The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges

 , and
, and {kind=link}
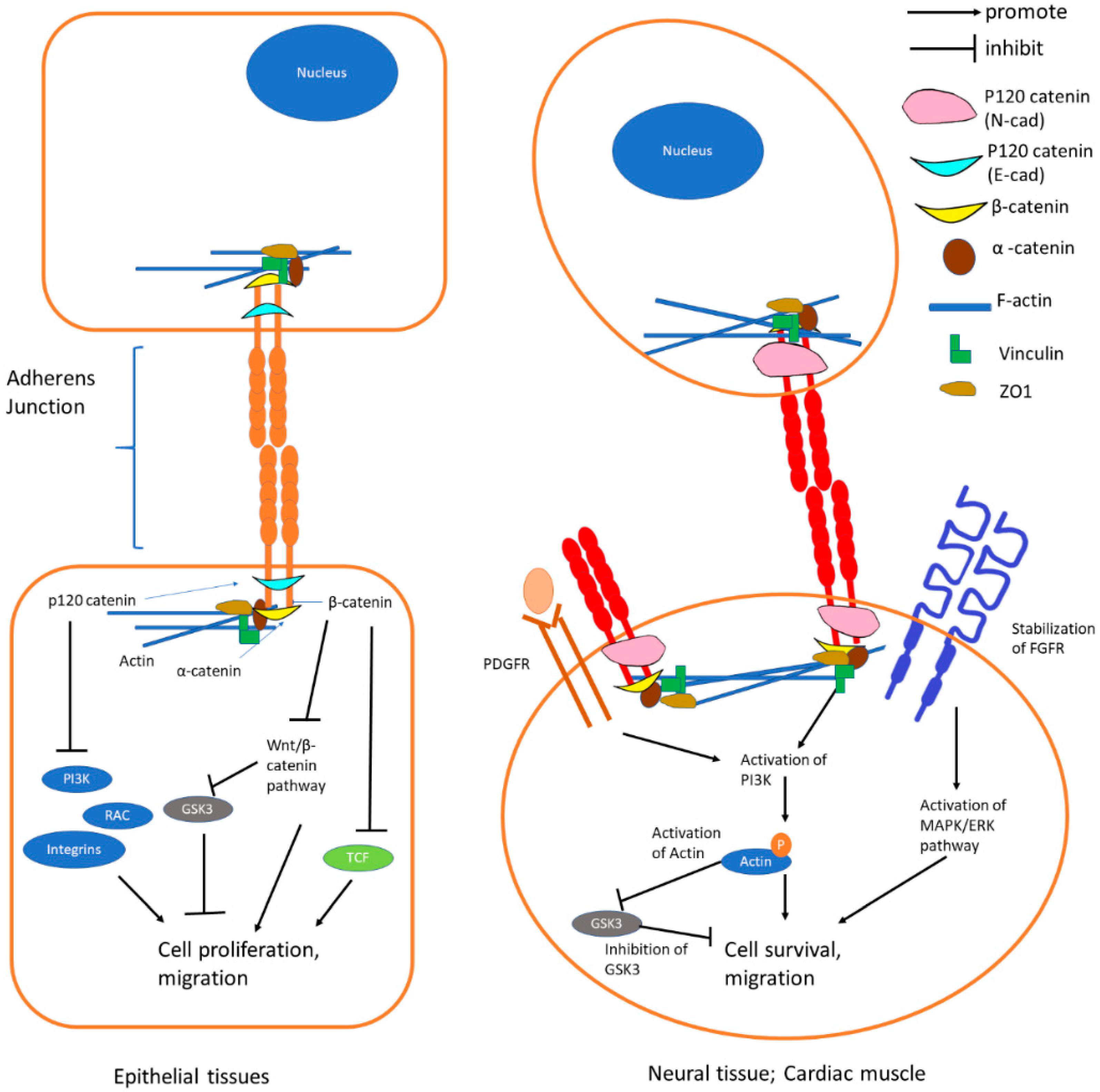{kind=link}
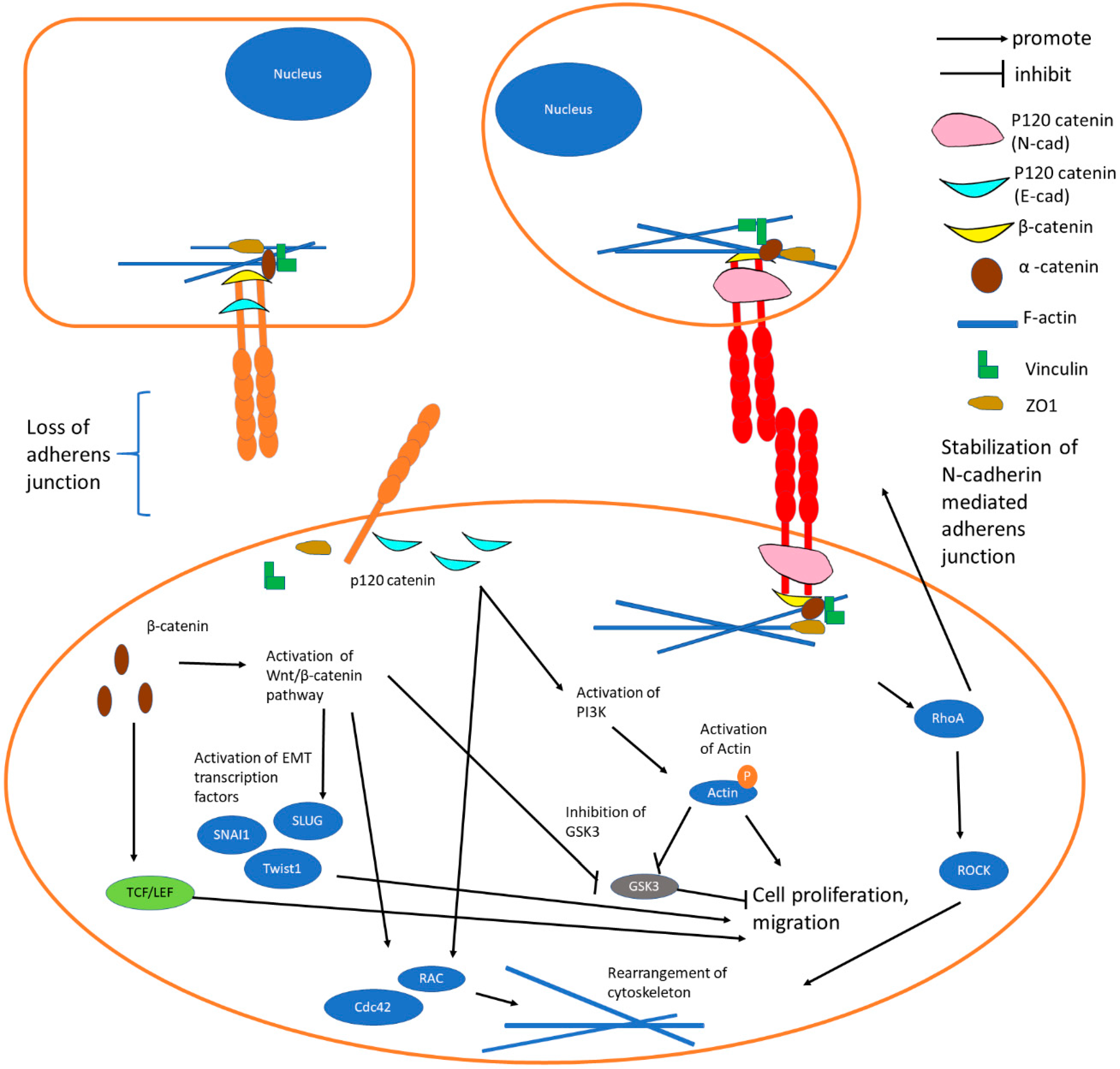{kind=link}
{kind=link}
Abstract
:1. Introduction
2. E-Cadherin and N-Cadherin in Epithelial-to-Mesenchymal Transition
3. Crosstalk of E-Cadherin and N-Cadherin with EMT-Related Signaling Pathways
3.1. TGF-β Pathway
3.2. MAPK Pathway
3.3. JAK/STAT Pathway
3.4. Hedgehog Pathway
3.5. Wnt Pathway
3.6. Hippo-YAP/TAZ Pathway
4. Therapeutic Implication Targeting EMT
4.1. shRNA and miRNA
4.2. Small Molecules and Tyrosine Kinase Inhibitors
4.3. Monoclonal Antibodies
4.4. Natural Compounds
4.4.1. Curcumin
4.4.2. Resveratrol
4.4.3. Honokiol
4.4.4. Other Natural Compounds
5. Challenges to Translating the Preclinical Research from Bench to Bedside
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takeichi, M. Functional correlation between cell adhesive properties and some cell surface proteins. J. Cell Biol. 1977, 75, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, M. Cadherins: A Molecular Family Important in Selective Cell-Cell Adhesion. Annu. Rev. Biochem. 1990, 59, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Hulpiau, P.; Gul, I.S.; Van Roy, F. Evolution of cadherins and associated catenins. In The Cadherin Superfamily; Suzuki, S.T., Hirano, S., Eds.; Springer: Tokyo, Japan, 2016; pp. 13–37. [Google Scholar]
- Gul, I.S.; Hulpiau, P.; Saeys, Y.; Van Roy, F. Evolution and diversity of cadherins and catenins. Exp. Cell Res. 2017, 358, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, M.P. Cadherins in development and cancer. Mol. Biosyst. 2008, 4, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. Cadherins in tissue architecture and disease. J. Mol. Med. 2015, 93, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Colás-Algora, N.; Millán, J. How many cadherins do human endothelial cells express? Cell. Mol. Life Sci. 2019, 76, 1299–1317. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Classical Cadherins. In The Cadherin Superfamily; Springer: Tokyo, Japan, 2016; pp. 41–69. [Google Scholar]
- Perez, T.D.; Nelson, W.J. Cadherin adhesion: Mechanisms and molecular interactions. Handb. Exp. Pharm. 2004, 3–21. [Google Scholar] [CrossRef]
- Parisini, E.; Higgins, J.M.G.; Liu, J.-h.; Brenner, M.B.; Wang, J.-h. The crystal structure of human E-cadherin domains 1 and 2, and comparison with other cadherins in the context of adhesion mechanism. J. Mol. Biol. 2007, 373, 401–411. [Google Scholar] [CrossRef]
- Seddiki, R.; Narayana, G.H.N.S.; Strale, P.-O.; Balcioglu, H.E.; Peyret, G.; Yao, M.; Le, A.P.; Teck Lim, C.; Yan, J.; Ladoux, B. Force-dependent binding of vinculin to α-catenin regulates cell–cell contact stability and collective cell behavior. Mol. Biol. Cell 2018, 29, 380–388. [Google Scholar] [CrossRef]
- Zaidel-Bar, R. Cadherin adhesome at a glance. J. Cell Sci. 2013, 126, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Bertocchi, C.; Wang, Y.; Ravasio, A.; Hara, Y.; Wu, Y.; Sailov, T.; Baird, M.A.; Davidson, M.W.; Zaidel-Bar, R.; Toyama, Y.; et al. Nanoscale architecture of cadherin-based cell adhesions. Nat. Cell Biol. 2017, 19, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.A.; Boscher, C.; Chu, Y.-S.; Cuvelier, D.; Martinez-Rico, C.; Seddiki, R.; Heysch, J.; Ladoux, B.; Thiery, J.P.; Mege, R.-M. α-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 2013, 288, 4957–4969. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, W.; Behrens, J. Cadherin expression in carcinomas: Role in the formation of cell junctions and the prevention of invasiveness. BBA Rev. Cancer 1994, 1198, 11–26. [Google Scholar] [CrossRef]
- Schneider, M.R.; Kolligs, F.T. E-cadherin’s role in development, tissue homeostasis and disease: Insights from mouse models: Tissue-specific inactivation of the adhesion protein E-cadherin in mice reveals its functions in health and disease. Bioessays 2015, 37, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.; Majem, B.; Devis, L.; Lapyckyj, L.; Besso, M.J.; Llauradó, M.; Abascal, M.F.; Matos, M.L.; Lanau, L.; Castellví, J.; et al. E-cadherin: A determinant molecule associated with ovarian cancer progression, dissemination and aggressiveness. PLoS ONE 2017, 12, e0184439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Berx, G.; Van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef]
- Winter, J.M.; Ting, A.H.; Vilardell, F.; Gallmeier, E.; Baylin, S.B.; Hruban, R.H.; Kern, S.E.; Iacobuzio-Donahue, C.A. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin. Cancer Res. 2008, 14, 412–418. [Google Scholar] [CrossRef]
- Hirohashi, S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am. J. Pathol. 1998, 153, 333–339. [Google Scholar] [CrossRef]
- Berx, G.; Becker, K.F.; Höfler, H.; Van Roy, F. Mutations of the human E-cadherin (CDH1) gene. Hum. Mutat. 1998, 12, 226–237. [Google Scholar] [CrossRef]
- Strathdee, G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin. Cancer Biol. 2002, 12, 373–379. [Google Scholar] [CrossRef]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A.J.C.r. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Shah, P.; Barcellos-Hoff, M.H.; He, Y.-Y. TGF-β signaling links E-cadherin loss to suppression of nucleotide excision repair. Oncogene 2016, 35, 3293. [Google Scholar] [CrossRef] [PubMed]
- Mayor, R.; Carmona-Fontaine, C. Keeping in touch with contact inhibition of locomotion. Trends Cell Biol. 2010, 20, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Perrais, M.; Chen, X.; Perez-Moreno, M.; Gumbiner, B.M. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell 2007, 18, 2013–2025. [Google Scholar] [CrossRef]
- Park, S.Y.; Shin, J.-H.; Kee, S.-H. E-cadherin expression increases cell proliferation by regulating energy metabolism through nuclear factor-κB in AGS cells. Cancer Sci. 2017, 108, 1769–1777. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, G.; Akhtar, N.; Kannius-Janson, M.; Baeckström, D. Loss of E-cadherin expression is not a prerequisite for c-erbB2-induced epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.; Godwin, T.D.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Çeliktaş, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S. Metastatic progression of prostate cancer and e-cadherin: Regulation by Zeb1 and Src family kinases. Am. J. Pathol. 2011, 179, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.P.; Kuang, J.Y.; Yang, Q.K.; Bian, X.W.; Yu, S.C. Beyond a tumor suppressor: Soluble E-cadherin promotes the progression of cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef]
- Wheelock, M.J.; Buck, C.A.; Bechtol, K.B.; Damsky, C.H. Soluble 80-kd fragment of cell-CAM 120/80 disrupts cell-cell adhesion. J. Cell. Biochem. 1987, 34, 187–202. [Google Scholar] [CrossRef]
- Grabowska, M.M.; Day, M.L. Soluble E-cadherin: More than a symptom of disease. Front. Biosci. (Landmark Ed.) 2012, 17, 1948. [Google Scholar] [CrossRef]
