Mechanisms of Post-Replication DNA Repair

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
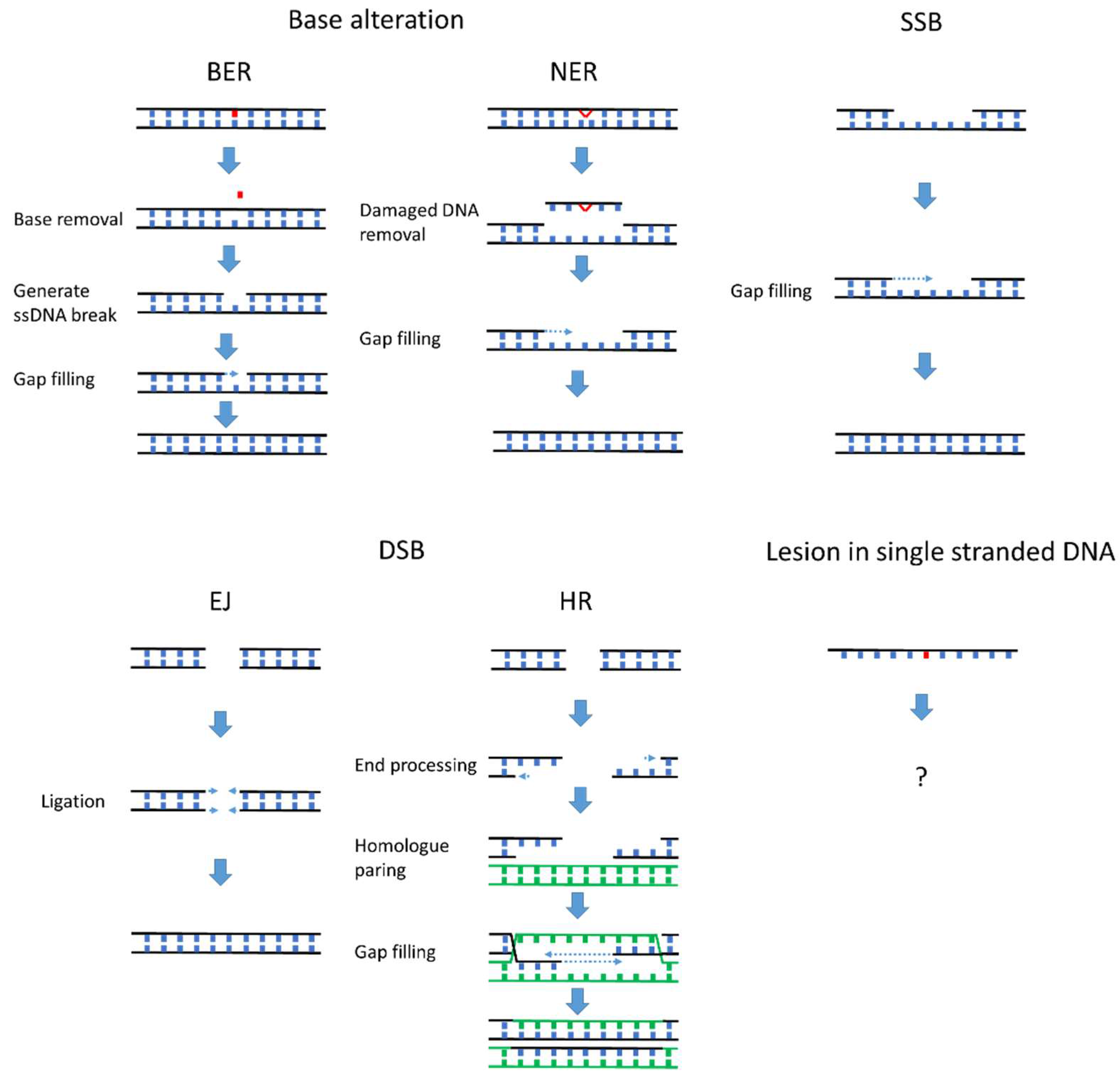
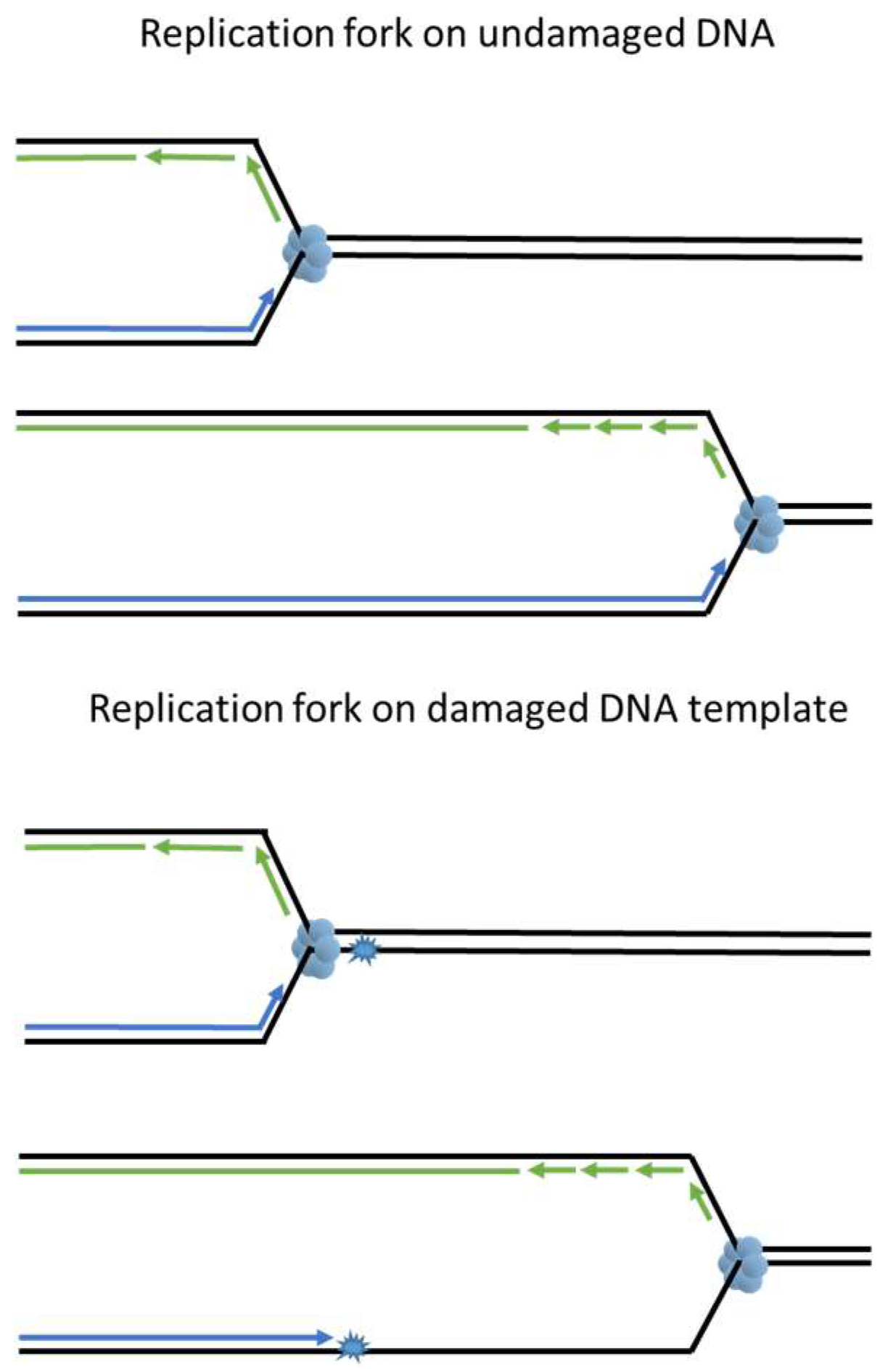
2. DNA Damage Repair and Complications at the Replication Fork
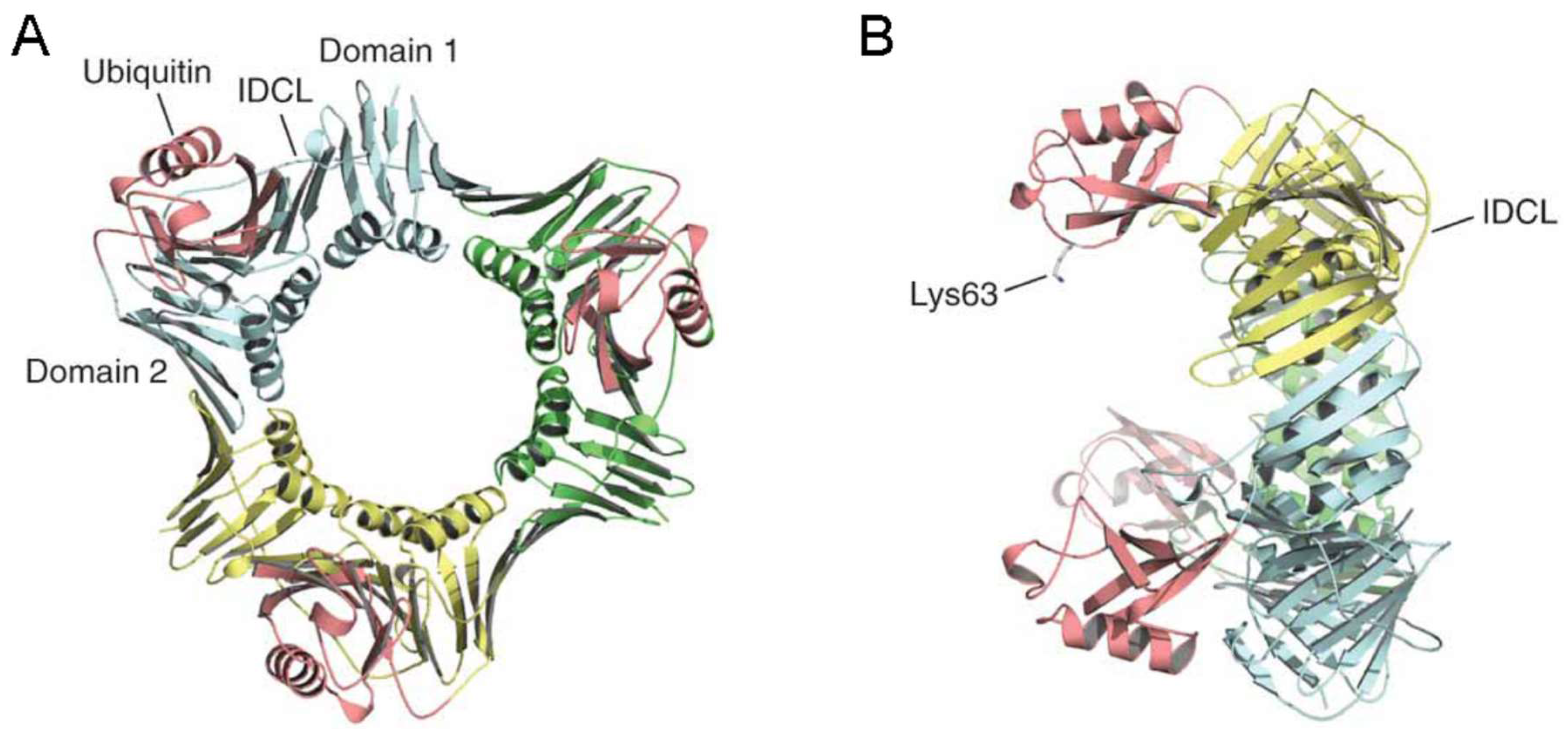
3. Activation of Post Replication Repair
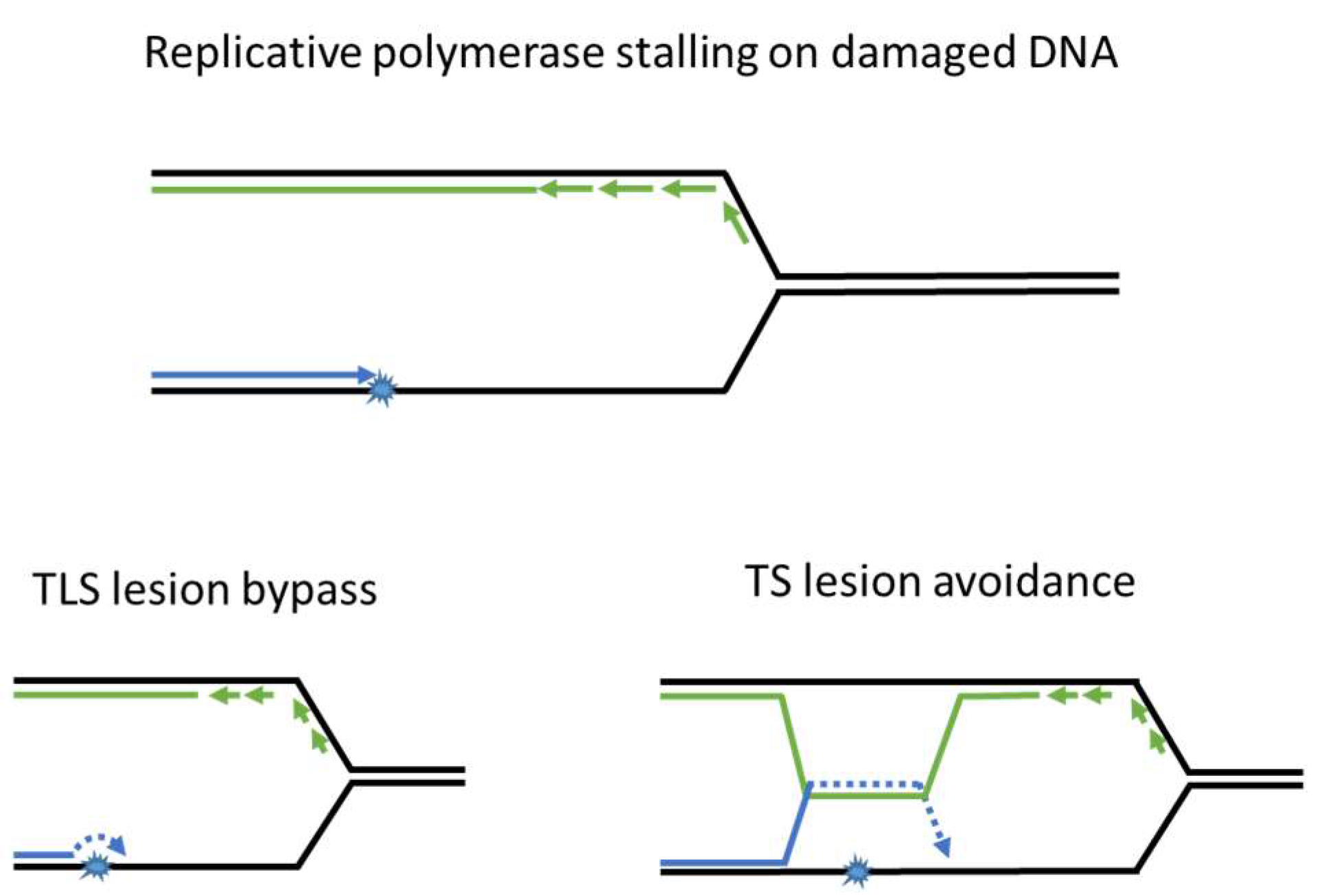
4. Trans-Lesion Synthesis
5. Template Switching
6. Timing of Post Replication Repair
7. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Burgers, P.M.J. Polymerase Dynamics at the Eukaryotic DNA Replication Fork. J. Biol. Chem. 2009, 284, 4041–4045. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. Replication of UV-damaged DNA: New insights into links between DNA polymerases, mutagenesis and human disease. Gene 2000, 253, 1–12. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; He, C. DNA Repair by Reversal of DNA Damage. Cold Spring Harb. Perspect. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Masai, H.; Matsumoto, S.; You, Z.; Yoshizawa-Sugata, N.; Oda, M. Eukaryotic Chromosome DNA Replication: Where, When, and How? Ann. Rev. Biochem. 2010, 79, 89–130. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, J.; Mutlu, E.; Sharma, V.; Collins, L.; Bodnar, W.; Yu, R.; Lai, Y.; Moeller, B.; Lu, K.; Swenberg, J. The Endogenous Exposome. DNA Repair 2014, 19, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D. Ubiquitin family modifications and template switching. FEBS Lett. 2011, 585, 2810–2817. [Google Scholar] [CrossRef] [PubMed]
- Majka, J.; Burgers, P.M.J. The PCNA–RFC Families of DNA Clamps and Clamp Loaders. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 227–260. [Google Scholar] [PubMed]
- Lee, K.Y.; Fu, H.; Aladjem, M.I.; Myung, K. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J. Cell Biol. 2013, 200, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Nishimura, K.; Kanemaki, M.T.; Donaldson, A.D. The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Mol. Cell 2013, 50, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O'Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Stelter, P.; Ulrich, H.D. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003, 425, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Byun, T.S.; Pacek, M.; Yee, M.-c.; Walter, J.C.; Cimprich, K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005, 19, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.A.; Huttner, D.; Daigaku, Y.; Chen, S.; Ulrich, H.D. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol. Cell 2008, 29, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Huttner, D.; Ulrich, H.D. Cooperation of replication protein A with the ubiquitin ligase Rad18 in DNA damage bypass. Cell Cycle 2008, 7, 3629–3633. [Google Scholar] [CrossRef]
