
CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting


1
Innovative Center for Aging Research, Yeungnam University Medical Center, Daegu 42415, Korea
2
Yeungnam University College of Medicine, Daegu 42415, Korea
*
Authors to whom correspondence should be addressed.
†
These authors equally contributed to this work.
Cells 2021, 10(7), 1833; https://doi.org/10.3390/cells10071833
Submission received: 30 April 2021
/
Revised: 14 June 2021
/
Accepted: 17 July 2021
/
Published: 20 July 2021
(This article belongs to the Special Issue New Insights into Oxidative Stress and Inflammation in Diabetes)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The cluster of differentiation 36 (CD36) is a scavenger receptor present on various types of cells and has multiple biological functions that may be important in inflammation and in the pathogenesis of metabolic diseases, including diabetes. Here, we consider recent insights into how the CD36 response becomes deregulated under metabolic conditions, as well as the therapeutic benefits of CD36 inhibition, which may provide clues for developing strategies aimed at the treatment or prevention of diabetes associated with metabolic diseases. To facilitate this process further, it is important to pinpoint regulatory mechanisms that are relevant under physiological and pathological conditions. In particular, understanding the mechanisms involved in dictating specific CD36 downstream cellular outcomes will aid in the discovery of potent compounds that target specific CD36 downstream signaling cascades.
1. Introduction
CD36 is a transmembrane glycoprotein found in platelets, mononuclear phagocytes, adipocytes, hepatocytes, myocytes, taste bud cells, and a variety of other cell types. It plays a role in lipid accumulation, inflammatory signaling, energy reprogramming, oxidative stress, and apoptosis, all of which contribute to metabolic dysfunction [1,2,3,4,5]. Many physiological and pathological factors, such as long-chain fatty acids and proteins containing thrombospondin structural homology domains and oxidized phospholipids, including oxidized LDL (oxLDL), perturb CD36 function and induce metabolic diseases [6]. The scavenger receptor functions of CD36 have been widely researched due to the possibility of CD36 ligands in disease etiology. For example, higher sCD36, a non-cell-bound CD36 found in human plasma that indirectly reflects CD36 expression in tissues, has been linked to obesity, insulin resistance, and diabetes according to recent community-based cohort research [7,8,9]. Yang et al. also found that inhibiting the integral membrane protein CD36 with pharmacological agents lowers body weight growth and improves glucose tolerance [10]. Furthermore, a rise in CD36 levels contributes to the advancement of obesity-related metabolic dysfunctions by increasing lipid accumulation and inflammation. These studies also showed that CD36 is a critical player in the development of obesity and type 2 diabetes caused by a high-fat diet [11]. Despite the fact that the pathophysiology of these metabolic illnesses is complicated, there is evidence that CD36 is implicated in the abnormal signaling and tissue damage observed in this context. Since CD36 is expressed in a variety of tissues, it is impossible to discuss every aspect within the scope of this review. In this review, we look at the molecular links between CD36 and metabolic disease in a broader framework, focusing on pancreatic β-cells and other cell systems during the progression of metabolic disorders.
2. Comment on CD36-Assisted Fatty Acid Uptake
The cellular fatty acid uptake rate is determined by the presence of CD36 at the cell surface, which is regulated by subcellular vesicular recycling from endosomes to the sarcolemma (lipid raft). More specifically, VAMP2, a vesicle-associated membrane protein (VAMP) isoform, participates in CD36 translocation to the plasma membrane upon stimulation with insulin [12,13,14,15]. Upon removal of insulin, CD36 is rapidly internalized, leading to a concomitant decrease in the fatty acid uptake rate. Under normal conditions, the endosomal lumen is slightly acidic (pH ~5.5), which allows optimal endosomal retention of CD36 and concomitant low rates of fatty acid uptake under non-challenging conditions. Moreover, protein palmitoylation governs several steps of the insulin cascade and consequently contributes to the regulation of substrate transporter recycling between intracellular compartments and the plasma membrane. However, palmitoylation of VAMP2 might regulate the membrane localization of VAMP2, and the inhibition of palmitoylation has been shown to decrease glucose uptake in adipocytes [16]. Given the well-established relationship between palmitoylation and protein targeting of membranes, it is possible that the palmitoylation of VAMP2 enhances association with CD36, thus facilitating the arrangement of CD36 translocation to the cell surface. Most likely, lipid rafts/caveolae and their marker protein, caveolin-1, may play a crucial role in the post-translational stabilization of CD36 at the cell membrane surface. Caveolae represent a morphologically identifiable subset of lipid rafts, and caveolin-1 is essential for the formation of the characteristic flask-shaped invaginations of the plasma membrane [17]. The depletion of caveolin-1 or deletion of the caveolin-1 gene is known to result in the complete loss of caveolae and CD36 surface availability and thereby, the regulation of fatty acid uptake [18,19,20,21]. These results indicate the importance of intracellular signaling/trafficking events in CD36 function, as well as the metabolic fates of FAs following uptake. In addition to residing on the cell surface, CD36 localizes to intracellular vesicles and mitochondria, where it interacts with carnitine palmitoyl transferase 1, which is the key mitochondrial enzyme regulating FA transport into mitochondria and oxidation and is especially important for meeting increased metabolic demands during muscle contraction [22,23,24]. The detailed signaling pathways mediating CD36 trafficking are still unclear, but there is emerging evidence indicating a role for AMPK activation [25]. Independent of its physiological function, it is obvious that CD36 may participate in abnormal FA utilization and its deleterious consequences during insulin resistance and obesity (reviewed in Refs. [26,27,28,29,30,31]).
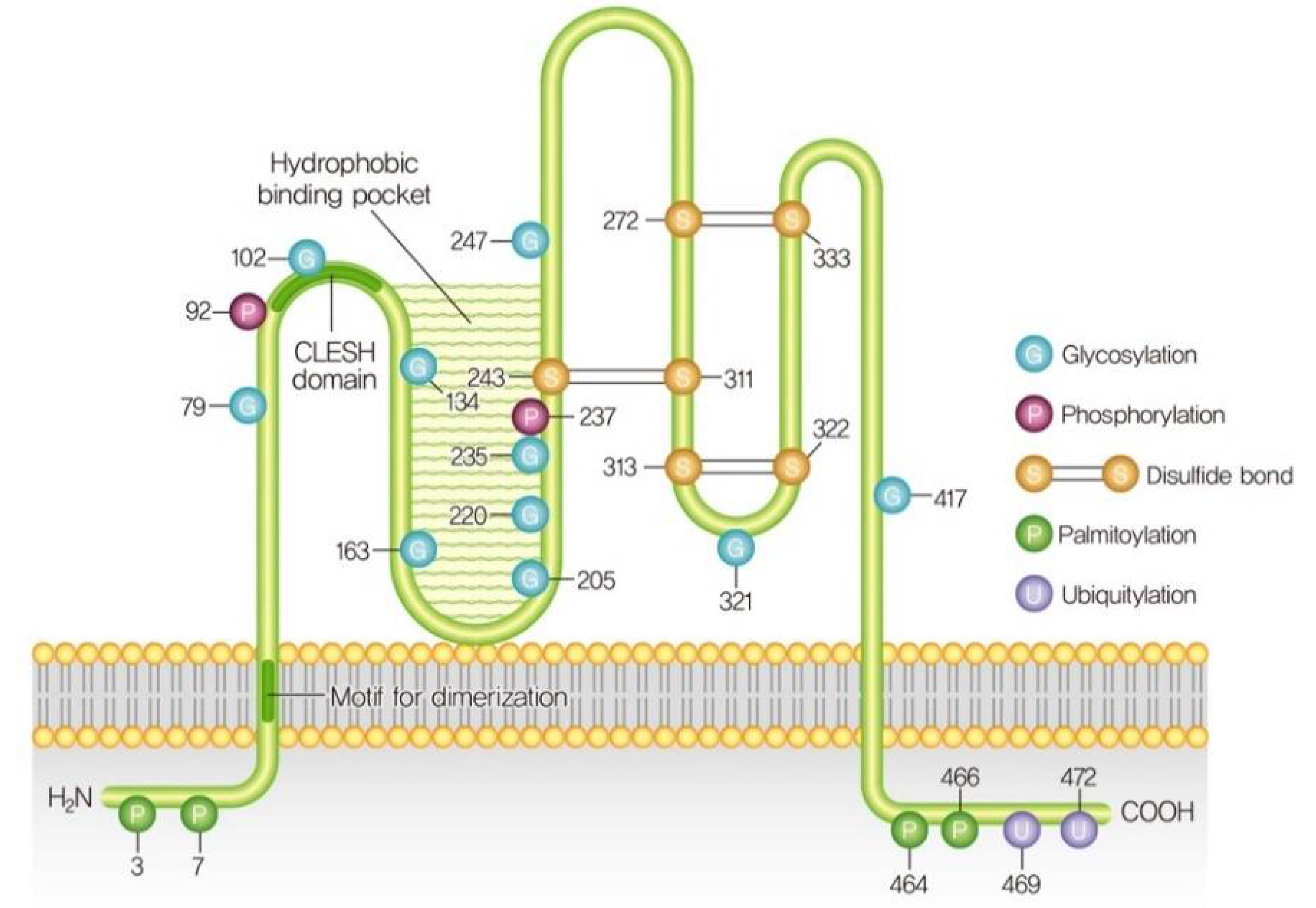
3. Structure and Post-Translational Modifications of CD36
