Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors

 , , , and
, , , and 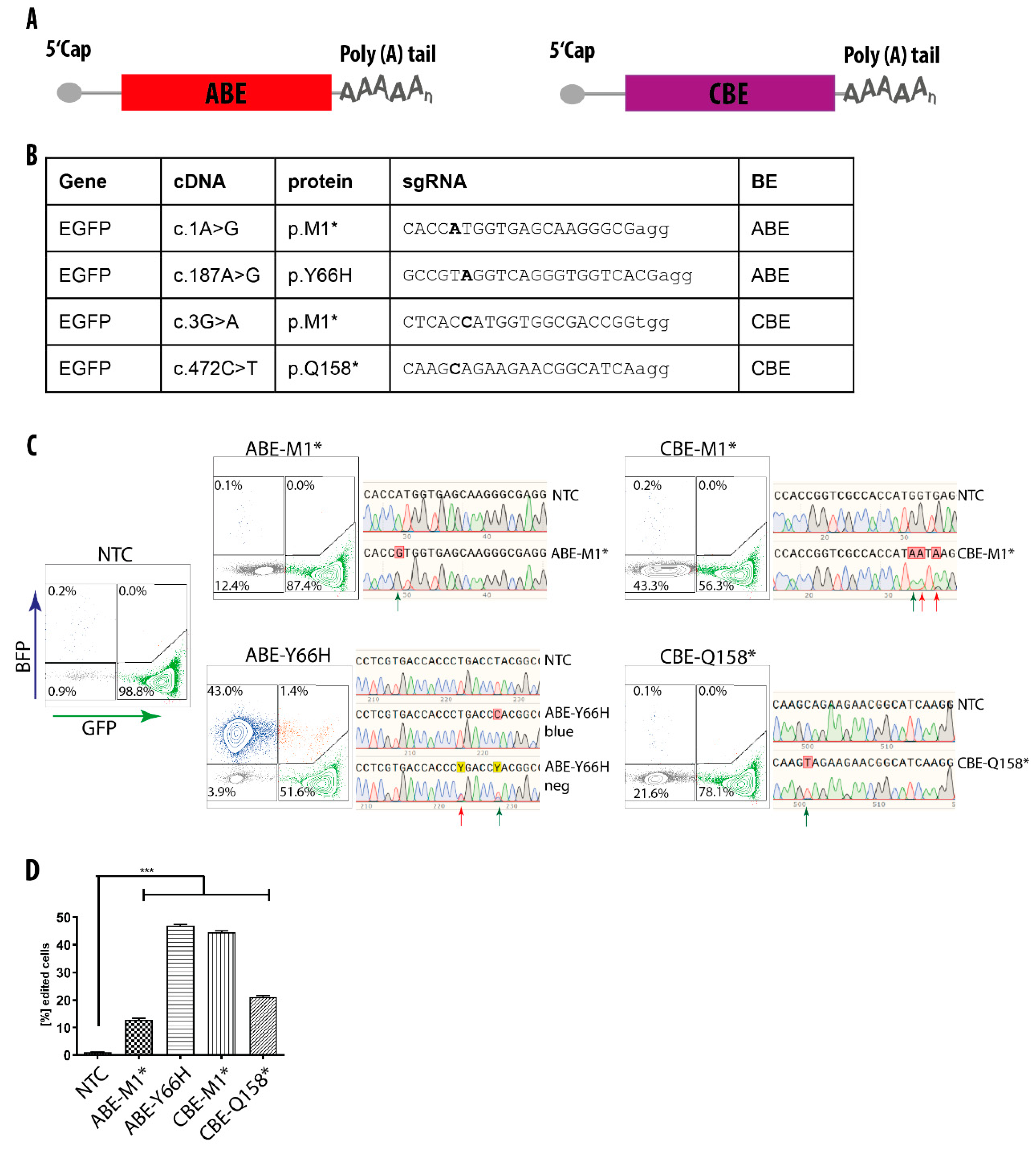{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Lentiviral Vector Production
2.2. In Vitro mRNA Transcription
2.3. HEK293T Cells Transfection with mRNA
2.4. Flow Cytometry and Cell Sorting
2.5. Reprogramming, hiPSC Culture and Characterization
2.6. Nucleofection
2.7. Transfection
2.8. EdU Staining of hiPSCs under Nutlin Treatment
2.9. Statistical Analysis
3. Results
3.1. Attempt for Correction of a Disease-Causing Mutation by HDR
3.2. mRNA-Mediated Base Editing in HEK293T Cells
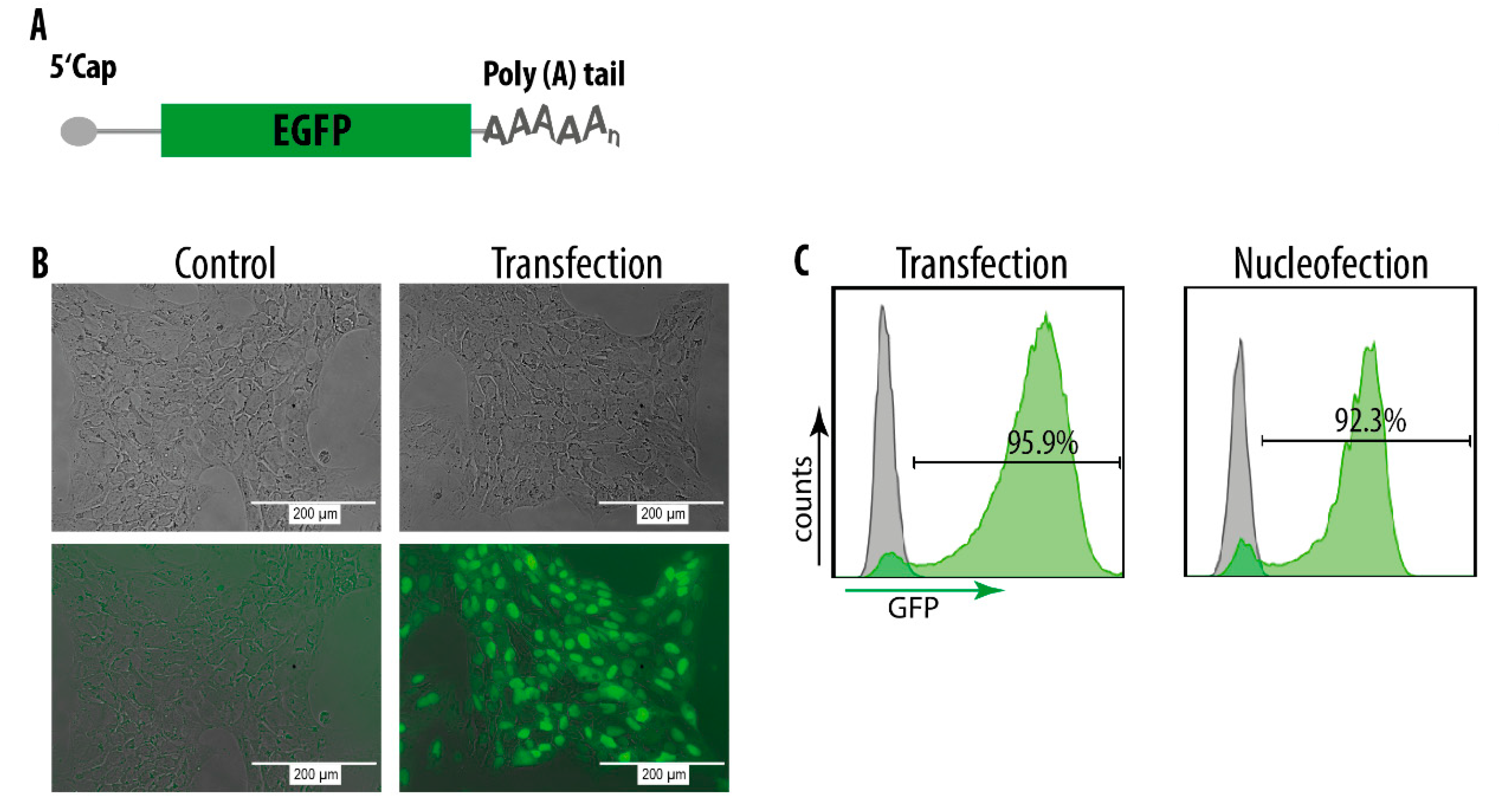
3.3. mRNA Delivery into hiPSCs
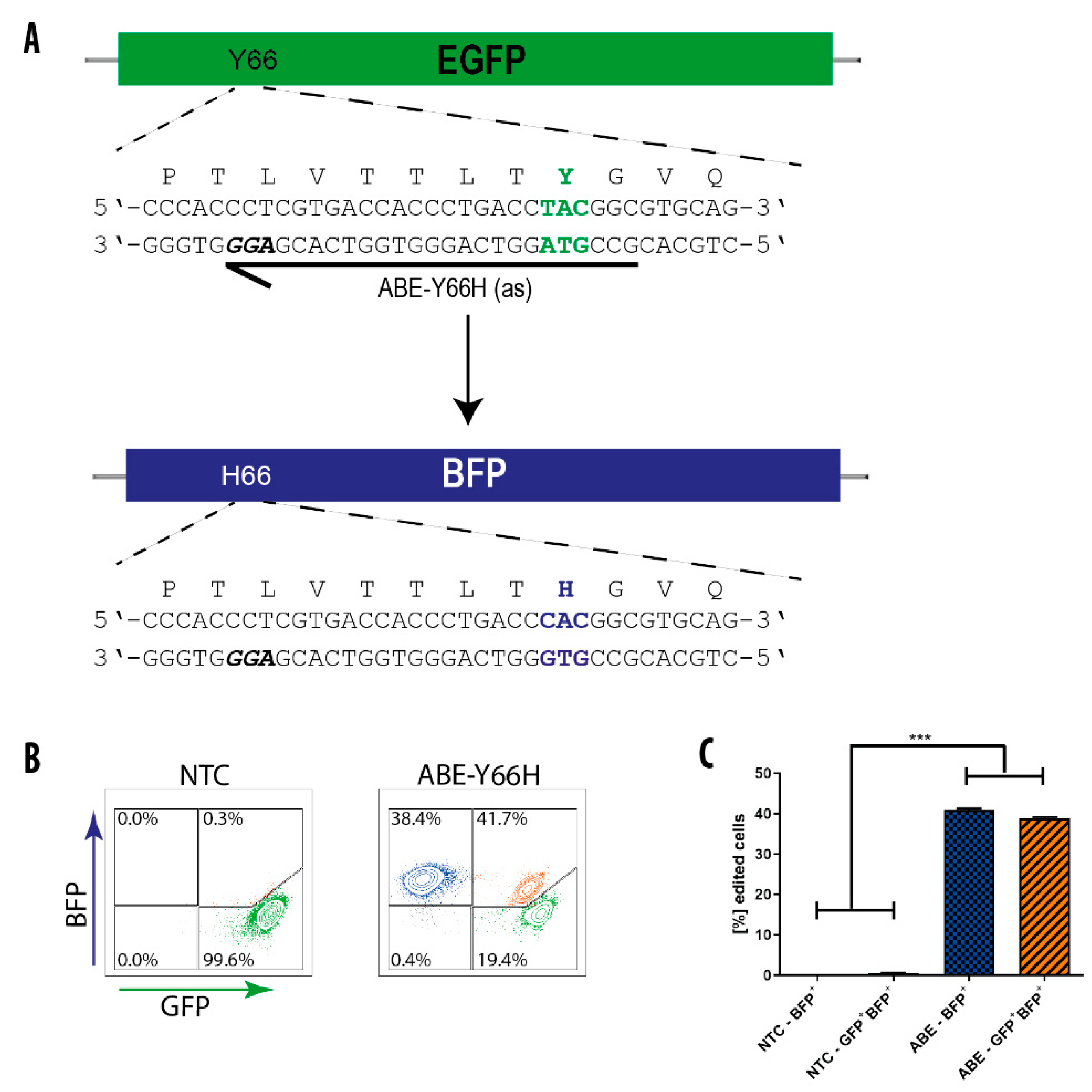
3.4. BE-Mediated Conversion of GFP into BFP in hiPS Cells
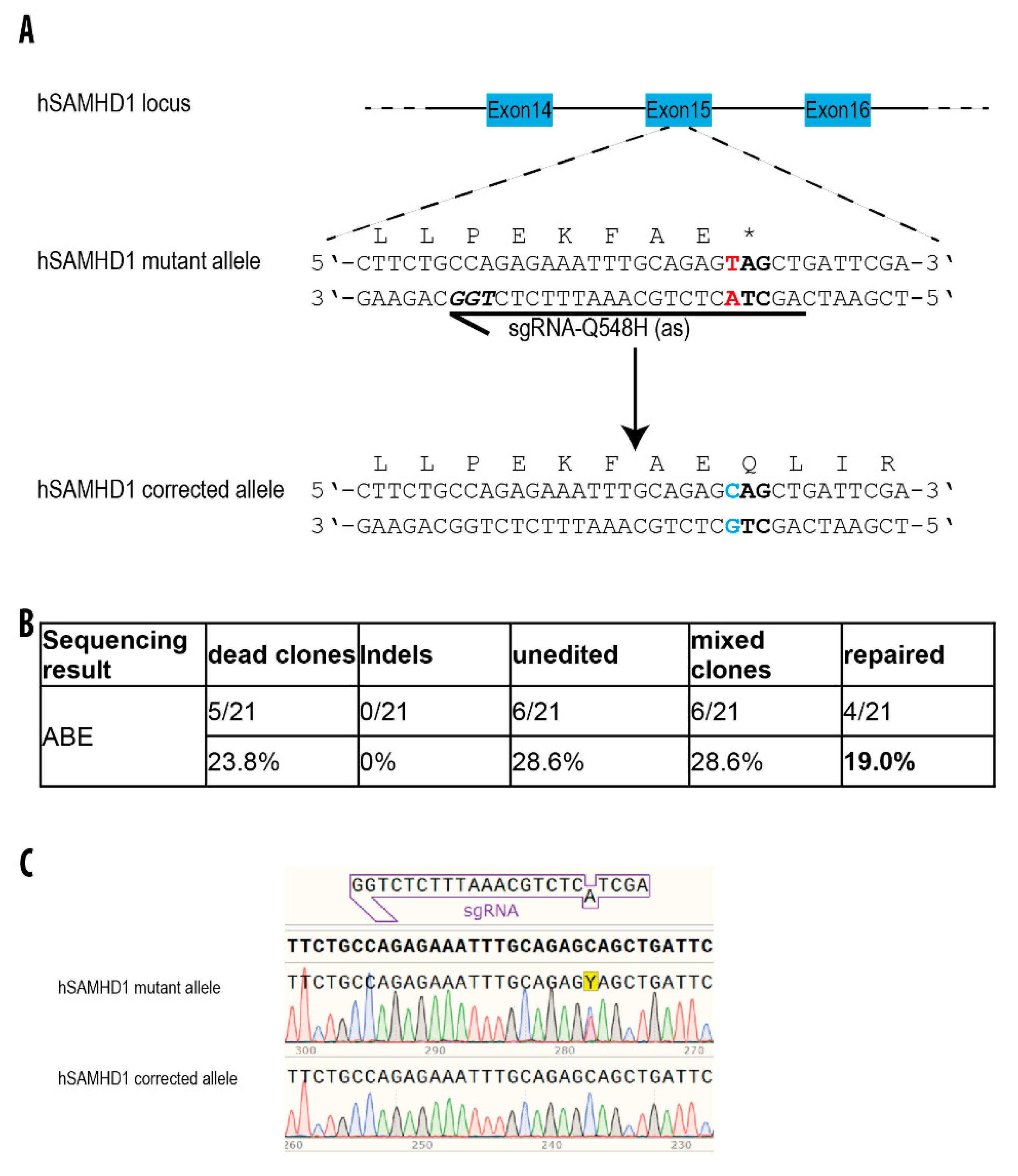
3.5. Repair of a Disease-Causing Mutation in Patient-Derived hiPSCs
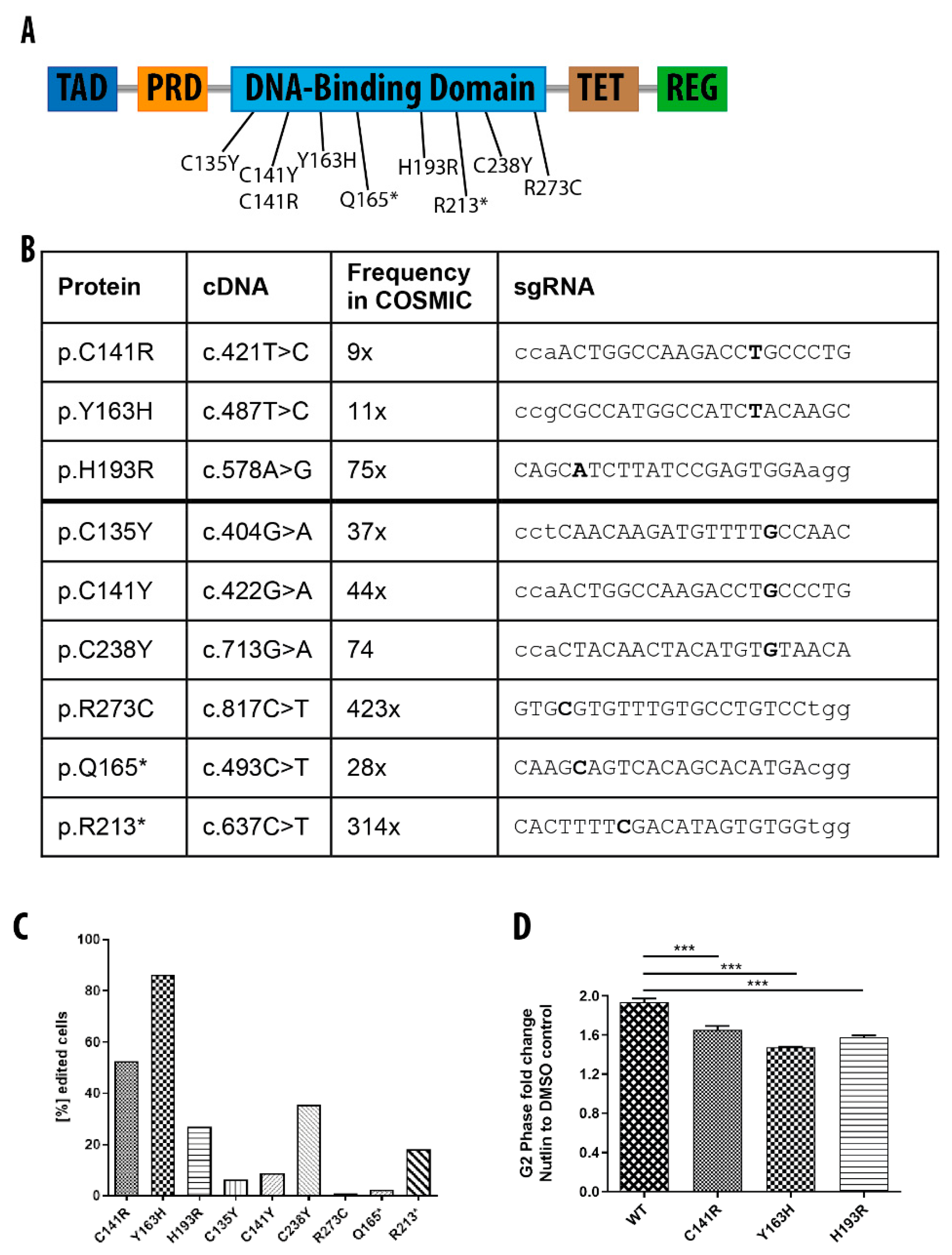
3.6. Generation of Isogenic hiPSCs with Different TP53 Mutations
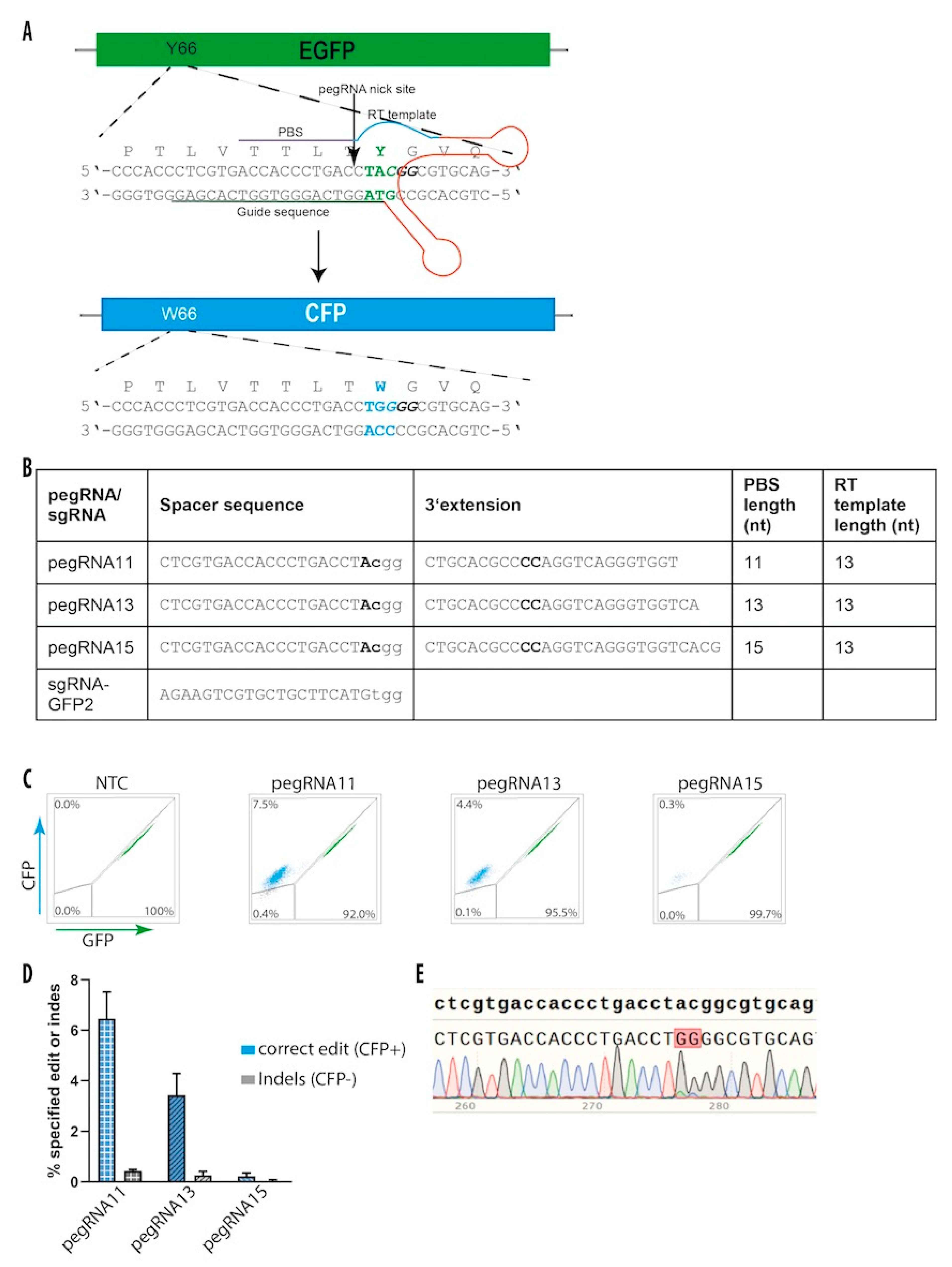
3.7. Prime Editing in hiPS Cells