- Reckamp, K.L.; Gardner, B.K.; Figlin, R.A.; Elashoff, D.; Krysan, K.; Dohadwala, M.; Mao, J.; Sharma, S.; Inge, L.; Rajasekaran, A. Tumor response to combination celecoxib and erlotinib therapy in non-small cell lung cancer is associated with a low baseline matrix metalloproteinase-9 and a decline in serum-soluble E-cadherin. J. Thorac. Oncol. 2008, 3, 117–124. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Lee, K.-W.; Cho, J.Y.; Kang, W.K.; Im, S.-A.; Kim, J.W.; Bang, Y.-J. Phase II trial of dacomitinib in patients with HER2-positive gastric cancer. Gastric Cancer 2016, 19, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.; Stec, R.; Cierniak, S.; Synowiec, A.; Wcisło, G.; Jesiotr, M.; Koktysz, R.; Chrom, P.; Szczylik, C. Role of WNT/β-Catenin Pathway as Potential Prognostic and Predictive Factors in Renal Cell Cancer Patients Treated With Everolimus in the Second and Subsequent Lines. Clin. Genitourin. Cancer 2018, 16, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.K.; Yue, P.Y.; Ip, P.P.; Huang, R.-L.; Lai, H.-C.; Cheung, A.N.; Tse, K.Y.; Ngan, H.Y.; Wong, A.S. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat. Commun. 2018, 9, 2270. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F. Beyond E-cadherin: Roles of other cadherin superfamily members in cancer. Nat. Rev. Cancer 2014, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Sakane, F.; Hashimoto, K. N-cadherin-based adherens junction regulates the maintenance, proliferation, and differentiation of neural progenitor cells during development. Cell Adh. Migr. 2015, 9, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Ren, D.; Guo, W.; Huang, S.; Wang, Z.; Li, Q.; Du, H.; Song, L.; Peng, X. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int. J. Oncol. 2016, 48, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Jennbacken, K.; Tešan, T.; Wang, W.; Gustavsson, H.; Damber, J.-E.; Welen, K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr. Relat. Cancer 2010, 17, 469–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulit, J.; Suyama, K.; Chung, S.; Keren, R.; Agiostratidou, G.; Shan, W.; Dong, X.; Williams, T.M.; Lisanti, M.P.; Knudsen, K. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007, 67, 3106–3116. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic significance of twist and N-cadherin expression in NSCLC. PLoS ONE 2013, 8, e62171. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Wolburg, H.; Redies, C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2000, 218, 472–479. [Google Scholar] [CrossRef]
- Paik, J.-H.; Skoura, A.; Chae, S.-S.; Cowan, A.E.; Han, D.K.; Proia, R.L.; Hla, T. Sphingosine 1-phosphate receptor regulation of N-cadherin mediates vascular stabilization. Genes Dev. 2004, 18, 2392–2403. [Google Scholar] [CrossRef] [PubMed]
- Blaschuk, O.W. N-cadherin antagonists as oncology therapeutics. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140039. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Li, J.; Shi, C.; Hruban, R.H.; Radice, G.L. N-cadherin functions as a growth suppressor in a model of K-ras-induced PanIN. Oncogene 2016, 35, 3335. [Google Scholar] [CrossRef] [PubMed]
- Lammens, T.; Swerts, K.; Derycke, L.; De Craemer, A.; De Brouwer, S.; De Preter, K.; Van Roy, N.; Vandesompele, J.; Speleman, F.; Philippe, J. N-cadherin in neuroblastoma disease: Expression and clinical significance. PLoS ONE 2012, 7, e31206. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Berx, G. Cadherins and epithelial-to-mesenchymal transition. In Progress in Molecular Biology and Translational Science; Van Roy, F., Ed.; Elsevier: Tokyo, Japan, 2013; Volume 116, pp. 317–336. [Google Scholar]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, A. Molecular architecture of adherens junctions. Curr. Opin. Cell Biol. 2001, 13, 600–603. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Somarelli, J.A.; Hanash, S.M.; Levine, H. Epithelial/mesenchymal plasticity: How have quantitative mathematical models helped improve our understanding? Mol. Oncol. 2017, 11, 739–754. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Shimura, T.; Suzuki, H.; Tsutsumi, S.; Wada, W.; Yajima, T.; Kobayahi, T.; Kubo, N.; Kuwano, H. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 2011, 105, 1885. [Google Scholar] [CrossRef] [PubMed]
- Aleskandarany, M.A.; Negm, O.H.; Green, A.R.; Ahmed, M.A.; Nolan, C.C.; Tighe, P.J.; Ellis, I.O.; Rakha, E.A. Epithelial mesenchymal transition in early invasive breast cancer: An immunohistochemical and reverse phase protein array study. Breast Cancer Res. Treat. 2014, 145, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Chen, N.; Wang, J.; Siu, C.-H. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the β-catenin signaling pathway. Mol. Biol. Cell 2005, 16, 4386–4397. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-S.; Gumbiner, B.M. Cadherin 6B induces BMP signaling and de-epithelialization during the epithelial mesenchymal transition of the neural crest. Develepment 2010, 137, 2691–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Marsters, S.; Ye, X.; Luis, E.; Gonzalez, L.; Ashkenazi, A. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell 2014, 54, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [CrossRef]
- Loreto, C.; La Rocca, G.; Anzalone, R.; Caltabiano, R.; Vespasiani, G.; Castorina, S.; Ralph, D.J.; Cellek, S.; Musumeci, G.; Giunta, S.; et al. The role of intrinsic pathway in apoptosis activation and progression in Peyronie’s disease. Biomed. Res. Int. 2014, 2014, 616149. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; Van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Ding, W.; You, H.; Dang, H.; LeBlanc, F.; Galicia, V.; Lu, S.C.; Stiles, B.; Rountree, C.B. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology 2010, 52, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Grotegut, S.; von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahm, I.; Barriga, E.H.; Frolov, A.; Theveneau, E.; Frankel, P.; Mayor, R. PDGF controls contact inhibition of locomotion by regulating N-cadherin during neural crest migration. Development 2017, 144, 2456–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef]
- Suyama, K.; Shapiro, I.; Guttman, M.; Hazan, R.B. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell 2002, 2, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-B.; Islam, S.; Kim, Y.J.; Prudoff, R.S.; Sass, K.M.; Wheelock, M.J.; Johnson, K.R. N-Cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J. Cell Biol. 2000, 151, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Pan, J.; Grove, B.D.; Peterson, T.; Fisher, J.; Maines-Bandiera, S.; Somasiri, A.; Roskelley, C.D. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc. Natl. Acad. Sci. USA 1999, 96, 6249–6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal–epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Jolly, M.K.; Lu, M.; Onuchic, J.N.; Clementi, C.; Ben-Jacob, E. Jagged-Delta asymmetry in Notch signaling can give rise to a Sender/Receiver hybrid phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E402–E409. [Google Scholar] [CrossRef] [PubMed]
- Shaya, O.; Sprinzak, D. From Notch signaling to fine-grained patterning: Modeling meets experiments. Curr. Opin. Genet. Dev. 2011, 21, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.-H.; Derynck, R. Specificity and versatility in TGF-β signaling through Smads. Annu. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial–mesenchymal transition induced by TGF-β. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.-H.; Moustakas, A. Transforming growth factor-β employs HMGA2 to elicit epithelial–mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef]
- Yang, H.; Wang, L.; Zhao, J.; Chen, Y.; Lei, Z.; Liu, X.; Xia, W.; Guo, L.; Zhang, H.-T. TGF-β-activated SMAD3/4 complex transcriptionally upregulates N-cadherin expression in non-small cell lung cancer. Lung Cancer 2015, 87, 249–257. [Google Scholar] [CrossRef]
