 Søs Grønbæk Mathiassen
Søs Grønbæk Mathiassen Daniela De Zio
Daniela De Zio Francesco Cecconi
Francesco Cecconi- 1Cell Stress and Survival Unit, Danish Cancer Society Research Center, Copenhagen, Denmark
- 2Department of Biology, University of Rome Tor Vergata, Rome, Italy
- 3Department of Pediatric Hematology and Oncology, Istituto di Ricovero e Cura a Carattere Scientifico Bambino Gesù Children’s Hospital, Rome, Italy
Autophagy is a self-degradation pathway, in which cytoplasmic material is sequestered in double-membrane vesicles and delivered to the lysosome for degradation. Under basal conditions, autophagy plays a homeostatic function. However, in response to various stresses, the pathway can be further induced to mediate cytoprotection. Defective autophagy has been linked to a number of human pathologies, including neoplastic transformation, even though autophagy can also sustain the growth of tumor cells in certain contexts. In recent years, a considerable correlation has emerged between autophagy induction and stress-related cell-cycle responses, as well as unexpected roles for autophagy factors and selective autophagic degradation in the process of cell division. These advances have obvious implications for our understanding of the intricate relationship between autophagy and cancer. In this review, we will discuss our current knowledge of the reciprocal regulation connecting the autophagy pathway and cell-cycle progression. Furthermore, key findings involving nonautophagic functions for autophagy-related factors in cell-cycle regulation will be addressed.
Introduction
Macroautophagy (herein referred to as autophagy) is a highly conserved catabolic pathway that mediates the sequestration and delivery of cytoplasmic material to the lysosome for degradation. This is achieved by the formation and expansion of an isolation membrane (or phagophore) that fuses to engulf cytoplasmic constituents in a double-membrane autophagic vacuole (the autophagosome). The autophagosome finally undergoes fusion with lysosomes whereby the enclosed cargo is degraded and subsequently released and recycled to support cellular metabolism. In physiological conditions, autophagy proceeds at a basal level to ensure the turnover of superfluous or damaged components, including organelles and long-lived proteins, to maintain cellular homeostasis. Moreover, the autophagic flux can be upregulated in response to a wide range of stresses, such as nutrient deprivation, reactive oxygen species, DNA damage, protein aggregates, damaged organelles, or intracellular pathogens, whereby it functions as an adaptive cytoprotective response (1, 2).
The molecular pathway that orchestrates the initiation and execution of autophagy has been comprehensively reviewed elsewhere (3–5). In short, the initiation phase of autophagy is governed by two main complexes: the unc-51-like autophagy-activating kinase (ULK) complex and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex (Figure 1A). The PtdIns3K complex produces phosphatidylinositol 3-phosphate (PtdIns3P) for recruitment of additional autophagy factors to the phagophore and is partially comprised of the key autophagy regulators vacuolar protein sorting 34 (Vps34), Beclin 1, vacuolar protein sorting 15 (Vps15), and activating molecule in Beclin 1-regulated autophagy (AMBRA1). Downstream of these complexes are two ubiquitin-like conjugation systems that mediate vesicle expansion [the autophagy-related gene 8 (Atg8) and autophagy-related gene 12 (Atg12) systems]. Both systems require the E1-like protein autophagy-related gene 7 (Atg7) for activation of the ubiquitin-like proteins Atg8 [light chain 3 (LC3) in mammals] and Atg12. In the Atg8 system, Atg8/LC3 undergoes proteolytic processing and covalent attachment to the lipid phosphatidylethanolamine (in mammalian cells, the precursor form is termed LC3-I and the lipidated form LC3-II), by which it becomes associated with the phagophore membrane. Consequently, autophagy can be detected biochemically (by assessing the generation of LC3-II) or microscopically (by observing the formation of LC3 puncta, representative of LC3 redistribution to the developing autophagosomes). Apart from these systems, the pathway includes the transmembrane protein autophagy-related gene 9 (Atg9), as well as factors involved in autophagosome–lysosome fusion [e.g., lysosomal-associated membrane protein 2 (LAMP2)], vacuolar permeases mediating the efflux of amino acids from the lysosome, and lysosomal enzymes required for cargo degradation (3–7). Furthermore, while originally considered a largely unspecific process, recent years have revealed the existence of selective autophagy pathways, in which specific cargoes can be targeted to the emerging autophagosomes for engulfment and degradation. Cargoes destined for selective autophagy are often ubiquitinated and recognized by autophagy receptors [i.e., p62/sequestosome 1, neighbor of BRCA1 gene (NBR1), nuclear dot protein 52 kDa (NDP52), optineurin, or C-Cbl] that contain ubiquitin-binding domains as well as LC3-interacting region (LIR) motifs for recruitment to the inner phagophore membrane (8, 9) (Figure 1A).
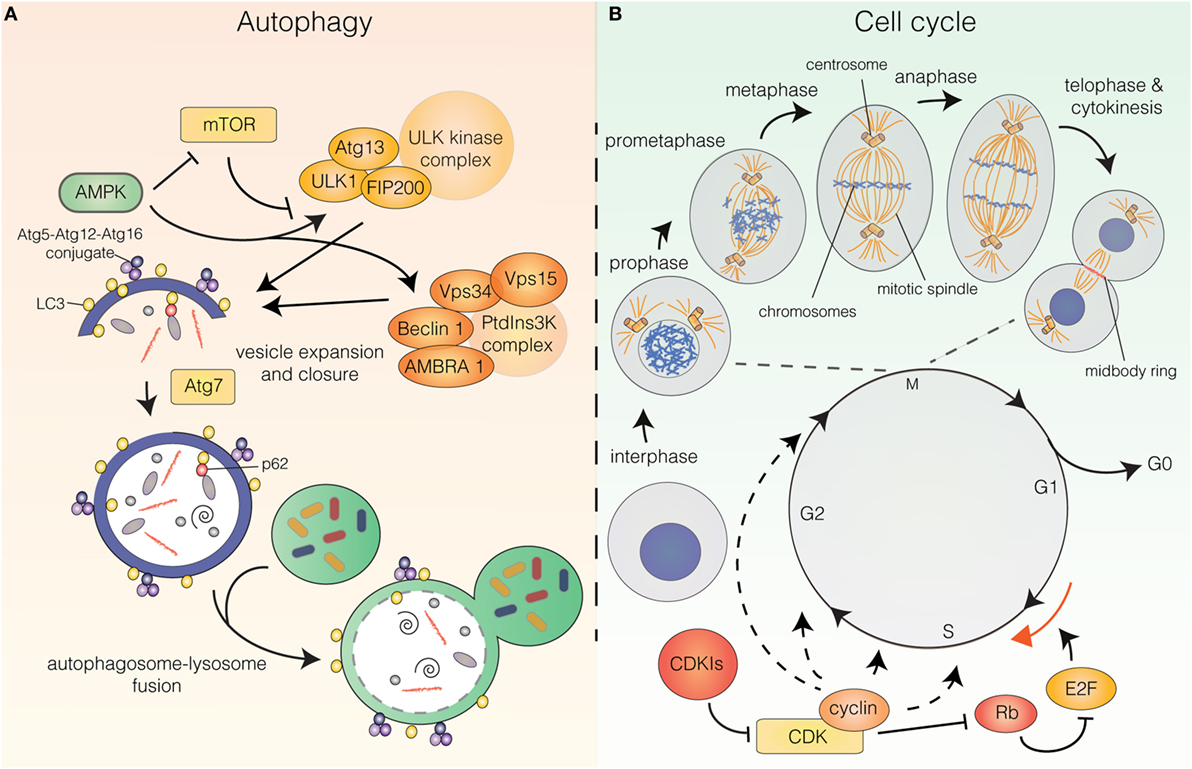
Figure 1. (A) The autophagy pathway. Autophagy induction is controlled upstream by energy sensors, mammalian target of rapamycin (mTOR), and AMP-activated protein kinase (AMPK). mTOR shuts off autophagy in the presence of abundant nutrients, while AMPK is activated upon energy stress. AMPK induces autophagy by inhibiting mTOR and stimulating upstream autophagy factors of the unc-51-like autophagy-activating kinase (ULK) and class III phosphatidylinositol 3-kinase (PtdIns3K) complexes. Vesicle expansion requires the autophagy-related gene 8 (Atg8)/light chain 3 (LC3) and autophagy-related gene 12 (Atg12) ubiquitin-like conjugation systems. Autophagy receptors (e.g., p62) can mediate selective recruitment of cargo to the inner vesicle membrane. Following vesicle closure, the autophagosome fuses with the lysosome whereby the engulfed material is degraded. (B) The cell cycle. The cell cycle can be divided into G0, G1, S, G2 (interphase), and M-phase (mitosis and cytokinesis). Mitosis can be subdivided into prophase (DNA condensation is initiated), prometaphase (the mitotic spindle starts to form and the nuclear envelope has been dissolved), metaphase (the chromosomes are aligned at the metaphase plate), anaphase (separation of the sisterchromatids) and telophase (DNA decondenses, the nuclear envelope reforms, the contractile ring starts forming) and is followed by cytokinesis (physical separation of the daughter cells). Cell-cycle progression is governed by cyclin-dependent kinase (CDK) holoenzymes. CDK activity can be inhibited by cyclin-dependent kinase inhibitors. For G1/S transition cyclin-CDKs phosphorylate retinoblastoma protein (Rb), which releases E2 factor (E2F) transcription factors from inhibitory binding, leading them to induce transcription of targets for G1/S transition.
