene expression [149]. Conversely, Wnt signaling contributes to microenvironments that favor re-colonization by CSCs. Wnt 7a is a canonical ligand of the Wnt pathway [150,151] and is required for the recruitment of cancer-associated fibroblasts that enhance invasion and metastatic potential of cancer cells [152]. Interestingly, the common ECM protein fibronectin (FN1) is directly upregulated by Wnt signaling through LEF/TCF-binding sites on its promoter region [34]. Moreover, FN1 promotes organ-specific metastasis in which hematopoietic progenitor cells expressing the FN1 receptor integrin α4β1 migrate and adhere to FN1-rich regions through ligand–receptor bonds and subsequently produce MMP-9 [153].
Lastly, Wnt signaling activation facilitates immune surveillance evasion through Cyclooxygenase-2 (COX2), which is directly upregulated by Wnt through its LEF/TCF promoter-binding site [35] and produces prostaglandin E2 (PGE2) to convert CD4+ T cells to regulatory T cells, thereby inducing apoptosis of CD8+ cytotoxic T cells [154]. Through this molecular mechanism, Wnt activation can promote bone metastasis of breast cancers by aggregating dead CD8+ cytotoxic T cells [30]. Collectively, current data indicate that Wnt signaling is a potent inducer of metastasis, induces CSC dormancy during circulation in the blood, re-initiates outgrowth of CSCs at secondary organs, and modulates microenvironments to favor CSCs.
7. Conclusions
Wnt signaling is considered as a major contributor to CSC biology. In this review, we summarized and characterized Wnt signaling mechanisms that regulate CSCs from initiation to metastasis. Initially, continuously upregulated Wnt signaling converts normal stem cells and differentiated cells into CSCs. Subsequently, abnormally hyperactivated Wnt signaling allows entry into the early stages of metastasis and then facilitates persistent growth, invasion, migration, and homing. Negative feedback of Wnt signaling pathway then induces CSC dormancy, and subsequent hyperactivated Wnt signaling is central to re-initiation and colonization of metastatic sites. Although Wnt signaling inhibitors have been developed in multiple studies, their use is limited by the involvement of Wnt signaling in homeostasis and development, leading to potential side effects. Moreover, Wnt signaling is dynamic throughout the process from initiation to metastasis, complicating the timing of therapeutic interventions that target Wnt. Therefore, further comprehensive studies on the downstream mechanisms of Wnt are required to develop novel therapeutic agents.
Supplementary Materials
Supplementary materials can be found at www.mdpi.com/1422-0067/18/8/1604/s1.
Acknowledgments
This work was supported by a grant from Cell Logistics Research Center, National Research Foundation of Korea (NFR-2016R1A5A1007318). And this research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI15C2056). This work was also supported by Gwangju Institute of Science and Technology (GIST) Research Institute (GRI) grant funded by the GIST in 2017.
Author Contributions
Jee-Heun Kim and So-Yeon Park performed systematic literature review, and wrote and edited manuscript; Youngsoo Jun and Ji-Young Kim critically revised the manuscript; Jeong-Seok Nam organized and provided the frame for the manuscript and critically revised the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| LEF1 | Lymphoid Enhancer-binding factor 1 |
| CSC | Cancer stem cell |
| FZD | Frizzled receptor |
| TCF | T-cell factor |
| APC | Adenomatous polyposis coli |
| LGR5 | Leucine-rich repeat-containing G-protein-coupled receptor 5 |
| DCLK1 | Doublecortin-like kinase 1 |
| EMT | Epithelial-to-mesenchymal transition |
| JNK | c-Jun N-terminal kinases |
| RYK | Receptor tyrosine kinase |
| ROR | Receptor tyrosine kinase-like orphan receptor |
| GSEA | Gene set enrichment analysis |
| ROS | Reactive oxygen species |
| NF-κB | Nuclear transcription factor-κB |
| CD44 | Cluster of differentiation 44 |
| CD133 | Cluster of differentiation 133 |
| ALDH | Aldehyde dehydrogenase |
| KLF5 | Kruppel-like factor |
| HDAC1 | Histone deacetylase 1 |
| EDN1 | Endothelin 1 |
| ASCL2 | Achaete-scute homolog 2 |
| FRA1 | Fos-related antigen-1 |
| CD47 | Cluster of differentiation 47 |
| PD-L1 | Programmed death-ligand 1 |
| CCND1 | Cyclin D1 |
| ABCB1 | ATP-binding cassette subfamily B member 1l |
| MDR | Multiple drug resistance |
