1. Introduction
DMD is one of the longest genes in the human genome, spanning 2.2 Mb [
1]. It is composed of 79 exons coding for a full-length transcript of 14 kb. The protein encoded by this transcript is called dystrophin since mutations lead to the Becker and Duchenne muscular dystrophies [
1]. DMD is located within a common fragile site (CFS), which is a region of profound genomic instability. CFS regions often comprise genes that are frequently deleted or altered in cancers and act as tumor-suppressor genes [
2].
Recently, Wang et al. (2014) reported DMD deletions in 25 of 40 myogenic tumors (29 gastrointestinal tumors (GIST), 4 rhabdomyosarcomas, and 7 leiomyosarcomas (LMS)), and observed that forced re-expression of the miniDMD construct (lacking exons 17–48) inhibited cell migration, cell invasion, anchorage independence, and invadopodia formation [
3]. The role of DMD as a tumor suppressor in sarcomas is strengthened by the observation that dystrophin-deficient mdx mice, which are murine models for Duchenne muscular dystrophy, spontaneously develop rhabdomyosarcomas in 6–9% of cases [
4]. More strikingly, Schmidt et al. (2011) noticed that the mdx mutation led in 39% of cases to the spontaneous formation of skeletal-muscle-derived malignant tumors in mice, presenting as mixed rhabdomyo-, fibro-, and liposarcomas (LPS) [
5]. In line with this observation, inactivation of other genes implicated in muscular dystrophy often accompanies age-related soft-tissue sarcomas (STS), thereby suggesting a common mechanism for their oncogenesis (Schmidt et al. 2011). Interestingly, these tumors harbor aneuploidy and genomic instability, which is illustrated by numerous double-strand breaks and the activation of DNA-repair mechanisms, but also recurrent genetic lesions (Trp53, Nf1, Cdkn2a, etc.) that provide links to human mesenchymal cancers and especially to sarcomas with complex genomic profiles [
5].
However, overexpression of the DMD gene has also been reported in leukemias, renal carcinomas, ependymomas, and astrocytomas [
6], which strengthens the argument for its role in oncogenesis. In addition, high DMD expression was associated with poorer overall survival in B-cell chronic lymphocytic leukemia [
7]. None of these observations suggest that DMD acts as a tumor suppressor. As a possible way to account for these discrepancies, the DMD locus comprises at least five internal promoters that give rise to shorter dystrophin transcripts, which encode smaller isoforms sharing the same COOH-terminal domains [