- Tsuji, Y.; Watanabe, K.; Araki, K.; Shinohara, M.; Yamagata, Y.; Tsurimoto, T.; Hanaoka, F.; Yamamura, K.; Yamaizumi, M.; Tateishi, S. Recognition of forked and single-stranded DNA structures by human RAD18 complexed with RAD6B protein triggers its recruitment to stalled replication forks. Genes Cells Devot. Mol. Cell. Mech. 2008, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of Human DNA Polymerase η with Monoubiquitinated PCNA: A Possible Mechanism for the Polymerase Switch in Response to DNA Damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Freudenthal, B.D.; Gakhar, L.; Ramaswamy, S.; Washington, M.T. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat. Struct. Mol. Biol. 2010, 17, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Durando, M.; Tateishi, S.; Vaziri, C. A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 2013, 41, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, H.; Kobayashi, J.; Tateishi, S.; Kato, A.; Matsuura, S.; Tauchi, H.; Yamada, K.; Takezawa, J.; Sugasawa, K.; Masutani, C.; et al. NBS1 recruits RAD18 via a RAD6-like domain and regulates Pol eta-dependent translesion DNA synthesis. Mol. Cell 2011, 43, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, F.; Sharma, S.; Zou, J.; Lin, S.Y.; Wang, B.; Rezvani, K.; Wang, H.; Parvin, J.D.; Ludwig, T.; Canman, C.E.; et al. BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc. Natl. Acad. Sci. USA 2013, 110, 13558–13563. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, T.; Huen, M.S.; Hu, L.; Chen, Z.; Huang, J. SIVA1 directs the E3 ubiquitin ligase RAD18 for PCNA monoubiquitination. J. Cell Biol. 2014, 205, 811–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centore, R.C.; Yazinski, S.A.; Tse, A.; Zou, L. Spartan/C1orf124, a Reader of PCNA Ubiquitylation and a Regulator of UV-Induced DNA Damage Response. Mol. Cell 2012, 46, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Mosbech, A.; Gibbs-Seymour, I.; Kagias, K.; Thorslund, T.; Beli, P.; Povlsen, L.; Nielsen, S.V.; Smedegaard, S.; Sedgwick, G.; Lukas, C.; et al. DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nat. Struct. Mol. Biol. 2012, 19, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, S.; Balogh, D.; Hajdu, I.; Burkovics, P.; Villamil, M.A.; Zhuang, Z.; Haracska, L. Characterization of human Spartan/C1orf124, an ubiquitin-PCNA interacting regulator of DNA damage tolerance. Nucleic Acids Res. 2012, 40, 10795–10808. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Lachaud, C.; Appleton, P.; Macartney, T.J.; Nathke, I.; Rouse, J. DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat. Struct. Mol. Biol. 2012, 19, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Machida, Y.; Kim, M.S.; Machida, Y.J. Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle 2012, 11, 3395–3402. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Moldovan, G.-L.; Saribasak, H.; Saribasak, N.N.; Jentsch, S.; Buerstedde, J.-M. A Role for PCNA Ubiquitination in Immunoglobulin Hypermutation. PLoS Biol. 2006, 4, e366. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.J.; Ross, A.L.; Szuts, D.; Alviani, C.A.; Oestergaard, V.H.; Patel, K.J.; Sale, J.E. RAD18-independent ubiquitination of proliferating-cell nuclear antigen in the avian cell line DT40. EMBO Rep. 2006, 7, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Tateishi, S.; Tanoue, Y.; Azuma, T.; Ohmori, H. Somatic hypermutation of immunoglobulin genes in Rad18 knockout mice. DNA Repair (Amst) 2017, 50, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Jentsch, S. Two RING finger proteins mediate cooperation between ubiquitin-conjugating enzymes in DNA repair. Embo. J. 2000, 19, 3388–3397. [Google Scholar] [CrossRef]