- Tuli, R.; Tuli, S.; Nandi, S.; Huang, X.; Manner, P.A.; Hozack, W.J.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. Transforming growth factor-β-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J. Biol. Chem. 2003, 278, 41227–41236. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Bergamini, C.; Fransvea, E.; Sgarra, C.; Antonaci, S. Laminin-5 with transforming growth factor-β1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology 2005, 129, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Fransvea, E.; Angelotti, U.; Antonaci, S.; Giannelli, G. Blocking transforming growth factor–beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008, 47, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-h.; Zhou, X.-l.; Shi, Y.-l.; Yang, J. Roles of p38 MAPK and JNK in TGF-β1-induced human alveolar epithelial to mesenchymal transition. Arch. Med Res. 2013, 44, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Uemura, K.; Kuzuya, A.; Maesako, M.; Asada-Utsugi, M.; Kubota, M.; Aoyagi, N.; Yoshioka, K.; Okawa, K.; Inoue, H. N-cadherin Regulates p38 MAPK Signaling via Association with JNK-associated Leucine Zipper Protein Implications for Neurodegeneration in Alzheimer Disease. J. Biol. Chem. 2011, 286, 7619–7628. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Wang, Y.; Wu, H.; Frank, J.A.; Zhang, Z.; Luo, J. Role of p38γ MAPK in regulation of EMT and cancer stem cells. BBA Mol. Basis Dis. 2018, 1864, 3605–3617. [Google Scholar] [CrossRef]
- Li, Q.; Mattingly, R.R. Restoration of E-cadherin cell-cell junctions requires both expression of E-cadherin and suppression of ERK MAP kinase activation in Ras-transformed breast epithelial cells. Neoplasia 2008, 10, 1444–1458. [Google Scholar] [CrossRef]
- Pece, S.; Gutkind, J.S. Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J. Biol. Chem. 2000, 275, 41227–41233. [Google Scholar] [CrossRef]
- Tang, G.; Du, R.; Tang, Z.; Kuang, Y. MiRNALet-7a mediates prostate cancer PC-3 cell invasion, migration by inducing epithelial-mesenchymal transition through CCR7/MAPK pathway. J. Cell. Biochem. 2018, 119, 3725–3731. [Google Scholar] [CrossRef]
- Wang, L.; Mukhopadhyay, D.; Xu, X.; Wang, L.; Mukhopadhyay, D.; Xu, X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB J. 2006, 20, 1513–1515. [Google Scholar] [CrossRef]
- Wild, J.R.; Staton, C.A.; Chapple, K.; Corfe, B.M. Neuropilins: Expression and roles in the epithelium. Int. J. Exp. Pathol. 2012, 93, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Song, X.; Yang, X.; Ma, L.; Zhu, J.; He, M.; Wang, Z.; Wu, Y. Neuropilin-1 promotes epithelial-to-mesenchymal transition by stimulating nuclear factor-kappa B and is associated with poor prognosis in human oral squamous cell carcinoma. PLoS ONE 2014, 9, e101931. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Choi, Y.; Oh, S.; Jeong, J.; Choi, D.; Seo, H.; Kim, C. Mucin 1 enhances the tumor angiogenic response by activation of the AKT signaling pathway. Oncogene 2012, 31, 2187. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Curry, J.M.; Roy, L.D.; Grover, P.; Haider, J.; Moore, L.J.; Wu, S.; Kamesh, A.; Yazdanifar, M.; Ahrens, W.A. A novel association of neuropilin-1 and MUC1 in pancreatic ductal adenocarcinoma: Role in induction of VEGF signaling and angiogenesis. Oncogene 2016, 35, 5608. [Google Scholar] [CrossRef]
- Fung, T.M.; Ng, K.Y.; Tong, M.; Chen, J.N.; Chai, S.; Chan, K.T.; Law, S.; Lee, N.P.; Choi, M.Y.; Li, B. Neuropilin-2 promotes tumourigenicity and metastasis in oesophageal squamous cell carcinoma through ERK–MAPK–ETV4–MMP–E-cadherin deregulation. J. Pathol. 2016, 239, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhou, J.; Dai, H.; Liu, F.; Li, W.; Wang, W.; Guo, F. CHIP functions as an oncogene by promoting colorectal cancer metastasis via activation of MAPK and AKT signaling and suppression of E-cadherin. J. Transl. Med. 2018, 16, 169. [Google Scholar] [CrossRef]
- Han, H.-B.; Gu, J.; Ji, D.-B.; Li, Z.-W.; Zhang, Y.; Zhao, W.; Wang, L.-M.; Zhang, Z.-Q. PBX3 promotes migration and invasion of colorectal cancer cells via activation of MAPK/ERK signaling pathway. World J. Gastroenterol. 2014, 20, 18260. [Google Scholar] [CrossRef]
- Lamprecht, S.; Kaller, M.; Schmidt, E.M.; Blaj, C.; Schiergens, T.S.; Engel, J.; Jung, A.; Hermeking, H.; Grünewald, T.G.; Kirchner, T. PBX3 is part of an EMT regulatory network and indicates poor outcome in colorectal cancer. Clin. Cancer Res. 2018, 24, 1974–1986. [Google Scholar] [CrossRef]
- Wang, S.; Li, C.; Wang, W.; Xing, C. PBX3 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition. Oncol. Lett. 2016, 12, 3485–3491. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.-Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, A.; Shanmugam, M.K.; Ong, T.H.; Li, F.; Perumal, E.; Chen, L.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; et al. Emodin inhibits growth and induces apoptosis in an orthotopic hepatocellular carcinoma model by blocking activation of STAT3. Br. J. Pharmacol. 2013, 170, 807–821. [Google Scholar] [CrossRef] [Green Version]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kumar, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M.; et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Geletu, M.; Arulanandam, R.; Chevalier, S.; Saez, B.; Larue, L.; Feracci, H.; Raptis, L. Classical cadherins control survival through the gp130/Stat3 axis. BBA Mol. Cell Res. 2013, 1833, 1947–1959. [Google Scholar] [CrossRef] [Green Version]
- Arulanandam, R.; Vultur, A.; Cao, J.; Carefoot, E.; Elliott, B.E.; Truesdell, P.F.; Larue, L.; Feracci, H.; Raptis, L. Cadherin-cadherin engagement promotes cell survival via Rac1/Cdc42 and signal transducer and activator of transcription-3. Mol. Cancer Res. 2009, 7, 1310–1327. [Google Scholar] [CrossRef]
- Onishi, A.; Chen, Q.; Humtsoe, J.O.; Kramer, R.H. STAT3 signaling is induced by intercellular adhesion in squamous cell carcinoma cells. Exp. Cell Res. 2008, 314, 377–386. [Google Scholar] [CrossRef] [Green Version]
- del Valle, I.; Rudloff, S.; Carles, A.; Li, Y.; Liszewska, E.; Vogt, R.; Kemler, R. E-cadherin is required for the proper activation of the Lifr/Gp130 signaling pathway in mouse embryonic stem cells. Development 2013, 140, 1684–1692. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, K.; Mohamet, L.; Ritson, S.; Merry, C.L.; Ward, C.M. E-cadherin and, in its absence, N-cadherin promotes Nanog expression in mouse embryonic stem cells via STAT3 phosphorylation. Stem Cells 2012, 30, 1842–1851. [Google Scholar] [CrossRef]
- Xiong, H.; Hong, J.; Du, W.; Lin, Y.-w.; Ren, L.-l.; Wang, Y.-c.; Su, W.-y.; Wang, J.-l.; Cui, Y.; Wang, Z.-h. Roles of STAT3 and ZEB1 proteins in E-cadherin down-regulation and human colorectal cancer epithelial-mesenchymal transition. J. Biol. Chem. 2012, 287, 5819–5832. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, P.; Lee, M.-H.; Zhang, L.; Baratelli, F.; Lee, G.; Srivastava, M.K.; Wang, G.; Walser, T.C.; Krysan, K.; Sharma, S. IL-27 inhibits epithelial-mesenchymal transition and angiogenic factor production in a STAT1-dominant pathway in human non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2013, 32, 97. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Cheng, X.; Wang, Y.; Han, R.; Li, L.; Xiang, T.; He, L.; Long, H.; Zhu, B.; He, Y. Metformin inhibits the IL-6-induced epithelial-mesenchymal transition and lung adenocarcinoma growth and metastasis. PLoS ONE 2014, 9, e95884. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lu, Y.; Li, Y.; Prinz, R.A. Sonic Hedgehog signaling in thyroid cancer. Front. Endocrinol. 2017, 8, 284. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, B.; Xie, C.; Wei, S.; Zhou, Y.; Liu, H.; Dai, W.; Cheng, P.; Wang, F.; Xu, X. Sonic hedgehog-Gli1 signaling pathway regulates the epithelial mesenchymal transition (EMT) by mediating a new target gene, S100A4, in pancreatic cancer cells. PLoS ONE 2014, 9, e96441. [Google Scholar] [CrossRef]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: A small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef]