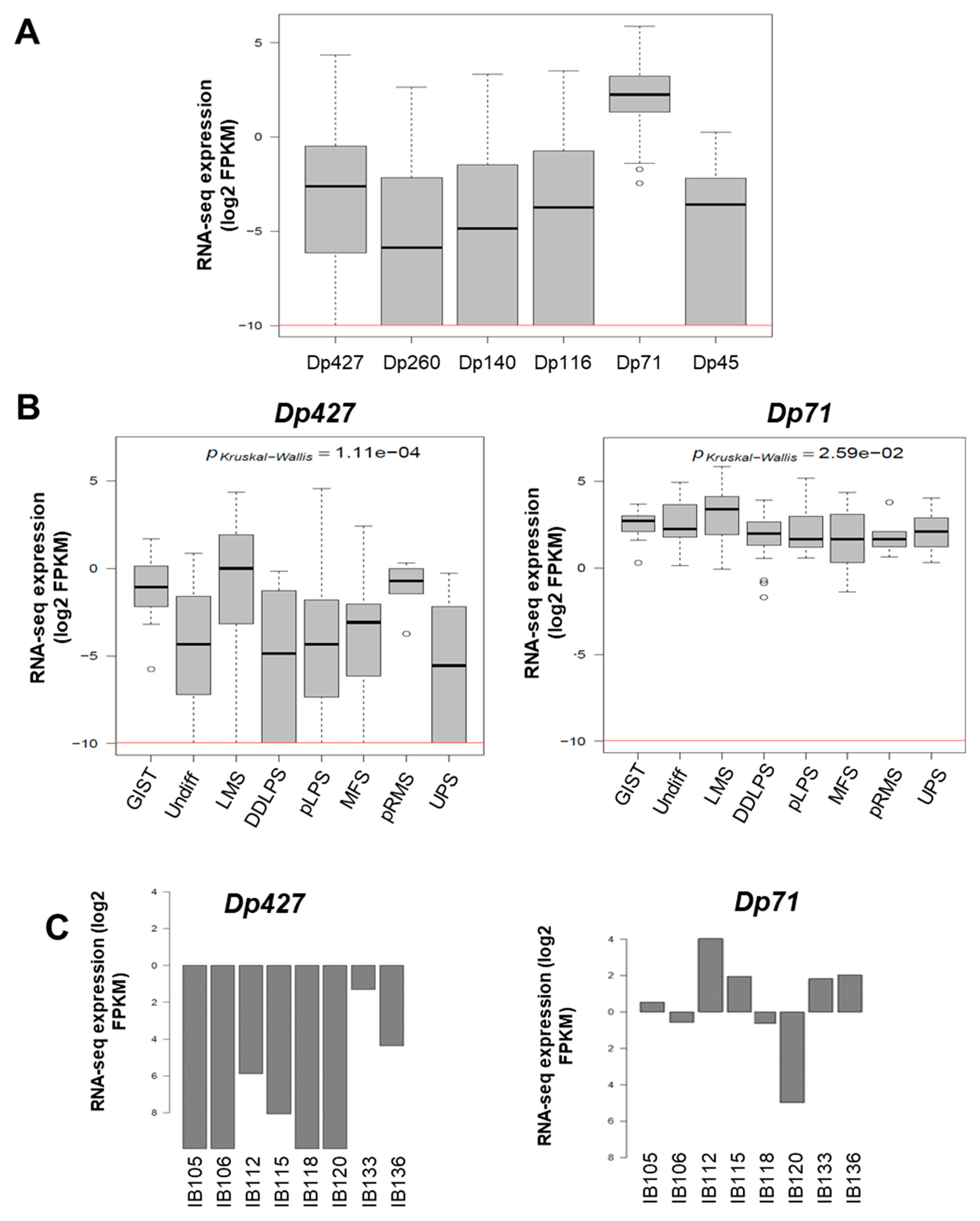
8]. Each of these promoters initiates the translation of a unique first exon. This leads to the production of different-sized proteins: Dp427 (427 kDa), Dp260 (260 kDa), Dp140 (140 kDa), Dp116 (116 kDa), Dp71 (71 kDa), and Dp45 (45 kDa). Whereas Dp427 is the one that is altered in muscular dystrophies, Dp71 is ubiquitously expressed, except in differentiated muscle cells, and is involved in cognition, cell adhesion, cell cycle, and in nuclear-envelope-related functions [
9]. Evidence is accumulating for the versatile role of Dp71 in cancer: on one hand, it seems to act as a tumor-suppressor gene in gastric adenocarcinoma [
10], yet on the other, its inhibition decreases the growth and malignancy of the A549 cell line both in vitro and in vivo. Furthermore, Wang et al. (2014) observed that inhibition of Dp71 dramatically decreased the growth of RMS cell lines [
3]. They also showed that inhibition of DMD enhanced migration and invasion, whereas its re-expression reduced migration and induced senescence in melanoma cell lines.
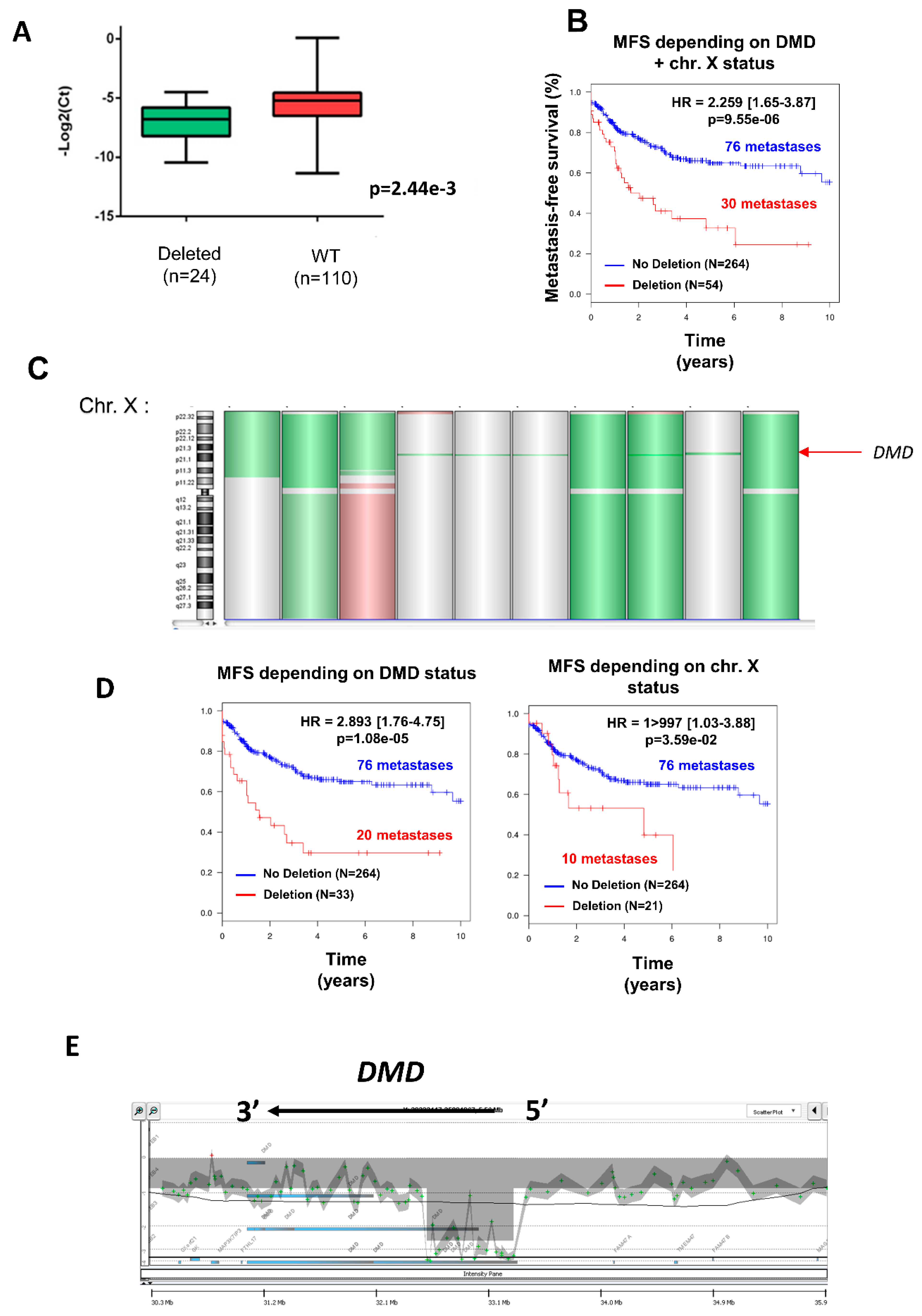
To elucidate the oncogenic impact of DMD in sarcoma oncogenesis, we studied the genomic status of DMD in 318 sarcomas, mainly those with complex genomic profiles but alsosynovial sarcomas and GIST. We also investigated isoform expression and performed functional analyses to decipher the role of Dp427 and Dp71, which are the two isoforms expressed in these sarcomas.
3. Discussion
Pleomorphic sarcomas are characterized more by frequent chromosome losses or gains than by recurrent single nucleotide variations [
12]. The deleted genes most frequently reported are p53, RB1 and PTEN, with frequencies ranging from 10% to 50% [
12]. Here we report that DMD was deleted in 16.5% of pleomorphic sarcomas with complex genomics (myxofibrosarcoma, UPS, LMS, and pleomorphic liposarcoma (pLPS)), 14.2% of GIST, and 20.7% of synovial sarcomas (SS). These proportions are particularly high in comparison with other studies that report a deletion frequency ranging from 3.4% to 5.5% in melanomas [
5] and carcinomas [
9]. In contrast, Wang and collaborators observed intragenic deletions in 25 of 40 (63%) high-grade myogenic tumors, which suggests that DMD deletion could be associated with aggressiveness [