Autophagy induction is controlled upstream by energy-sensing proteins, a key regulator being the mammalian target of rapamycin (mTOR), which provides the major inhibitory signal that shuts off autophagy in the presence of abundant nutrients. A key inhibitor of mTOR AMP-activated protein kinase (AMPK) is activated upon energy stress that increases the AMP/ATP ratio. Once activated, AMPK downregulates ATP-consuming (anabolic) pathways and upregulates ATP-generating (catabolic) pathways, such as autophagy, to maintain cellular energy homeostasis. Besides inhibiting the catalytic activity of mTOR, AMPK also directly stimulates autophagy by phosphorylating upstream autophagy factors [e.g., unc-51-like autophagy-activating kinase 1 (ULK1) and Beclin 1] (1, 3, 4) (Figure 1A).
In recent years, the notion that autophagy may represent a bona fide tumor suppressor pathway has obtained increasing support. Autophagy-deficient animal models are often prone to tumor formation (10–16) and autophagy deficiency is associated with increased DNA damage and chromosomal instability (CIN) (17). Thus, autophagy is thought to constitute a barrier against malignant transformation by preserving intracellular homeostasis, even though the exact mechanism of autophagy-mediated oncosuppression is not well-understood. Autophagy can conversely sustain the survival and proliferation of neoplastic cells exposed to intracellular and environmental stresses, such as hypoxia and chemotherapy, and thereby supports tumor growth and progression. Hence, depending on the context, autophagy can act either as a tumor-suppressive or a tumor-promoting pathway (2, 18, 19).
As many signaling pathways exhibit opposing effects on autophagy and cell-cycle progression (20), these are often considered mutually exclusive processes. Accumulating evidence suggests that this opposing regulation may be coordinated and that an interplay between the two processes exists. This is exemplified by the scaffold protein AMBRA1, a pro-autophagic protein that is also able to negatively regulate the oncogene c-Myc (10). AMBRA1 interacts with the catalytic subunit of the protein phosphatase 2 A (PP2A) and facilitates PP2A-mediated dephosphorylation and subsequent proteasomal degradation of c-Myc, thus resulting in inhibition of proliferation and in tumor suppression (10). Both the role of AMBRA1 in promoting c-Myc degradation, as well as in AMBRA1-dependent autophagy, is controlled upstream by mTOR (10, 21), which argues for a coordinated regulation of autophagy and cell-cycle progression.
In the present review, we will focus on various aspects of the reciprocal regulation connecting autophagy and the cell cycle. Cell-cycle progression is governed by cyclin-dependent kinases (CDKs). CDK activity is coordinated by binding of their essential regulatory subunits, cyclins, which are synthesized and degraded at specified times during the cell cycle to coordinate timely CDK activation and drive cell-cycle progression (Figure 1B). The decision to enter or exit the cell cycle depends on the nutrient and mitogen availability and is also affected by stress-stimuli that may block the cell cycle transiently or irreversibly. Once committed to cell-cycle progression, the cell undergoes a series of regulated events (i.e., cell growth, DNA replication, and quality control checkpoints) culminating in the highly orchestrated process of cell division. Dysregulation of proteins controlling the frequency and fidelity of proliferation is inextricably linked to neoplastic transformation (22–24).
Herein, we will address the activation of autophagy during normal and abnormal cell-cycle progression as well as the coordinated induction of autophagy and cell-cycle responses following exposure to various stresses. Finally, the involvement of autophagy and autophagy-related proteins in the regulation of cell division will be discussed.
Autophagy Status During Cell-Cycle Progression
Only few studies have focused on a putative correlation between autophagy flux and cell-cycle progression. The cell cycle can be divided into five major phases: G0, G1, S, G2, and M-phase (Figure 1B). G0, G1, S, and G2 are collectively referred to as interphase, while M-phase is comprised of mitosis and cytokinesis, the processes by which the duplicated genome and other cellular constituents are distributed to the two daughter cells and the subsequent separation of these. Mitosis is traditionally subdivided into five phases: prophase (DNA condensation is initiated), prometaphase (the mitotic spindle starts to form and the nuclear envelope is dissolved), metaphase (the chromosomes are aligned at the metaphase plate), anaphase (separation of the sister chromatids to separate chromosomes) and telophase (DNA decondenses, the nuclear envelope is reformed and the contractile ring at the intercellular bridge between the two nuclei starts forming). This is followed by cytokinesis, in which the two daughter cells are physically separated (25) (Figure 1B).
Autophagy and Interphase
The question of differential regulation of autophagy during cell-cycle progression was initially addressed by Tasdemir et al., prompted by their observation that autophagy-inducing treatment of unsynchronized cell populations only induced green fluorescence protein (GFP)–LC3 aggregation in approximately 50% of cells (26). To understand if autophagy preferentially occurs in certain cell-cycle phases, immunocytochemical approaches were employed to monitor cytoplasmic GFP–LC3 aggregation in connection with cell-cycle progression (26). Using a panel of autophagy activators, including the BH3 mimetic ABT737, lithium, rapamycin, tunicamycin, or starvation, autophagy induction was observed to preferentially occur in the G1 and S phases of the cell cycle (26). More recently, Kaminskyy et al. developed another strategy to monitor autophagosome accumulation by extracting membrane-unbound LC3-I from cells, followed by flow cytometric detection of the remaining autophagosomal membrane-associated fraction of LC3-II. This was combined with propidium iodide staining for detection of cell-cycle status (27). By using this approach, basal autophagy was detected in G1, S, and G2/M phases. Furthermore, autophagy induction by starvation or rapamycin treatment resulted in LC3-II accumulation in all stages (27), suggesting the absence of cell-cycle-dependent autophagy regulation. The contradictory findings may be the result of the variant experimental approaches. Thus, further studies are required to determine if autophagy activation is preferentially linked to specific cell-cycle phases.
Autophagy and Mitosis
As the above studies do not allow discrimination between G2 and M phase, this leaves the question of autophagy status during mitosis. Two elegant studies have reported a striking decrease in autophagic activity during mitosis (28, 29). By means of electron microscopy and stereology to quantify the presence of autophagic vacuoles in mitotic cells, Eskelinen et al. found a strong reduction in autophagosomal content in both (pro)metaphase and anaphase cells (28). Furuya et al. expanded on these findings revealing that mitotic autophagy inhibition depends on cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of Vps34 on Thr159 during mitosis (29). This phosphorylation event negatively regulates the interaction between Vps34 and Beclin 1, thereby inhibiting PtdIns3K activity, PtdIns3P production, and autophagy induction (29). Of note, during mitosis, cells undergo extensive structural rearrangements and the inhibition of autophagy has been speculated to function as a protective mechanism to prevent unintended loss of organelles and chromosomes. Indeed, break down of the nuclear envelope during mitosis leaves the condensed chromosomes potentially vulnerable to the cytoplasmic autophagy machinery. Accordingly, Eskelinen et al. observed that re-appearance of autophagosomes occurred in telophase/G1 after formation of the new nuclear envelopes (28). Furthermore, autophagosomal engulfment of mitotic chromosomes was reported in mitotic cells undergoing programmed cell death (30), suggesting that autophagy inhibition may, indeed, protect the condensed genome from accidental autophagic engulfment. Moreover, during cell division, mitochondria and the Golgi apparatus become fragmented to facilitate their distribution between the two daughter cells (31, 32). While elongated mitochondria are spared from autophagic degradation (33, 34), the smaller size of fragmented mitochondria facilitates their uptake by autophagosomes (35, 36). Mitotic fragmentation of mitochondria is mediated by CDK1-dependent phosphorylation and activation of the dynamin-like protein (Drp1), involved in mitochondrial fission (37). Interestingly, cells arrested in mitosis by abrogated Cyclin B1 degradation, exhibit a gradual decline in mitochondrial mass due to ongoing mitophagic degradation (38). Prevention of mitophagy by depletion of Drp1 or key autophagy proteins delayed cell death by mitotic arrest; thus, mitophagy may facilitate mitotic cell death during prolonged mitotic block (38). The resistance to mitotic cell death acquired upon Drp1 knock-down supports the speculated vulnerability of fragmented mitotic mitochondria to autophagic degradation. Ongoing mitophagy during mitotic arrest may simply represent leaky degradation from incompletely blocked autophagy, which is functionally relevant during prolonged mitotic arrest but likely negligible during normal mitotic progression. However, this mechanism may also participate in pushing cells with mitotic abnormalities toward cell death.
In accordance with the reported ongoing mitophagy in arrested mitotic cells (38), LC3 puncta have been observed in mitotic cells, although at a significantly decreased level compared to interphase cells (28, 29, 39, 40). While these may also represent inefficient autophagy inhibition, Loukil et al. observed LC3, p62, and lysosomal markers colocalizing with Cyclin A2 foci during mitosis and found that autophagy partially contributes to mediating mitotic Cyclin A2 degradation (40). Thus, an intriguing although highly controversial theory is the existence of distinct sites of active autophagy during cell division. Treatment with autophagy inducers or lysosomal inhibitors has been shown to result in accumulation of LC3 puncta in mitotic cells, which was suggested as an indication of active autophagy flux in mitosis (39, 41). The short duration of mitosis, however, poses technical challenges in employing these treatments, as it is difficult to rule out autophagosome accumulation from interphase. Live-cell imaging using GFP–LC3 cell lines or preferably cell lines carrying endogenously tagged autophagy proteins may help in determinining the degree of autophagy inhibition as well as the potential presence of active autophagic compartments in mitosis.
Interplay Between Autophagy and Cell-Cycle Arrest
In response to unfavorable or stressful conditions, cells are able to arrest the cell cycle transiently or irreversibly. This ability helps regulate proliferation during development and differentiation, and prevents the expansion of potentially harmful cell populations (23, 42). Autophagy, like cell cycle arrest, is induced in response to a variety of stress conditions, where it plays a pivotal role in preserving cellular viability (2). While the correlative induction of autophagy and cell-cycle arrest has been extensively documented, the molecular mechanisms linking them together are still debated and largely unknown.