- Zhang, J.; Cui, Y.; Sun, S.; Cao, J.; Fang, X. Casticin inhibits the epithelial-mesenchymal transition in ovarian carcinoma via the hedgehog signaling pathway. Oncol. Lett. 2018, 15, 4495–4502. [Google Scholar] [CrossRef]
- Gao, Q.; Yuan, Y.; Gan, H.Z.; Peng, Q. Resveratrol inhibits the hedgehog signaling pathway and epithelial-mesenchymal transition and suppresses gastric cancer invasion and metastasis. Oncol. Lett. 2015, 9, 2381–2387. [Google Scholar] [CrossRef]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. Hedgehog signaling regulates epithelial-mesenchymal transition in pancreatic cancer stem-like cells. J. Cancer 2016, 7, 408. [Google Scholar] [CrossRef]
- Wang, L.; Jin, J.Q.; Zhou, Y.; Tian, Z.; Jablons, D.M.; He, B. Gli is activated and promotes epithelial-mesenchymal transition in human esophageal adenocarcinoma. Oncotarget 2018, 9, 853. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Ogle, S.A.; Schumacher, M.A.; Schilling, N.; Tokhunts, R.A.; Orr-Asman, M.A.; Miller, M.L.; Robbins, D.J.; Hollande, F.; Zavros, Y. Hedgehog signaling regulates E-cadherin expression for the maintenance of the actin cytoskeleton and tight junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1252–G1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neill, G.W.; Harrison, W.J.; Ikram, M.S.; Williams, T.D.; Bianchi, L.S.; Nadendla, S.K.; Green, J.L.; Ghali, L.; Frischauf, A.-M.; O’toole, E.A. GLI1 repression of ERK activity correlates with colony formation and impaired migration in human epidermal keratinocytes. Carcinogenesis 2008, 29, 738–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2008, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N. Tumor suppressor gene E-cadherin and its role in normal and malignant cells. Cancer Cell Int. 2003, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Guilford, P. E-cadherin downregulation in cancer: Fuel on the fire? Mol. Med. Today 1999, 5, 172–177. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Haÿ, E.; Buczkowski, T.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Peptide-based mediated disruption of N-cadherin-LRP5/6 interaction promotes Wnt signaling and bone formation. J. Bone Miner. Res. 2012, 27, 1852–1863. [Google Scholar] [CrossRef]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef]
- Maher, M.T.; Flozak, A.S.; Stocker, A.M.; Chenn, A.; Gottardi, C.J. Activity of the β-catenin phosphodestruction complex at cell–cell contacts is enhanced by cadherin-based adhesion. J. Cell Biol. 2009, 186, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem. Soc. Trans. 2008, 36, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.H.; Weis, W.I. The structure of the β-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by β-catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef]
- Qi, J.; Wang, J.; Romanyuk, O.; Siu, C.-H. Involvement of Src family kinases in N-cadherin phosphorylation and β-catenin dissociation during transendothelial migration of melanoma cells. Mol. Biol. Cell 2006, 17, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Thievessen, I.; Seifert, H.; Swiatkowski, S.; Florl, A.; Schulz, W. E-cadherin involved in inactivation of WNT/β-catenin signalling in urothelial carcinoma and normal urothelial cells. Br. J. Cancer 2003, 88, 1932. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Lobo-Ruppert, S.; Ruppert, J. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene 2007, 26, 4489. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.; Deroo, T.; Fujita, Y.; Itasaki, N. A positive role of cadherin in Wnt/β-catenin signalling during epithelial-mesenchymal transition. PLoS ONE 2011, 6, e23899. [Google Scholar] [CrossRef]
- Haÿ, E.; Laplantine, E.; Geoffroy, V.; Frain, M.; Kohler, T.; Müller, R.; Marie, P.J. N-cadherin interacts with axin and LRP5 to negatively regulate Wnt/β-catenin signaling, osteoblast function, and bone formation. Mol. Cell. Biol. 2009, 29, 953–964. [Google Scholar] [CrossRef]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R. Yap1 acts downstream of α-catenin to control epidermal proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef]
- Dobrokhotov, O.; Samsonov, M.; Sokabe, M.; Hirata, H. Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin. Transl. Med. 2018, 7, 23. [Google Scholar] [CrossRef]
- Klezovitch, O.; Vasioukhin, V. Cadherin signaling: Keeping cells in touch. F1000Research 2015, 4, 550. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, K.T.; Yamashita, K.; Sakurai, N.; Ohno, S. The epithelial circumferential actin belt regulates YAP/TAZ through nucleocytoplasmic shuttling of merlin. Cell Rep. 2017, 20, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Calvisi, D.F.; Ranganathan, S.; Cigliano, A.; Zhou, L.; Singh, S.; Jiang, L.; Fan, B.; Terracciano, L.; Armeanu–Ebinger, S. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014, 147, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.R.; Kreger, B.T.; Lien, W.-H.; Klezovitch, O.; Rudakova, G.M.; Camargo, F.D.; Lantz, D.M.; Seykora, J.T.; Vasioukhin, V. α-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci. Signal. 2011, 4, ra33. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, E.; Vite, A.; Yi, R.; Gomez, L.; Goossens, S.; Van Roy, F.; Radice, G.L. Alpha-catenins control cardiomyocyte proliferation by regulating Yap activity. Circ. Res. 2015, 116, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [Green Version]
- Gumbiner, B.M.; Kim, N.-G. The Hippo-YAP signaling pathway and contact inhibition of growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef]
- Sansores-Garcia, L.; Bossuyt, W.; Wada, K.I.; Yonemura, S.; Tao, C.; Sasaki, H.; Halder, G. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. Embo J. 2011, 30, 2325–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Li, G.; Huang, S.; Feng, Y.; Zhou, A. SOX9 promotes epithelial-mesenchymal transition via the Hippo-YAP signaling pathway in gastric carcinoma cells. Oncol. Lett. 2019, 18, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Kremerskothen, J.; Lin, X.; Zhang, X.; Rong, X.; Zhang, D.; Wang, E. WWc3 inhibits epithelial–mesenchymal transition of lung cancer by activating hippo-YaP signaling. Oncotargets Ther. 2018, 11, 2581–2591. [Google Scholar] [CrossRef] [PubMed]
- Pei, T.; Li, Y.; Wang, J.; Wang, H.; Liang, Y.; Shi, H.; Sun, B.; Yin, D.; Sun, J.; Song, R. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015, 6, 17206–17220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.-Y.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.; Yang, D.; Xiao, X.; Sun, R.; Huang, L.; Xu, J. MiRNA-145 suppresses lung adenocarcinoma cell invasion and migration by targeting N-cadherin. Biotechnol. Lett. 2017, 39, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, Z.; Wu, S.; Zhuo, Y.; Otsetov, A.G.; Cai, C.; Zhong, W.; Wu, C.-L.; Olumi, A.F. Metformin represses cancer cells via alternate pathways in N-cadherin expressing vs. N-cadherin deficient cells. Oncotarget 2015, 6, 28973–28987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology 2017, 22, 589–597. [Google Scholar] [CrossRef]