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Drews, K.; Jozefczuk, J.; Prigione, A.; Adjaye, J. Human induced pluripotent stem cells-from mechanisms to clinical applications. J. Mol. Med. 2012, 90, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.Y.; Weick, J.P.; Yu, J.; Ma, L.X.; Zhang, X.Q.; Thomson, J.A.; Zhang, S.C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.-D.; Yu, J.; Smuga-Otto, K.; Salvagiotto, G.; Rehrauer, W.; Vodyanik, M.; Thomson, J.; Slukvin, I. Hematopoietic and Endothelial Differentiation of Human Induced Pluripotent Stem Cells. Stem Cells 2009, 27, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.-H.; Gepstein, L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, C.; Koonce, C.; Aoyama, N.; Einhorn, S.; Fiene, S.; Thompson, A.; Swanson, B.; Anson, B.; Kattman, S. Phenotypic Screening with Human iPS Cell–Derived Cardiomyocytes. J. Biomol. Screen. 2013, 18, 1203–1211. [Google Scholar] [CrossRef] [Green Version]
- Drawnel, F.M.; Boccardo, S.; Prummer, M.; Delobel, F.; Graff, A.; Weber, M.; Gérard, R.; Badi, L.; Kam-Thong, T.; Bu, L.; et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014, 9, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.M.; Kim, Y.; Shim, J.S.; Park, J.T.; Wang, R.H.; Leach, S.D.; Liu, J.O.; Deng, C.; Ye, Z.; Jang, Y.Y. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology 2013, 57, 2458–2468. [Google Scholar] [CrossRef] [Green Version]
- Laugsch, M.; Rostovskaya, M.; Velychko, S.; Richter, C.; Zimmer, A.; Klink, B.; Schröck, E.; Haase, M.; Neumann, K.; Thieme, S.; et al. Functional restoration of gp91phox-oxidase activity by BAC transgenesis and gene targeting in X-linked chronic granulomatous disease iPSCs. Mol. Ther. 2016, 24, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Raya, Á.; Rodríguez-Piz, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castell, M.; Río, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells using CRISPR/Cas9. Stem Cell Rev. 2018, 14, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.; Kumasaka, N.; de Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef] [PubMed]