| GSK-3β | Glycogen synthase kinase-3β |
| MMP7 | Matrix metalloproteinase-7 |
| ECM | Extracellular matrix |
| MT1-MMP | Membrane type 1-matrix metalloproteinase 1 |
| HA | Hyaluronan |
| HAS-2 | Hyaluronan synthase-2 |
| CXCR4 | C-X-C chemokine receptor 4 |
| CXCL12 | C-X-C motif chemokine ligand 12 |
| Src | Proto-oncogene tyrosine-protein kinase |
| DKK1 | Dickkopf-related protein |
| LRP6 | LDL receptor related protein 6 |
| CLDN1 | Claudin1 |
| TJP | Tight junction protein |
| CD44v | CD44 variant |
| HGF | Hepatocellular growth factor |
| OPN | Ostepontin |
| VEGF | Vascular endothelial growth factor |
| FGF | Fibroblast growth factors |
| FN1 | Fibronectin |
| COX2 | Cyclooxygenase 2 |
| PGE2 | Prostaglandin E2 |
References
- Kahn, M. Can we safely target the WNT pathway? Nature reviews. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Ormanns, S.; Neumann, J.; Horst, D.; Kirchner, T.; Jung, A. WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear β-Catenin depend on active PI3K signaling. Oncotarget 2014, 5, 2999–3011. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.; Akiri, G.; Chin, C.; Wisnivesky, J.P.; Beasley, M.B.; Weiser, T.S.; Swanson, S.J.; Aaronson, S.A. Wnt pathway activation predicts increased risk of tumor recurrence in patients with stage I nonsmall cell lung cancer. Ann. Surg. 2013, 257, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Yoshioka, Y.; Isohashi, F.; Seo, Y.; Yoshida, K.; Yamazaki, H. Radiotherapy targeting cancer stem cells: Current views and future perspectives. Anticancer Res. 2013, 33, 747–754. [Google Scholar] [PubMed]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Skoda, J.; Hermanova, M.; Loja, T.; Nemec, P.; Neradil, J.; Karasek, P.; Veselska, R. Co-Expression of Cancer Stem Cell Markers Corresponds to a Pro-Tumorigenic Expression Profile in Pancreatic Adenocarcinoma. PLoS ONE 2016, 11, e0159255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, H.Y.; Park, K.K.; Choi, Y.K.; Nam, J.S.; Hong, I.S. CWP232228 targets liver cancer stem cells through Wnt/β-catenin signaling: A novel therapeutic approach for liver cancer treatment. Oncotarget 2016, 7, 20395–20409. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on β-catenin signalling. Nature 2008, 452, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Zhang, B.; Ho, Y.; Cook, A.; Li, L.; Wang, Y.; McLaughlin, M.; Bhatia, R. Inhibition of CML Stem Cell Growth By Targeting WNT Signaling Using a Porcupine Inhibitor. Blood 2014, 124, 3130. [Google Scholar]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Hong, I.S.; Kim, R.J.; Lee, S.Y.; Park, S.J.; Lee, E.S.; Park, J.H.; Yun, C.H.; Chung, J.U.; Lee, K.J.; et al. Wnt/β-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Felipe De Sousa, E.M.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhu, Q.; Neuenschwander, M.; Specker, E.; Wulf-Goldenberg, A.; Weis, W.I.; Von Kries, J.P.; Birchmeier, W. A small-molecule antagonist of the β-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016, 76, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Prasetyanti, P.R.; Zimberlin, C.D.; Bots, M.; Vermeulen, L.; Felipe De Sousa, E.M.; Medema, J.P. Regulation of stem cell self-renewal and differentiation by Wnt and Notch are conserved throughout the adenoma-carcinoma sequence in the colon. Mol. Cancer 2013, 12, 126. [Google Scholar] [CrossRef] [PubMed]
- Vadakkan, T.J.; Landua, J.D.; Bu, W.; Wei, W.; Li, F.; Wong, S.T.; Dickinson, M.E.; Rosen, J.M.; Lewis, M.T.; Zhang, M. Wnt-responsive cancer stem cells are located close to distorted blood vessels and not in hypoxic regions in a p53-null mouse model of human breast cancer. Stem Cells Transl. Med. 2014, 3, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Jurinovic, V.; Krebs, S.; Thieme, S.E.; Blum, H.; Goke, B.; Kolligs, F.T. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signalling. BMC Genom. 2014, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Bottomly, D.; Kyler, S.L.; McWeeney, S.K.; Yochum, G.S. Identification of β-catenin binding regions in colon cancer cells using ChIP-Seq. Nucleic Acids Res. 2010, 38, 5735–5745. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Sarkar, S.; Luthra, G.K.; Okugawa, Y.; Toiyama, Y.; Gajjar, A.H.; Qiu, S.; Goel, A.; Singh, P. Epigenetic changes and alternate promoter usage by human colon cancers for expressing DCLK1-isoforms: Clinical Implications. Sci. Rep. 2015, 5, 14983. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Xiong, H.; Zhang, Z.; Ren, B. β-Catenin activates the growth factor endothelin-1 in colon cancer cells. Oncogene 2005, 24, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Schuijers, J.; Junker, J.P.; Mokry, M.; Hatzis, P.; Koo, B.K.; Sasselli, V.; van der Flier, L.G.; Cuppen, E.; van Oudenaarden, A.; Clevers, H. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015, 16, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takaoka, A.S.; Naishiro, Y.; Hayashi, R.; Maruyama, K.; Maesawa, C.; Ochiai, A.; Hirohashi, S. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/β-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000, 60, 4761–4766. [Google Scholar] [PubMed]
- Gustavson, M.D.; Crawford, H.C.; Fingleton, B.; Matrisian, L.M. Tcf binding sequence and position determines β-catenin and Lef-1 responsiveness of MMP-7 promoters. Mol. Carcinog. 2004, 41, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, I.; Freudenberger, T.; Twarock, S.; Yamaguchi, Y.; Grandoch, M.; Fischer, J.W. Esophageal squamous cell carcinoma cells modulate chemokine expression and hyaluronan synthesis in fibroblasts. J. Biol. Chem. 2016, 291, 4091–4106. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; Smits, R.; Korinek, V.; Smit, L.; Kielman, M.; Fodde, R.; Clevers, H.; Pals, S.T. Expression of CD44 in Apc and Tcfmutant mice implies regulation by the WNT pathway. Am. J. Pathol. 1999, 154, 515–523. [Google Scholar] [CrossRef]
- Holland, J.D.; Györffy, B.; Vogel, R.; Eckert, K.; Valenti, G.; Fang, L.; Lohneis, P.; Elezkurtaj, S.; Ziebold, U.; Birchmeier, W. Combined Wnt/β-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep. 2013, 5, 1214–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, Y.; Pleasure, S.J. Wnt signaling regulates intermediate precursor production in the postnatal dentate gyrus by regulating cxcr4 expression. Dev. Neurosci. 2012, 34, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef] [PubMed]
- Miwa, N.; Furuse, M.; Tsukita, S.; Niikawa, N.; Nakamura, Y.; Furukawa, Y. Involvement of claudin-1 in the β-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol. Res. 2001, 12, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradl, D.; Kühl, M.; Wedlich, D. The Wnt/Wg signal transducer β-catenin controls fibronectin expression. Mol. Cell. Biol. 1999, 19, 5576–5587. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, F.; Bravo, S.; Cruzat, F.; Montecino, M.; De Ferrari, G.V. Wnt/β-catenin signaling enhances cyclooxygenase-2 (COX2) transcriptional activity in gastric cancer cells. PLoS ONE 2011, 6, e18562. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Chen, L.; Li, C.; Zhu, Y. Heterogeneity in cancer stem cells. Cancer Lett. 2015, 357, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature 2017, 545, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage Tracing Reveals Lgr5+ Stem Cell Activity in Mouse Intestinal Adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.B.; Houchen, C.W.; Ali, N. APSA Awardee Submission: Tumor/cancer stem cell marker doublecortin-like kinase 1 in liver diseases. Exp. Biol. Med. 2017, 242, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Wang, M.; Xu, L.; Wen, T.; Liu, J.; An, G. DCLK1 is up-regulated and associated with metastasis and prognosis in colorectal cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Tanaka, S.; Akiyama, Y.; Shimada, S.; Adikrisna, R.; Matsumura, S.; Aihara, A.; Mitsunori, Y.; Ban, D.; Ochiai, T.; et al. Dominant Expression of DCLK1 in Human Pancreatic Cancer Stem Cells Accelerates Tumor Invasion and Metastasis. PLoS ONE 2016, 11, e0146564. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Yao, J.; Qu, D.; May, R.; Weygant, N.; Ge, Y.; Ali, N.; Sureban, S.M.; Gude, M.; Vega, K.; et al. Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells. Mol. Cancer 2017, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Invest. 2014, 124, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Panneerselvam, J.; Qu, D.; Weygant, N.; May, R.; Bronze, M.S.; Houchen, C.W. Regulatory Roles of Dclk1 in Epithelial Mesenchymal Transition and Cancer Stem Cells. J. Carcinog. Mutagen. 2016, 7, 257. [Google Scholar] [PubMed]