- Haracska, L.; Torres-Ramos, C.A.; Johnson, R.E.; Prakash, S.; Prakash, L. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol. Cell Biol. 2004, 24, 4267–4274. [Google Scholar] [CrossRef] [PubMed]
- Leach, C.A.; Michael, W.M. Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J. Cell Biol. 2005, 171, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Motegi, A.; Sood, R.; Moinova, H.; Markowitz, S.D.; Liu, P.P.; Myung, K. Human SHPRH suppresses genomic instability through proliferating cell nuclear antigen polyubiquitination. J. Cell Biol. 2006, 175, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdu, I.; Fatyol, K.; Szakal, B.; Blastyak, A.; Bermudez, V.; Hurwitz, J.; Prakash, L.; Prakash, S.; Haracska, L. Human SHPRH is a ubiquitin ligase for Mms2-Ubc13-dependent polyubiquitylation of proliferating cell nuclear antigen. Proc. Natl. Acad. Sci. USA 2006, 103, 18107–18112. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Hajdu, I.; Fatyol, K.; Hurwitz, J.; Yoon, J.H.; Prakash, L.; Prakash, S.; Haracska, L. Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc. Natl. Acad. Sci. USA 2008, 105, 3768–3773. [Google Scholar] [CrossRef] [PubMed]
- Motegi, A.; Liaw, H.J.; Lee, K.Y.; Roest, H.P.; Maas, A.; Wu, X.; Moinova, H.; Markowitz, S.D.; Ding, H.; Hoeijmakers, J.H.; et al. Polyubiquitination of proliferating cell nuclear antigen by HLTF and SHPRH prevents genomic instability from stalled replication forks. Proc. Natl. Acad. Sci. USA 2008, 105, 12411–12416. [Google Scholar] [CrossRef] [PubMed]
- Kannouche, P.L.; Lehmann, A.R. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle 2004, 3, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Chiu, R.K.; Brun, J.; Ramaekers, C.; Theys, J.; Weng, L.; Lambin, P.; Gray, D.A.; Wouters, B.G. Lysine 63-polyubiquitination guards against translesion synthesis-induced mutations. PLoS Genet. 2006, 2, e116. [Google Scholar] [CrossRef]
- Gangavarapu, V.; Haracska, L.; Unk, I.; Johnson, R.E.; Prakash, S.; Prakash, L. Mms2-Ubc13-dependent and -independent roles of Rad5 ubiquitin ligase in postreplication repair and translesion DNA synthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 7783–7790. [Google Scholar] [CrossRef]
- Pages, V.; Bresson, A.; Acharya, N.; Prakash, S.; Fuchs, R.P.; Prakash, L. Requirement of Rad5 for DNA polymerase zeta-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics 2008, 180, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lin, A.; Zhou, C.; Blackwell, S.R.; Zhang, Y.; Wang, Z.; Feng, Q.; Guan, R.; Hanna, M.D.; Chen, Z.; et al. Involvement of budding yeast Rad5 in translesion DNA synthesis through physical interaction with Rev1. Nucleic Acids Res. 2016, 44, 5231–5245. [Google Scholar] [CrossRef] [PubMed]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Papouli, E.; Chen, S.; Davies, A.A.; Huttner, D.; Krejci, L.; Sung, P.; Ulrich, H.D. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 2005, 19, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Schiestl, R.H.; Prakash, S.; Prakash, L. The SRS2 suppressor of rad6 mutations of Saccharomyces cerevisiae acts by channeling DNA lesions into the RAD52 DNA repair pathway. Genetics 1990, 124, 817–831. [Google Scholar] [PubMed]