- Hotta, A.; Ellis, J. Retroviral vector silencing during iPS cell induction: An epigenetic beacon that signals distinct pluripotent states. J. Cell Biochem. 2008, 105, 940–948. [Google Scholar] [CrossRef]
- Geis, F.K.; Galla, M.; Hoffmann, D.; Kuehle, J.; Zychlinski, D.; Maetzig, T.; Schott, J.W.; Schwarzer, A.; Goffinet, C.; Goff, S.P.; et al. Potent and reversible lentiviral vector restriction in murine induced pluripotent stem cells. Retrovirology 2017, 14, 48. [Google Scholar] [CrossRef] [Green Version]
- Guo, D.; Liu, H.; Gao, G.; Liu, Y.; Zhuang, Y.; Yang, F.; Wang, K.; Zhou, T.; Qin, D.; Hong, L.; et al. Creating a patient carried Men1 gene point mutation on wild type iPSCs locus mediated by CRISPR/Cas9 and ssODN. Stem Cell Res. 2017, 18, 67–69. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.-S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Webber, B.R.; Osborn, M.J.; McElroy, A.N.; Twaroski, K.; Lonetree, C.; DeFeo, A.P.; Xia, L.; Eide, C.; Lees, C.J.; McElmurry, R.T.; et al. CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. npj Regen. Med. 2016, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.; Grundmann, A.; Renz, P.; Hänseler, W.; James, W.S.; Cowley, S.A.; Moore, M.D. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp. Hematol. 2015, 43, 838–848.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef] [PubMed]
- Brookhouser, N.; Tekel, S.J.; Standage-Beier, K.; Nguyen, T.; Schwarz, G.; Wang, X.; Brafman, D.A. BIG-TREE: Base-Edited Isogenic hPSC Line Generation Using a Transient Reporter for Editing Enrichment. Stem Cell Reports 2020, 14, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-J.; Xu, C.L.; Cui, X.; Bassuk, A.G.; Mahajan, V.B.; Tsai, Y.-T.; Tsang, S.H. CRISPR Base Editing in Induced Pluripotent Stem Cells. Methods Mol. Biol. 2019, 2045, 337–346. [Google Scholar]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Jin, S.; Zong, Y.; Gao, Q.; Zhu, Z.; Wang, Y.; Qin, P.; Liang, C.; Wang, D.; Qiu, J.-L.; Zhang, F.; et al. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science 2019, 8, eaaw7166. [Google Scholar] [CrossRef]
- McGrath, E.; Shin, H.; Zhang, L.; Phue, J.N.; Wu, W.W.; Shen, R.F.; Jang, Y.Y.; Revollo, J.; Ye, Z. Targeting specificity of APOBEC-based cytosine base editor in human iPSCs determined by whole genome sequencing. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Tan, J.; Zhang, F.; Karcher, D.; Bock, R. Engineering of high-precision base editors for site-specific single nucleotide replacement. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 2019, 126, 21. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Komor, A.C.; Yeh, W.-H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Shaw, M.W. Uses of banding techniques for the identification of human diseases of cytogenetic origin. Environ. Health Perspect. 1973, 6, 151–156. [Google Scholar] [CrossRef]
- Ramantani, G.; Häusler, M.; Niggemann, P.; Wessling, B.; Guttmann, H.; Mull, M.; Tenbrock, K.; Lee-Kirsch, M.A. Aicardi-goutières syndrome and systemic lupus erythematosus (SLE) in a 12-year-old boy with SAMHD1 mutations. J. Child Neurol. 2011, 26, 1425–1428. [Google Scholar] [CrossRef]