- Williams, E.; Williams, G.; Gour, B.J.; Blaschuk, O.W.; Doherty, P. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J. Biol. Chem. 2000, 275, 4007–4012. [Google Scholar] [CrossRef]
- Peluso, J.; Pappalardo, A.; Trolice, M. N-cadherin-mediated cell contact inhibits granulosa cell apoptosis in a progesterone-independent manner. Endocrinology 1996, 137, 1196–1203. [Google Scholar] [CrossRef]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Kannaiyan, R.; Sethi, G. Targeting cell signaling and apoptotic pathways by dietary agents: Role in the prevention and treatment of cancer. Nutr. Cancer 2011, 63, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Bordoloi, D.; Harsha, C.; Banik, K.; Gupta, S.C.; Aggarwal, B.B. Curcumin mediates anticancer effects by modulating multiple cell signaling pathways. Clin. Sci. 2017, 131, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T. Targeting cancer stem cells by curcumin and clinical applications. Cancer Lett. 2014, 346, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Lee, J.H.; Chai, E.Z.; Kanchi, M.M.; Kar, S.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Ramar, P.S.; Looi, C.Y.; et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. Semin. Cancer Biol. 2016, 40–41, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Warrier, S.; Kumar, A.P.; Sethi, G.; Arfuso, F. Potential role of natural compounds as anti-angiogenic agents in cancer. Curr. Vasc. Pharm. 2017, 10, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Mazumdar, M.; Chakraborty, S.; Manna, A.; Saha, S.; Khan, P.; Bhattacharjee, P.; Guha, D.; Adhikary, A.; Mukhjerjee, S.; et al. Curcumin inhibits breast cancer stem cell migration by amplifying the E-cadherin/beta-catenin negative feedback loop. Stem Cell Res. 2014, 5, 116. [Google Scholar] [CrossRef]
- Thacker, P.C.; Karunagaran, D. Curcumin and emodin down-regulate TGF-beta signaling pathway in human cervical cancer cells. PLoS ONE 2015, 10, e0120045. [Google Scholar] [CrossRef]
- Sun, X.D.; Liu, X.E.; Huang, D.S. Curcumin reverses the epithelial-mesenchymal transition of pancreatic cancer cells by inhibiting the Hedgehog signaling pathway. Oncol. Rep. 2013, 29, 2401–2407. [Google Scholar] [CrossRef]
- Chen, W.C.; Lai, Y.A.; Lin, Y.C.; Ma, J.W.; Huang, L.F.; Yang, N.S.; Ho, C.T.; Kuo, S.C.; Way, T.D. Curcumin suppresses doxorubicin-induced epithelial-mesenchymal transition via the inhibition of TGF-beta and PI3K/AKT signaling pathways in triple-negative breast cancer cells. J. Agric. Food Chem. 2013, 61, 11817–11824. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Yang, H.P.; Zhou, X.D.; Wang, H.J.; Gong, L.; Tang, C.L. Role of Wnt Inhibitory Factor-1 in Inhibition of Bisdemethoxycurcumin Mediated Epithelial-to-Mesenchymal Transition in Highly Metastatic Lung Cancer 95D Cells. Chin. Med. J. (Engl.) 2015, 128, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, X.; Gao, Y.; Zhang, C.; Bao, J.; Guan, H.; Yu, H.; Lu, R.; Xu, Q.; Sun, Y. Curcumin inhibits metastasis in human papillary thyroid carcinoma BCPAP cells via down-regulation of the TGF-beta/Smad2/3 signaling pathway. Exp. Cell Res. 2016, 341, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Long, Q.; Zhang, L.; Shi, Y.; Liu, X.; Li, X.; Guan, B.; Tian, Y.; Wang, X.; Li, L.; et al. Curcumin inhibits cancer-associated fibroblast-driven prostate cancer invasion through MAOA/mTOR/HIF-1alpha signaling. Int. J. Oncol. 2015, 47, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Jiao, D.; Wang, J.; Lu, W.; Tang, X.; Chen, J.; Mou, H.; Chen, Q.Y. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Mol. Oncolytics 2016, 3, 16018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, M.; Ray, L.; Purohit, M.P.; Patnaik, S.; Pant, A.B.; Shukla, Y.; Kumar, P.; Gupta, K.C. Curcumin loading potentiates the chemotherapeutic efficacy of selenium nanoparticles in HCT116 cells and Ehrlich’s ascites carcinoma bearing mice. Eur. J. Pharm. Biopharm. 2017, 117, 346–362. [Google Scholar] [CrossRef]
- Kumari, M.; Purohit, M.P.; Patnaik, S.; Shukla, Y.; Kumar, P.; Gupta, K.C. Curcumin loaded selenium nanoparticles synergize the anticancer potential of doxorubicin contained in self-assembled, cell receptor targeted nanoparticles. Eur. J. Pharm. Biopharm. 2018, 130, 185–199. [Google Scholar] [CrossRef]
- Meng, X.; Cai, J.; Liu, J.; Han, B.; Gao, F.; Gao, W.; Zhang, Y.; Zhang, J.; Zhao, Z.; Jiang, C. Curcumin increases efficiency of gamma-irradiation in gliomas by inhibiting Hedgehog signaling pathway. Cell Cycle 2017, 16, 1181–1192. [Google Scholar] [CrossRef]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- He, S.-J.; Xiang, C.-Q.; Zhang, Y.; Lu, X.-T.; Chen, H.-W.; Xiong, L.-X. Recent progress on the effects of microRNAs and natural products on tumor epithelial–mesenchymal transition. Oncotargets Ther. 2017, 10, 3435–3451. [Google Scholar] [CrossRef]
- Yoshida, K.; Toden, S.; Ravindranathan, P.; Han, H.; Goel, A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis 2017, 38, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Howail, H.A.; Hakami, H.A.; Al-Otaibi, B.; Al-Mazrou, A.; Daghestani, M.H.; Al-Jammaz, I.; Al-Khalaf, H.H.; Aboussekhra, A. PAC down-regulates estrogen receptor alpha and suppresses epithelial-to-mesenchymal transition in breast cancer cells. BMC Cancer 2016, 16, 540. [Google Scholar] [CrossRef] [PubMed]
- Al-Hujaily, E.M.; Mohamed, A.G.; Al-Sharif, I.; Youssef, K.M.; Manogaran, P.S.; Al-Otaibi, B.; Al-Haza’a, A.; Al-Jammaz, I.; Al-Hussein, K.; Aboussekhra, A. PAC, a novel curcumin analogue, has anti-breast cancer properties with higher efficiency on ER-negative cells. Breast Cancer Res. Treat. 2011, 128, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Al-Qasem, A.; Al-Howail, H.A.; Al-Swailem, M.; Al-Mazrou, A.; Al-Otaibi, B.; Al-Jammaz, I.; Al-Khalaf, H.H.; Aboussekhra, A. PAC exhibits potent anti-colon cancer properties through targeting cyclin D1 and suppressing epithelial-to-mesenchymal transition. Mol. Carcinog. 2016, 55, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.-M.; Xie, W.-Y.; Wang, H.-S.; Du, J.; Wu, B.-P.; Xu, W.; Liu, H.-F.; Xiao, P.; Liu, Z.-G.; Li, H.-Y.; et al. Curcumin combined with FAPαc vaccine elicits effective antitumor response by targeting indolamine-2,3-dioxygenase and inhibiting EMT induced by TNF-α in melanoma. Oncotarget 2015, 6, 25932–25942. [Google Scholar] [CrossRef] [PubMed]