CD36 is also known as fatty acid translocase (FAT), glycoprotein IIIb (GPIIIb), or platelet glycoprotein IV, PAS4, and SR-B2 [32]. The human CD36 gene consists of 472 amino acids with a molecular weight of 53 kDa and is located on chromosome 7 (7q11.2) [33]. Analysis of the amino acid sequence of CD36 predicts a hairpin-like configuration with two transmembrane domains and two short cytoplasmic tails at the N- and C-termini. The extracellular loop contains a large hydrophobic cavity responsible for the recognition of ligand binding to moieties such as advanced glycated end products, cholesterol, and fatty acids [34,35,36]. The ligand-binding site also contains multiple glycosylation sites and three disulfide bridges essential for the intracellular processing and targeting of the protein to the outer leaflet of the plasma membrane [37]. In contrast, partially glycosylated CD36 mutants were also distributed to the cell surface without alterations in ligand binding [38]. However, the regulatory mechanism of CD36 glycosylation that leads to fatty acid absorption or fatty acid transport is not known. Contrary to glycosylation, myocellular fatty acid uptake is enhanced by the O-GlcNAcylation of CD36 via translocation to the sarcolemma [39]. O-GlcNAcylation is the dynamic regulatory post-translational modification of proteins stimulated by the addition of O-linked β-N-acetyl glucosamine (O-GlcNAc) to serine and threonine residues on target proteins [40,41]. However, increased O-GlcNAcylation levels of CD36 promote high-fat uptake by gastric cancer cells, which is needed for their metastasis [42]. Increased CD36-O-GlcNAcylation has also been linked to the reprogramming of cardiac fatty acid and glucose metabolism during acute or chronic stresses [43], although further studies are necessary to define the alteration of metabolic reprogramming by CD36 O-GlcNAcylation. CD36 is also palmitoylated and depalmitoylated by palmitoyl-transferases (PATs) and palmitoyl-protein thioesterases respectively. CD36, N-, and C-terminal tails contain cysteine residues that are palmitoylated by PATs, which anchors the protein to the plasma membrane in a reversible process. Under palmitic acid stimulation, CD36 is palmitoylated by PATs in the endoplasmic reticulum (ER) in a reversible thioester linkage between palmitate and cysteine residues [44,45]. However, the inhibition of CD36 palmitoylation causes CD36 ubiquitylation and degradation in the ER [46]. CD36 has two polyubiquitylation Lys469 and Lys472 sites in the C-terminus. However, CD36 ubiquitination does not affect CD36 distribution between cell surfaces from intracellular storage compartments [47]. Recently, CD36 was found to be a target of ubiquitin-specific peptidase 10 (USP10), which is a mammalian deubiquitinase. USP10 directly stabilizes CD36 via deubiquitination, and the inhibition of USP10 increases the polyubiquitylation of CD36 and enhances its proteasomal degradation, leading to reduced CD36-mediated fatty acid uptake by macrophages [48]. CD36 can also be monoubiquitinated by the E3 ubiquitin-protein ligase Parkin, which warrants further investigation [49]. Notably, CD36 is also phosphorylated at Thr92 by protein kinase C (PKC) and Ser237 by protein kinase A (PKA) within the extracellular loop, which is linked to the inhibition of fatty acid uptake [50,51]. Along these lines, published evidence suggests that CD36 has four acetylation sites (Lys52, Lys166, Lys231, and Lys403), and the functional effects of these acetylations are not yet known [52]. Sulfosuccinimidyl oleate (SSO), an irreversible inhibitor of CD36, binds to CD36 at Lys164 via the formation of N-hydroxysuccinimidyl esters in the fatty acid-binding pocket, leading to the inhibition of fatty acid intake and/or fatty acid-induced signaling [53]. Thus, further investigations are needed to know the role of CD36 acetylation at Lys52, Lys166, Lys231, and Lys403 and its downstream signaling. Taken together, CD36 could be targeted by different post-translational modifications in different tissues in a context-dependent manner and have been incompletely resolved, which could be a topic for future studies (Figure 1).
4. Role of CD36 in Pancreatic β-Cell Pathophysiology
4.1. Glucotoxicity
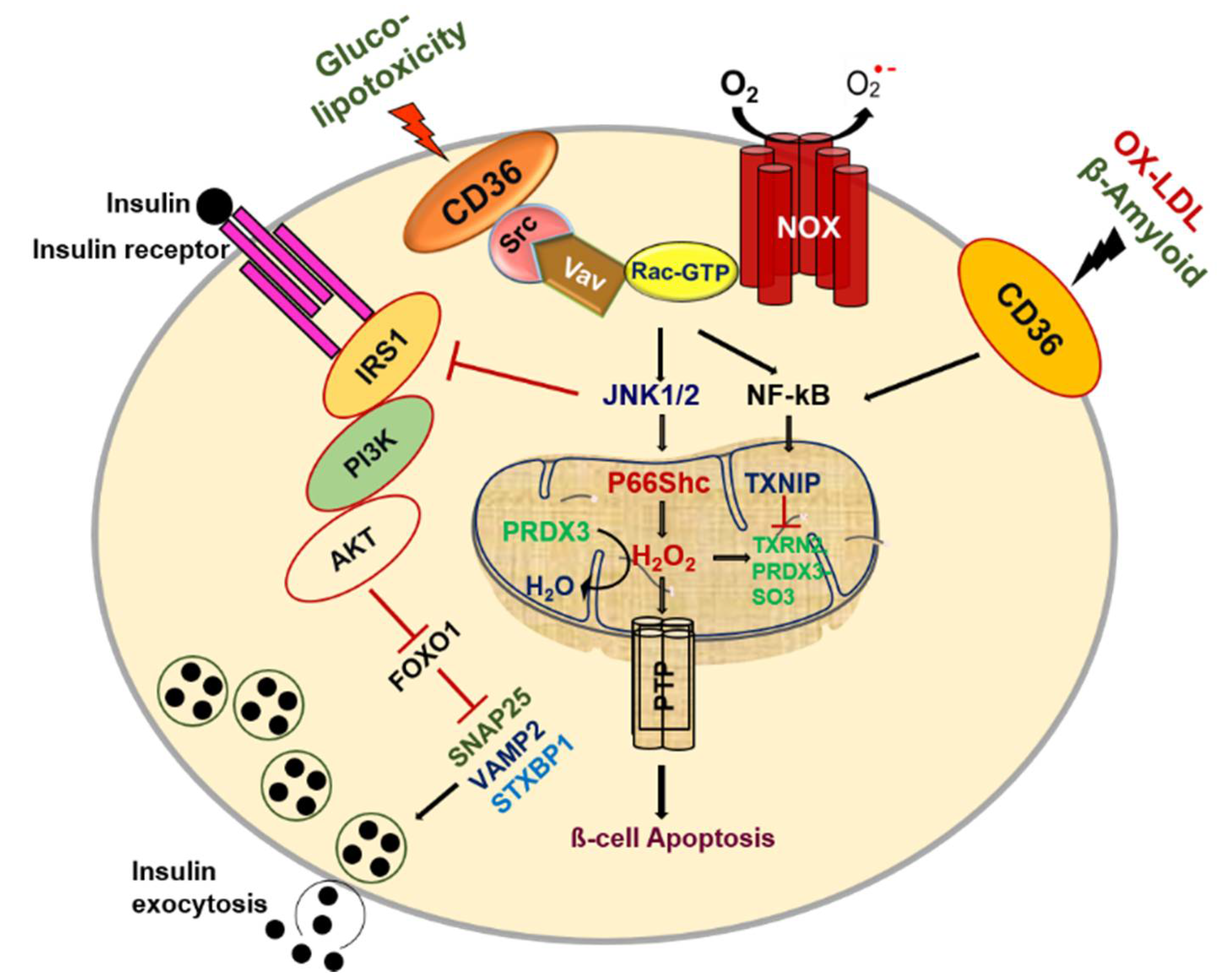
CD36 exerts fundamental biological functions at the cellular and tissue levels in multiple homeostatic and pathological processes by its distinct binding sites. Pancreatic β-cells play a central role in regulating glucose metabolism to sustain energy homeostasis by mediating a balance between insulin, an anabolic hormone, and glucagon, a catabolic hormone. Pancreatic β-cells require suitable sensors and signaling molecules that are integrated to modulate insulin secretion and maintain homeostasis. However, type 2 diabetes (T2D) is based on the inability of pancreatic β-cells to sustain a compensatory secretory response, leading to insulin secretory dysfunction and the pathogenesis of T2D. CD36 is the most generously expressed transporter among fatty acid transporters in human islets. It is located in the plasma membrane and co-localizes with insulin granules [54]. Interestingly, CD36 was shown to traffic between intracellular compartments and the cell surface in a vesicle-mediated process [55]. It has been well established that glucose potentiates fatty acid-induced β-cell death via apoptosis [56,57]. Similarly, the overexpression of CD36 in β-cells increases the uptake of fatty acids and leads to metabolic and functional dysfunction [58]. We also reported that glucotoxicity influences pancreatic β-cell dysfunction by increasing the influx of free fatty acids (FFAs) via CD36 [59]. To evaluate the mechanisms by which glucotoxicity affects β-cell dysfunction, we investigated CD36 expression and trafficking in β-cells and observed that Rac1, a small Rho family protein, displays increased glucose-mediated CD36 expression on the membrane surface in pancreatic β-cells [60]. The importance of Rac1 signaling in early-phase insulin secretion was previously demonstrated in β-cell-specific Rac1-deficient mice via the inhibition of F-actin depolymerization [61,62]. Glucose stimulates the recruitment of insulin granules to the cell membrane through actin remodeling, which is necessary for glucose-stimulated insulin secretion [63,64]. The F-actin function is coupled to SNARE-associated proteins such as syntaxin 1, syntaxin 4, and SNAP-25 in β-cells, and many F-actin-binding proteins interact with the SNARE machinery [63,65,66,67]. Several studies have shown that deficiencies in SNARE proteins are likely caused by high glucose and might contribute to cell dysfunction in disease states [68,69,70]. More considerations concerning SNARE protein function are discussed in the review article by Gaisano et al. [71]. Recently, it was shown that CD36 overexpression attenuates insulin secretion in human islets through the reduction of the exocytotic genes Snap25 and Vamp2, resulting in a decreased number of docked granules. It was further demonstrated that CD36 overexpression attenuates insulin signaling, resulting in the accumulation of the transcription factor FoxO1 in the nucleus as a potent transcriptional repressor of exocytotic genes. Interestingly, the inhibition of CD36 was shown to upregulate exocytotic gene expression in human islets, improving granule docking and resulting in increased insulin secretion without affecting insulin content [72]. Further research is required to identify the roles of CD36 in exocytotic gene function as well as whether F-actin function is involved in suppressing insulin secretion by CD36. Such studies will provide greater insights into the mechanisms of how CD36 induces metabolic dysfunction in pancreatic β-cells.