Autophagy Regulation by Cyclin-Dependent Kinase Inhibitors (CDKIs) and Retinoblastoma Protein (Rb)/E2 Factor (E2F) Activity
Cell-cycle arrest often relies on the action of various cell-cycle inhibitors. An important class of those are CDKIs that inhibit CDK activity by direct interaction with CDKs or cyclin-CDK holoenzymes (43) (Figure 1B). CDKIs can be categorized into two main families: the inhibitors of CDK4 (INK4) family consisting of p15INK4B, p16INK4A, p18INK4C, and p19INK4D; and the Cip/Kip family composed of p21Cip1, p27Kip1, and p57Kip2 (23, 42). In spite of their similar modes of action, CDKIs are speculated to have functionally distinct roles and appear to be activated by different stimuli (42). Thus, while p21 is most strongly linked to stress and DNA damage signaling, downstream of p53-mediated pathways, p27 is more often associated with cell-cycle arrest in response to low nutrient and mitogen conditions (42). CDKIs were originally strictly linked to proliferation control, but they are now demonstrated to have a wide range of alternative functions in processes including transcription, apoptosis, migration (42), as well as autophagy induction (44–46). Cell-cycle arrest can also occur by repression of E2F transcription factors that mediate transcriptional induction of a plethora of targets, including cyclins and replication regulators required for G1/S transition and cell-cycle progression (47, 48) (Figure 1B). E2F activity is controlled by binding of the Rb protein or other Rb family members (49). Upon mitogenic stimuli, Rb is gradually phosphorylated by cyclin-CDK complexes whereby E2F is released to induce transcription of its target genes, pushing cells to pass the G1/S boundary (47, 49). CDKIs, through their ability to inhibit CDKs, are also important indirect promoters of Rb/E2F interactions (48) (Figure 1B).
A number of CDKIs, including p16, p21, and p27 have been reported to induce autophagy (44–46), suggesting the existence of coordinated stress responses linking autophagy induction and cell-cycle arrest. Liang et al. showed that in response to starvation, p27 is activated by the liver kinase B1 (LKB1)–AMPK nutrient-sensing pathway through phosphorylation of Thr198, thus resulting in p27 stabilization (46). Interestingly, p27 was required for efficient starvation-induced autophagy in murine embryonic fibroblast and protected from cell death resulting from metabolic stress (46), indicating a critical role for p27 in autophagy activation under starvation conditions. The increased stability of p-p27Thr198 implies a function for the LKB1–AMPK pathway in mediating p27-dependent cell-cycle arrest. Accordingly, a nonphosphorylatable p27T198A mutant was less efficient than wild type p27 or a phospho-mimicking p27T198D mutant at inhibiting colony formation (46). This is in line with previous reports arguing for a central role for p27 in starvation-induced cell-cycle arrest (50, 51). p27 is upregulated in response to serum starvation (50) and its depletion allows serum-starved cells to evade cell-cycle arrest and continue proliferation (50, 51). Thus, p27 may be a key effector of the cellular response to metabolic stress, functioning downstream of the LKB1–AMPK axis to mediate both cell-cycle arrest and autophagy induction. Accordingly, p27 is degraded by caspases during growth-factor deprivation-induced apoptosis (52).
The mechanism by which p27 mediates autophagy induction and the relevance of its CDK inhibitory function in this context is, however, not clear. Nonetheless, it has been reported that the cyclin-binding region of p27 is required for autophagy induction (46, 53) and that depletion of CDK2 and CDK4 partially reproduces p27-induced effects on autophagy and apoptosis (46). In this context, indirect activation of Rb by p27 could be a contributing factor, as this has been reported for p16 (45). Overexpression of p16 is able to induce autophagy in an Rb-dependent manner through promoting Rb/E2F interaction (45), which suggests negative regulation of autophagy by E2Fs. This supports a model in which p16-mediated CDK inhibition facilitates Rb/E2F interaction and consequent E2F inhibition, resulting in activation of autophagy through an unspecified mechanism. However, while autophagy induction by p16 appears to largely depend on Rb/E2F regulation, p27-induced autophagy was only mildly affected by Rb status (45), suggesting varying mechanisms of autophagy activation between CDKIs. Intriguingly, in budding yeast, the CDK Pho85 is able to both induce or inhibit autophagy, depending on its associated cyclin partner (54).
The literature linking Rb/E2F and autophagy is complex, as positive regulation of autophagy by E2Fs has also been reported. Using an inducible E2F activation system, Polager et al. demonstrated that several autophagy genes such as LC3, ULK1, and DRAM were direct targets of E2F transcription factors (55). Moreover, E2Fs were shown to bind the promoter region of Beclin 1 (56), even though the functional significance of this binding remains to be demonstrated. E2F downstream targets such as smARF or the hypoxia-inducible B-cell lymphoma 2 (Bcl-2) family member BCL2 interacting protein 3 (BNIP3) have also been shown to induce autophagy (44, 57, 58). BNIP3 was demonstrated to be required for efficient hypoxia-induced autophagy activation (58) and E2F1 to be required for efficient DNA-damage-induced autophagy (55). This evidence indicates a potential role for E2Fs in mediating autophagy during acute stress responses, rather than during normal cell-cycle progression. E2F-mediated autophagy induction may therefore depend on the context and stimuli. Furthermore, as the E2F family comprises eight family members that can both transactivate and repress gene expression (47), E2F contribution to autophagy regulation likely depends on the involved E2F factor.
Autophagy and p53
The most well-documented connection between autophagy and stress-induced cell-cycle responses is likely the link between p53 and autophagy regulation. p53 is one of the most extensively characterized tumor suppressor proteins and a central coordinator of the cellular response to acute stress (59, 60). Under basal conditions, p53 levels are strictly controlled by mouse double minute 2 homolog (Mdm2)-mediated ubiquitination and proteasomal degradation, while in response to a wide range of stresses (e.g., DNA damage, oncogene expression or nutrient deprivation), p53 undergoes rapid post-translational modifications that allow for its stabilization and activation (59) (Figure 2). Upon activation, p53 orchestrates the induction of appropriate cellular responses, be it apoptosis, cell-cycle arrest, DNA repair, metabolic adaptation, or autophagy, with the purpose of limiting the expansion of damaged and potentially harmful cells (59, 60) (Figure 2). The shared involvement of p53 and autophagy in stress-related processes, as well as their relevance for neoplastic transformation has motivated great efforts to understand the role of autophagy ablation in the context of p53-deficient and -proficient animal models of human cancers, reviewed in Ref. (61). In this article, we will focus our attention on the molecular mechanisms linking p53 to autophagy regulation.
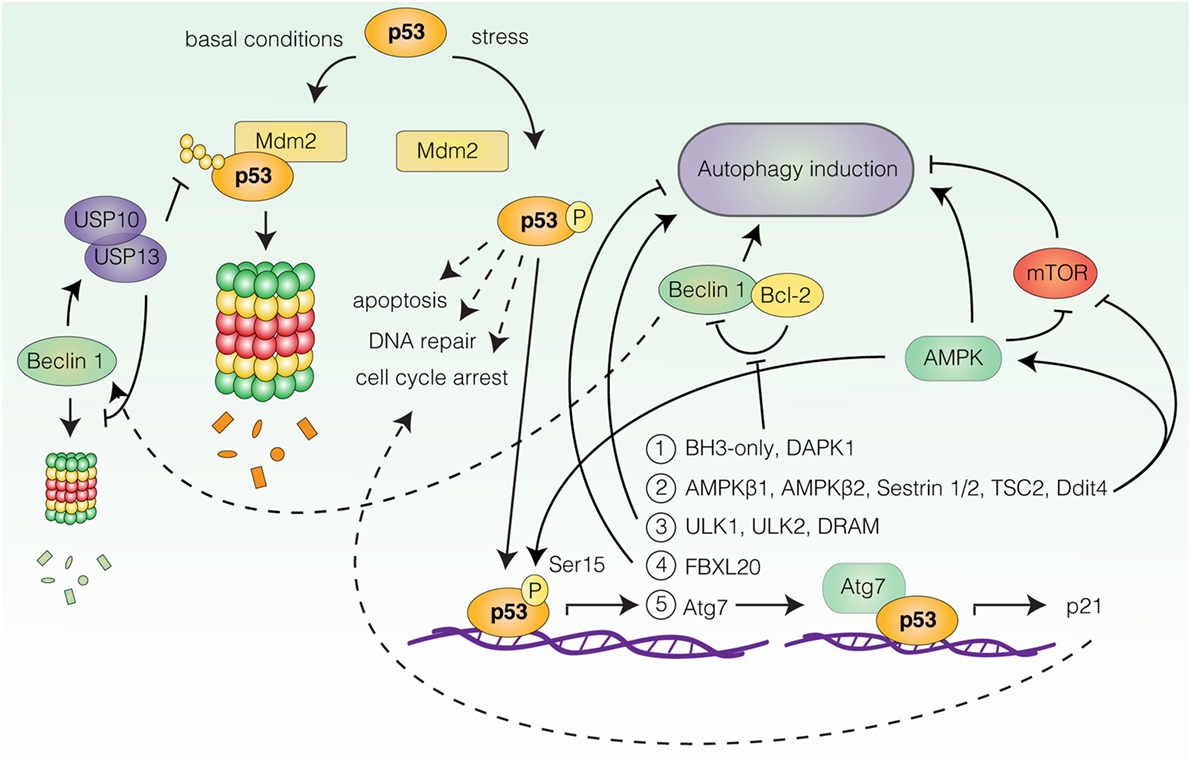
Figure 2. Transcriptional regulation of autophagy by p53. Under basal conditions p53 is degraded by mouse double minute 2 homolog (Mdm2)-mediated proteasomal degradation. In response to stress, p53 undergoes post-translational modifications leading to its stabilization and activation. Upon activation, p53 can induce transcription of autophagy-related genes (only a selection is represented here). Group 1: BH3-only proteins and death-associated protein kinase 1 (DAPK1), all stimulate autophagy by favoring Beclin 1 displacement from B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma extra large (Bcl-XL). Beclin 1 can contribute to p53 stabilization by stabilizing the deubiquitinating enzymes ubiquitin-specific peptidase 10/13 (USP10/13). Group 2: AMP-activated protein kinase (AMPK) subunits β1 and β2, AMPK activators Sestrin 1/2, negative regulators of mammalian target of rapamycin (mTOR), tuberous sclerosis 2 (TSC2), and DNA damage-inducible transcript 4 (Ddit4), all promote autophagy induction. AMPK can in turn phosphorylate and activate p53. Group 3: unc-51-like autophagy-activating kinase 1 (ULK1), unc-51-like autophagy-activating kinase 2 (ULK2), and damage-regulated autophagy modulator. Target 4: F-box/LRR-repeat protein 20 (FBXL20) negatively regulates autophagy by promoting vacuolar protein sorting 34 (Vps34) degradation. Target 5: Key autophagy protein autophagy-related gene 7 (Atg7) cooperates with p53 for p21 induction.