- Parker, J.L.; Ulrich, H.D. A SUMO-interacting motif activates budding yeast ubiquitin ligase Rad18 towards SUMO-modified PCNA. Nucleic Acids Res. 2012, 40, 11380–11388. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Johnson, R.E.; Prakash, L. Eukaryotic translesion synthesis DNA polymerases: Specificity of structure and function. Annu. Rev. Biochem. 2005, 74, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.E.; Prakash, S.; Prakash, L. Efficient Bypass of a Thymine-Thymine Dimer by Yeast DNA Polymerase, Polη. Science 1999, 283, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Wang, M.; Rechkoblit, O.; Taylor, J.-S.; Geacintov, N.E.; Wang, Z. Error-free and error-prone lesion bypass by human DNA polymerase κ in vitro. Nucleic Acids Res. 2000, 28, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Slater, D.M.; Ohmori, H.; Vaziri, C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J. Biol. Chem. 2005, 280, 22343–22355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Wu, X.; Taylor, J.-S.; Wang, Z. Response of human DNA polymerase ι to DNA lesions. Nucleic Acids Res. 2001, 29, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase [eta]. Nature 1999, 399, 700–704. [Google Scholar] [PubMed]
- Wang, Y.; Woodgate, R.; McManus, T.P.; Mead, S.; McCormick, J.J.; Maher, V.M. Evidence that in Xeroderma Pigmentosum Variant Cells, which Lack DNA Polymerase η, DNA Polymerase ι Causes the Very High Frequency and Unique Spectrum of UV-Induced Mutations. Cancer Res. 2007, 67, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Ziv, O.; Geacintov, N.; Nakajima, S.; Yasui, A.; Livneh, Z. DNA polymerase zeta cooperates with polymerases kappa and iota in translesion DNA synthesis across pyrimidine photodimers in cells from XPV patients. Proc. Natl. Acad. Sci. USA 2009, 106, 11552–11557. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Johnson, R.E.; Haracska, L.; Prakash, L.; Prakash, S.; Benkovic, S.J. Regulation of polymerase exchange between Polη and Polδ by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 5361–5366. [Google Scholar] [CrossRef] [PubMed]
- Bienko, M.; Green, C.M.; Crosetto, N.; Rudolf, F.; Zapart, G.; Coull, B.; Kannouche, P.; Wider, G.; Peter, M.; Lehmann, A.R.; et al. Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Indiani, C.; McInerney, P.; Georgescu, R.; Goodman, M.F.; O'Donnell, M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell 2005, 19, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Hara, K.; Shimizu, T.; Sato, M.; Hashimoto, H. Structural basis of recruitment of DNA polymerase zeta by interaction between REV1 and REV7 proteins. J. Biol. Chem. 2012, 287, 33847–33852. [Google Scholar] [CrossRef] [PubMed]