On the other hand, evidence suggests that hyperglycemia leads to the generation of reactive oxygen species (ROS), resulting in increased oxidative stress in β-cells [73,74]. The activation of Rac1 increases the production of oxidants, such as H2O2, via the activation of NADPH oxidase (NOX), which might trigger oxidative stress linked to β-cell death in T2D [75]. It was previously observed that CD36 deficiency reduces NOX activity and attenuates obesity-associated oxidative stress in the heart [76]. We also observed that Rac1 mediates NOX activity, leading to an increase in CD36 at the plasma membrane and that Rac1 and NOX inhibition can abrogate CD36 downstream signaling damage in response to high glucose [60]. However, it remains unknown how CD36 translocation to the plasma membrane is detected after Rac1-NOX activation by high glucose. One possible explanation may involve the palmitoylation of CD36 by supraphysiologic glucose levels. High-glucose-induced Rac1-palmitoylation has been suggested to be a driving force behind the activation of NOX, which in turn would alter the localization of Rac1 remodeling in diabetic retinopathy [77]. Nonetheless, a current topic of research is to elucidate how protein palmitoylation influences the function of proteins under high glucose conditions in pancreatic β-cells. It should be noted that increasing ROS production can alter cellular dysfunction stimulated by the activation of stress kinases by changing the balance of antioxidant enzymes. Previous findings also reported that CD36 altered cellular signaling under metabolic stress conditions by downregulating the redox-sensitive nuclear factor Nrf2 via Fyn kinase in murine vascular smooth muscle cells [78]. In addition, CD36 signaling in response to scavenger ligands leads to the activation of Src and MAPK family kinases, such as Lyn and c-Jun N-terminal kinase (JNK) in macrophages and platelets, whereas Fyn and p38 are the primary mediators of endothelial cells [79,80,81]. We also reported that Rac1-CD36 signaling by high-glucose-induced JNK and p38MAPK activation and the inhibition of CD36 inhibition blocks high-glucose-induced oxidative stress. Lots of evidence has suggested that ER stress is linked to insulin resistance, and pancreatic β-cell ER expansion was detected in patients with T2D [82,83]. Cells activate adaptive, self-protective mechanisms in response to ER stress, which are collectively referred to as the ER stress response (also named UPR). These include enhanced ER size, increased ER folding capacity through the manipulation of chaperones and foldases, decreased biosynthetic load, and the increased clearance of unfolded proteins through the stimulation of ER-related degradation. When these systems fail to alleviate the stress, apoptosis is triggered. Subsequent work from our lab has demonstrated that chronic glucose exposure or thapsigargin treatment induces ER stress through the reduced expression and activity of insulin and PDX1 with CD36 induction. Inhibition of CD36 in β-cells by metformin treatment or by using CD36 siRNA was shown to prevent the generation of ER stress markers and stress kinase activation [84]. It is clear that signaling related to CD36 regulation and its dynamics has impacts on oxidative stress, and understanding this linkage warrants further investigation. Given that CD36 signaling is related to numerous pathological events, β-cell CD36 downstream targets need to be further investigated.
4.2. Lipotoxicity
Hyperglycemia with elevated FFAs plays a significant role in insulin resistance and β-cell dysfunction in T2D [85,86,87]. Many studies have shown that glucose enhances fatty acid-induced β-cell death via apoptosis [56,57]. Fatty acid–glucose balance is essential for maintaining normal β-cell function, but lipotoxicity-induced β-cell dysfunction occurs with increased ROS, ceramide and nitric oxide levels, and mitochondrial perturbations [88,89]. Studies in Zucker diabetic fatty (ZDF) rats, an obesity-induced diabetic animal model, confirmed FFA-induced ceramide accumulation leading to β-cell apoptosis [90]. Another study demonstrated that superoxide production was elevated in islets isolated from Zucker lean fatty (ZLF) and Zucker diabetic fatty (ZDF) rats in the presence of glucose [91]. The resting superoxide content of ZDF rat islets was higher than that of Zucker lean control islets and was accompanied by the alteration of mitochondrial morphology. The FFA-induced formation of ceramide also induces the generation of ROS and DNA fragmentation [92]. Collectively, oxidative stress and mitochondrial dysfunction result in endogenous antioxidant impairment. However, plasma from patients with obesity and T2D shows enhanced levels of ceramides, which may serve as biomarkers for the diagnosis and treatment of obesity and diabetes [93,94,95]. Based on its role in FFA uptake, our lab showed that CD36 might promote ceramide-induced β-cell dysfunction by the Src-mediated tyrosine phosphorylation of Vav, a guanine nucleotide exchange factor (GEF), and also elevate metabolic pathways via its GEFs activity [96]. Evidence suggests that saturated fatty acids impair insulin secretion and induce insulin resistance via Src signaling in T2D [97]. Accordingly, we hypothesize that the induction of CD36 could be recapitulated in cells with functional vav tyrosine phosphorylation by Src promoting Rac1 signaling that generates ROS by NOX. Our results indicate that CD36-mediated Src-Vav activation is necessary for optimal Rac1-NADPH-induced superoxide production. To determine whether or not CD36 is linked to Vav-Rac1-NOX activation, we performed pharmacological inhibition of Src activity or CD36 siRNA, which significantly reduced ceramide-induced RAC1-NOX and inhibited ROS formation [98]. Holzer et al. demonstrated that saturated fatty acids stimulate stress-signaling activation by Src via transfer to a membrane micro domain [99]. In addition, the FFA-mediated Src-dependent Vav phosphorylation coordinates the engagement of Rac1-NOX-JNK signaling, which contributes to insulin resistance, obesity, and the production of inflammatory cytokines [100,101]. In podocytes, CD36-dependent uptake of palmitic acid leads to impaired mitochondrial energy metabolism, the alteration of mitochondrial and ER morphology, increased levels of mitochondrial ROS, the depolarization of mitochondria, ATP depletion, and apoptosis [102,103,104].
A recent study showed that p66Shc mediates lipotoxicity-induced impaired metabolic changes that promote pancreatic β-cell dysfunction and apoptosis in diabetes [74]. Earlier studies have suggested that p66Shc serine36 phosphorylation by JNK leads to ROS production and cell death [105]. Activated JNK combined with p66Shc serine36 phosphorylation activation to induce mitochondrial ROS in response to CD36 signaling promote cellular dysfunction. Cells lacking this pathway, as a consequence of CD36 inhibition, significantly block ceramide-induced β-cell dysfunction. Evidence points to CD36 signaling-generated H2O2, which promotes cysteine sulfenylation, a post-translational modification important to the augmentation of platelet activation and aggregation [106]. Peroxiredoxins, a thioredoxin-dependent peroxide reductase family of antioxidant proteins, catalyze the reduction of both hydrogen peroxide and alkyl peroxides to water and their corresponding alcohols [107,108]. The expression of peroxiredoxin-3 (PRDX3) is restricted to β-cells in pancreatic tissue. The oxidation of peroxidase cysteine to sulfonic acid (peroxiredoxin-SO3) promotes the accumulation of oxidized PRDX3 in mitochondria, which favors mitochondrial permeability transition pore (MPTP) opening and mitochondrial swelling [109]. Importantly, ceramide-induced sulfenylation is reduced in the presence of CD36 inhibition, which is consistent with a CD36-dependent mechanism. However, reduced PRDX3 is regulated by the thioredoxin–thioredoxin reductase system. Accordingly, we hypothesize that the inhibition of thioredoxin could be recapitulated in cells with functional thioredoxin-interacting protein (TXNIP) by preventing peroxiredoxin-3 activity in response to ceramide. We observed that TXNIP translocates to mitochondria and inhibits the antioxidative protein thioredoxin in response to ceramide. Moreover, ceramide-induced nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation has been shown to increase TXNIP expression in β-cells [110]. This finding suggests that CD36 plays an important role in the initiation of oxidative stress induced by ceramide under conditions of β-cell failure. Thus, exploration of CD36 warrants further investigation.