Autophagy Modulation by Nuclear p53
A number of reports have demonstrated autophagy induction by p53 (18, 62, 63). The ability of p53 to stimulate autophagy appears to rely on its function as a stress-induced transcription factor, as p53 can transactivate a wide range of autophagy-related genes (18, 62, 63) (Figure 2). Activation of some of these genes converges on activation of AMPK and inhibition of mTOR. These include genes encoding the AMPKβ1 and β2 subunits (64), the AMPK activators Sestrin 1 and Sestrin 2 (65, 66), as well as negative regulators of mTORC1, tuberous sclerosis 2 (TSC2), phosphatase and tensin homolog (PTEN), and DNA damage-inducible transcript 4 (Ddit4) (64, 67, 68). Accordingly, Feng et al. reported that p53-induced autophagy following DNA damage relied on AMPK-mediated inhibition of mTOR (69). Other p53 responsive genes include ULK1 and unc-51-like autophagy-activating kinase 2 (ULK2) (70), genes encoding various BH3-only proteins and death-associated protein kinase 1 (DAPK1), all of which stimulate autophagy by favoring the displacement of Beclin 1 from inhibitory interactions with Bcl-2 and Bcl-XL (71–74), as well as the gene coding for DRAM (75), a highly conserved lysosomal protein, which was also suggested to be required for p53-dependent autophagy induction in response to DNA damage (75). Furthermore, Kenzelmann Broz et al. utilized a high-throughput approach to uncover novel p53 transcriptional targets in response to DNA damage (67). This approach identified extensive transactivation of the autophagy network, encompassing both upstream autophagy regulators, members of the autophagy core machinery, and lysosomal proteins by all three p53 family members; p53, p63, and p73 (67). Interestingly, one of the identified targets, Atg7 (67), has been reported to bind the promoter of p21, collaborating with p53 for efficient p21 upregulation in a nonautophagy-dependent manner (76) (Figure 2). Thus, p53-dependent upregulation of Atg7 may function as an effector mechanism boosting the p53 response through p21 production. Similarly, AMPK can activate p53 upon glucose deprivation by phosphorylation of Ser15, which is required for AMPK-mediated cell-cycle arrest in this context (77) (Figure 2). Surprisingly, Beclin 1 can also contribute to p53 stabilization by promoting the stabilization of deubiquitinating enzymes ubiquitin-specific peptidase 10/13 (USP10/13) (78), which counteract the Mdm2-mediated degradation of p53 (78, 79), as well as degradation of Beclin 1 itself (78) (Figure 2). Thus, autophagy and p53 pathways may potentiate and sustain each other in establishing efficient stress-related cell-cycle programs.
Interestingly, activated p53 is also able to decrease autophagy, as the p53-responsive gene F-box/LRR-repeat protein 20 (FBXL20) is able to mediate the degradation of Vps34 following DNA damage, resulting in autophagy inhibition (80). In which context p53 activation results in autophagy stimulation and inhibition, respectively, is not understood. Furthermore, the effect of p53-induced autophagy is not clear, but in several contexts autophagy surprisingly appears to function as an effector of p53-mediated cell death rather than as a survival mechanism (67, 70, 75).
Autophagy Inhibition by Cytosolic p53
Contrasting the proautophagic transcriptional activity of nuclear p53, the cytoplasmic pool of p53 has been demonstrated to suppress autophagy (81). Knockout, depletion, or pharmacological inhibition of p53 in human, mouse as well as nematode cells, can induce autophagy in a manner appearing to depend on the AMPK/mTOR pathway (81). Correspondingly, p53 restricted to the cytosol but not nucleus-restricted p53 inhibited autophagy, a regulation that also persisted in enucleated cells (81). Accordingly, suppression of autophagy by p53 correlated with its nuclear-to-cytosolic distribution in a panel of cancer-associated p53 mutants (82). Surprisingly, several distinct proautophagic stimuli, including nutrient deprivation and mTOR inhibition by rapamycin were found to induce Mdm2-dependent proteasomal degradation of p53. Inhibition of proteasomal activity, Mdm2 depletion, or pharmacological inhibition of Mdm2 reduced autophagy induction in response to these stimuli (81), suggesting the requirement of p53 degradation for efficient autophagy activation. The molecular mechanism underlying this p53-mediated autophagy suppression is not understood, but has been suggested to involve negative regulation of the upstream autophagy factor RB1 inducible coiled-coil 1/FAK family kinase-interacting protein of 200 kDa (RB1CC1/FIP200) through a physical interaction with p53 (83). How the contradictory regimes of cytoplasmic versus nuclear p53-mediated autophagy regulation can be reconciled remains to be determined.
Autophagy and Senescence
While several lines of evidence suggest coordinated induction of autophagy and cell-cycle arrest pathways, another issue remains the involvement of autophagy in the execution of cell-cycle exit programs, in particular, senescence. The terms quiescence and senescence are often used interchangeably to describe cell-cycle arrest, although they refer to distinct cell states (84). Quiescence represents a reversible cell-cycle arrest often caused by lack of nutrients and/or mitogens and growth factors, while senescence is an irreversible state of cell-cycle arrest that is more often induced in abnormal (potentially cancerous), DNA-damaged, or aging cells as a stress response (84–86). While it is clear that autophagy and senescence are often parallel processes, the question of their interdependence is a subject of much debate. It is beyond the scope of the present review to comprehensively recapitulate the literature involving this topic, and for more on this subject, we refer to Ref. (84, 87, 88). In this article, we will focus our attention on key findings and recent publications that offer mechanistic insight to the relationship between autophagy and senescence.
Autophagy and Senescence Transition
In recent years, a number of studies have argued for a more direct link between autophagy and senescence that goes beyond their correlative induction, by showing that inhibition of autophagy delays senescence transition (89–93). Young et al. employed models of oncogene-induced and DNA damage-induced senescence to study autophagy activation during senescence transition (93). In the applied model of oncogene-induced senescence (OIS), an initial “mitotic phase” of proliferative burst occurs around day 1. This is followed by a “transition phase,” preceding the “senescence phase,” which is achieved after 5–6 days. Autophagy was induced specifically in the senescence transition phase in a manner that correlated with inhibition of mTOR activity. Importantly, Young et al. observed that depletion of the autophagy proteins autophagy-related gene 5 (Atg5) or Atg7 resulted in delayed senescence transition (93), thus indicating that autophagy contributes to the establishment of senescence. Similar results were obtained in a system of therapy-induced senescence, in which pharmacological or genetic inhibition of autophagy delayed senescence acquisition in response to treatment with the chemotherapeutic drugs adriamycin or camptothecin (90). In accordance with these findings, a recent study expands on a putative mechanism of autophagy-mediated senescence transition, as Dou et al. found that autophagy facilitates OIS by degrading the nuclear lamina constituent, Lamin B1, and associated heterochromatin domains called lamin-associated domains (LADs) (89). Degradation was a result of nuclear blebbing of Lamin B1 regions and a direct interaction between Lamin B1 and LC3, and preferentially occurred in response to oncogenic transformation, oxidative stress, and DNA damage, but not starvation (89), indicating that the degradation event is specific to a subset of stresses. Senescence was delayed upon expression of Lamin B1 mutants unable to bind LC3 and undergo autophagic degradation (89). Thus, autophagic Lamin B1 degradation may be of key importance during senescence transition. Interestingly, senescent cells have previously been shown to exhibit a gradual decline in histone mass that was dependent on lysosomal activity (94). Whether the degradation of Lamin B1-associated chromatin is of relevance for senescence transition is an interesting point for further investigation. Furthermore, autophagy was found to mediate the selective degradation of Δ133p53α (95), a p53 isoform suppressing the action of full-length p53 (96, 97), for induction of replicative senescence but not OIS (95, 97). Interestingly, overexpression of autophagy proteins is, in some cases, sufficient to stimulate coordinated induction of autophagy and premature senescence (93, 98). Nonetheless, as autophagy inhibition, in most cases, delays rather than fully abrogates senescence, it has been argued that autophagy is not required for senescence transition, but may function in potentiating and accelerating the response (87).
It should also be noted that active mTOR is demonstrated to have a key role in favoring senescence over quiescence and may even be a requirement for senescence transition and/or maintenance in many contexts (99–104). In fact, the main characteristics of senescent cells include hyperactive features such as cellular hypertrophy and the senescence-associated secretion phenotype, which require high metabolic activity (84, 104), and have been speculated to be in part the result of uncoupling proliferation and mTOR activity (85, 105). It should therefore follow that an intrinsic feature of senescent cells would be decreased autophagic activity, as has indeed been demonstrated in some reports (106). However, Narita et al. intriguingly described the formation of a compartment termed the mTOR-autophagy spatial coupling compartment (TASCC) upon OIS, in which mTOR and lysosomes are enriched in the vicinity of the rough endoplasmic reticulum–Golgi apparatus (107). The TASCC was speculated to shield mTOR from the upstream autophagy factors it usually inhibits (4, 107), allowing for concurrence of protein synthesis and degradation, while strategically situating mTOR and lysosomes in a favorable context for mTOR activation on the lysosomal surface (107, 108). In addition, an increasing number of reports have identified pathways and molecules that regulate autophagy independently of mTOR status, as reviewed in Ref. (109). Thus, mTOR activation and autophagy induction are likely not mutually exclusive processes in all contexts.