- Qu, D.; May, R.; Sureban, S.M.; Weygant, N.; Chandrakesan, P.; Ali, N.; Li, L.; Barrett, T.; Houchen, C.W. Inhibition of Notch signaling reduces the number of surviving Dclk1+ reserve crypt epithelial stem cells following radiation injury. American journal of physiology. Gastrointest. Liver Physiol. 2014, 306, G404–G411. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, P.; Subramaniam, D.; Paul, S.; Kwatra, D.; Palaniyandi, K.; Islam, S.; Harihar, S.; Ramalingam, S.; Gutheil, W.; Putty, S.; et al. Crocetinic acid inhibits hedgehog signaling to inhibit pancreatic cancer stem cells. Oncotarget 2015, 6, 27661–27673. [Google Scholar] [CrossRef] [PubMed]
- May, R.; Riehl, T.E.; Hunt, C.; Sureban, S.M.; Anant, S.; Houchen, C.W. Identification of a novel putative gastrointestinal stem cell and adenoma stem cell marker, doublecortin and CaM kinase-like-1, following radiation injury and in adenomatous polyposis coli/multiple intestinal neoplasia mice. Stem Cells 2008, 26, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Jakkula, L.U.M.R.; Ahmed, I.; Roy, B.; Anant, S.; Umar, S. Differential Effects of β-catenin and NF-κB Interplay in the Regulation of Cell Proliferation, Inflammation and Tumorigenesis in Response to Bacterial Infection. PLoS ONE 2013, 8, e79432. [Google Scholar] [CrossRef] [PubMed]
- Chandrakesan, P.; Weygant, N.; May, R.; Qu, D.; Chinthalapally, H.R.; Sureban, S.M.; Ali, N.; Lightfoot, S.A.; Umar, S.; Houchen, C.W. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014, 5, 9269–9280. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.-B.; Kim, J.-Y.; Cho, S.-D.; Park, K.-S.; Jung, J.-Y.; Lee, H.-Y.; Hong, I.-S.; Nam, J.-S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Lei, X.; GuAN, Y.; MOu, L.S.; Yuan, Y.-F.; Yue, H.; Lu, Y.; Xu, G.-T.; Qian, J. Maintenance of retinal cancer stem cell-like properties through long-term serum-free culture from human retinoblastoma. Oncol. Rep. 2011, 26, 135. [Google Scholar] [PubMed]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Cidado, J.; Wong, H.Y.; Rosen, D.M.; Cimino-Mathews, A.; Garay, J.P.; Fessler, A.G.; Rasheed, Z.A.; Hicks, J.; Cochran, R.L.; Croessmann, S. Ki-67 is required for maintenance of cancer stem cells but not cell proliferation. Oncotarget 2016, 7, 6281. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, T.; Ogawa, S.; Manabe, I.; Tanaka, M.; Sanada, M.; Sato, T.; Taketo, M.M.; Nakao, K.; Clevers, H.; Fukayama, M.; et al. KLF5 Regulates the Integrity and Oncogenicity of Intestinal Stem Cells. Cancer Res. 2014, 74, 2882–2891. [Google Scholar] [CrossRef] [PubMed]
- Maehara, O.; Sato, F.; Natsuizaka, M.; Asano, A.; Kubota, Y.; Itoh, J.; Tsunematsu, S.; Terashita, K.; Tsukuda, Y.; Nakai, M.; et al. A pivotal role of Kruppel-like factor 5 in regulation of cancer stem-like cells in hepatocellular carcinoma. Cancer Biol. Ther. 2015, 16, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-W.; Liao, C.-Y.; Yang, W.-Y.; Lin, Y.-M.; Jin, S.-L.C.; Wang, H.-D.; Yuh, C.-H. Overexpression of Endothelin 1 Triggers Hepatocarcinogenesis in Zebrafish and Promotes Cell Proliferation and Migration through the AKT Pathway. PLoS ONE 2014, 9, e85318. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; van Gijn, M.E.; Hatzis, P.; Kujala, P.; Haegebarth, A.; Stange, D.E.; Begthel, H.; van den Born, M.; Guryev, V.; Oving, I.; et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell 2009, 136, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Chalasani, S.; Frantz, G.D.; Smits, R.; Grabsch, H.I.; Kavi, V.; Maughan, N.J.; Hillan, K.J.; Quirke, P.; Koeppen, H. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene 2006, 25, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Stange, D.E.; Engel, F.; Longerich, T.; Koo, B.K.; Koch, M.; Delhomme, N.; Aigner, M.; Toedt, G.; Schirmacher, P.; Lichter, P.; et al. Expression of an ASCL2 related stem cell signature and IGF2 in colorectal cancer liver metastases with 11p15.5 gain. Gut 2010, 59, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Yang, Y.; Tian, Y.; Bai, J.; Zhang, X.; Li, X.; Peng, Z.; He, Y.; Chen, L.; Pan, Q.; et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PLoS ONE 2012, 7, e32170. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Pan, Q.; Shang, Y.; Zhu, R.; Ye, J.; Liu, Y.; Zhong, X.; Li, S.; He, Y.; Chen, L.; et al. MicroRNA-200 (miR-200) cluster regulation by achaete scute-like 2 (Ascl2): Impact on the epithelial-mesenchymal transition in colon cancer cells. J. Biol. Chem. 2014, 289, 36101–36115. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.H.; Cui, Y.H.; Guo, Q.N.; Zhou, Y. Elevated ASCL2 expression is associated with metastasis of osteosarcoma and predicts poor prognosis of the patients. Am. J. Cancer Res. 2016, 6, 1431–1440. [Google Scholar] [PubMed]