3]. This is consistent with our data demonstrating that DMD deletions—especially those restricted to this gene—are associated with a poorer prognosis. Some authors have suggested that DMD deletion is restricted to sarcomas with a myogenic differentiation. However, we also noticed such a deletion in myxofibrosarcoma, UPS, pLPS, and SS. This observation may have two major implications. On one hand, these tumors might share a muscular origin, as has been proposed for SS [
13]. On the other hand, DMD loss might affect not only skeletal muscle cells but also other mesenchymal tissues. Indeed, DMD-deleted sarcomas could arise from multipotent mesenchymal or muscle-derived stem cells. This hypothesis is strengthened by the study of Schmidt et al. (2011), who reported the spontaneous development of mixed rhabdomyo-, fibro-, and lipo-sarcomas harboring genomic instability in 39% of dystrophic mdx mice (characterized by a DMD mutation) [
5]. Whatever the hypothesis, these deletions might arise from anomalies during DMD transcription. Wang et al. also reported that DMD deletions in women affect only the active X chromosome [
3] (i.e., the one that is transcribed).
Indeed, DMD is one of the longest genes (2.2 Mb) in the human genome, and is located within a CFS [
14]. CFS are regions of genomic instability that are highly susceptible to homologous recombination in normal cells [
15], and have been described as hot spots for chromosomal rearrangements in the initial phase of oncogenesis [
16], leading to the amplification of oncogenes and/or inactivation of tumor suppressor genes. This genomic instability can be explained by the collision between transcription and replication complexes of the long genes. Helmrich et al. (2011) noticed that transcription of these long genes starts in the G2/M phase of the cell cycle and extends to the next S phase [
17]. This observation has numerous implications: first, the presence of pre-mRNAs on mitotic chromosomes can interfere with proper chromatin condensation and therefore correspond to regions of genomic fragility. Secondly, this means that transcription pausing or stalling of replication forks leads to the creation of stable RNA:DNA hybrids that form R-loops, which are responsible for DNA double-strand breaks and, therefore, for genomic instability [
17]. In the same study, they also showed that the frequency of CFS breaks correlates with its transcription level, thereby confirming that DMD deletion is particularly relevant in myogenic sarcomas or more widely in those derived from DMD-expressing cells. Altogether, this could explain why we observed only deletions and no mutation, and especially deletions targeting only the 5’ part of DMD, which accounted for 61% of deleted cases.