Decreased Autophagy can Favor Senescence
At variance with the above studies, it has also been reported that inhibition of autophagy promotes senescence (87, 110, 111). Autophagy was reported to counteract senescence by mediating the selective degradation of the transcription factor GATA binding protein 4 (GATA4), which is linked to acquisition of a senescent phenotype in response to DNA damage (112). GATA4 degradation depends on GATA4 recognition by the autophagy receptor p62. Following DNA damage, the p62/GATA4 interaction is reduced, leading to GATA4 stabilization and activation (112). Interestingly, GATA4 activation depends on the DNA damage response regulators, ataxia telangiectasia mutated (ATM), and ataxia telangiectasia and Rad3-related protein (ATR), but not on the traditional senescence effector molecules, p53, and p16 (112). GATA4 may therefore function in DNA damage-induced senescence rather than being a universal senescence-effector molecule.
In addition, a study by Wang et al. adds complexity to the role of autophagy during OIS, as it was reported that genetic ablation of autophagy was permissive rather than restrictive for senescence acquisition during oncogenic RAS-induced senescence (113). In this system, overexpression of Atg5 but not of an autophagy-deficient Atg5 point mutant promoted senescence by-pass, while depletion of Atg5 or Atg3 was permissive for senescence acquisition (113). Induction of OIS was regulated by apoptosis-stimulating of p53 protein 2 (ASPP2) that promoted senescence and inhibited oncogene-induced autophagy through direct disruption of the Atg16–Atg5–Atg12 complex (113), the assembly of which is required for autophagosome formation (114). This suggests a role for ASPP2 in modulating autophagy levels to control the cellular response to oncogene activation. Whether ASPP2 functions in senescence regulation in response to other stimuli remains to be determined. Of note, the ability of autophagy to inhibit OIS appeared not to involve protection from reactive oxygen species or abrogation of p53-activation (113). Understanding the mechanism by which autophagy counteracts senescence in this system may hold the key to combine the contradictory findings on the impact of autophagy on OIS.
Autophagy may also counteract senescence in the context of aging-related senescence and stem-cell maintenance. A study focusing on the regenerative capacity of muscle stem cells using physiologically aged mice, demonstrated that quiescent muscle stem cells preserve their integrity over time through active maintenance of organelle and protein homeostasis by continuous basal autophagy (110). The physiological decline of autophagy in old satellite cells or its genetic impairment in young cells, resulted in accumulation of toxic cellular waste and entry into senescence (110). Similarly, Kang et al. reported that depletion of essential autophagy components resulted in senescence due to build-up of toxic material in primary human fibroblasts (111). The latter studies represent a markedly different experimental system than stress-induced senescence, as they are devoid of external stimuli. Thus, while long-term autophagy inhibition may cause senescence due to accumulation of toxic constituents, autophagy may also function in acute responses to facilitate cellular remodeling in senescence transition in response to conditions such as oncogenic stress or DNA damage.
Cell Division and Autophagy
Apart from the complex interplay between autophagy and cell-cycle arrest pathways, several studies have reported more specialized regulatory functions for autophagy or autophagy-related factors in the cell division process. Correct segregation of the duplicated genome during cell division is a prerequisite for preventing CIN and aneuploidy, well-described contributors to cellular transformation (115, 116). Involvement of autophagy factors in regulating the progression or fidelity of cell division may thus be an additional component to consider when discussing the intricate relationship between autophagy and tumorigenesis.
Autophagy and Cytokinesis
In accordance with studies reporting decreased autophagy during mitosis (28, 29), autophagy proteins have primarily been linked to the final phase of cell division, cytokinesis. Cytokinesis is the process in which the two daughter cells are physically separated following chromosome segregation. This is achieved by the formation of a contractile actomyosin ring that constricts the cytoplasm between the segregated reforming nuclei, thereby generating a narrow intercellular bridge. In the center of the bridge is a dense proteinaceous structure termed the midbody ring (MR), which is thought to function as a targeting platform for cleavage factors. Cytokinesis is completed by plasma membrane fission at the intercellular bridge in a process called abscission (117).
A number of studies have reported cytokinesis failure following knock-down of members of the Vps34 complex including Vps34, Beclin 1, Vps15, Bax-interacting factor 1 (BIF-1), and UV irradiation resistance-associated gene (UVRAG) (118–120). The role of the Vps34 complex in cytokinesis regulation is distinct from its function in autophagy induction as it depends on Vps34-mediated production of PtdIns3P at the MR, which functions as a recruitment signal for the FYVE domain-containing cytokinesis regulator FYVE-CENT (120, 121). Accordingly, PI3Kinase inhibition by 3-methyladenine, but not inhibition of autophagy by the lysosome inhibitor bafilomycin A1 or Atg14 depletion, results in abscission failure (120).
The initiation of cytokinesis and mitotic exit is signaled by the anaphase-promoting complex/cyclosome (APC/C) that promotes proteasomal degradation of mitotic regulators including cyclin B; this, in turn, results in CDK1 inactivation and dephosphorylation of its substrates by counteracting phosphatases (122). Vps34 may be one of such CDK1 substrates that are re-activated during the late stages of mitosis after initially being inhibited (29), to participate in the regulation of mitotic exit, although the timing of Vps34 re-activation is not known. Cytokinesis failure can result in the generation of tetraploid cells with supernumerary centrosomes (123). Such tetraploid cells display CIN due to chromosome segregation defects in subsequent cell divisions and are suggested to exhibit increased tumorigenic potential (123–125). Interestingly, the Vps34 complex members Beclin 1, BIF-1, and UVRAG are amongst the autophagy-related proteins with the most well-substantiated tumor suppressor properties (11–14, 16). A detailed dissection of how the individual roles of the Vps34 complex in regulating cytokinesis and autophagy as well as growth factor receptor degradation (126) each contribute to the tumor suppressor function of these proteins, is an important issue for further investigation.
At variance with the studies discussed above, Belaid et al. reported abscission failure upon depletion of Atg5 and in cells derived from lysosomal vacuolar-type H+-ATPase a3-null mice (127), indicating a function for autophagy in cytokinesis. The cytokinesis defects observed in these systems were attributed to defective turn-over of active RhoA (127), a member of the Rho GTPase family that orchestrates cytokinesis through its ability to regulate the actomyosin contractile network at the cleavage zone (128). Depletion of Atg5 resulted in RhoA enrichment at the intercellular bridge leading to approximately three times broader RhoA activity zones. Consequently, Atg5-depleted cells progressing through mitosis exhibited loose and unstable cleavage furrows and increased generation of multinucleated cells (127). RhoA activity depends on GDP–GTP exchange factors (GEFs) including Ect2, which localizes at the mitotic midbody zone to mediate local RhoA activation and cleavage furrow formation (128, 129). Furthermore, an alternative function for cyclin A2 in potentiating RhoA GTP loading by its GEFs has also been described (130). While the majority of cyclin A2 is degraded by the proteasome in prometaphase (131–133), a small fraction of cyclin A2 was shown to persist in foci later in mitosis and appeared to be subjected to autophagic degradation (40). It is therefore possible that autophagy may have a composite function in controlling appropriate RhoA protein levels and activity at the cytokinesis midzone, by mediating RhoA and Cyclin A2 degradation in late mitosis.
The apparent discrepancies between the reported Vps34 and autophagy-mediated cytokinesis regulation may be most efficiently addressed by expanding these studies to a wider panel of cell systems and autophagy-related proteins. Understanding the contribution of these pathways to cytokinesis completion also in vivo is vital for evaluating the potential relevance of these mechanisms in the context of autophagy-related tumor suppression.
In addition, an autophagy-independent function for unc-51-like autophagy-activating kinase 3 (ULK3) as an abscission regulator has been reported (134). Abscission is mediated by the endosomal sorting complexes required for transport (ESCRT) machinery, which mediates membrane remodeling in a number of processes including cytokinesis, viral budding, and autophagy (135). The timing of abscission is regulated by the abscission checkpoint, which delays abscission in response to a number of mitotic abnormalities (136). Interestingly, ULK3 was shown to function in the abscission checkpoint to delay abscission by phosphorylating and binding ESCRT-III subunits in response to lagging chromosomes, nuclear pore defects, and tension forces at the midbody (134). Thus, ULK3 appears to function as an integral part of the abscission checkpoint machinery.
Autophagy and Cell Division Cleanup
In accordance with the more traditional function for autophagy in cellular maintenance, autophagy may also have a role in returning the cell to its interphase state by clearing leftover structures from normal and abnormal cell divisions.
Removal of the MR
Following cytokinesis, the MR is inherited asymmetrically by one of the two daughter cells, and is hereafter often referred to as a MR derivative (MRd). MRds can be eliminated by extrusion to the extracellular space (137–139) or by p62/NBR1-mediated selective autophagy (140–142). The NBR1-dependent pathway relies on the interaction between NBR1 and the midbody protein centrosomal protein 55 (CEP55) (141), while the mechanism of p62-mediated MRd degradation and the varying requirement for the two autophagy receptors is not understood. Intriguingly, the MRd extrusion pathway may also involve CEP55 recognition (138). Which elimination pathway predominates varies between cell lines (138), but how MRds are allocated for extrusion or retention and subsequent autophagic degradation is not known. Midbody extrusion likely leads to disposal of both the cytoplasmic and membraneous midbody components, which is not necessarily the case for autophagic degradation; thus, there could be a functional difference between the two midbody disposal pathways. Accumulation of MRds preferentially occurs in stem cells and cancer cells and was suggested to contribute to an undifferentiated phenotype (138, 141). Cells accumulating MRds show decreased autophagic activity and an ability to evade MRd encapsulation and autophagic degradation (141), suggesting a link between autophagy status and MRd accumulation. Nonetheless, MRds remain poorly described structures. How they influence cellular differentiation and their potential tumorigenic relevance is an interesting open question.