Under normal conditions, mitochondria in β-cells persistently undergo fusion and fission. These processes may function to refute the negative impacts of the long-term presentation of β-cells to palmitate under high glucose conditions, causing mitochondrial fragmentation and impeding network dynamics by abolishing fusion and fission activity [111]. There are two powerfully contradictory processes that determine mitochondrial shape and morphology: fusion and fission. The ablation of both fusion and fission produces a significant effect on the progression of cells to apoptosis [112]. It has been reported that the mitochondria of β-cells from Zucker diabetic rats are divided, suggesting an imbalance in the mitochondrial fusion and fission process [91]. The exposure of β-cells to high-fat glucose conditions causes the discharge of Ca2+ from the ER to the cytoplasm, driving a rise in the cytosolic Ca2+ concentration that reflects expanded mitochondrial Ca2+ uptake. Increased mitochondrial Ca2+ uptake improves local buffering capacity and the discharge of proteins competent in apoptosis induction. Hence, Ca2+ activates the phosphatase calcineurin, which dephosphorylates and inactivates dynamin-related protein 1 (Drp1), a master controller of mitochondrial fission [113]. It can be assumed that crosstalk between the ER and mitochondria may promote cellular commitment to apoptosis through Ca2+. Recently, the presence of inositol trisphosphate receptor (IP3Rs) has been implicated in proapoptotic Ca2+ transfer between the ER and mitochondria [114]. An important remark is that Akt restrains the ER-to-mitochondria Ca2+ exchange by means of IP3R3 and ensures against Ca2+ intervened apoptosis [115]. Interestingly, CD36 was found to be overexpressed in obese diabetic islets and suppressed the insulin-signaling PI3K/AKT pathway, as well as its downstream transcription factors [72]. These effects result in ER-mitochondrial reprogramming, which contributes to the development of β-cell death and failure. The different pathways enacted by CD36 require further confirmation of the precise roles of CD36 signaling pathways in β-cell failure.
4.3. OX-LDL and Amyloid Deposition
CD36 can generate cell-specific reactions to multiple ligands through the binding of context-specific binding partners that contribute to the development of β-cell dysfunction. As described above, it has been shown that ER stress is connected to insulin resistance in diabetes, and, conjointly, an expansion of ER was recognized in β-cells from patients with T2D [81,82]. Oxidized-LDL (oxLDL)-induced ER stress activation is coupled with oxidative stress, leading to β-cell dysfunction and death [116]. It has been shown that oxLDL induces β-cell dysfunction and apoptosis via the activation of ROS and that radical lipid hydroperoxides contribute to JNK activation [117,118,119]. However, the downstream mechanism by which JNK leads to apoptosis is not yet clear, and the crosslink between oxLDL and CD36 may promote cellular commitment to apoptosis through JNK enactment. A previous study reported that oxLDL intervenes with the JNK-dependent phosphorylation of p66Shc in endothelial cells, which contributes to oxidative stress and the atherogenic progression [120], Thus, we cannot preclude a role for PRDX3 oxidation in CD36 signaling. In this way, p66Shc can result in the overproduction of H2O2, which in turn can react with PRDX3 to cause toxic mitochondrial dysfunction and apoptosis.
Regarding the molecular mechanisms involved, CD36 overexpression partners with the increased uptake of oxLDL without exerting additive effects on oxLDL toxicity [121]. Evidence suggests that CD36 causes a mitochondrial metabolic switch from oxidative phosphorylation to superoxide generation in reaction to oxLDL, which subsequently promotes NF-κB activation and the generation of pro-inflammatory cytokines [122]. Hence, redox status is subordinate on the degree to which a cell’s components exist in an oxidative state, whereby a reducing environment inside cells can prevent oxidative stress. oxLDL also initiated the ASM/ceramide signaling pathway, which is involved in macrophage apoptosis via the ER stress pathway [123]. However, the downstream targets of oxLDL in β-cells are not well known, and additional studies are needed.
On the other hand, β-cells have a lower abundance of antioxidant defense enzymes, such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx) [124,125,126]. As such, the administration of antioxidant supplements can increase the defense capacity of islet cells to cope with oxidative stress [127]. Vitamin E is a redox-active natural compound that downregulates levels of ROS under different experimental conditions [128,129,130,131]. Interestingly, vitamin E reduces the uptake of OX-LDL by inhibiting CD36 expression via the PPARγ signaling pathway [132,133,134]. Furthermore, vitamin E facilitates the activation of PI3Kγ/AKT, leading to increased VEGF expression as well as elevation of cell survival and angiogenesis via its ability to increase tissue remodeling [135]. In addition, genetic data indicate that VEGF is a major regulator of islet vascularization and the revascularization of transplanted islets, and reduced beta-cell VEGF expression impairs glucose-stimulated insulin secretion [136]. Furthermore, the addition of vitamin E induces insulin secretion and islet-cell survival and functionality by enhancing PDX1, a master regulator of insulin gene expression [137,138]. Thus, the elevated expression of CD36 in beta cells exposed to elevated OX-LDL results in increased ROS expression that could induce discrete oxidative stress. Therefore, we suggest that the vitamin E-induced elevation of insulin expression may be mediated by CD36 inhibition, which may explain, at least in part, the reported protection against oxidative stress. Further studies are needed to elucidate the relationship between CD36, vitamin E, and OX-LDL in the pancreatic β-cell dysfunction.
Among the variety of proapoptotic factors present in pancreatic β-cells, islet amyloid polypeptide (IAPP) is thought to play a crucial role in β-cell apoptosis. The proapoptotic effects of IAPP are mediated through a complex sequence of signaling events that lead to defects in mitochondrial dysfunction, autophagy, local inflammation, oxidative stress, cytokine production, and the enactment of signaling pathways driving to apoptosis [139,140,141,142]. Interestingly, CD36 can create a solid pro-inflammatory reaction through its interaction with secreted amyloid-beta 1–42 (Aβ) in macrophages [143]. However, the presence of this CD36-dependent pro-inflammatory signaling hub within the pancreatic β-cells has not been studied and warrants further investigation (Figure 2).
5. Role of CD36 in Peripheral Insulin Resistance and Metainflammation
Insulin resistance could be a trademark of metabolic defects, and it paves the way for the development of T2D. Impaired glucose tolerance, alcoholism, smoking, hypercholesterolemia, hypertriglyceridemia, low HDL, and hypertension are some of the risk factors involved in insulin resistance [144,145]. Modifications of lipid metabolism upon chronic FA oversupply interceded by CD36 lead to the accumulation of specific lipid species that are especially critical for the development of insulin resistance [146,147]. Increasing evidence shows that dysfunctional adipose tissue is associated with insulin resistance. This mechanism could be related to the recruitment of macrophages and other immune cells, which aggravate systemic inflammation and ectopic fat accumulation [148,149,150]. Accumulating evidence suggests that insulin resistance is not only induced by fat accumulation in adipose tissues but also by pro-inflammation caused by ectopic fat toxic lipids, such as ceramides, which alter the insulin signaling pathway, generates ROS-induced ER stress, and induces inflammation during the development of T2D [151]. It has also been reported that CD36-dependent inflammation and the apoptosis of adipocytes in response to diet-induced obesity reduces insulin sensitivity [152]. These changes are related to the activation of stress signaling pathways, such as the JNK, NF-κB, and ER stress cascades [153,154]. Moreover, CD36-associated JNK activation correlated with the impaired tyrosine phosphorylation of IRS-1, and IRS-2 blocks interaction with phosphoinositide 3-kinase (PI3K), thereby inducing insulin resistance. A recent study by Vandanmagsar et al. [155] detailed that lipotoxicity-associated increments in intracellular ceramide induce caspase-1 cleavage in macrophages and fat tissue through nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain-containing protein 3 (NLRP3) inflammasome activation. The development of insulin resistance and T2D has been connected with the expanded release of proinflammatory cytokines, which may impair insulin signaling, and this is supported by a clinical study in which T2D elevated FFAs and IL-1β in plasma, promoting insulin resistance [156]. Other evidence suggests that NOX-TXNIP-dependent ROS production drives caspase1-dependent IL-1β secretion by NLRP3 activators [157,158]. We also showed that CD36 signaling leads to ROS production due to TXNIP translocation to the mitochondria and represses the antioxidative protein thioredoxin in reaction to ceramide [110]. Indeed, TXNIP plays a role in producing IL-1β through NLRP3 inflammasome enactment beneath ER stress within pancreatic β-cells [159]. Inflammasome activation impedes insulin signaling in several target tissues to diminish glucose tolerance and insulin sensitivity [160]. Likewise, it has been reported that CD36-mediated pathogenic inflammasome activation is linked to cholesterol accumulation in the inflammatory process of atherosclerosis [161].