- Toden, S.; Okugawa, Y.; Jascur, T.; Wodarz, D.; Komarova, N.L.; Buhrmann, C.; Shakibaei, M.; Boland, C.R.; Goel, A. Curcumin mediates chemosensitization to 5-fluorouracil through miRNA-induced suppression of epithelial-to-mesenchymal transition in chemoresistant colorectal cancer. Carcinogenesis 2015, 36, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves Vde, P.; Ortega, A.A.; Guimaraes, M.R.; Curylofo, F.A.; Rossa Junior, C.; Ribeiro, D.A.; Spolidorio, L.C. Chemopreventive activity of systemically administered curcumin on oral cancer in the 4-nitroquinoline 1-oxide model. J. Cell Biochem. 2015, 116, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, J.; Ma, Q.; Li, B.; Han, L.; Liu, J.; Xu, Q.; Duan, W.; Yu, S.; Wang, F.; et al. Resveratrol inhibits the epithelial-mesenchymal transition of pancreatic cancer cells via suppression of the PI-3K/Akt/NF-kappaB pathway. Curr. Med. Chem. 2013, 20, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-H.; Sethi, G.; Um, J.-Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Tang, L.; Chen, H.; Wu, C.; Zhao, M.; Yang, Y.; Chen, X.; Liu, G. Resveratrol inhibits TGF-beta1-induced epithelial-to-mesenchymal transition and suppresses lung cancer invasion and metastasis. Toxicology 2013, 303, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.A.; Lin, C.H.; Chi, W.H.; Wang, C.Y.; Hsieh, Y.T.; Wei, Y.H.; Chen, Y.J. Resveratrol Impedes the Stemness, Epithelial-Mesenchymal Transition, and Metabolic Reprogramming of Cancer Stem Cells in Nasopharyngeal Carcinoma through p53 Activation. Evid. Based Complement. Altern. Med. 2013, 2013, 590393. [Google Scholar] [CrossRef] [PubMed]
- Karimi Dermani, F.; Saidijam, M.; Amini, R.; Mahdavinezhad, A.; Heydari, K.; Najafi, R. Resveratrol Inhibits Proliferation, Invasion, and Epithelial-Mesenchymal Transition by Increasing miR-200c Expression in HCT-116 Colorectal Cancer Cells. J. Cell Biochem. 2017, 118, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.W.; Tsai, L.L.; Yu, C.H.; Chen, P.N.; Chou, M.Y.; Yu, C.C. Impairment of tumor-initiating stem-like property and reversal of epithelial-mesenchymal transdifferentiation in head and neck cancer by resveratrol treatment. Mol. Nutr. Food Res. 2012, 56, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Valente, C.M.; Tinelli, A.; Siciliano, C.; Lorusso, V.; Acierno, R.; Giovinazzo, G.; Santino, A.; Storelli, C.; Maffia, M. Resveratrol inhibits the epidermal growth factor-induced epithelial mesenchymal transition in MCF-7 cells. Cancer Lett. 2011, 310, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Moshiri, A.; Puppo, M.; Rossi, M.; Gherzi, R.; Briata, P. Resveratrol limits epithelial to mesenchymal transition through modulation of KHSRP/hnRNPA1-dependent alternative splicing in mammary gland cells. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kohno, N.; Kondo, K.; Yokoyama, A.; Hamada, H.; Hiwada, K.; Miyake, M. Adriamycin activates E-cadherin-mediated cell-cell adhesion in human breast cancer cells. Int. J. Oncol. 1999, 15, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Wei, Y.; Liu, Y.; Lu, X.; Ding, F.; Wang, J.; Yang, S. Resveratrol promotes sensitization to Doxorubicin by inhibiting epithelial-mesenchymal transition and modulating SIRT1/beta-catenin signaling pathway in breast cancer. Cancer Med. 2019, 8, 1246–1257. [Google Scholar] [CrossRef]
- Venkatadri, R.; Iyer, A.K.V.; Kaushik, V.; Azad, N. A novel resveratrol-salinomycin combination sensitizes ER-positive breast cancer cells to apoptosis. Pharm. Rep. 2017, 69, 788–797. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, W.; Lim, S.O.; Hsu, J.L.; Huo, L.; Wu, Y.; Li, L.Y.; Lai, C.C.; Chang, S.S.; Hsu, Y.H.; et al. AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation. Cancer Res. 2016, 76, 1451–1462. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Xie, Q.; Chen, Z.; Ni, H.; Xia, L.; Zhao, Q.; Chen, Z.; Chen, P. Resveratrol suppresses the invasion and migration of human gastric cancer cells via inhibition of MALAT1-mediated epithelial-to-mesenchymal transition. Exp. Med. 2019, 17, 1569–1578. [Google Scholar] [CrossRef]
- Kim, S.E.; Shin, S.H.; Lee, J.Y.; Kim, C.H.; Chung, I.K.; Kang, H.M.; Park, H.R.; Park, B.S.; Kim, I.R. Resveratrol Induces Mitochondrial Apoptosis and Inhibits Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma Cells. Nutr. Cancer 2018, 70, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; De Domenico, S.; Tinelli, A.; Stanca, E.; Del Mercato, L.L.; Giudetti, A.M.; Simeone, P.; Guazzelli, N.; Lessi, M.; Manzini, C.; et al. Anticancer effects of novel resveratrol analogues on human ovarian cancer cells. Mol. Biosyst. 2017, 13, 1131–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Liu, D.; Niu, H.; Zhu, G.; Xu, Y.; Ye, D.; Li, J.; Zhang, Q. Resveratrol reverses Doxorubicin resistance by inhibiting epithelial-mesenchymal transition (EMT) through modulating PTEN/Akt signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2017, 36, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilibrasi, C.; Riva, G.; Romano, G.; Cadamuro, M.; Bazzoni, R.; Butta, V.; Paoletta, L.; Dalpra, L.; Strazzabosco, M.; Lavitrano, M.; et al. Resveratrol Impairs Glioma Stem Cells Proliferation and Motility by Modulating the Wnt Signaling Pathway. PLoS ONE 2017, 12, e0169854. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Hwang, K.A.; Choi, K.C. Anti-metastatic potential of resveratrol and its metabolites by the inhibition of epithelial-mesenchymal transition, migration, and invasion of malignant cancer cells. Phytomedicine 2016, 23, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J.; Debnath, T. Role of phytochemicals in the inhibition of epithelial-mesenchymal transition in cancer metastasis. Food Funct. 2016, 7, 3677–3685. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.A.; Hwang, K.A.; Choi, K.C. Roles of Dietary Phytoestrogens on the Regulation of Epithelial-Mesenchymal Transition in Diverse Cancer Metastasis. Toxins 2016, 8, 162. [Google Scholar] [CrossRef]
- Buhrmann, C.; Shayan, P.; Kraehe, P.; Popper, B.; Goel, A.; Shakibaei, M. Resveratrol induces chemosensitization to 5-fluorouracil through up-regulation of intercellular junctions, Epithelial-to-mesenchymal transition and apoptosis in colorectal cancer. Biochem. Pharm. 2015, 98, 51–68. [Google Scholar] [CrossRef]
- Ji, Q.; Liu, X.; Han, Z.; Zhou, L.; Sui, H.; Yan, L.; Jiang, H.; Ren, J.; Cai, J.; Li, Q. Resveratrol suppresses epithelial-to-mesenchymal transition in colorectal cancer through TGF-beta1/Smads signaling pathway mediated Snail/E-cadherin expression. BMC Cancer 2015, 15, 97. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, H.; Liu, M.; Lin, F.; Hua, J. Resveratrol abrogates the effects of hypoxia on cell proliferation, invasion and EMT in osteosarcoma cells through downregulation of the HIF-1alpha protein. Mol. Med. Rep. 2015, 11, 1975–1981. [Google Scholar] [CrossRef]
- Li, J.; Chong, T.; Wang, Z.; Chen, H.; Li, H.; Cao, J.; Zhang, P.; Li, H. A novel anticancer effect of resveratrol: Reversal of epithelialmesenchymal transition in prostate cancer cells. Mol. Med. Rep. 2014, 10, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Baribeau, S.; Chaudhry, P.; Parent, S.; Asselin, E. Resveratrol inhibits cisplatin-induced epithelial-to-mesenchymal transition in ovarian cancer cell lines. PLoS ONE 2014, 9, e86987. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Patel, S.; Imran, M.; Maalik, A.; Arshad, M.U.; Saeed, F.; Mabkhot, Y.N.; Al-Showiman, S.S.; Ahmad, N.; Elsharkawy, E. Honokiol: An anticancer lignan. Biomed. Pharm. 2018, 107, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Shanmugam, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Ahn, K.S.; Kumar, A.P.; Sethi, G. Honokiol inhibits signal transducer and activator of transcription-3 signaling, proliferation, and survival of hepatocellular carcinoma cells via the protein tyrosine phosphatase SHP-1. J. Cell. Physiol. 2012, 227, 2184–2195. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.S.; Tsai, C.H.; Hsieh, M.S.; Tsai, S.C.; Jan, Y.J.; Lin, W.Y.; Lai, D.W.; Wu, S.M.; Hsing, H.Y.; Arbiser, J.L.; et al. Exploiting Honokiol-induced ER stress CHOP activation inhibits the growth and metastasis of melanoma by suppressing the MITF and beta-catenin pathways. Cancer Lett. 2019, 442, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zhou, Y.; Jin, Y.; He, C.; Deng, Y.; Han, S.; Zhou, C.; Li, X.; Zhou, Y.; Liu, Y. Synergistically Enhanced Antimetastasis Effects by Honokiol-Loaded pH-Sensitive Polymer-Doxorubicin Conjugate Micelles. ACS Appl. Mater. Interfaces 2018, 10, 18585–18600. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, F.; Huang, R.; Yan, J.; Shen, B. Honokiol inhibits bladder cancer cell invasion through repressing SRC-3 expression and epithelial-mesenchymal transition. Oncol. Lett. 2017, 14, 4294–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.Q.; Qiao, X.R.; Su, L.; Chen, S.Z. Honokiol inhibits EMT-mediated motility and migration of human non-small cell lung cancer cells in vitro by targeting c-FLIP. Acta Pharm. Sin. 2016, 37, 1574–1586. [Google Scholar] [CrossRef]