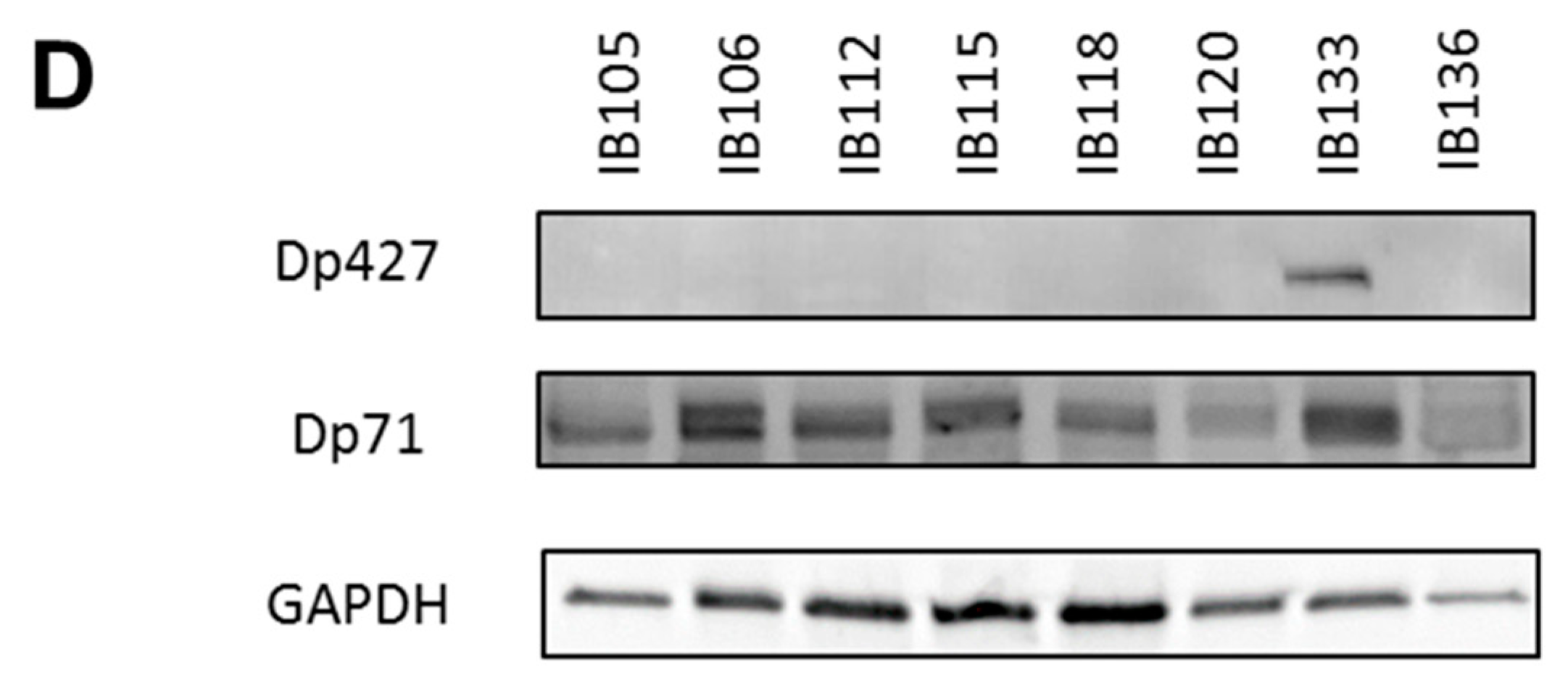
In a cohort of 145 sarcomas, Dp427 and Dp71 were the two isoforms that were mainly expressed in tumors, while the DMD-5’ deletion affected only Dp427 but not Dp71. The Dp427 transcript encodes the full-length dystrophin protein, whose altered expression or loss of function is responsible for muscular dystrophies like Duchenne and Becker myopathies [
10]. In addition to its implication in muscular dystrophies, evidence is accumulating for a tumor-suppressor role of Dp427 in melanoma, myogenic sarcomas [
3,
18], and murine models [
4]. Regarding Dp71, its involvement in cancer is less clear, with conflicting results depending on the cancer model [
10]. In our sarcoma cell line, Dp427 downregulation did not lead to any modification of cell growth or clonogenic capacities. However, this result should be taken cautiously since we did not exactly reproduce the alterations detected in tumors (i.e., large deletions and not deletions of one or two nucleotides). On the other hand, Dp71 inhibition dramatically reduced cell proliferation and clonogenicity in three sarcoma cell lines. This observation is consistent with the study by Villarreal-Silva and collaborators that showed that Dp71 is involved in cell division [
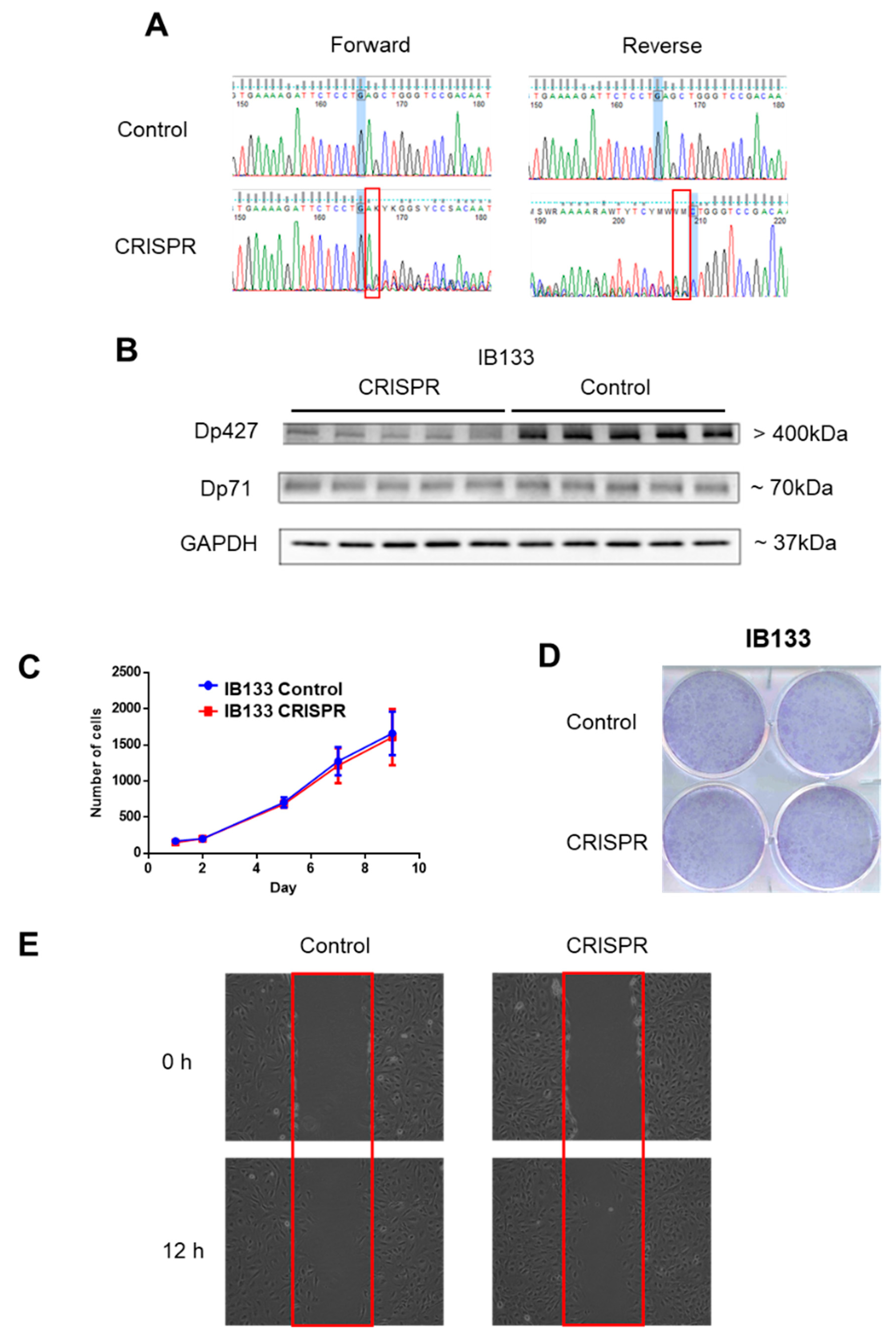