Removal of Micronuclei
If a cell fails to incorporate all chromosomes and chromosome fragments in the reforming nuclei during cell division, micronuclei can be generated (143). Two studies have observed micronuclei associated with LC3 and LAMP2-stained structures (144, 145), and also colocalizing with charged multivesicular body protein 4B (CHMP4B) (145), a member of the ESCRT machinery. Rello-Varona et al. treated U2OS cells with various cell-cycle inhibitors to increase formation of micronuclei, 2–5% of which colocalized with LC3 and p62, and partially with the lysosome marker LAMP2 (144). Importantly, LC3 colocalization was abrogated upon depletion of Atg5 and Atg7, and electron microscopy further confirmed the presence of micronuclei sequestered within double-membrane vesicles (autophagosomes). LC3-positive micronuclei contained less dense chromatin and discontinuous Lamin B1-stained nuclear envelopes (144), suggesting ongoing digestion. How the autophagy machinery is recruited to micronuclei is, however, not known. Furthermore, as only a small fraction of micronuclei appears to be targeted by autophagy, it remains to be investigated to what extent autophagy contributes to their elimination in comparison to other mechanisms of micronuclei removal, such as extrusion (143).
Of note, the formation of extranuclear chromatin entities does not strictly occur as a result of abnormal mitosis (143). Indeed, Ivanov et al. observed the formation of what was referred to as cytoplasmic chromatin fragments (CCFs) in senescent cells (94). CCFs, in contrast to micronuclei generated from malfunctioning mitosis, were negative for the nuclear lamin A/C and positive for the DNA damage marker γ-Histone 2AX and were generated by nuclear blebbing. CCFs were suggested to be identical to the Lamin B1-associated LADs that were later identified in senescent cells by Dou et al. (89), and intriguingly, both are degraded by autophagy (89, 94). These studies suggest a more general role for autophagy in disposing of extranuclear chromatin.
Autophagy in Mitotic Arrest and Mitotic Life/Death Decisions
Upon starvation, eukaryotic cells usually arrest in G1 (22). Nonetheless, it has been reported that nitrogen starved budding yeast, lacking essential autophagy genes arrest at the G2/M transition or in mitosis (146, 147). Matsui et al. reported that also nitrogen-starved wild type yeast exhibits a transient G2/M arrest (147). Recovery and progression from this arrest for subsequent G1 block requires autophagy-dependent supplementation of selected amino acids required for cell growth (147). Following replenishment with a nitrogen source, the previously arrested autophagy-deficient cells showed abnormal mitosis associated with a higher incidence of aneuploidy (147). This suggests a role for autophagy in maintaining genome stability by securing arrest in G1 during starvation, at least in budding yeast. Surprisingly, budding yeast may also require autophagy for completing cytokinesis and mitotic exit during nitrogen starvation (146, 147), even though the importance of amino acid supplementation in this context and the relevance of this phenotype in relation to the described mammalian autophagy-related cytokinesis regulation is not fully understood.
In mammalian cells, autophagy may have an important role in determining cell survival during mitotic arrest and mitotic catastrophe. Mitotic catastrophe is a complex oncosuppressive mechanism that is thought to sense mitotic failure and respond by driving cells toward an irreversible fate, be it apoptosis, necrosis, or senescence (148). Autophagy has been shown to facilitate cell survival during mitotic catastrophe (149, 150). Interestingly, during DNA damage-activated mitotic arrest, the previously identified mitosis-related CDK1-mediated phosphorylation of Vps34 on Thr159 (118) promotes Vps34 ubiquitination and proteasomal degradation (80). Degradation is mediated by the p53-responsive gene FBXL20 and the associated Skp1-Cullin-1 complex, and leads to inhibition of autophagy and receptor endocytosis (80). Thus, mitotic Vps34 phosphorylation in the context of p53 activation appears to promote Vps34 degradation (80). Such a mechanism may prevent survival of defective mitotic cells in a dual fashion, by potentially impeding both cytokinesis completion (118) as well as autophagy-dependent cell survival during mitotic arrest.
An alternative function for the autophagy-related protein Atg5 in mitotic catastrophe has also been demonstrated (150). Atg5 was found to be both necessary and sufficient for induction of mitotic catastrophe resulting from sublethal concentrations of DNA-damaging drugs (150). Following these insults, Atg5 translocates to the nucleus, where it physically interacts with survivin and causes the displacement of elements of the chromosomal passenger complex during mitosis, thus resulting in chromosome misalignment and segregation defects, representative of mitotic catastrophe (150). Atg5-mediated mitotic catastrophe does not depend on Atg5–Atg12 conjugation and is unaffected by pharmacological inhibition of autophagy (150); thus, Atg5-mediated mitotic catastrophe occurs independently of its role in autophagy regulation. While the applied drug concentrations only resulted in modest cell death, pharmacological inhibition of the autophagy pathway shifted the response to early caspase-dependent cell death (150), suggesting that in response to DNA damage, cytoplasmic Atg5 and nuclear Atg5 have distinct roles in autophagy induction and mitotic catastrophe, respectively.
Autophagy may under some conditions also participate in promoting mitotic cell death. Doménech et al. reported that, during mitotic arrest caused by abrogation of cyclin B1 degradation, autophagy promotes cell death through ongoing mitophagy (38). The gradual decline in the mitochondrial mass and oxidative respiration, however, resulted in a metabolic switch through activation of AMPK and subsequent induction of glycolysis in a 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase3-(PFKFB3)-dependent manner. Inhibition of glycolysis in breast cancer cells resulted in accelerated death of mitotic cells caused by microtubule poisons (38). This system represents a nonstress-induced mitotic arrest and is likely devoid of p53 activation. Thus, while autophagy induction occurred both during prolonged mitotic arrest (38) as well as in response to DNA damage-induced mitotic catastrophe (150), the resulting effect on cell survival may depend on the nature and severity of the stimulus leading to mitotic block. Of note, exploring how metabolic pathways influence life and death decisions of mitotically arrested cells is of particular interest in the context of cancer treatments, such as DNA-damaging agents or microtubule poisons, which affect the progression and fidelity of mitosis.
Nutrient Sensing and Cell Division: Involvement of AMPK in Mitosis Regulation
Surprisingly, in recent years, an unexpected mitotic role for the nutrient sensing and autophagy-inducing factor, AMPK, has been discovered. AMPK depletion results in mitotic abnormalities, including spindle misorientation and cytokinesis failure in Drosophila Melanogaster S2 cells and human cell lines (151–153). Furthermore, Drosophila AMPK-null embryos display severe abnormalities in cell polarity and mitosis (154). AMPK activation, evaluated by AMPK Thr172 phosphorylation (p-AMPKThr172), correlates with induction of mitosis (151, 153), during which p-AMPKThr172 is enriched specifically at centrosomes and at the cleavage furrow (153, 155). Furthermore, an elegant chemical genetics screen designed to identify novel substrates of AMPKα2 provided additional emphasis to a mitotic function for AMPK as it revealed 28 previously unidentified putative AMPK substrates enriched for proteins involved in chromosomal segregation, mitosis, cytokinesis, and cytoskeletal reorganization (151). These evidence indicate a role for AMPK in regulating mitosis through phosphorylation of mitosis-specific substrates. Nonetheless, there appears to be a considerable overlap between the pathways governing AMPK induction and responses during mitosis and during nutrient stress.
Several reports have implicated myosin regulatory light chain (MRLC) as a key target of AMPK-mediated mitosis regulation (151, 153, 154). AMPK facilitates the phosphorylation of MRLC at Serine 19 (151, 154), a phosphorylation event known to stimulate the Mg2+-ATPase activity of myosin II leading to actin-based regulation of mitosis, cell migration, and cell polarity (156–159). Accordingly, AMPK depletion decreases the level of p-MRLCSer19 at spindle poles and reduces overall mitotic p-MRLCSer19 levels. MRLC has been suggested to be a direct target of AMPK in Drosophila (154), but mammalian cells may employ alternative strategies for AMPK-mediated p-MRLCSer19 regulation. Protein phosphatase 1 regulatory subunit 12C (PPP1R12C) and p21-activated protein kinase (PAK2), both regulators of MRLC phosphorylation status (160–163), were identified as direct targets of AMPK (151). AMPK phosphorylation of these substrates indirectly induces MRLC Ser19 phosphorylation (151). Of note, the AMPK substrate and upstream autophagy regulator ULK1 has also been implicated in the regulation of MRLC phosphorylation (164). Thus, AMPK-induced MRLC phosphorylation may also involve autophagy factors. While Banko et al. identified a number of well-known mitotic regulators as putative AMPK substrates (151), MRLC regulation appears to be a major contributing factor, as depletion of MRLC partially reproduces AMPK depletion phenotypes (153). Moreover, the expression of a phosphomimetic mutant of MRLC is able to rescue AMPK-null-related cell polarity and mitosis defects in Drosophila (154). Whether AMPK regulates other substrates during mitosis remains to be determined.