- Avtanski, D.B.; Nagalingam, A.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol inhibits epithelial-mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E-cadherin axis. Mol. Oncol. 2014, 8, 565–580. [Google Scholar] [CrossRef]
- Avtanski, D.B.; Nagalingam, A.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol activates LKB1-miR-34a axis and antagonizes the oncogenic actions of leptin in breast cancer. Oncotarget 2015, 6, 29947–29962. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.H.; Lee, W.J.; Lai, D.W.; Wu, S.M.; Liu, C.Y.; Tien, H.R.; Chiu, C.S.; Peng, Y.C.; Jan, Y.J.; Chao, T.H.; et al. Honokiol confers immunogenicity by dictating calreticulin exposure, activating ER stress and inhibiting epithelial-to-mesenchymal transition. Mol. Oncol. 2015, 9, 834–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H.C.; Lai, D.W.; Lan, K.H.; Shen, C.C.; Wu, S.M.; Chiu, C.S.; Wang, K.B.; Sheu, M.L. Honokiol thwarts gastric tumor growth and peritoneal dissemination by inhibiting Tpl2 in an orthotopic model. Carcinogenesis 2013, 34, 2568–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wang, Q.; Su, Q.; Ma, D.; An, C.; Ma, L.; Liang, H. Honokiol suppresses renal cancer cells’ metastasis via dual-blocking epithelial-mesenchymal transition and cancer stem cell properties through modulating miR-141/ZEB2 signaling. Mol. Cells 2014, 37, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Duan, X.; Yang, G.; Zhang, X.; Deng, L.; Zheng, H.; Deng, C.; Wen, J.; Wang, N.; Peng, C. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS ONE 2011, 6, e18490. [Google Scholar] [CrossRef] [PubMed]
- Joo, Y.N.; Eun, S.Y.; Park, S.W.; Lee, J.H.; Chang, K.C.; Kim, H.J. Honokiol inhibits U87MG human glioblastoma cell invasion through endothelial cells by regulating membrane permeability and the epithelial-mesenchymal transition. Int. J. Oncol. 2014, 44, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Mereles, D.; Hunstein, W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int. J. Mol. Sci. 2011, 12, 5592–5603. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.D.; Chen, S.H.; Lin, C.L.; Tsai, S.H.; Liang, Y.C. Inhibition of melanoma growth and metastasis by combination with (−)-epigallocatechin-3-gallate and dacarbazine in mice. J. Cell. Biochem. 2001, 83, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.D.; Ellis, L.M. Inhibition of tumour invasion and angiogenesis by epigallocatechin gallate (EGCG), a major component of green tea. Int. J. Exp. Pathol. 2001, 82, 309–316. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, Y.; Liu, H.; Li, J. Inhibition of invasion and up-regulation of E-cadherin expression in human malignant melanoma cell line A375 by (-)-epigallocatechin-3-gallate. J. Huazhong Univ. Sci. Technol. Med. Sci. 2008, 28, 356. [Google Scholar] [CrossRef]
- Ju, J.; Hong, J.; Zhou, J.-n.; Pan, Z.; Bose, M.; Liao, J.; Yang, G.-Y.; Liu, Y.Y.; Hou, Z.; Lin, Y. Inhibition of intestinal tumorigenesis in Apcmin/+ mice by (−)-epigallocatechin-3-gallate, the major catechin in green tea. Cancer Res. 2005, 65, 10623–10631. [Google Scholar] [CrossRef]
- Rieger-Christ, K.M.; Hanley, R.; Lodowsky, C.; Bernier, T.; Vemulapalli, P.; Roth, M.; Kim, J.; Yee, A.S.; Le, S.M.; Marie, P.J. The green tea compound,(−)-epigallocatechin-3-gallate downregulates N-cadherin and suppresses migration of bladder carcinoma cells. J. Cell. Biochem. 2007, 102, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Lei, Y.; Chen, J.; Zhou, X. Potential ameliorative effects of epigallocatechin-3-gallate against testosterone-induced benign prostatic hyperplasia and fibrosis in rats. Int. Immunopharmacol. 2018, 64, 162–169. [Google Scholar] [CrossRef]
- Jiang, P.; Xu, C.; Chen, L.; Chen, A.; Wu, X.; Zhou, M.; Haq, I.U.; Mariyam, Z.; Feng, Q. Epigallocatechin-3-gallate inhibited cancer stem cell–like properties by targeting hsa-mir-485-5p/RXRα in lung cancer. J. Cell. Biochem. 2018, 119, 8623–8635. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Feng, H.; Ling, M.-T.; Chu, Q.; Tsao, S.W.; Wang, X.; Wong, Y.C. A novel anticancer effect of garlic derivatives: Inhibition of cancer cell invasion through restoration of E-cadherin expression. Carcinogenesis 2006, 27, 2180–2189. [Google Scholar] [CrossRef]
- Howard, E.W.; Ling, M.-T.; Chua, C.W.; Cheung, H.W.; Wang, X.; Wong, Y.C. Garlic-derived S-allylmercaptocysteine is a novel in vivo antimetastatic agent for androgen-independent prostate cancer. Clin. Cancer Res. 2007, 13, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Liu, L.-J.; Yan, Y.-M.; Fu, R.; Li, Y.; Xu, Y.; Cheng, Y.-X.; Wu, Z.-Q. Discovery of a natural small-molecule compound that suppresses tumor EMT, stemness and metastasis by inhibiting TGFβ/BMP signaling in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 134. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, B.; Li, X.Z.; Qi, L.F.; Liang, Y.Z. Preparative separation and purification of liensinine, isoliensinine and neferine from seed embryo of Nelumbo nucifera GAERTN using high-speed counter-current chromatography. J. Sep. Sci. 2009, 32, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Asokan, S.M.; Mariappan, R.; Muthusamy, S.; Velmurugan, B.K. Pharmacological benefits of neferine-A comprehensive review. Life Sci. 2018, 199, 60–70. [Google Scholar] [CrossRef]
- Deng, G.; Zeng, S.; Ma, J.; Zhang, Y.; Qu, Y.; Han, Y.; Yin, L.; Cai, C.; Guo, C.; Shen, H. The anti-tumor activities of Neferine on cell invasion and oxaliplatin sensitivity regulated by EMT via Snail signaling in hepatocellular carcinoma. Sci. Rep. 2017, 7, 41616. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Kong, S.; Song, F.; Li, L.; Jiang, H. Pharmacokinetic study of luteolin, apigenin, chrysoeriol and diosmetin after oral administration of Flos Chrysanthemi extract in rats. Fitoterapia 2012, 83, 1616–1622. [Google Scholar] [CrossRef]