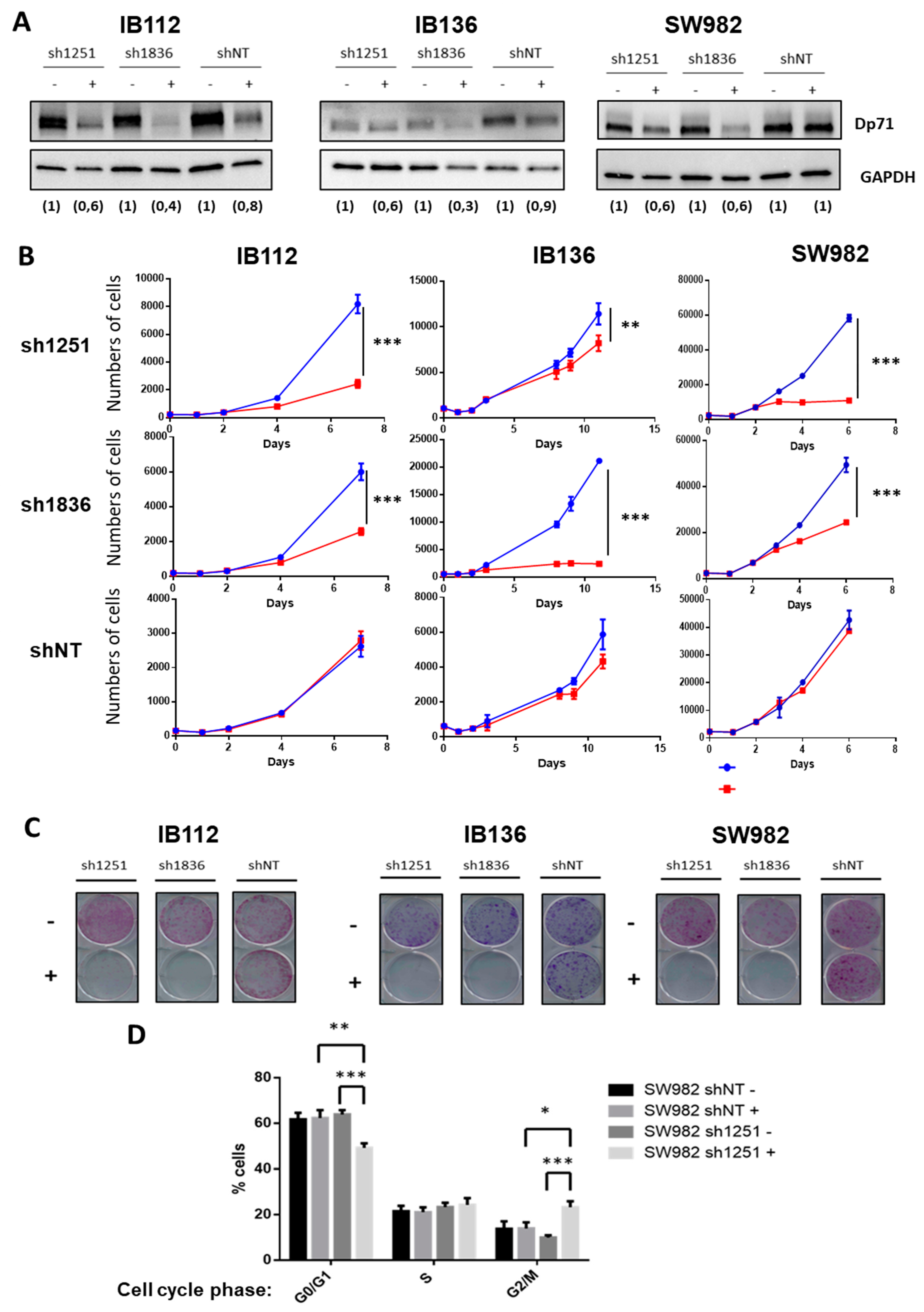
19]. They postulated that Dp71 is localized at the mitotic spindle-cleavage furrow and midbody of PC12 cells and binds to lamin B1 and β-dystroglycan, thereby providing proper localization and stability for these cytokinesis multiprotein apparatuses. This supports our observation that Dp71 inhibition seems to induce a cell cycle arrest during the G2/M phase in the SW982 cell line. This is further evidence that Dp71 plays an essential role in cell growth and tumor development in STS. This may explain why it is widely expressed in tumors and why specific DMD deletions systematically preserve the Dp71 reading frame.
In conclusion, DMD deletion was observed in 15% of this STS cohort and was significantly associated with metastatic evolution. The fact that Dp427 or Dp71 expression level are not prognostic for metastatic progression is not very surprising given that Dp427 and Dp71 are respectively barely and widely expressed among tumors and so, Dp71 or Dp427 expression levels taken apart may not be the determining parameter for metastatic progression. On the other hand, this strengthens the idea that the impact of DMD on metastatic progression is rather determined by the overall expression pattern of the different isoforms—that is to say, Dp71 expression combined with Dp427 deletion—and possibly relies on their protein interactions and/or subcellular localizations. Finally, our work points to Dp71 as a highly interesting therapeutic target, at least in Dp427-deleted sarcoma tumors. This would highlight the point elegantly made by Muller et al. in 2012, when they stated that deletions can expose cancer-specific therapeutic vulnerabilities when the deleted gene is a member of a functionally redundant family of genes carrying out an essential function [
20].
4. Materials and Methods
4.1. Ethics Statement
The samples used in this study are part of the Biological Resources Center of Institut Bergonié (CRB-IB). In accordance with the French Public Health Code (articles L. 1243-4 and R. 1243-61), the CRB-IB received agreement from the French authorities to deliver samples for scientific research (number AC-2008-812). Pleomorphic sarcoma, synovial sarcoma, and GIST cohorts have already been reported in previous studies from our team [
21,
22,
23,
24]. Every case was histologically reviewed by the pathologist subgroup of the French Sarcoma Group and classified according to the 2013 World Health Organization classification by histology, immunohistochemistry, and molecular genetics and cytogenetics when needed (Cohort description in
Table 1).