- Gueranger, Q.; Stary, A.; Aoufouchi, S.; Faili, A.; Sarasin, A.; Reynaud, C.-A.; Weill, J.-C. Role of DNA polymerases η, ι and ζ in UV resistance and UV-induced mutagenesis in a human cell line. DNA Repair 2008, 7, 1551–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, N.; Yoon, J.H.; Gali, H.; Unk, I.; Haracska, L.; Johnson, R.E.; Hurwitz, J.; Prakash, L.; Prakash, S. Roles of PCNA-binding and ubiquitin-binding domains in human DNA polymerase eta in translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 17724–17729. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Yoon, J.H.; Hurwitz, J.; Prakash, L.; Prakash, S. DNA polymerase eta lacking the ubiquitin-binding domain promotes replicative lesion bypass in humans cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10401–10405. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Krijger, P.H.L.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reißner, T.; Lee, K.-Y.; Geacintov, N.E.; Carell, T.; et al. PCNA Ubiquitination Is Important, But Not Essential for Translesion DNA Synthesis in Mammalian Cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. Elife 2016. [Google Scholar] [CrossRef] [PubMed]
- Prakash, L. Characterization of postreplication repair in Saccharomyces cerevisiae and effects of rad6, rad18, rev3 and rad52 mutations. Mol. Gen. Genet. MGG 1981, 184, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Sung, P.; Prakash, L. DNA repair genes and proteins of Saccharomyces cerevisiae. Annu. Rev. Genet. 1993, 27, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Chow, B.L.; Broomfield, S.; Hanna, M. The Saccharomyces cerevisiae RAD6 group is composed of an error-prone and two error-free postreplication repair pathways. Genetics 2000, 155, 1633–1641. [Google Scholar] [PubMed]
- Minca, E.C.; Kowalski, D. Multiple Rad5 Activities Mediate Sister Chromatid Recombination to Bypass DNA Damage at Stalled Replication Forks. Mol. Cell 2010, 38, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, R.M.; Pickart, C.M. Noncanonical MMS2-Encoded Ubiquitin-Conjugating Enzyme Functions in Assembly of Novel Polyubiquitin Chains for DNA Repair. Cell 1999, 96, 645–653. [Google Scholar] [CrossRef]
- Broomfield, S.; Chow, B.L.; Xiao, W. MMS2, encoding a ubiquitin-conjugating-enzyme-like protein, is a member of the yeast error-free postreplication repair pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5678–5683. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ramos, C.A.; Prakash, S.; Prakash, L. Requirement of Yeast DNA Polymerase δ in Post-replicational Repair of UV-damaged DNA. J. Biol. Chem. 1997, 272, 25445–25448. [Google Scholar] [CrossRef] [PubMed]
- Gangavarapu, V.; Prakash, S.; Prakash, L. Requirement of RAD52 group genes for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell Biol. 2007, 27, 7758–7764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lawrence, C.W. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc. Natl. Acad. Sci. USA 2005, 102, 15954–15959. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Vanoli, F.; Foiani, M. SUMOylation regulates Rad18-mediated template switch. Nature 2008, 456, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 2003, 426, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, M.; Zwicky, K.; Follonier, C.; Foiani, M.; Lopes, M.; Branzei, D. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014, 21, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Fumasoni, M.; Sienski, G.; Vanoli, F.; Branzei, D.; Jentsch, S. Noncanonical Role of the 9-1-1 Clamp in the Error-Free DNA Damage Tolerance Pathway. Mol. Cell 2013, 49, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Fumasoni, M.; Zwicky, K.; Vanoli, F.; Lopes, M.; Branzei, D. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol. Cell 2015, 57, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.O.; Yoon, H.; Park, S.O.; Lee, M.; Shin, J.-S.; Ryu, K.-S.; Lee, J.-O.; Seo, Y.-S.; Jung, H.S.; Choi, B.-S. Srs2 possesses a non-canonical PIP box in front of its SBM for precise recognition of SUMOylated PCNA. J. Mol. Cell Biol. 2012, 4, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Urulangodi, M.; Sebesta, M.; Menolfi, D.; Szakal, B.; Sollier, J.; Sisakova, A.; Krejci, L.; Branzei, D. Local regulation of the Srs2 helicase by the SUMO-like domain protein Esc2 promotes recombination at sites of stalled replication. Genes Dev. 2015, 29, 2067–2080. [Google Scholar] [CrossRef] [PubMed]
- Izhar, L.; Ziv, O.; Cohen, I.S.; Geacintov, N.E.; Livneh, Z. Genomic assay reveals tolerance of DNA damage by both translesion DNA synthesis and homology-dependent repair in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1462–E1469. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Dejsuphong, D.; Petalcorin, M.I.R.; Hofmann, K.; Takeda, S.; Boulton, S.J.; D’Andrea, A.D. Inhibition of Homologous Recombination by the PCNA-Interacting Protein PARI. Mol. Cell 2012, 45, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Dome, L.; Juhasz, S.; Altmannova, V.; Sebesta, M.; Pacesa, M.; Fugger, K.; Sorensen, C.S.; Lee, M.Y.W.T.; Haracska, L.; et al. The PCNA-associated protein PARI negatively regulates homologous recombination via the inhibition of DNA repair synthesis. Nucleic Acids Res. 2016, 44, 3176–3189. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Suzuki, M.; Kawai, H.; Hishiki, A.; Hashimoto, H.; Masutani, C.; Hishida, T.; Suzuki, F.; Kamiya, K. En bloc transfer of polyubiquitin chains to PCNA in vitro is mediated by two different human E2-E3 pairs. Nucleic Acids Res. 2012, 40, 10394–10407. [Google Scholar] [CrossRef] [PubMed]
- Krijger, P.H.; Lee, K.Y.; Wit, N.; van den Berk, P.C.; Wu, X.; Roest, H.P.; Maas, A.; Ding, H.; Hoeijmakers, J.H.; Myung, K.; et al. HLTF and SHPRH are not essential for PCNA polyubiquitination, survival and somatic hypermutation: Existence of an alternative E3 ligase. DNA Repair (Amst) 2011, 10, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-R.; Zeman, M.K.; Chen, J.-Y.; Yee, M.-C.; Cimprich, K.A. SHPRH and HLTF Act in a Damage-Specific Manner to Coordinate Different Forms of Postreplication Repair and Prevent Mutagenesis. Mol. Cell 2011, 42, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Sebesta, M.; Balogh, D.; Haracska, L.; Krejci, L. Strand invasion by HLTF as a mechanism for template switch in fork rescue. Nucleic Acids Res. 2014, 42, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Adar, S.; Izhar, L.; Hendel, A.; Geacintov, N.; Livneh, Z. Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res. 2009, 37, 5737–5748. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Nimonkar, A.V.; Hu, Y.; Hajdu, I.; Achar, Y.J.; Izhar, L.; Petit, S.A.; Adamson, B.; Yoon, J.C.; Kowalczykowski, S.C.; et al. Polyubiquitinated PCNA Recruits the ZRANB3 Translocase to Maintain Genomic Integrity after Replication Stress. Mol. Cell 2012, 47, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Weston, R.; Peeters, H.; Ahel, D. ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 2012, 26, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Kile, A.C.; Chavez, D.A.; Bacal, J.; Eldirany, S.; Korzhnev, D.M.; Bezsonova, I.; Eichman, B.F.; Cimprich, K.A. HLTF’s Ancient HIRAN Domain Binds 3′ DNA Ends to Drive Replication Fork Reversal. Mol. Cell 2015, 58, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Achar, Y.J.; Balogh, D.; Neculai, D.; Juhasz, S.; Morocz, M.; Gali, H.; Dhe-Paganon, S.; Venclovas, Č.; Haracska, L. Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 2015, 43, 10277–10291. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.S.; Walker, G.C. The critical mutagenic translesion DNA polymerase Rev1 is highly expressed during G(2)/M phase rather than S phase. Proc. Natl. Acad. Sci. USA 2006, 103, 8971–8976. [Google Scholar] [CrossRef] [PubMed]
- Edmunds, C.E.; Simpson, L.J.; Sale, J.E. PCNA Ubiquitination and REV1 Define Temporally Distinct Mechanisms for Controlling Translesion Synthesis in the Avian Cell Line DT40. Mol. Cell 2008, 30, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Bazán, M.Á.; Gallo-Fernández, M.; Saugar, I.; Jiménez-Martín, A.; Vázquez, M.V.; Tercero, J.A. Rad5 Plays a Major Role in the Cellular Response to DNA Damage during Chromosome Replication. Cell Rep. 2014, 9, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Daigaku, Y.; Davies, A.A.; Ulrich, H.D. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010, 465, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Jentsch, S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010, 141, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.; Foiani, M.; Sogo, J.M. Multiple Mechanisms Control Chromosome Integrity after Replication Fork Uncoupling and Restart at Irreparable UV Lesions. Mol. Cell 2006, 21, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Yeeles, J.T.P.; Marians, K.J. The Escherichia coli replisome is inherently DNA damage tolerant. Science 2011, 334, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, S.; Reyes, A.; Martínez-Jiménez, M.I.; Chocrón, E.S.; Mourón, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Mol. Cell 2013, 52, 541–553. [Google Scholar]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Mourón, S.; Rodriguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct. Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Elvers, I.; Johansson, F.; Groth, P.; Erixon, K.; Helleday, T. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 2011, 39, 7049–7057. [Google Scholar] [CrossRef] [PubMed]