Perhaps, the most intriguing questions in this context remains whether AMPK activation during mitosis is coordinated with its nutrient sensing ability, and if not, which mitosis-specific signals facilitate AMPK activation. Starvation or stress-induced AMPK activation involves allosteric activation by AMP and phosphorylation by upstream kinases on Thr172 in the activation loop of the catalytic α subunit (165). In mammals, the primary kinases performing this task are LKB1 (166–168) and calcium/calmodulin-dependent protein kinase kinase (CAMKK) (169, 170). LKB1 deficiency reproduces the mitotic abnormalities of AMPK deficiency (152, 153, 171), although CAMKK can also promote mitotic AMPK activation in LKB1-deficient systems (153). Thus, mitotic AMPK activation appears to rely on mechanisms resembling those governing starvation-induced AMPK activation. Interestingly, phosphorylation of PPP1R12C, PAK2, and MRLC also occurs in response to energy deprivation (151, 154, 164), indicating that regulation of these factors may be a general response to AMPK-activating stimuli rather than mitosis-specific. Intriguingly, myosin II activation, as indicated by MRLC phosphorylation, is reported to participate in autophagy induction by modulating Atg9 trafficking during starvation (164). Whether autophagy is induced in response to mitotic AMPK activation remains to be investigated.
It is entirely possible that AMPK regulation of mitosis represents a novel function that is unaffected by the cellular energy status, potentially involving selective AMPK activation at specific subcellular localizations during cell division. However, it has also been speculated that AMPK could alternatively promote the completion of already initiated cell cycles in response to energy deprivation to secure proper cell-cycle arrest in the ensuing G1 phase (165). This theory would imply a role for AMPK in initiating responses similar to those reported in yeast, in which autophagy supplies amino acids required for mitotic completion during starvation (147). Thus, intriguing questions for further investigation include understanding the exact mechanism governing mitotic AMPK activation and the requirement for AMPK (and possibly autophagy) for mitotic progression in response to diverse nutrient conditions.
Concluding Remarks
Autophagy, being traditionally viewed as a bulk process, was initially rarely linked to strictly structured processes, such as cell-cycle progression. Recent advances in the field, however, clearly suggest a strong correlation between autophagy activation and the induction and possibly execution of cell-cycle arrest programs, as well as autophagy (factor) regulation of the cell division process. Cell-cycle stress responses and resulting senescence acquisition constitute important anticancer barriers. Therefore, the relevance of autophagy in executing these responses and the role of autophagy in determining cellular life and death decisions in these contexts are of discernible interest. The role of autophagy and autophagy-related factors in regulating the fidelity of cell division is also potentially of substantial relevance, as findings on this topic suggest that the genomic instability observed upon ablation of autophagy (or specific autophagy components) may be partially attributed to dysregulation of this process. Furthermore, as an increasing number of autophagy proteins are being demonstrated to mediate alternative nonautophagic functions (i.e., PtdIns3K components, Atg5, Atg7, AMPK, AMBRA1, ULK1), we may need to more frequently consider autophagy factors individually. Of note, most evidence linking autophagy and cell-cycle regulation has been obtained in yeast and mammalian cell culture systems and remains to be tested in vivo. Thus, an important topic for future investigation includes evaluating the contribution of cell-cycle arrest programs and mitosis regulation to tumor progression or prevention in autophagy-manipulated animal models. While considering cell-cycle (dys)regulation as a factor surely adds to the complexity, it may also open up new avenues for improving our understanding of the intricate relationship between autophagy and tumorigenesis.
Author Contributions
SM wrote the manuscript and drafted the figures. FC and DZ provided senior supervision and critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Emiliano Maiani for helping with figures and to Laila Fischer for secretarial work.
Funding
The lab of Francesco Cecconi is supported by grants from the Telethon Foundation (GGP14202), AIRC (IG2016-18906), FISM (2013), KBVU from the Danish Cancer Society (R146-A9364), the Bjarne Saxhof Foundation, the Novo Nordisk Foundation (7559, 22544), and the European Union (Horizon 2020 MEL-PLEX, grant agreement 642295). Further, FC lab in Copenhagen is part of the Center of Excellence for Autophagy, Recycling and Disease (CARD), funded by the Danish National Research Foundation.
Abbreviations
γH2AX, γ-Histone 2AX; 3-MA, 3-methyladenine; AMBRA1, activating molecule in Beclin 1-regulated autophagy; AMPK, AMP-activated protein kinase; APC/C, anaphase-promoting complex/cyclosome; ASPP2, apoptosis-stimulating of p53 protein 2; Atg 3, 5, 7, 8, 9, 12, 14, autophagy-related gene 3, 5, 7, 8, 9, 12, 14; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3-related protein; Bcl-2, B-cell lymphoma 2; Bcl-XL, B-cell lymphoma extra large; BIF-1, Bax-interacting factor 1; BNIP3, BCL2-interacting protein 3; CAMKK, calcium/calmodulin-dependent protein kinase kinase; C-Cbl, Casitas B-lineage lymphoma; CCFs, cytoplasmic chromatin fragments; CDK 1, 2, 4, cyclin-dependent kinase 1, 2, 4; CDKIs, cyclin-dependent kinase inhibitors; CDKs, cyclin-dependent kinases; CEP55, centrosomal protein 55; CHMP4B, charged multivesicular body protein 4B; CIN, chromosomal instability; Cip/Kip, CDK interacting protein/kinase inhibitory protein; c-Myc, myelocytomatosis oncogene cellular homolog; DAPK1, death-associated protein kinase 1; Ddit4/Redd1, DNA damage-inducible transcript 4; DRAM, damage-regulated autophagy modulator; Drp1, dynamin-like protein; E2F, E2 factor; Ect2, epithelial cell-transforming sequence 2; ESCRT, endosomal sorting complex required for transport; FBXL20, F-box/LRR-repeat protein 20; GATA4, GATA binding protein 4; GEFs, GDP–GTP exchange factors; GFP, green fluorescence protein; INK4, inhibitors of CDK4; LADs, lamin-associated domains; LAMP2, lysosomal-associated membrane protein 2; LC3, light chain 3; LIR, LC3-interacting region; LKB1, liver kinase B1; MAP1LC3B, microtubule-associated protein 1 light chain 3 beta; Mdm2, mouse double minute 2 homolog; MEFs, murine embryonic fibroblast; MR, midbody ring; MRd, midbody ring derivative; MRLC, myosin regulatory light chain; mTOR, mammalian target of rapamycin; NBR1, neighbor of BRCA1 gene; NDP52, nuclear dot protein 52 kDa; OIS, oncogene-induced senescence; PAK2, p21-activated protein kinase; PE, phosphatidylethanolamine; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase3; PP2A, protein phosphatase 2 A; PPP1R12C, protein phosphatase 1 regulatory subunit 12C; PRKAB1, 2, protein kinase AMP-activated noncatalytic subunit beta 1, 2; PtdIns3K, class III phosphatidylinositol 3-kinase; PtdIns3P, phosphatidylinositol 3-phosphate; PTEN, phosphatase and tensin homolog; RAS, rat sarcoma; Rb, retinoblastoma protein; RB1CC1/FIP200, RB1 inducible coiled-coil 1/FAK family kinase-interacting protein of 200 kDa; RhoA, Ras homolog family member A; SASP, senescence-associated secretion phenotype; Skp1, S-phase kinase-associated protein 1; smARF, small mitochondrial ARF; SQSTM1, sequestosome 1; TASCC, mTOR-autophagy spatial coupling compartment; TIS, therapy-induced senescence; TSC2, tuberous sclerosis 2; ULK1, 2, 3, unc-51-like autophagy-activating kinase 1, 2, 3; USP 10, 13, ubiquitin-specific peptidase 10, 13; V-ATPase, vacuolar-type H+-ATPase; UVRAG, UV irradiation resistance-associated gene; Vps 15, 34, vacuolar protein sorting 15, 34.