4.2. Cell Lines
Cell lines (IB112 and IB136) were established as previously described [
25]. Authentication of cell lines was performed by CGH array and compared with the corresponding original tumor. Sarcoma cell lines were cultured in RPMI 1640 + GlutaMAX I (Life Technologies, Inc., brand of ThermoFisher Scientific, Waltham, MA, USA) supplemented with 10% FCS and 1% penicillin-streptomycin (Life Technologies). The HEK-293T cell line was cultured in DMEM + GlutaMAX I (Life Technologies) supplemented with 10% FCS and 1% penicillin-streptomycin (Life Technologies). Cells were grown at 37 °C in a humidified atmosphere containing 5% CO
2.
Sarcoma cell lines IB112, IB136, and SW982 were infected with a lentiviral vector containing the shRNA sequence against DMD under the control of a Tet-on promoter. Control cell lines were established with lentiviral transduction of a vector containing a non-targeting shRNA. VSV-G-pseudotyped lentiviral particles were produced by co-transfection of 293T cells with previous vectors and the compatible packaging plasmids psPAX2 and pVSVg. Cell lines were incubated overnight with lentiviral supernatants and 8 μg/mL polybrene (Sigma H9268). Stably transduced cells were selected with the addition of puromycin (1 µg/mL, Sigma P9620) to culture medium. Downregulation of the protein was verified by Western blotting after induction of shRNA expression by doxycycline (2 µg/mL).
IB133 was infected with a lentiviral vector containing the CRISPR/CAS9 system and the gRNA specifically targeting Dp427 isoforms (Hs0000003709, Sigma Aldrich, St. Quentin Fallavier, France). A control cell line was established with lentiviral transduction of a vector containing a gRNA CNeg targeting no sequence of the human genome (CRISPR/CAS gRNA-NEGATIVE CONTROL 1, CRISPR06, Sigma Aldrich). Cell infection was done as previously. Lentiviral transduction of the CRISPR/Cas9 system was performed in five replicates that consequently produced five IB133 cell lines potentially carrying DMD deletion and five control cell lines (Cneg). Deletion and inhibition of Dp427 were verified by Sanger sequencing and Western blot, respectively.
4.3. Bioinformatics Analysis Pipeline for Genomic and Transcriptomic Data
Genomic DNA was extracted using the standard phenol-chloroform extraction protocol [
24]. The Affymetrix SNP array 6.0 (Affymetrix, Santa Clara, CA, USA) was used according to the manufacturer’s instructions. One hundred and six samples were normalized with the Genotyping console 2.0 software (Affymetrix). Total RNAs were extracted as previously described [
21]. RNA quality was checked on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples were then analyzed on Human Genome U133 Plus 2.0 array (Affymetrix), according to the manufacturer’s procedures. All expression data obtained are publicly available on
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE71118.
4.4. RNA Sequencing
The process from RNA extraction to final BAM files was previously described [
11]. We used SAMtools and BCFtools (v0.1.19) [
26,
27] with custom depths (at least two alternate bases and five total bases) for reporting a candidate variant. These variants were then annotated by ANNOVAR (October 2013) [
28] with the hg19 genome version, transcriptome annotations (November 2013), and using the databases of the observed variants: dbSNP (v138) [
29], 1000G project (April 2012) [
30], ClinVar (September 2014) [
31], and COSMIC (v70) [
32].
4.5. Sanger Sequencing
To observe the effect of CRISPR/Cas9 on gDNA, the primers used were: CATTGAAAGCTAGAAGGTGAG (Forward) and GTTCATTCCAATGGAACGTTAG (Reverse). The Touchdown 60 °C program was used (TD 60 °C; two cycles at 60 °C, followed by two cycles at 59 °C, two cycles at 58 °C, three cycles at 57 °C, three cycles at 56 °C, four cycles at 55 °C, four cycles at 54 °C, five cycles at 53 °C, and finally 10 cycles at 52 °C). PCR was performed on 25 ng of DNA using AmpliTaqGold DNA polymerase (Applied Biosystems, brand of ThermoFisher Scientific, Waltham, MA, USA). PCR products were then purified using the ExoSAP-IT PCR purification kit (GE Healthcare), and sequencing reactions were performed with the Big Dye Terminator V1.1 kit (Applied Biosystems) according to the manufacturer’s recommendations. Samples were then purified using the Big Dye XTerminator purification kit (Applied Biosystems) according to the manufacturer’s instructions, and sequencing was performed on a 3130xl Genetic Analyzer (Applied Biosystems). Sequence analysis was performed with SeqScape software v2.5 (Applied Biosystems).