- Zlatanou, A.; Despras, E.; Braz-Petta, T.; Boubakour-Azzouz, I.; Pouvelle, C.; Stewart, G.S.; Nakajima, S.; Yasui, A.; Ishchenko, A.A.; Kannouche, P.L. The hMsh2-hMsh6 Complex Acts in Concert with Monoubiquitinated PCNA and Pol η in Response to Oxidative DNA Damage in Human Cells. Mol. Cell 2011, 43, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Durando, M.; Smith-Roe, S.L.; Sproul, C.; Greenwalt, A.M.; Kaufmann, W.; Oh, S.; Hendrickson, E.A.; Vaziri, C. Cell cycle stage-specific roles of Rad18 in tolerance and repair of oxidative DNA damage. Nucleic Acids Res. 2013, 41, 2296–2312. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Lan, L.; Kanno, S.-i.; Usami, N.; Kobayashi, K.; Mori, M.; Shiomi, T.; Yasui, A. Replication-dependent and -independent Responses of RAD18 to DNA Damage in Human Cells. J. Biol. Chem. 2006, 281, 34687–34695. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.; Carr, A.M. Replication stress and genome rearrangements: Lessons from yeast models. Curr. Opin. Genet. Dev. 2013, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Doles, J.; Hemann, M.T.; Walker, G.C. Error-prone translesion synthesis mediates acquired chemoresistance. Proc. Natl. Acad. Sci. USA 2010, 107, 20792–20797. [Google Scholar] [CrossRef] [PubMed]
- Bavoux, C.; Leopoldino, A.M.; Bergoglio, V.; O-Wang, J.; Ogi, T.; Bieth, A.; Judde, J.-G.; Pena, S.D.J.; Poupon, M.-F.; Helleday, T.; et al. Up-Regulation of the Error-Prone DNA Polymerase κ Promotes Pleiotropic Genetic Alterations and Tumorigenesis. Cancer Res. 2005, 65, 325–330. [Google Scholar] [PubMed]
- Yang, Y.; Poe, J.C.; Yang, L.; Fedoriw, A.; Desai, S.; Magnuson, T.; Li, Z.; Fedoriw, Y.; Araki, K.; Gao, Y.; et al. Rad18 confers hematopoietic progenitor cell DNA damage tolerance independently of the Fanconi Anemia pathway in vivo. Nucleic Acids Res. 2016, 44, 4174–4188. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, P.; Novello, S.; Cambieri, A.; Longo, M.; Monica, V.; Lo Iacono, M.; Giaj-Levra, M.; Saviozzi, S.; Volante, M.; Papotti, M.; et al. Polymerase eta mRNA expression predicts survival of non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res. 2009, 15, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Doles, J.; Oliver, T.G.; Cameron, E.R.; Hsu, G.; Jacks, T.; Walker, G.C.; Hemann, M.T. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl. Acad. Sci. USA 2010, 107, 20786–20791. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Mutter-Rottmayer, E.; Greenwalt, A.M.; Goldfarb, D.; Yan, F.; Yang, Y.; Martinez-Chacin, R.C.; Pearce, K.H.; Tateishi, S.; Major, M.B.; et al. A neomorphic cancer cell-specific role of MAGE-A4 in trans-lesion synthesis. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Mutter-Rottmayer, E.; Zlatanou, A.; Vaziri, C.; Yang, Y. Mechanisms of Post-Replication DNA Repair. Genes 2017, 8, 64. https://doi.org/10.3390/genes8020064
Gao Y, Mutter-Rottmayer E, Zlatanou A, Vaziri C, Yang Y. Mechanisms of Post-Replication DNA Repair. Genes. 2017; 8(2):64. https://doi.org/10.3390/genes8020064
Chicago/Turabian StyleGao, Yanzhe, Elizabeth Mutter-Rottmayer, Anastasia Zlatanou, Cyrus Vaziri, and Yang Yang. 2017. "Mechanisms of Post-Replication DNA Repair" Genes 8, no. 2: 64. https://doi.org/10.3390/genes8020064