- Lim, S.H.; Jung, S.K.; Byun, S.; Lee, E.J.; Hwang, J.A.; Seo, S.G.; Kim, Y.A.; Yu, J.G.; Lee, K.W.; Lee, H.J. Luteolin suppresses UVB-induced photoageing by targeting JNK1 and p90RSK2. J. Cell. Mol. Med. 2013, 17, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharmacother. 2019, 112, 108612. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yan, B.; Hu, X.; Li, X.-B.; Zhang, J.; Fang, J. Luteolin inhibits invasion of prostate cancer PC3 cells through E-cadherin. Mol. Cancer Ther. 2009, 8, 1684–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.; Ge, R.; Li, Y.; Liu, S. Luteolin exhibits anti-breast cancer property through up-regulating miR-203. Artif. CellsNanomed. Biotechnol. 2019, 47, 3265–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvi, R.B.; Swaminathan, A.; Chatterjee, S.; Shanmugam, M.K.; Li, F.; Ramakrishnan, G.B.; Siveen, K.S.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; et al. Inhibition of p300 lysine acetyltransferase activity by luteolin reduces tumor growth in head and neck squamous cell carcinoma (HNSCC) xenograft mouse model. Oncotarget 2015, 6, 43806–43818. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Fan, Z.-y.; Wang, H.-x.; Zhu, Z.-l.; Cao, S.; Wu, X.-y.; Li, J.-f.; Su, L.-p.; Li, C.; Zhu, Z.-g. Luteolin suppresses gastric cancer progression by reversing epithelial-mesenchymal transition via suppression of the Notch signaling pathway. J. Transl. Med. 2017, 15, 52. [Google Scholar]
- Li, C.; Wang, Q.; Shen, S.; Wei, X.; Li, G. HIF-1α/VEGF signaling-mediated epithelial–mesenchymal transition and angiogenesis is critically involved in anti-metastasis effect of luteolin in melanoma cells. Phytother. Res. 2019, 33, 798–807. [Google Scholar] [CrossRef]
- Chen, K.-C.; Chen, C.-Y.; Lin, C.-J.; Yang, T.-Y.; Chen, T.-H.; Wu, L.-C.; Wu, C.-C. Luteolin attenuates TGF-β1-induced epithelial–mesenchymal transition of lung cancer cells by interfering in the PI3K/Akt–NF-κB–Snail pathway. Life Sci. 2013, 93, 924–933. [Google Scholar] [CrossRef]
- Devi, K.P.; Rajavel, T.; Nabavi, S.F.; Setzer, W.N.; Ahmadi, A.; Mansouri, K.; Nabavi, S.M. Hesperidin: A promising anticancer agent from nature. Ind. Crop. Prod. 2015, 76, 582–589. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Shah, Z.A.; Saeed, F.; Imran, A.; Arshad, M.U.; Ahmad, B.; Bawazeer, S.; Atif, M.; Peters, D.G. Chemo-preventive and therapeutic effect of the dietary flavonoid kaempferol: A comprehensive review. Phytother. Res. 2019, 33, 263–275. [Google Scholar] [CrossRef]
- Luo, H.; Rankin, G.O.; Liu, L.; Daddysman, M.K.; Jiang, B.-H.; Chen, Y.C. Kaempferol inhibits angiogenesis and VEGF expression through both HIF dependent and independent pathways in human ovarian cancer cells. Nutr. Cancer 2009, 61, 554–563. [Google Scholar] [CrossRef]
- Jo, E.; Park, S.J.; Choi, Y.S.; Jeon, W.-K.; Kim, B.-C. Kaempferol Suppresses Transforming Growth Factor-β1-Induced Epithelial-to-Mesenchymal Transition and Migration of A549 Lung Cancer Cells by Inhibiting Akt1-Mediated Phosphorylation of Smad3 at Threonine-179. Neoplasia 2015, 17, 525–537. [Google Scholar] [CrossRef]
- Liang, S.; Marti, T.; Dorn, P.; Froment, L.; Hall, S.; Berezowska, S.; Kocher, G.; Schmid, R.; Peng, R. Blocking the epithelial-to-mesenchymal transition pathway abrogates resistance to anti-folate chemotherapy in lung cancer. Cell Death Dis. 2015, 6, e1824. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Ferlini, C.; Raspaglio, G.; Mozzetti, S.; Distefano, M.; Filippetti, F.; Martinelli, E.; Ferrandina, G.; Gallo, D.; Ranelletti, F.O.; Scambia, G. Bcl-2 Down-Regulation Is a Novel Mechanism of Paclitaxel Resistance. Mol. Pharmacol. 2003, 64, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Barbuti, A.M.; Chen, Z.-S. Paclitaxel Through the Ages of Anticancer Therapy: Exploring Its Role in Chemoresistance and Radiation Therapy. Cancers 2015, 7, 2360–2371. [Google Scholar] [CrossRef]
- Avendano, C.; Menendez, J.C. Chapter 8−Anticancer Drugs Targeting Tubulin and Microtubules. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Oxford, UK, 2008; pp. 229–249. [Google Scholar]
- Butler, M.S.; Robertson, A.A.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef]
- Gao, S.; Hu, M. Bioavailability challenges associated with development of anti-cancer phenolics. Mini. Rev. Med. Chem. 2010, 10, 550–567. [Google Scholar] [CrossRef]
- Lv, P.; Huang, X.; Lv, Q. Advances in studies on absorption, distribution, metabolism of flavonoids. China J. Chin. Mater. Med. 2007, 32, 1961–1964. [Google Scholar]
- Sun, M.; Su, X.; Ding, B.; He, X.; Liu, X.; Yu, A.; Lou, H.; Zhai, G. Advances in nanotechnology-based delivery systems for curcumin. Nanomedicine 2012, 7, 1085–1100. [Google Scholar] [CrossRef]
- Manach, C.; Donovan, J.L. Pharmacokinetics and metabolism of dietary flavonoids in humans. Free Radic. Res. 2004, 38, 771–786. [Google Scholar] [CrossRef]
- Mohanty, C.; Das, M.; Sahoo, S.K. Emerging role of nanocarriers to increase the solubility and bioavailability of curcumin. Expert Opin. Drug Deliv. 2012, 9, 1347–1364. [Google Scholar] [CrossRef]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef]
- Shen, T.; Jiang, T.; Long, M.; Chen, J.; Ren, D.-M.; Wong, P.K.; Chapman, E.; Zhou, B.; Zhang, D.D. A curcumin derivative that inhibits vinyl carbamate-induced lung carcinogenesis via activation of the Nrf2 protective response. Antioxid. Redox Signal. 2015, 23, 651–664. [Google Scholar] [CrossRef]
- Bukhari, S.N.A.; Jantan, I.B.; Jasamai, M.; Ahmad, W.; Amjad, M.W.B. Synthesis and biological evaluation of curcumin analogues. J. Med. Sci. 2013, 13, 501–513. [Google Scholar] [CrossRef]
- Wenzel, E.; Somoza, V. Metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 2005, 49, 472–481. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Chimento, A.; De Amicis, F.; Sirianni, R.; Sinicropi, M.S.; Puoci, F.; Casaburi, I.; Saturnino, C.; Pezzi, V. Progress to Improve Oral Bioavailability and Beneficial Effects of Resveratrol. Int. J. Mol. Sci. 2019, 20, 1381. [Google Scholar] [CrossRef]
- Jain, N.; Jain, R.; Thakur, N.; Gupta, B.P.; Jain, D.K.; Banveer, J.; Jain, S. Nanotechnology: A safe and effective drug delivery system. Asian J. Pharm. Clin. Res. 2010, 3, 159–165. [Google Scholar]
- Khalil, N.M.; Nascimento, T.C.F.d.; Casa, D.M.; Dalmolin, L.F.; Mattos, A.C.d.; Hoss, I.; Romano, M.A.; Mainardes, R.M. Pharmacokinetics of curcumin-loaded PLGA and PLGA–PEG blend nanoparticles after oral administration in rats. Colloids Surf. B Biointerfaces 2013, 101, 353–360. [Google Scholar] [CrossRef]
- Umerska, A.; Gaucher, C.; Oyarzun-Ampuero, F.; Fries-Raeth, I.; Colin, F.; Villamizar-Sarmiento, M.; Maincent, P.; Sapin-Minet, A. Polymeric nanoparticles for increasing oral bioavailability of curcumin. Antioxidants 2018, 7, 46. [Google Scholar] [CrossRef]
- Thipe, V.C.; Panjtan Amiri, K.; Bloebaum, P.; Raphael Karikachery, A.; Khoobchandani, M.; Katti, K.K.; Jurisson, S.S.; Katti, K.V. Development of resveratrol-conjugated gold nanoparticles: Interrelationship of increased resveratrol corona on anti-tumor efficacy against breast, pancreatic and prostate cancers. Int. J. Nanomed. 2019, 14, 4413–4428. [Google Scholar] [CrossRef]
- Park, S.Y.; Chae, S.Y.; Park, J.O.; Lee, K.J.; Park, G. Gold-conjugated resveratrol nanoparticles attenuate the invasion and MMP-9 and COX-2 expression in breast cancer cells. Oncol. Rep. 2016, 35, 3248–3256. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. https://doi.org/10.3390/cells8101118
Loh C-Y, Chai JY, Tang TF, Wong WF, Sethi G, Shanmugam MK, Chong PP, Looi CY. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells. 2019; 8(10):1118. https://doi.org/10.3390/cells8101118
Chicago/Turabian StyleLoh, Chin-Yap, Jian Yi Chai, Ting Fang Tang, Won Fen Wong, Gautam Sethi, Muthu Kumaraswamy Shanmugam, Pei Pei Chong, and Chung Yeng Looi. 2019. "The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges" Cells 8, no. 10: 1118. https://doi.org/10.3390/cells8101118