4.6. Western Blot
Cells were rinsed with ice-cold PBS and lysed for 20 min at 4 °C in RIPA lysis and extraction buffer (R0278, Sigma) supplemented with a protease/phosphatase inhibitor cocktail (11697498001, Roche, Basel, Switzerland). Lysates were pelleted for 10 min at 15,000× g at 4 °C and supernatants were collected for protein quantitation (DC protein assay kit, Bio-Rad, Hercules, CA, USA). The total proteins of each sample (40 µg) were loaded on gels and separated by SDS-PAGE. Following transfer onto a PVDF membrane using a dry transfer system, membranes were blocked in non-fat dry milk in PBS-Tween 0.1% and then incubated with the primary antibody: Mouse anti-Dp427 (NCL-DYS1, Leica Biosystem, Nussloch, Germany) or Rabbit anti-Dp71 (Ab15277, Abcam) at 4 °C overnight. After washing, blots were incubated for 1 h with a horseradish-peroxidase-linked anti-rabbit antibody (Amersham, brand of GE Healthcare Europe GmbH, Velizy-Villacoublay, France) and processed for chemiluminescent substrate (Amersham ECL Select detection reagent kit, Sigma) according to the manufacturer’s instructions. Signal was detected using the Fusion Fx7 (Thermo Fisher Scientific, Waltham, MA, USA) imaging system. GAPDH (Santa Cruz Biotechnology, sc-166574, Dallas, TX, USA) was used as a loading and the quantification of protein abundance was performed by blot densitometry using the ImageJ 1.48v software (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, MD, USA).
4.7. TaqMan Expression
cDNAs were synthesized from 1 μg of RNA using the GeneAmp RNA PCR core kit (Applied Biosystem, Courtaboeuf, France). Quantitative PCR analyses were performed using TaqMan Assays-on-demand Gene expression reagents (Applied Biosystem) with qPCR Mastermix Plus without UNG (Eurogentec, Belgium). We used the TaqMan Gene Expression assays provided by Applied Biosystems. The assay IDs were as follows: Hs00758098_m1 for DMD, and Hs99999902_m1 for RPLP0. To normalize the results, we used the RPLP0 gene as a reference gene.
4.8. Proliferation Assay
For this, 5000 cells were seeded in five replicates in 96-well plates. Culture medium was changed every 2 days with or without doxycycline. Every 2 to 3 days, cells were washed, trypsinized, and harvested in a final volume of 200 µL of PBS1X plus 10% of FBS. The number of viable cells was evaluated by flow cytometry (FACS Calibur, BD Biosciences, San Jose, CA, USA) based on their morphological features. Data were acquired using CellQuestPro software (BD FACS Systems, Sunnyvale, CA, USA) and analyzed using FlowJo (Tree Star, Celeza GmbH, Ashland, OR, USA) and GraphPad (La Jolla, CA, USA) software.
4.9. Clonogenic Assay
To assess cell clonogenic activity, 1000 cells were plated on 6-well plates in standard culture medium. Cells were incubated for 10 days at 37 °C and 5% of CO2. Medium was changed every 2 days with or without doxycycline. Then, cells were washed with PBS 1X, fixed for 5 min with 70% ethanol and then stained with crystal violet and scanned.
4.10. Migration Assay
For the wound healing assay, 4.105 cells were plated on a 6-well plate. Twenty-four hours later, a strip of cells was removed from the monolayer of cells using a pipette tip. Phase contrast images were acquired with a 10× objective at the time of the scratch and 12 hours later using a Nikon Eclipse TS100 microscope.
4.11. Cell Cycle
Cells were fixed after 48 h incubation with or without doxycycline. Then, cells were stained with propidium iodide with the FxCycle™ PI/RNase Staining Solution (F10797, Life Technologies) according to the manufacturer’s instructions. Data were acquired using CellQuestPro software (BD FACS Systems, Sunnyvale, CA, USA) and at least 10,000 cells per sample were analyzed using FlowJo (Tree Star, Celeza GmbH).
4.12. Statistical Analysis
Metastasis-free survival (MFS) was defined as the interval between diagnosis and the time of distant recurrence or the last follow-up. Survival rates were estimated using the Kaplan–Meier method and compared using the log-rank test and hazard ratios (HRs).
Each experiment was repeated at least three times. To examine the statistical significance of the results, analyses were performed with Prism6 v6.01 (GraphPad software Inc., La Jolla, CA, USA) software. Normal distribution of data sets was examined with a Shapiro–Wilk normality test. If data passed the test, the statistical significance between two conditions was assessed with an unpaired t-test, and results were represented as mean ± standard deviation (SD). Otherwise, a Mann–Whitney test was used. Significant differences are represented as * p-value p < 0.05, ** p < 0.01, and *** p < 0.001.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}