- Kretschmer, S.; Wolf, C.; König, N.; Staroske, W.; Guck, J.; Häusler, M.; Luksch, H.; Nguyen, L.A.; Kim, B.; Alexopoulou, D.; et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis. 2015, 74, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liang, Z.; Zong, Y.; Wang, Y.; Liu, J.; Chen, K.; Qiu, J.L.; Gao, C. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Vassilev, L.T. Targeting the p53-MDM2 interaction to treat cancer. Br. J. Cancer 2004, 91, 1415–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.P.; Zhao, K.T.; Miller, S.M.; Gaudelli, N.M.; Oakes, B.L.; Fellmann, C.; Savage, D.F.; Liu, D.R. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 2019, 37, 626–631. [Google Scholar] [CrossRef]
- Gehrke, J.M.; Cervantes, O.; Clement, M.K.; Wu, Y.; Zeng, J.; Bauer, D.E.; Pinello, L.; Joung, J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Publ. Gr. 2018, 36, 977–982. [Google Scholar] [CrossRef]
- Shi, J.; Ma, Y.; Zhu, J.; Chen, Y.; Sun, Y.; Yao, Y.; Yang, Z.; Xie, J. A Review on Electroporation-Based Intracellular Delivery. Molecules 2018, 23, 3044. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-Step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.M.; Wang, T.; Randolph, P.B.; Arbab, M.; Shen, M.W.; Huang, T.P.; Matuszek, Z.; Newby, G.A.; Rees, H.A.; Liu, D.R. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 2020, 38, 471–481. [Google Scholar] [CrossRef]
- Doman, J.L.; Raguram, A.; Newby, G.A.; Liu, D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sürün, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hänchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes 2020, 11, 511. https://doi.org/10.3390/genes11050511
Sürün D, Schneider A, Mircetic J, Neumann K, Lansing F, Paszkowski-Rogacz M, Hänchen V, Lee-Kirsch MA, Buchholz F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes. 2020; 11(5):511. https://doi.org/10.3390/genes11050511
Chicago/Turabian StyleSürün, Duran, Aksana Schneider, Jovan Mircetic, Katrin Neumann, Felix Lansing, Maciej Paszkowski-Rogacz, Vanessa Hänchen, Min Ae Lee-Kirsch, and Frank Buchholz. 2020. "Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors" Genes 11, no. 5: 511. https://doi.org/10.3390/genes11050511