- Kwon, O.H.; Park, J.L.; Baek, S.J.; Noh, S.M.; Song, K.S.; Kim, S.Y.; Kim, Y.S. Aberrant upregulation of ASCL2 by promoter demethylation promotes the growth and resistance to 5-fluorouracil of gastric cancer cells. Cancer Sci. 2013, 104, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Chen, X.; Fang, X.; Wang, D.; Xie, M.; Chen, Q. Fra-1 is upregulated in lung cancer tissues and inhibits the apoptosis of lung cancer cells by the P53 signaling pathway. Oncol. Rep. 2016, 35, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial-mesenchymal transition. Biosci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Motrich, R.D.; Castro, G.M.; Caputto, B.L. Old players with a newly defined function: Fra-1 and c-Fos support growth of human malignant breast tumors by activating membrane biogenesis at the cytoplasm. PLoS ONE 2013, 8, e53211. [Google Scholar] [CrossRef] [PubMed]
- Sayan, A.E.; Stanford, R.; Vickery, R.; Grigorenko, E.; Diesch, J.; Kulbicki, K.; Edwards, R.; Pal, R.; Greaves, P.; Jariel-Encontre, I.; et al. Fra-1 controls motility of bladder cancer cells via transcriptional upregulation of the receptor tyrosine kinase AXL. Oncogene 2012, 31, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371–34383. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.V. The role of c-myc in cellular growth control. Oncogene 1999, 18, 2988–2996. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- de Alboran, I.M.; O’Hagan, R.C.; Gärtner, F.; Malynn, B.; Davidson, L.; Rickert, R.; Rajewsky, K.; DePinho, R.A.; Alt, F.W. Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 2001, 14, 45–55. [Google Scholar] [CrossRef]
- Schuhmacher, M.; Staege, M.S.; Pajic, A.; Polack, A.; Weidle, U.H.; Bornkamm, G.W.; Eick, D.; Kohlhuber, F. Control of cell growth by c-Myc in the absence of cell division. Curr. Biol. 1999, 9, 1255–1258. [Google Scholar] [CrossRef]
- Kim, S.; Li, Q.; Dang, C.V.; Lee, L.A. Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 11198–11202. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016, 14, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Hasan, M.R. Cancer metabolism and drug resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F.; Kline, J. Myc—A thorn in the side of cancer immunity. Cell Res. 2016, 26, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Stacey, D.W. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 2003, 15, 158–163. [Google Scholar] [CrossRef]
- Zhang, J.; Gill, A.J.; Issacs, J.D.; Atmore, B.; Johns, A.; Delbridge, L.W.; Lai, R.; McMullen, T.P. The Wnt/β-catenin pathway drives increased cyclin D1 levels in lymph node metastasis in papillary thyroid cancer. Hum. Pathol. 2012, 43, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-L.; Jiang, L.-M.; Han, W.-D. Wnt/β-catenin signaling pathway in lung cancer stem cells is a potential target for the development of novel anticancer drugs. J. BUON 2015, 20, 1094–1100. [Google Scholar] [PubMed]
- Musgrove, E.A.; Hunter, L.J.; Lee, C.S.; Swarbrick, A.; Hui, R.; Sutherland, R.L. Cyclin D1 overexpression induces progestin resistance in T-47D breast cancer cells despite p27Kip1 association with cyclin E-Cdk2. J. Biol. Chem. 2001, 276, 47675–47683. [Google Scholar] [CrossRef] [PubMed]
- Biliran, H., Jr.; Wang, Y.; Banerjee, S.; Xu, H.; Heng, H.; Thakur, A.; Bollig, A.; Sarkar, F.H.; Liao, J.D. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin. Cancer Res. 2005, 11, 6075–6086. [Google Scholar] [CrossRef] [PubMed]
- Flahaut, M.; Meier, R.; Coulon, A.; Nardou, K.A.; Niggli, F.K.; Martinet, D.; Beckmann, J.S.; Joseph, J.M.; Muhlethaler-Mottet, A.; Gross, N. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/β-catenin pathway. Oncogene 2009, 28, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Xia, W.; Wong, G. Hyaluronan-mediated CD44 interaction with p300 and SIRT1 regulates β-catenin signaling and NFκB-specific transcription activity leading to MDR1 and Bcl-xL gene expression and chemoresistance in breast tumor cells. J. Biol. Chem. 2009, 284, 2657–2671. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.; Binato, R.; Du Rocher, B.; Castelo-Branco, M.T.; Pizzatti, L.; Abdelhay, E. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer 2012, 12, 303. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Weinberg, R.A. In Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol. 2012, 22, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Du, L.; Chen, D.; Ye, Z.; Duan, H.; Tu, T.; Feng, J.; Yang, Y.; Chen, Q.; Yan, X. Reduced CD146 expression promotes tumorigenesis and cancer stemness in colorectal cancer through activating Wnt/β-catenin signaling. Oncotarget 2016, 7, 40704. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Iljin, K.; Sara, H.; Mpindi, J.P.; Mirtti, T.; Vainio, P.; Rantala, J.; Alanen, K.; Nees, M.; Kallioniemi, O. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2010, 70, 6735–6745. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-W.; Su, Y.-J.; Hsiao, M.; Wei, K.-C.; Lin, W.-H.; Liang, C.-J.; Chen, S.-C.; Lee, J.-L. Diverse targets of β-catenin during the epithelial–mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res. 2015, 75, 3398–3410. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Thiagarajan, P.S.; Rai, V.; Reizes, O.; Lathia, J.; Egelhoff, T. Increased cancer stem cell invasion is mediated by myosin IIB and nuclear translocation. Oncotarget 2016, 7, 47586. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hur, W.; Hong, S.W.; Kim, J.-H.; Kim, S.M.; Lee, E.B.; Yoon, S.K. ELK3 promotes the migration and invasion of liver cancer stem cells by targeting HIF-1α. Oncol. Rep. 2017, 37, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Yonekawa, A.; Harada, C.; Hamada, M.; Katagiri, W.; Nakazawa, M.; Yura, Y. Involvement of the Wnt-β-catenin pathway in invasion and migration of oral squamous carcinoma cells. Int. J. Oncol. 2010, 37, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Said, A.H.; Raufman, J.-P.; Xie, G. The role of matrix metalloproteinases in colorectal cancer. Cancers 2014, 6, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Reunanen, N.; Kähäri, V. Matrix metalloproteinases in cancer cell invasion. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2002; pp. 1–19. [Google Scholar]
- Yamamoto, H.; Itoh, F.; Iku, S.; Adachi, Y.; Fukushima, H.; Sasaki, S.; Mukaiya, M.; Hirata, K.; Imai, K. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human pancreatic adenocarcinomas: Clinicopathologic and prognostic significance of matrilysin expression. J. Clin. Oncol. 2001, 19, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Yamamoto, H.; Itoh, F.; Hinoda, Y.; Okada, Y.; Imai, K. Contribution of matrilysin (MMP-7) to the metastatic pathway of human colorectal cancers. Gut 1999, 45, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Aihara, R.; Mochiki, E.; Nakabayashi, T.; Akazawa, K.; Asao, T.; Kuwano, H. Clinical significance of mucin phenotype, β-catenin and matrix metalloproteinase 7 in early undifferentiated gastric carcinoma. Br. J. Surg. 2005, 92, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Hlubek, F.; Spaderna, S.; Jung, A.; Kirchner, T.; Brabletz, T. β-Catenin activates a coordinated expression of the proinvasive factors laminin-5 γ2 chain and MT1-MMP in colorectal carcinomas. Int. J. Cancer 2004, 108, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and molecular mechanisms of MT1-MMP-dependent cancer cell invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Li, Q.; Li, S.; He, J.; Cao, W.; Lan, J.; Sun, B.; Zou, H.; Wang, C.; Liu, R.; et al. Membrane type 1-matrix metalloproteinase induces epithelial-to-mesenchymal transition in esophageal squamous cell carcinoma: Observations from clinical and in vitro analyses. Sci. Rep. 2016, 6, 22179. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Zhang, X.; Mizumoto, Y.; Maida, Y.; Bono, Y.; Takakura, M.; Kyo, S. Molecular characterization of CD133+ cancer stem-like cells in endometrial cancer. Int. J. Oncol. 2014, 44, 669–677. [Google Scholar] [PubMed]
- Dunn, K.M.B.; Lee, P.K.; Wilson, C.M.; Iida, J.; Wasiluk, K.R.; Hugger, M.; McCarthy, J.B. Inhibition of hyaluronan synthases decreases matrix metalloproteinase-7 (MMP-7) expression and activity. Surgery 2009, 145, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Bernert, B.; Porsch, H.; Heldin, P. Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1). J. Biol. Chem. 2011, 286, 42349–42359. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Kobayashi, A.; Xia, B.; Watabe, M.; Pai, S.K.; Hirota, S.; Xing, F.; Liu, W.; Pandey, P.R.; Fukuda, K. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012, 72, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Boil. 2003, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Singh, U.P.; Grizzle, W.E.; Lillard, J.W. CXCL12–CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab. Investig. 2004, 84, 1666–1676. [Google Scholar] [CrossRef] [PubMed]