In an unforeseen finding, CD36 signaling through lysosomal impairment was shown to promote inflammasome activation in adipose tissue from obese mice, suggesting that lysosomes may be vital in obesity-induced adipose tissue inflammation [162]. In addition to these proposed mechanisms, the central part of inflammasomes is specifically activated through lysosome-dependent pathways in macrophages and adipose tissue from obese mice [163,164]. This signaling pathway is subordinate to the activation of the CD36-FYN kinase-mediated Tyr353 phosphorylation of IP3R1, which leads to IP3R1-mediated Ca2+ exchange from the ER to the lysosome, and this causes lysosomal impairment and inflammation in adipocytes. The inhibition of CD36 palmitoylation reduces both FYN kinase activation and Tyr353 phosphorylation of IP3R1, which may contribute to reduced lysosomal Ca2+ overload and inflammation in adipocytes. A recent study by Wang et al. demonstrated that palmitoyl acyltransferases DHHC4 and DHHC5 control the palmitoylation of CD36 in directing fatty acid uptake in adipose tissues [165]. In addition to adipocytes, CD36 palmitoylation and the localization of CD36 on the plasma membrane of hepatocytes are significantly increased in patients with non-alcoholic steatohepatitis (NASH), as well as in livers from mice with NASH. The inhibition of the palmitoylation of CD36 protects mice from NASH and the inflammatory response [166]. In addition, membrane expression of CD36 was shown to be elevated in livers from mice and humans with non-alcoholic fatty liver disease during the aging process [167]. In support of this, a recent study by Chong et al. demonstrated that sustained exposure to amyloid-beta (Ab) drives senescence-associated secretory phenotype (SASP) via pro-inflammatory cytokine production and cell-cycle arrest in both epithelial cells and fibroblasts at the CD36–NF-κB signaling axis [168]. Therefore, CD36 activation has been proposed as a novel inflammatory agent to target and promote healthy aging and lifespan extension (Figure 3). Further research is required to identify CD36 targets in inflammatory aging disease. Based on its role in FFA uptake and the inflammatory process, it has been suggested that CD36 is involved in cardiovascular disease [169,170,171]. The pathophysiological role of CD36 in the cardiovascular system is discussed in the following recent reviews [172,173,174,175].
6. Conclusions
Experimental and clinical studies have fortified the noteworthiness of CD36 in metabolic disorders. CD36 has shown to act as a gatekeeper of various intracellular signaling systems that induce cell dysfunction under an excessive nutrition milieu. CD36 initiating redox signaling revealed the core pathophysiology of metabolic disorders such as diabetes, and promising therapeutic targets are expected. However, several questions regarding its impact on metabolic disease progression will need to be answered in future studies. Understanding this complex situation and the therapeutic agents that modulate the CD36 function could have a considerable impact on the treatment of metabolic diseases such as insulin resistance and diabetes.
Author Contributions
Conceptualization, U.K. and S.E.; resources, U.K. and J.-S.M.; data curation, U.K. and J.-S.M.; writing—original draft preparation, U.K. and S.E.; writing—review and editing, U.K., S.E., J.-S.M. and K.-C.W.; visualization, U.K. and S.E.; supervision, K.-C.W.; funding acquisition, K.-C.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the 2019 Yeungnam University Research Grant and National Research Foundation of Korea (NRF) grants funded by the Korean government (NRF-2020R1A2C1003649 Kyu Chang Won).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Harmon, C.M.; Abumrad, N.A. Binding of sulfosuccinimidyl fatty acids to adipocyte membrane proteins: Isolation and amino-terminal sequence of an 88-kD protein implicated in transport of long-chain fatty acids. J. Membr. Biol. 1993, 133, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Fukuwatari, T.; Kawada, T.; Tsuruta, M.; Hiraoka, T.; Iwanaga, T.; Sugimoto, E.; Fushiki, T. Expression of the putative membrane fatty acid transporter (FAT) in taste buds of the circumvallate papillae in rats. FEBS Lett. 1997, 414, 461–464. [Google Scholar] [PubMed] [Green Version]
- Van Nieuwenhoven, F.A.; Verstijnen, C.P.; Abumrad, N.A.; Willemsen, P.H.; Van Eys, G.J.; Van der Vusse, G.J.; Glatz, J.F. Putative membrane fatty acid translocase and cytoplasmic fatty acid-binding protein are co-expressed in rat heart and skeletal muscles. Biochem. Biophys. Res. Commun. 1995, 207, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Hogg, N.; Ren, Y.; Haslett, C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Investig. 1992, 90, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Li, W.; Febbraio, M.; Espinola, R.G.; McCrae, K.R.; Cockrell, E.; Silverstein, R.L. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. J. Clin. Investig. 2008, 118, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonen, D.P.; Jensen, M.K.; Handberg, A. Soluble CD36- a marker of the (pathophysiological) role of CD36 in the metabolic syndrome? Arch. Physiol. Biochem. 2011, 117, 57–63. [Google Scholar] [CrossRef]
- Wang, Y.; Koch, M.; di Giuseppe, R.; Evans, K.; Borggrefe, J.; Nöthlings, U.; Handberg, A.; Jensen, M.K.; Lieb, W. Associations of plasma CD36 and body fat distribution. J. Clin. Endocrinol. Metab. 2019, 104, 4016–4023. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Kashyap, S.R. Pathogenic role of scavenger receptor CD36 in the metabolic syndrome and diabetes. Metab. Syndr. Relat. Disord. 2011, 9, 239–245. [Google Scholar] [CrossRef]
- Yang, J.; Park, K.W.; Cho, S. Inhibition of the CD36 receptor reduces visceral fat accumulation and improves insulin resistance in obese mice carrying the BDNF-Val66Metvariant. J. Biol. Chem. 2018, 293, 13338–13348. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Tso, P.; Woods, S.C. Receptor CD36 links a risk-associated allele to obesity and metabolic disorders. J. Biol. Chem. 2018, 293, 13349–13350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abumrad, N.A.; el-Maghrabi, M.R.; Amri, E.Z.; Lopez, E.; Grimaldi, P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 1993, 268, 17665–17668. [Google Scholar] [CrossRef]
- Schwenk, R.W.; Dirkx, E.; Coumans, W.A.; Bonen, A.; Klip, A.; Glatz, J.F.; Luiken, J.J. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 2010, 53, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- van Oort, M.M.; van Doorn, J.M.; Bonen, A.; Glatz, J.F.; van der Horst, D.J.; Rodenburg, K.W.; Luiken, J.J. Insulin-induced translocation of CD36 to the plasma membrane is reversible and shows similarity to that of GLUT4. Biochim. Biophys. Acta 2008, 1781, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Murakami, S.; Sun, Y.; Kilpatrick, C.L.; Luscher, B. DHHC7 Palmitoylates Glucose Transporter 4 (Glut4) and Regulates Glut4 Membrane Translocation. J. Biol. Chem. 2017, 292, 2979–2991. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Pohl, J.; Ring, A.; Korkmaz, U.; Ehehalt, R.; Stremmel, W. FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol. Biol. Cell. 2005, 16, 24–31. [Google Scholar] [CrossRef]
- Ehehalt, R.; Sparla, R.; Kulaksiz, H.; Herrmann, T.; Füllekrug, J.; Stremmel, W. Uptake of long chain fatty acids is regulated by dynamic interaction of FAT/CD36 with cholesterol/sphingolipid enriched microdomains (lipid rafts). BMC Cell Biol. 2008, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.; Le Lay, S.; Pohl, J.; Verkade, P.; Stremmel, W. Caveolin-1 is required for fatty acid translocase (FAT/CD36) localization and function at the plasma membrane of mouse embryonic fibroblasts. Biochim. Biophys. Acta. 2006, 1761, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Pohl, J.; Ring, A.; Ehehalt, R.; Schulze-Bergkamen, H.; Schad, A.; Verkade, P.; Stremmel, W. Long-chain fatty acid uptake into adipocytes depends on lipid raft function. Biochemistry 2004, 43, 4179–4187. [Google Scholar] [CrossRef]
- Campbell, S.E.; Tandon, N.N.; Woldegiorgis, G.; Luiken, J.J.; Glatz, J.F.; Bonen, A. A novel function for fatty acid translocase (FAT)/CD36: Involvement in long chain fatty acid transfer into the mitochondria. J. Biol. Chem. 2004, 279, 36235–36241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holloway, G.P.; Luiken, J.J.; Glatz, J.F.; Spriet, L.L.; Bonen, A. Contribution of FAT/CD36 to the regulation of skeletal muscle fatty acid oxidation: An overview. Acta Physiol. 2008, 194, 293–309. [Google Scholar] [CrossRef]
- Holloway, G.P.; Jain, S.S.; Bezaire, V.; Han, X.X.; Glatz, J.F.; Luiken, J.J.; Harper, M.E.; Bonen, A. FAT/CD36-null mice reveal that mitochondrial FAT/CD36 is required to upregulate mitochondrial fatty acid oxidation in contracting muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R960–R967. [Google Scholar] [CrossRef]