- Orian-Rousseau, V. CD44 Acts as a Signaling Platform Controlling Tumor Progression and Metastasis. Front. Immunol. 2015, 6, 154. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.; Chen, J.H.; Wang, B.; Zhang, R. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Liekens, S.; Schols, D.; Hatse, S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr. Pharm. Des. 2010, 16, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Bajetto, A.; Harrison, J.K.; Barbieri, F.; Florio, T. CXCL12 modulation of CXCR4 and CXCR7 activity in human glioblastoma stem-like cells and regulation of the tumor microenvironment. Front. Cell. Neurosci. 2014, 8, 144. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, H.; Watanabe, T.; Mimori, K.; Adachi, M.; Hayashi, N.; Tamura, J.; Matsuda, K.; Fukushima, R.; Okinaga, K.; Sasako, M. Clinical significance of circulating tumor cells, including cancer stem-like cells, in peripheral blood for recurrence and prognosis in patients with Dukes’ stage B and C colorectal cancer. J. Clin. Oncol. 2011, 29, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.-F.; Wang, Q.; Gerald, W.; Hudis, C.A.; Norton, L.; Smid, M.; Foekens, J.A.; Massagué, J. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 2009, 16, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, W.; King, T.D.; Liu, C.C.; Bijur, G.N.; Bu, G. Dkk1 stabilizes Wnt co-receptor LRP6: Implication for Wnt ligand-induced LRP6 down-regulation. PLoS ONE 2010, 5, e11014. [Google Scholar] [CrossRef] [PubMed]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Chiang, A.C.; Zhang, X.H.-F.; Kim, J.Y.; Kris, M.G.; Ladanyi, M.; Gerald, W.L.; Massagué, J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 2009, 138, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.G.; Aschenbach, J.R.; Amasheh, S. Claudin clusters as determinants of epithelial barrier function. IUBMB Life 2015, 67, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Salvador, E.; Burek, M.; Förster, C.Y. Tight Junctions and the Tumor Microenvironment. Curr. Pathobiol. Rep. 2016, 4, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Tabariès, S.; Siegel, P. The role of claudins in cancer metastasis. Oncogene 2016, 36, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.-C.; Pan, S.-H.; Yang, S.-C.; Yu, S.-L.; Che, T.-F.; Lin, C.-W.; Tsai, M.-S.; Chang, G.-C.; Wu, C.-H.; Wu, Y.-Y. Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am. J. Respir. Crit. Care Med. 2009, 179, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Filipović, A.; Giamas, G. Claudin-1 as a promoter of EMT in hepatocellular carcinoma. Oncogene 2013, 32, 4871–4872. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; He, C.; Qu, Y.; Li, J.; Zhang, J.; Du T, C.X.; Yu, Y.; Liu, B.; Zhu, Z. Claudin-1 enhances tumor proliferation and metastasis by regulating cell anoikis in gastric cancer. Oncotarget 2015, 6, 1652. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.-R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, T.; Akagi, Y.; Ochi, T.; Tanaka, N.; Kawahara, A.; Ishibashi, Y.; Gotanda, Y.; Yamaguchi, K.; Shiratuchi, I.; Oka, Y. Increased claudin-1 protein expression in hepatic metastatic lesions of colorectal cancer. Anticancer Res. 2012, 32, 2309–2314. [Google Scholar] [PubMed]
- Liu, T.; Cheng, W.; Lai, D.; Huang, Y.; Guo, L. Characterization of primary ovarian cancer cells in different culture systems. Oncol. Rep. 2010, 23, 1277. [Google Scholar] [PubMed]
- Bhat, A.A.; Sharma, A.; Pope, J.; Krishnan, M.; Washington, M.K.; Singh, A.B.; Dhawan, P. Caudal homeobox protein Cdx-2 cooperates with Wnt pathway to regulate claudin-1 expression in colon cancer cells. PLoS ONE 2012, 7, e37174. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-T.; Ye, Y.-P.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q. Metastatic cancer stem cells: From the concept to therapeutics. Am. J. Stem Cells 2014, 3, 46–62. [Google Scholar] [PubMed]
- Diehn, M.; Majeti, R. Metastatic cancer stem cells: An opportunity for improving cancer treatment? Cell Stem Cell 2010, 6, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Motiani, K.; Giridhar, P.V.; Kasper, S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp. Biol. Med. 2013, 238, 324–338. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived IL-1β stimulates Wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin D3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.K.; Zou, Y.; Ghosh, A. Wnts acting through canonical and noncanonical signaling pathways exert opposite effects on hippocampal synapse formation. Neural Dev. 2008, 3, 32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shu, W.; Lu, M.M.; Morrisey, E.E. Wnt7b activates canonical signaling in epithelial and vascular smooth muscle cells through interactions with Fzd1, Fzd10, and LRP5. Mol. Cell. Biol. 2005, 25, 5022–5030. [Google Scholar] [CrossRef] [PubMed]
- Avgustinova, A.; Iravani, M.; Robertson, D.; Fearns, A.; Gao, Q.; Klingbeil, P.; Hanby, A.M.; Speirs, V.; Sahai, E.; Calvo, F. Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat. Commun. 2016, 7, 10305. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Karavitis, J.; Zhang, M. COX2 regulation of breast cancer bone metastasis. Oncoimmunology 2013, 2, e23129. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Enrichment of Wnt target genes in multiple metastatic cancer types. A group of genes that are induced by β-catenin/LEF1/TCF4 complex were identified through performed gene set enrichment analysis (GSEA). Microarray data of four different cancer, liver (LIHC), colon (GSE14333), gastric (STAD) and ovarian cancer (GSE2109), were analyzed. Genes upregulated by LEF1 were significantly enriched in metastatic cancers. The detailed methods are described in Supplementary Materials. TCGA; The Cancer Genome Atlas, LEF1; lymphoid enhancer-binding factor 1, GEO; Gene Expression Omnibus.
Figure 1.
Enrichment of Wnt target genes in multiple metastatic cancer types. A group of genes that are induced by β-catenin/LEF1/TCF4 complex were identified through performed gene set enrichment analysis (GSEA). Microarray data of four different cancer, liver (LIHC), colon (GSE14333), gastric (STAD) and ovarian cancer (GSE2109), were analyzed. Genes upregulated by LEF1 were significantly enriched in metastatic cancers. The detailed methods are described in Supplementary Materials. TCGA; The Cancer Genome Atlas, LEF1; lymphoid enhancer-binding factor 1, GEO; Gene Expression Omnibus.


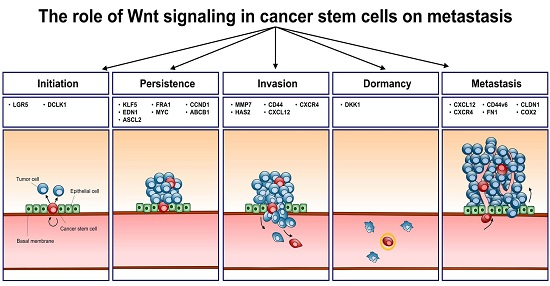{kind=link}
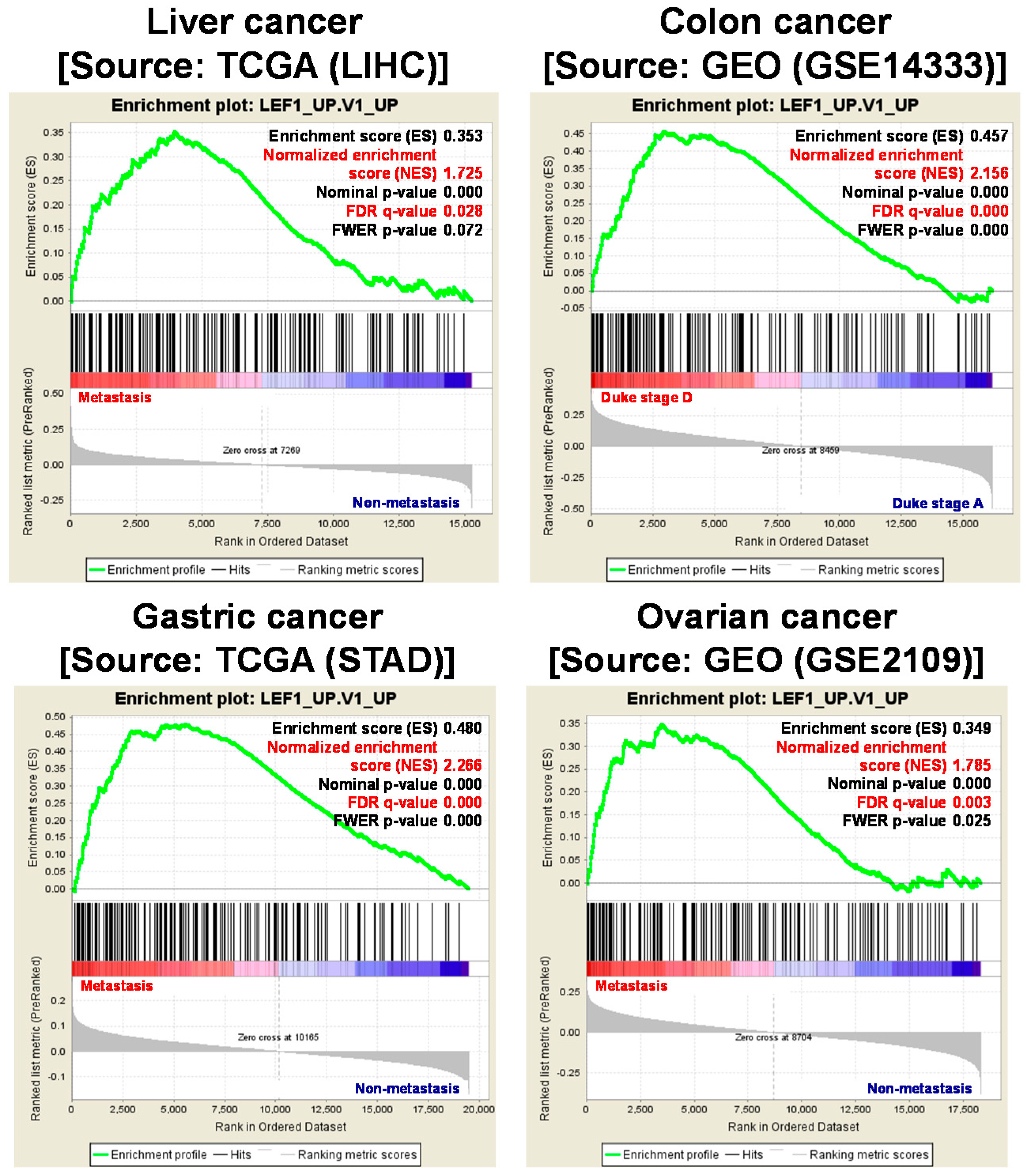{kind=link}
Table 1.
Summary of Wnt target genes and their biological functions in cancer stem cells (CSCs).
| Biological Function | Gene | Full Name | Direct/Indirect Target | Reference |
|---|---|---|---|---|
| Initiation | LGR5 | Leucine-rich repeat-containing G-protein-coupled receptor 5 | Direct | [21] |
| DCLK1 | Doublecortin-like kinase | Direct | [22] | |
| Persistence | KLF5 | Krueppel-like factor 5 | Direct | [21] |
| EDN1 | Endothelin-1 | Direct | [23] | |
| ASCL2 | Achaete-scute homolog 2 | Direct | [24] | |
| FRA1 | Fos-related antigen 1 | Direct | [21] | |
| MYC | Myc proto-oncogene protein | Direct | [21] | |
| CCND1 | CyclinD1 | Direct | [21] | |
| ABCB1 | ABC multidrug transporter | Direct | [25] | |
| Invasion | MMP7 | Matrix Metallopeptidase 7 | Direct | [26] |
| HAS2 | Hyaluronan synthase-2 | Direct | [27] | |
| CD44 | Cluster of differentiation 44 | Indirect | [28] | |
| CXCL12 | C-X-C motif chemokine ligand 12 | Direct | [29] | |
| CXCR4 | Chemokine receptor type 4 | Direct | [30] | |
| Metastasis | CXCL12 | C-X-C motif chemokine ligand 12 | Direct | [29] |
| CXCR4 | Chemokine receptor type 4 | Direct | [30] | |
| DKK1 | Dickkopf-related protein 1 | Direct | [31] | |
| CLDN1 | Claudin-1 | Direct | [32] | |
| CD44v6 | Cluster of differentiation 44 variant exon 6 | Indirect | [33] | |
| FN1 | Fibronectin | Direct | [34] | |
| COX2 | Cyclooxygenase-2 | Direct | [35] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, J.-H.; Park, S.-Y.; Jun, Y.; Kim, J.-Y.; Nam, J.-S. Roles of Wnt Target Genes in the Journey of Cancer Stem Cells. Int. J. Mol. Sci. 2017, 18, 1604. https://doi.org/10.3390/ijms18081604
AMA Style
Kim J-H, Park S-Y, Jun Y, Kim J-Y, Nam J-S. Roles of Wnt Target Genes in the Journey of Cancer Stem Cells. International Journal of Molecular Sciences. 2017; 18(8):1604. https://doi.org/10.3390/ijms18081604
Chicago/Turabian StyleKim, Jee-Heun, So-Yeon Park, Youngsoo Jun, Ji-Young Kim, and Jeong-Seok Nam. 2017. "Roles of Wnt Target Genes in the Journey of Cancer Stem Cells" International Journal of Molecular Sciences 18, no. 8: 1604. https://doi.org/10.3390/ijms18081604
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.