- Samovski, D.; Sun, J.; Pietka, T.; Gross, R.W.; Eckel, R.H.; Su, X.; Stahl, P.D.; Abumrad, N.A. Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes 2015, 64, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abumrad, N.A.; Goldberg, I.J. CD36 actions in the heart: Lipids, calcium, inflammation, repair and more? Biochim. Biophys. Acta 2016, 1861, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.S.; Karunakaran, U.; Suma, E.; Chung, S.M.; Won, K.C. The role of CD36 in type 2 diabetes nellitus: β-cell dysfunction and beyond. Diabetes Metab. J. 2020, 44, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J. From fat to FAT (CD36/SR-B2): Understanding the regulation of cellular fatty acid uptake. Biochimie 2017, 136, 21–26. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. Time for a détente in the war on the mechanism of cellular fatty acid uptake. J. Lipid. Res. 2020, 61, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Ruiz, E.; Armesilla, A.L.; Sanchez-Madrid, F.; Vega, M.A. Gene encoding the collagen type I and thrombospondin receptor CD36 is located on chromosome 7q11.2. Genomics 1993, 17, 759–761. [Google Scholar] [CrossRef] [Green Version]
- Armesilla, A.L.; Vega, M.A. Structural organization of the gene for human CD36 glycoprotein. J. Biol. Chem. 1994, 269, 18985–18991. [Google Scholar] [CrossRef]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Kuniyasu, A.; Horiuchi, S.; Nakayama, H. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J. Biol. Chem. 2001, 276, 3195–3202. [Google Scholar] [CrossRef] [Green Version]
- Puente Navazo, M.D.; Daviet, L.; Ninio, E.; McGregor, J.L. Identification on human CD36 of a domain (155–183) implicated in binding oxidized low-density lipoproteins (Ox-LDL). Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1033–1039. [Google Scholar] [CrossRef]
- Neculai, D.; Schwake, M.; Ravichandran, M.; Zunke, F.; Collins, R.F.; Peters, J.; Neculai, M.; Plumb, J.; Loppnau, P.; Pizarro, J.C.; et al. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013, 504, 172–176. [Google Scholar] [CrossRef]
- Hoosdally, S.J.; Andress, E.J.; Wooding, C.; Martin, C.A.; Linton, K.J. The Human Scavenger Receptor CD36: Glycosylation status and its role in trafficking and function. J. Biol. Chem. 2009, 284, 16277–16288. [Google Scholar] [CrossRef] [Green Version]
- Laczy, B.; Fulop, N.; Onay-Besikci, A.; Des Rosiers, C.; Chatham, J.C. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PLoS ONE 2011, 6, e18417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinez, C.; Morelle, W.; Michalski, J.C.; Lefebvre, T. O-GlcNAc glycosylation: A signal for the nuclear transport of cytosolic proteins? Int. J. Biochem. Cell Biol. 2005, 37, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Vosseller, K.; Hart, G.W. Glycosylation of nucleocytoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001, 291, 2376–2378. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Wu, N.; Xu, B.; Chu, Y.; Li, X.; Su, S.; Chen, D.; Li, W.; Shi, Y.; Gao, X.; et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics 2019, 9, 5359–5373. [Google Scholar] [CrossRef] [PubMed]
- Nabeebaccus, A.A.; Zoccarato, A.; Hafstad, A.D.; Santos, C.X.; Aasum, E.; Brewer, A.C.; Zhang, M.; Beretta, M.; Yin, X.; West, J.A.; et al. Nox4 reprograms cardiac substrate metabolism via protein O-GlcNAcylation to enhance stress adaptation. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Biophys. Acta 2011, 1808, 2981–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, N.; Wagner, S.J.; Lublin, D.M. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J. Biol. Chem. 1996, 271, 22315–22320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, R.F.; Ralston, K.J.; de Bock, C.E.; Mhaidat, N.M.; Zhang, X.D.; Boyd, A.W.; Burns, G.F. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Biophys. Acta 2010, 1803, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.; Su, X.; El-Maghrabi, R.; Stahl, P.D.; Abumrad, N.A. Opposite regulation of CD36 ubiquitination by fatty acids and insulin: Effects on fatty acid uptake. J. Biol. Chem. 2008, 283, 13578–13585. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Hu, T.; He, J.; Xu, Q.; Yu, C.; Liu, X.; Shao, Z.; Liao, Y.; Huang, H.; Liu, N. USP10 deletion inhibits macrophage-derived foam cell formation and cellular-oxidized low density lipoprotein uptake by promoting the degradation of CD36. Aging 2020, 12, 22892–22905. [Google Scholar] [CrossRef]
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712. [Google Scholar] [CrossRef] [PubMed]
- Asch, A.S.; Liu, I.; Briccetti, F.M.; Barnwell, J.W.; Kwakye-Berko, F.; Dokun, A.; Goldberger, J.; Pernambuco, M. Analysis of CD36 binding domains: Ligand specificity controlled by dephosphorylation of an ectodomain. Science 1993, 262, 1436–1440. [Google Scholar] [CrossRef]
- Hatmi, M.; Gavaret, J.M.; Elalamy, I.; Vargaftig, B.B.; Jacquemin, C. Evidence for cAMP-dependent platelet ectoprotein kinase activity that phosphorylates platelet glycoprotein IV (CD36). J. Biol. Chem. 1996, 271, 24776–24780. [Google Scholar] [CrossRef] [Green Version]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013, 288, 15547–15555. [Google Scholar] [CrossRef] [Green Version]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 149–154. [Google Scholar] [CrossRef]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maedler, K.; Spinas, G.A.; Dyntar, D.; Moritz, W.; Kaiser, N.; Donath, M.Y. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 2001, 50, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, T.; Ma, Z.; Ogata, H.; Jorgensen, I.H.; Iezzi, M.; Wang, H.; Wollheim, C.B.; Bjorklund, A. Facilitation of fatty acid uptake by CD36 in insulin-producing cells reduces fatty-acid-induced insulin secretion and glucose regulation of fatty acid oxidation. Biochim. Biophys. Acta 2010, 1801, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.W.; Moon, J.S.; Seo, Y.J.; Park, S.Y.; Kim, J.Y.; Yoon, J.S.; Lee, I.K.; Lee, H.W.; Won, K.C. Inhibition of fatty acid translocase cluster determinant 36 (CD36), stimulated by hyperglycemia, prevents glucotoxicity in INS-1 cells. Biochem. Biophys. Res. Commun. 2012, 420, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Elumalai, S.; Karunakaran, U.; Lee, I.K.; Moon, J.S.; Won, K.C. Rac1-NADPH oxidase signaling promotes CD36 activation under glucotoxic conditions in pancreatic beta cells. Redox Biol. 2017, 11, 126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahara, S.; Shibutani, Y.; Teruyama, K.; Inoue, H.Y.; Kawada, Y.; Etoh, H.; Matsuda, T.; Kimura-Koyanagi, M.; Hashimoto, N.; Sakahara, M.; et al. Ras-related C3 botulinum toxin substrate 1 (RAC1) regulates glucose-stimulated insulin secretion via modulation of F-actin. Diabetologia 2013, 56, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Luo, R.; Kowluru, A.; Li, G. Novel regulation by Rac1 of glucose- and forskolin-induced insulin secretion in INS-1 beta-cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E818–E827. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, D.C.; Gonelle-Gispert, C.; Furukawa, M.; Halban, P.A.; Pessin, J.E. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol. Endocrinol. 2003, 17, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Tomas, A.; Yermen, B.; Min, L.; Pessin, J.E.; Halban, P.A. Regulation of pancreatic beta-cell insulin secretion by actin cytoskeleton remodelling: Role of gelsolin and cooperation with the MAPK signalling pathway. J. Cell Sci. 2006, 119, 2156–2167. [Google Scholar] [CrossRef] [Green Version]
- Band, A.M.; Ali, H.; Vartiainen, M.K.; Welti, S.; Lappalainen, P.; Olkkonen, V.M.; Kuismanen, E. Endogenous plasma membrane t-SNARE syntaxin 4 is present in rab11 positive endosomal membranes and associates with cortical actin cytoskeleton. FEBS Lett. 2002, 531, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Luo, W.; Oh, E.; Wang, Z.; Thurmond, D.C. Filamentous actin regulates insulin exocytosis through direct interaction with Syntaxin 4. J. Biol. Chem. 2008, 283, 10716–10726. [Google Scholar] [CrossRef] [Green Version]
- Rondas, D.; Tomas, A.; Soto-Ribeiro, M.; Wehrle-Haller, B.; Halban, P.A. Novel mechanistic link between focal adhesion remodeling and glucose-stimulated insulin secretion. J. Biol. Chem. 2012, 287, 2423–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamatsu, S.; Nakamichi, Y.; Yamamura, C.; Matsushima, S.; Watanabe, T.; Ozawa, S.; Furukawa, H.; Ishida, H. Decreased expression of t-SNAR.E.; syntaxin 1, and SNAP-25 in pancreatic beta-cells is involved in impaired insulin secretion from diabetic GK rat islets: Restoration of decreased t-SNARE proteins improves impaired insulin secretion. Diabetes 1999, 48, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, D.C.; Gaisano, H.Y. Recent Insights into Beta-cell Exocytosis in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Torrejon-Escribano, B.; Escoriza, J.; Montanya, E.; Blasi, J. Glucose-dependent changes in SNARE protein levels in pancreatic beta-cells. Endocrinology 2011, 152, 1290–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaisano, H.Y. Recent new insights into the role of SNARE and associated proteins in insulin granule exocytosis. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 115–123. [Google Scholar] [CrossRef] [Green Version]
- Nagao, M.; Esguerra, J.L.S.; Asai, A.; Ofori, J.K.; Edlund, A.; Wendt, A.; Sugihara, H.; Wollheim, C.B.; Oikawa, S.; Eliasson, L. Potential Protection Against Type 2 Diabetes in Obesity Through Lower CD36 Expression and Improved Exocytosis in beta-Cells. Diabetes 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jorns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes 2005, 54 (Suppl. 2), S97–S107. [Google Scholar] [CrossRef] [Green Version]
- Ihara, Y.; Toyokuni, S.; Uchida, K.; Odaka, H.; Tanaka, T.; Ikeda, H.; Hiai, H.; Seino, Y.; Yamada, Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999, 48, 927–932. [Google Scholar] [CrossRef]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharib, M.; Tao, H.; Fungwe, T.V.; Hajri, T. Cluster Differentiating 36 (CD36) Deficiency Attenuates Obesity-Associated Oxidative Stress in the Heart. PLoS ONE 2016, 11, e0155611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veluthakal, R.; Kumar, B.; Mohammad, G.; Kowluru, A.; Kowluru, R.A. Tiam1-Rac1 Axis Promotes Activation of p38 MAP Kinase in the Development of Diabetic Retinopathy: Evidence for a Requisite Role for Protein Palmitoylation. Cell Physiol. Biochem. 2015, 36, 208–220. [Google Scholar] [CrossRef]
- Li, W.; Febbraio, M.; Reddy, S.P.; Yu, D.Y.; Yamamoto, M.; Silverstein, R.L. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J. Clin. Investig. 2010, 120, 3996–4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; El Khoury, J.; Medeiros, L.A.; Terada, K.; Geula, C.; Luster, A.D.; Freeman, M.W. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 2002, 277, 47373–47379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007, 50, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30. [Google Scholar] [CrossRef]
- Karunakaran, U.; Kim, H.J.; Kim, J.Y.; Lee, I.K. Guards and culprits in the endoplasmic reticulum: Glucolipotoxicity and beta-cell failure in type II diabetes. Exp. Diabetes Res. 2012, 2012, 639762. [Google Scholar] [CrossRef] [Green Version]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic beta-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maestre, I.; Jordan, J.; Calvo, S.; Reig, J.A.; Cena, V.; Soria, B.; Prentki, M.; Roche, E. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology 2003, 144, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimabukuro, M.; Ohneda, M.; Lee, Y.; Unger, R.H. Role of nitric oxide in obesity-induced beta cell disease. J. Clin. Investig. 1997, 100, 290–295. [Google Scholar] [CrossRef]
- Shimabukuro, M.; Zhou, Y.T.; Levi, M.; Unger, R.H. Fatty acid-induced beta cell apoptosis: A link between obesity and diabetes. Proc. Natl. Acad. Sci. USA 1998, 95, 2498–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindokas, V.P.; Kuznetsov, A.; Sreenan, S.; Polonsky, K.S.; Roe, M.W.; Philipson, L.H. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J. Biol. Chem. 2003, 278, 9796–9801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupi, R.; Dotta, F.; Marselli, L.; Del Guerra, S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [Green Version]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef] [Green Version]
- Wigger, L.; Cruciani-Guglielmacci, C.; Nicolas, A.; Denom, J.; Fernandez, N.; Fumeron, F.; Marques-Vidal, P.; Ktorza, A.; Kramer, W.; Schulte, A.; et al. Plasma Dihydroceramides Are Diabetes Susceptibility Biomarker Candidates in Mice and Humans. Cell Rep. 2017, 18, 2269–2279. [Google Scholar] [CrossRef]
- Bustelo, X.R. Vav family exchange factors: An integrated regulatory and functional view. Small GTPases 2014, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Kominato, R.; Fujimoto, S.; Mukai, E.; Nakamura, Y.; Nabe, K.; Shimodahira, M.; Nishi, Y.; Funakoshi, S.; Seino, Y.; Inagaki, N. Src activation generates reactive oxygen species and impairs metabolism-secretion coupling in diabetic Goto-Kakizaki and ouabain-treated rat pancreatic islets. Diabetologia 2008, 51, 1226–1235. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 dependent redoxosomes promotes ceramide-mediated pancreatic beta-cell failure via p66Shc activation. Free Radic. Biol. Med. 2019, 134, 505–515. [Google Scholar] [CrossRef]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein Scaffold Coordinates SRC-Mediated JNK Activation in Response to Metabolic Stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Hua, W.; Huang, H.Z.; Tan, L.T.; Wan, J.M.; Gui, H.B.; Zhao, L.; Ruan, X.Z.; Chen, X.M.; Du, X.G. CD36 Mediated Fatty Acid-Induced Podocyte Apoptosis via Oxidative Stress. PLoS ONE 2015, 10, e0127507. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [Green Version]
- Natalicchio, A.; Tortosa, F.; Labarbuta, R.; Biondi, G.; Marrano, N.; Carchia, E.; Leonardini, A.; Cignarelli, A.; Bugliani, M.; Marchetti, P.; et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia 2015, 58, 1260–1271. [Google Scholar] [CrossRef]
- Khalid, S.; Drasche, A.; Thurner, M.; Hermann, M.; Ashraf, M.I.; Fresser, F.; Baier, G.; Kremser, L.; Lindner, H.; Troppmair, J. cJun N-terminal kinase (JNK) phosphorylation of serine 36 is critical for p66Shc activation. Sci. Rep. 2016, 6, 20930. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, W.; Harberg, C.; Chen, W.; Yue, H.; Ferreira, R.B.; Wynia-Smith, S.L.; Carroll, K.S.; Zielonka, J.; Flaumenhaft, R.; et al. Cysteine sulfenylation by CD36 signaling promotes arterial thrombosis in dyslipidemia. Blood Adv. 2020, 4, 4494–4507. [Google Scholar] [CrossRef]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef]
- Lim, Y.S.; Cha, M.K.; Kim, H.K.; Uhm, T.B.; Park, J.W.; Kim, K.; Kim, I.H. Removals of hydrogen peroxide and hydroxyl radical by thiol-specific antioxidant protein as a possible role in vivo. Biochem. Biophys. Res. Commun. 1993, 192, 273–280. [Google Scholar] [CrossRef]
- Cox, A.G.; Pullar, J.M.; Hughes, G.; Ledgerwood, E.C.; Hampton, M.B. Oxidation of mitochondrial peroxiredoxin 3 during the initiation of receptor-mediated apoptosis. Free Radic. Biol. Med. 2008, 44, 1001–1009. [Google Scholar] [CrossRef]
- Karunakaran, U.; Moon, J.S.; Lee, H.W.; Won, K.C. CD36 initiated signaling mediates ceramide-induced TXNIP expression in pancreatic beta-cells. Biochim. Biophys. Acta 2015, 1852, 2414–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.S.; Wiederkehr, A.; Kirkpatrick, C.; Mattenberger, Y.; Martinou, J.C.; Marchetti, P.; Demaurex, N.; Wollheim, C.B. Selective actions of mitochondrial fission/fusion genes on metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2008, 283, 33347–33356. [Google Scholar] [CrossRef] [Green Version]
- Wasilewski, M.; Scorrano, L. The changing shape of mitochondrial apoptosis. Trends Endocrinol. Metab. 2009, 20, 287–294. [Google Scholar] [CrossRef]
- Koshkin, V.; Dai, F.F.; Robson-Doucette, C.A.; Chan, C.B.; Wheeler, M.B. Limited mitochondrial permeabilization is an early manifestation of palmitate-induced lipotoxicity in pancreatic beta-cells. J. Biol. Chem. 2008, 283, 7936–7948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Marinello, M.; Bononi, A.; Bonora, M.; Giorgi, C.; Rimessi, A.; Pinton, P. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 2012, 3, e304. [Google Scholar] [CrossRef] [Green Version]
- Plaisance, V.; Brajkovic, S.; Tenenbaum, M.; Favre, D.; Ezanno, H.; Bonnefond, A.; Bonner, C.; Gmyr, V.; Kerr-Conte, J.; Gauthier, B.R.; et al. Endoplasmic Reticulum Stress Links Oxidative Stress to Impaired Pancreatic Beta-Cell Function Caused by Human Oxidized LDL. PLoS ONE 2016, 11, e0163046. [Google Scholar] [CrossRef] [Green Version]
- Abderrahmani, A.; Niederhauser, G.; Favre, D.; Abdelli, S.; Ferdaoussi, M.; Yang, J.Y.; Regazzi, R.; Widmann, C.; Waeber, G. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia 2007, 50, 1304–1314. [Google Scholar] [CrossRef] [Green Version]
- Cnop, M.; Hannaert, J.C.; Grupping, A.Y.; Pipeleers, D.G. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology 2002, 143, 3449–3453. [Google Scholar] [CrossRef] [Green Version]
- Grupping, A.Y.; Cnop, M.; Van Schravendijk, C.F.; Hannaert, J.C.; Van Berkel, T.J.; Pipeleers, D.G. Low density lipoprotein binding and uptake by human and rat islet beta cells. Endocrinology 1997, 138, 4064–4068. [Google Scholar] [CrossRef]
- Shi, Y.; Cosentino, F.; Camici, G.G.; Akhmedov, A.; Vanhoutte, P.M.; Tanner, F.C.; Luscher, T.F. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2090–2097. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Ketelhuth, D.F.J.; Wirstrom, T.; Ohki, T.; Forteza, M.J.; Wang, H.; Grill, V.; Wollheim, C.B.; Bjorklund, A. Increased uptake of oxLDL does not exert lipotoxic effects in insulin-secreting cells. J. Mol. Endocrinol. 2019, 62, 159–168. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Huang, W.; Chen, W.; Zhao, Y.; Schulte, M.L.; Volberding, P.; Gerbec, Z.; Zimmermann, M.T.; Zeighami, A.; et al. Mitochondrial Metabolic Reprogramming by CD36 Signaling Drives Macrophage Inflammatory Responses. Circ. Res. 2019, 125, 1087–1102. [Google Scholar] [CrossRef]
- Zhao, M.; Pan, W.; Shi, R.Z.; Bai, Y.P.; You, B.Y.; Zhang, K.; Fu, Q.M.; Schuchman, E.H.; He, X.X.; Zhang, G.G. Acid Sphingomyelinase Mediates Oxidized-LDL Induced Apoptosis in Macrophage via Endoplasmic Reticulum Stress. J. Atheroscler. Thromb. 2016, 23, 1111–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benáková, Š.; Holendová, B.; Plecitá-Hlavatá, L. Redox Homeostasis in Pancreatic β-Cells: From Development to Failure. Antioxidants 2021, 10, 526. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Holendová, B.; Jabůrek, M.; Tauber, J.; Dlasková, A.; Plecitá-Hlavatá, L. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants 2021, 10, 197. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Podszun, M.C.; Alawad, A.S.; Lingala, S.; Morris, N.; Huang, W.A.; Yang, S.; Schoenfeld, M.; Rolt, A.; Ouwerkerk, R.; Valdez, K.; et al. Vitamin E treatment in NAFLD patients demonstrates that oxidative stress drives steatosis through upregulation of de-novo lipogenesis. Redox Biol. 2020, 37, 101710. [Google Scholar] [CrossRef]
- Ziegler, M.; Wallert, M.; Lorkowski, S.; Peter, K. Cardiovascular and Metabolic Protection by Vitamin E: A Matter of Treatment Strategy? Antioxidants 2020, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Ungurianu, A.; Zanfirescu, A.; Nițulescu, G.; Margină, D. Vitamin E beyond Its Antioxidant Label. Antioxidants 2021, 10, 634. [Google Scholar] [CrossRef] [PubMed]
- Di Vincenzo, A.; Tana, C.; El Hadi, H.; Pagano, C.; Vettor, R.; Rossato, M. Antioxidant, Anti-Inflammatory, and Metabolic Properties of Tocopherols and Tocotrienols: Clinical Implications for Vitamin E Supplementation in Diabetic Kidney Disease. Int. J. Mol. Sci. 2019, 20, 5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozer, N.K.; Negis, Y.; Aytan, N.; Villacorta, L.; Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E inhibits CD36 scavenger receptor expression in hypercholesterolemic rabbits. Atherosclerosis 2006, 184, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 2000, 102, 82–87. [Google Scholar] [CrossRef]
- Munteanu, A.; Taddei, M.; Tamburini, I.; Bergamini, E.; Azzi, A.; Zingg, J.M. Antagonistic effects of oxidized low density lipoprotein and alpha-tocopherol on CD36 scavenger receptor expression in monocytes: Involvement of protein kinase B and peroxisome proliferator-activated receptor-gamma. J. Biol. Chem. 2006, 281, 6489–6497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingg, J.M.; Azzi, A.; Meydani, M. α-Tocopheryl Phosphate Induces VEGF Expression via CD36/PI3Kγ in THP-1 Monocytes. J. Cell Biochem. 2017, 118, 1855–1867. [Google Scholar] [CrossRef]
- Brissova, M.; Shostak, A.; Shiota, M.; Wiebe, P.O.; Poffenberger, G.; Kantz, J.; Chen, Z.; Carr, C.; Jerome, W.G.; Chen, J.; et al. Pancreatic islet production of vascular endothelial growth factor—A is essential for islet vascularization, revascularization, and function. Diabetes 2006, 55, 2974–2985. [Google Scholar] [CrossRef] [Green Version]
- Chia, L.L.; Jantan, I.; Chua, K.H. Tocotrienols Stimulate Insulin Secretion of Rat Pancreatic Isolated Islets in a Dynamic Culture. Curr. Pharm. Biotechnol. 2017, 18, 560–568. [Google Scholar] [CrossRef]
- Hani, H.; Allaudin, Z.N.; Mohd-Lila, M.A.; Sarsaifi, K.; Rasouli, M.; Tam, Y.J.; Tengku-Ibrahim, T.A.; Othman, A.M. Improvement of isolated caprine islet survival and functionality in vitro by enhancing of PDX1 gene expression. Xenotransplantation 2017, 24, e12302. [Google Scholar] [CrossRef] [Green Version]
- Abedini, A.; Schmidt, A.M. Mechanisms of islet amyloidosis toxicity in type 2 diabetes. FEBS Lett. 2013, 587, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Bishoyi, A.K.; Roham, P.H.; Rachineni, K.; Save, S.; Hazari, M.A.; Sharma, S.; Kumar, A. Human islet amyloid polypeptide (hIAPP)—A curse in type II diabetes mellitus: Insights from structure and toxicity studies. Biol. Chem. 2021, 402, 133–153. [Google Scholar] [CrossRef]
- Subramanian, S.L.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Udayasankar, J.; Kahn, S.E. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2012, 55, 166–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Liu, J.; Dragunow, M.; Cooper, G.J. Fibrillogenic amylin evokes islet beta-cell apoptosis through linked activation of a caspase cascade and JNK1. J. Biol. Chem. 2003, 278, 52810–52819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.S.; et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Del Prato, S. Insulin resistance and diabetes mellitus. J. Diabetes Complicat. 1996, 10, 243–245. [Google Scholar] [CrossRef]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Lessard, J.; Laforest, S.; Pelletier, M.; Leboeuf, M.; Blackburn, L.; Tchernof, A. Low abdominal subcutaneous preadipocyte adipogenesis is associated with visceral obesity, visceral adipocyte hypertrophy, and a dysmetabolic state. Adipocyte 2014, 3, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.; Sultana, R.; Greene, M.W. Adipose tissue and insulin resistance in obese. Biomed. Pharmacother. 2021, 137, 111315. [Google Scholar] [CrossRef]
- Cai, L.; Wang, Z.; Ji, A.; Meyer, J.M.; van der Westhuyzen, D.R. Scavenger receptor CD36 expression contributes to adipose tissue inflammation and cell death in diet-induced obesity. PLoS ONE 2012, 7, e36785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Diehl, J.A.; Chin, Y.E.; Yan, W.; Xu, H. FFA-induced adipocyte inflammation and insulin resistance: Involvement of ER stress and IKKbeta pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Roden, M. Mechanisms of Disease: Hepatic steatosis in type 2 diabetes—Pathogenesis and clinical relevance. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Dostert, C.; Petrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Li, Y.; Yang, P.; Chen, Y.; Wei, L.; Yu, T.; Xia, J.; Ruan, X.Z.; Zhao, L.; Chen, Y. Obesity induces preadipocyte CD36 expression promoting inflammation via the disruption of lysosomal calcium homeostasis and lysosome function. EBioMedicine 2020, 56, 102797. [Google Scholar] [CrossRef] [PubMed]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hao, J.W.; Wang, X.; Guo, H.; Sun, H.H.; Lai, X.Y.; Liu, L.Y.; Zhu, M.; Wang, H.Y.; Li, Y.F.; et al. DHHC4 and DHHC5 Facilitate Fatty Acid Uptake by Palmitoylating and Targeting CD36 to the Plasma Membrane. Cell Rep. 2019, 26, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Sheedfar, F.; Sung, M.M.; Aparicio-Vergara, M.; Kloosterhuis, N.J.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Febbraio, M.; Jacobs, R.L.; de Bruin, A.; Vinciguerra, M.; et al. Increased hepatic CD36 expression with age is associated with enhanced susceptibility to nonalcoholic fatty liver disease. Aging 2014, 6, 281–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, M.; Yin, T.; Chen, R.; Xiang, H.; Yuan, L.; Ding, Y.; Pan, C.C.; Tang, Z.; Alexander, P.B.; Li, Q.J.; et al. CD36 initiates the secretory phenotype during the establishment of cellular senescence. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Zhao, S.; Braunstein, Z.; Mao, H.; Razavi, M.; Duan, L.; Wei, Y.; Toomey, A.C.; Rajagopalan, S.; Zhong, J. Oxidized LDL upregulates macrophage DPP4 expression via TLR4/TRIF/CD36 pathways. EBioMedicine 2019, 41, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.C.; Luiken, J.; Nabben, M. CD36 (SR-B2) as a Target to Treat Lipid Overload-Induced Cardiac Dysfunction. J. Lipid. Atheroscler. 2020, 9, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Silverstein, R.L. CD36 signaling in vascular redox stress. Free Radic. Biol. Med. 2019, 136, 159–171. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
CD36 structure and post-translational modifications. (Adopted from Ref: [28], copyright 2020 © Korea Diabetes Association).
Figure 1.
CD36 structure and post-translational modifications. (Adopted from Ref: [28], copyright 2020 © Korea Diabetes Association).

Figure 2.
CD36 signal transduction in pancreatic β-cell dysfunction.

Figure 3.
CD36 redox signaling mediates peripheral insulin resistance during metabolic disease.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karunakaran, U.; Elumalai, S.; Moon, J.-S.; Won, K.-C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells 2021, 10, 1833. https://doi.org/10.3390/cells10071833
AMA Style
Karunakaran U, Elumalai S, Moon J-S, Won K-C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells. 2021; 10(7):1833. https://doi.org/10.3390/cells10071833
Chicago/Turabian StyleKarunakaran, Udayakumar, Suma Elumalai, Jun-Sung Moon, and Kyu-Chang Won. 2021. "CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting" Cells 10, no. 7: 1833. https://doi.org/10.3390/cells10071833
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.