References
1. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell (2010) 40:280–93. doi:10.1016/j.molcel.2010.09.023
2. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell (2008) 132:27–42. doi:10.1016/j.cell.2007.12.018
3. Abada A, Elazar Z. Getting ready for building: signaling and autophagosome biogenesis. EMBO Rep (2014) 15:839–52. doi:10.15252/embr.201439076
4. He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet (2009) 43:67–93. doi:10.1146/annurev-genet-102808-114910
5. Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr (2007) 27:19–40. doi:10.1146/annurev.nutr.27.061406.093749
6. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy (2012) 8:445–544. doi:10.4161/auto.19496
7. Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol (2004) 15:231–6. doi:10.1016/j.semcdb.2003.12.004
8. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy (2011) 7:279–96. doi:10.4161/auto.7.3.14487
9. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ (2012) 20:21–30. doi:10.1038/cdd.2012.72
10. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol (2015) 17:20–30. doi:10.1038/ncb3072
11. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature (1999) 402:672–6. doi:10.1038/45257
12. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh B-H, et al. Autophagic and tumour suppressor activity of a novel Beclin 1-binding protein UVRAG. Nat Cell Biol (2006) 8:688–99. doi:10.1038/ncb1426
13. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the Beclin 1 autophagy gene. J Clin Invest (2003) 112:1809–20. doi:10.1172/JCI200320039
14. Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol (2007) 9:1142–51. doi:10.1038/ncb1634
15. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25:795–800. doi:10.1101/gad.2016211
16. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A (2003) 100:15077–82. doi:10.1073/pnas.2436255100
17. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Chromosomal instability autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev (2007) 21:1367–81. doi:10.1101/gad.1545107
18. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34:856–80. doi:10.15252/embj.201490784
19. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer (2012) 12:401–10. doi:10.1038/nrc3262
20. Neufeld TP. Autophagy and cell growth – the yin and yang of nutrient responses. J Cell Sci (2012) 125:2359–68. doi:10.1242/jcs.103333
21. Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol (2013) 15:406–16. doi:10.1038/ncb2708
22. Jorgensen P, Tyers M. How cells coordinate growth and division. Curr Biol (2004) 14:1014–27. doi:10.1016/j.cub.2004.11.027
23. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer (2009) 9:153–66. doi:10.1038/nrc2602
24. Massagué J. G1 cell-cycle control and cancer. Nature (2004) 432:298–306. doi:10.1038/nature03094
25. Harper JV, Brooks G. The mammalian cell cycle: an overview. Methods Mol Biol (2005) 296:113–53. doi:10.1385/1-59259-857-9:113
26. Tasdemir E, Maiuri MC, Tajeddine N, Vitale I, Criollo A, Vicencio JM, et al. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle (2007) 6:2263–7. doi:10.4161/cc.6.18.4681
27. Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy (2011) 7:83–90. doi:10.4161/auto.7.1.13893
28. Eskelinen E-L, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, et al. Inhibition of autophagy in mitotic animal cells. Traffic (2002) 3:878–93. doi:10.1034/j.1600-0854.2002.31204.x
29. Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell (2010) 38:500–11. doi:10.1016/j.molcel.2010.05.009
30. Sit KH, Paramanantham R, Bay BH, Chan HL, Wong KP, Thong P, et al. Sequestration of mitotic (M-phase) chromosomes in autophagosomes: mitotic programmed cell death in human Chang liver cells induced by an OH* burst from vanadyl(4). Anat Rec (1996) 245:1–8. doi:10.1002/(SICI)1097-0185(199605)245:1<1:AID-AR1>3.0.CO;2-2
31. Álvarez-Fernández M, Malumbres M. Preparing a cell for nuclear envelope breakdown: spatio-temporal control of phosphorylation during mitotic entry. Bioessays (2014) 36:757–65. doi:10.1002/bies.201400040
32. Nelson WJ. W(h)ither the Golgi during mitosis? J Cell Biol (2000) 149:243–8. doi:10.1083/jcb.149.2.243
33. Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol (2011) 13:589–98. doi:10.1038/ncb2220
34. Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA (2011) 108:10190–5. doi:10.1073/pnas.1107402108
35. Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta (2008) 1777:860–6. doi:10.1016/j.bbabio.2008.05.442
36. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12:9–14. doi:10.1038/nrm3028
37. Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem (2007) 282:11521–9. doi:10.1074/jbc.M607279200
38. Doménech E, Maestre C, Esteban-Martínez L, Partida D, Pascual R, Fernández-Miranda G, et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol (2015) 17:1304–16. doi:10.1038/ncb3231
39. Li Z, Ji X, Wang D, Liu J, Zhang X. Autophagic flux is highly active in early mitosis and differentially regulated throughout the cell cycle. Oncotarget (2016) 7:39705–18. doi:10.18632/oncotarget.9451
40. Loukil A, Zonca M, Rebouissou C, Baldin V, Coux O, Biard-Piechaczyk M, et al. High-resolution live-cell imaging reveals novel cyclin A2 degradation foci involving autophagy. J Cell Sci (2014) 127:2145–50. doi:10.1242/jcs.139188
41. Liu L, Xie R, Nguyen S, Ye M, McKeehan WL. Robust autophagy/mitophagy persists during mitosis. Cell Cycle (2009) 8:1616–20. doi:10.4161/cc.8.10.8577
42. Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell (2008) 14:159–69. doi:10.1016/j.devcel.2008.01.013
43. Pavletich NP. Mechanisms of cyclin-dependent kinase regulation: structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J Mol Biol (1999) 287:821–8. doi:10.1006/jmbi.1999.2640
44. Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle (2012) 11:3599–610. doi:10.4161/cc.21884
45. Jiang H, Martin V, Gomez-Manzano C, Johnson DG, Alonso M, White E, et al. The RB-E2F1 pathway regulates autophagy. Cancer Res (2010) 70:7882–93. doi:10.1158/0008-5472.CAN-10-1604
46. Liang J, Shao SH, Xu Z-X, Hennessy B, Ding Z, Larrea M, et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol (2007) 9:218–24. doi:10.1038/ncb1537
47. Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer (2009) 9:738–48. doi:10.1038/nrc2718
48. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev (1999) 13:1501–12. doi:10.1101/gad.13.12.1501
49. Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol (2013) 14:297–306. doi:10.1038/nrm3567
50. Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science (1996) 272:877–80. doi:10.1126/science.272.5263.877
51. Rivard N, L’Allemain G, Bartek J, Pouysségur J. Abrogation of p27Kip1 by cDNA antisense suppresses quiescence (G0 state) in fibroblasts. J Biol Chem (1996) 271:18337–41. doi:10.1074/jbc.271.31.18337
52. Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, et al. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a caspase cascade. Mol Cell (1998) 1:553–63. doi:10.1016/S1097-2765(00)80055-6
53. Campos T, Ziehe J, Palma M, Escobar D, Tapia JC, Pincheira R, et al. Rheb promotes cancer cell survival through p27Kip1-dependent activation of autophagy. Mol Carcinog (2016) 55:220–9. doi:10.1002/mc.22272
54. Yang Z, Geng J, Yen WL, Wang K, Klionsky DJ. Positive or negative roles of different cyclin-dependent kinase Pho85-cyclin complexes orchestrate induction of autophagy in Saccharomyces cerevisiae. Mol Cell (2010) 38:250–64. doi:10.1016/j.molcel.2010.02.033
55. Polager S, Ofir M, Ginsberg D. E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene (2008) 27:4860–4. doi:10.1038/onc.2008.117
56. Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol (2001) 21:6820–32. doi:10.1128/MCB.21.20.6820-6832.2001
57. Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell (2006) 22:463–75. doi:10.1016/j.molcel.2006.04.014
58. Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol (2007) 27:6229–42. doi:10.1128/MCB.02246-06
60. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol (2007) 8:275–83. doi:10.1038/nrm2147
61. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev (2016) 30:1913–30. doi:10.1101/gad.287524.116
62. Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol (2010) 22:181–5. doi:10.1016/j.ceb.2009.12.001
63. Pietrocola F, Izzo V, Niso-santano M, Kroemer G. Seminars in cancer biology regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol (2013) 23:310–22. doi:10.1016/j.semcancer.2013.05.008
64. Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, et al. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res (2007) 67:3043–53. doi:10.1158/0008-5472.CAN-06-4149
65. Budanov AV, Karin M. p53 target genes Sestrin1 and Sestrin2 connect genotoxic stress and mTOR signaling. Cell (2008) 134:451–60. doi:10.1016/j.cell.2008.06.028
66. Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, et al. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle (2009) 8:1571–6. doi:10.4161/cc.8.10.8498
67. Kenzelmann Broz D, Mello SS, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev (2013) 27:1016–31. doi:10.1101/gad.212282.112
68. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcriptional by p53. Mol Cell (2001) 8:317–25. doi:10.1016/S1097-2765(01)00323-9
69. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A (2005) 102:8204–9. doi:10.1073/pnas.0502857102
70. Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ (2011) 18:1598–607. doi:10.1038/cdd.2011.33
71. Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene (2008) 27(Suppl 1):S2–19. doi:10.1038/onc.2009.39
72. Maiuri MC, Le Toumelin G, Criollo A, Rain J-C, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J (2007) 26:2527–39. doi:10.1038/sj.emboj.7601689
73. Martoriati A, Doumont G, Alcalay M, Bellefroid E, Pelicci PG, Marine J-C. DAPK1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene (2005) 24:1461–6. doi:10.1038/sj.onc.1208256
74. Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of Beclin 1 promotes dissociation of Beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep (2009) 10:285–92. doi:10.1038/embor.2008.246
75. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell (2006) 126:121–34. doi:10.1016/j.cell.2006.05.034
76. Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJR, Motoyama N, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science (2012) 336:225–8. doi:10.1126/science.1218395
77. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell (2005) 18:283–93. doi:10.1016/j.molcel.2005.03.027
78. Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin 1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell (2011) 147:223–34. doi:10.1016/j.cell.2011.08.037
79. Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell (2010) 140:384–96. doi:10.1016/j.cell.2009.12.032
80. Xiao J, Zhang T, Xu D, Wang H, Cai Y, Jin T, et al. FBXL20-mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev (2015) 29:184–96. doi:10.1101/gad.252528.114
81. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol (2008) 10:676–87. doi:10.1038/ncb1730
82. Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A, et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle (2008) 7:3056–61. doi:10.4161/cc.7.19.6751
83. Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Mariño G, Galluzzi L, et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle (2011) 10:2763–9. doi:10.4161/cc.10.16.16868
84. Terzi MY, Izmirli M, Gogebakan B. The cell fate: senescence or quiescence. Mol Biol Rep (2016) 43(11):1213–20. doi:10.1007/s11033-016-4065-0
85. Blagosklonny MV. Cell cycle arrest is not senescence. Aging (2011) 3:94–101. doi:10.18632/aging.100281
86. Fridlyanskaya I, Alekseenko L, Nikolsky N. Senescence as a general cellular response to stress: a mini-review. Exp Gerontol (2015) 72:124–8. doi:10.1016/j.exger.2015.09.021
87. Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy (2013) 9:808–12. doi:10.4161/auto.23922
88. White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev (2009) 23:784–7. doi:10.1101/gad.1795309
89. Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J, et al. Autophagy mediates degradation of nuclear lamina. Nature (2015) 527:1–17. doi:10.1038/nature15548
90. Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, et al. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep? J Pharmacol Exp Ther (2012) 343:763–78. doi:10.1124/jpet.112.197590
91. Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon H-U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med (2013) 5:202ra123. doi:10.1126/scitranslmed.3005864
92. Qi M, Fan S, Yao G, Li Z, Zhou H, Tashiro S, et al. Pseudolaric acid B-induced autophagy contributes to senescence via enhancement of ROS generation and mitochondrial dysfunction in murine fibrosarcoma L929 cells. J Pharmacol Sci (2013) 121:200–11. doi:10.1254/jphs.12269FP
93. Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev (2009) 23:798–803. doi:10.1101/gad.519709
94. Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, et al. Lysosome-mediated processing of chromatin in senescence. J Cell Biol (2013) 202:129–43. doi:10.1083/jcb.201212110
95. Horikawa I, Fujita K, Jenkins LMM, Hiyoshi Y, Mondal AM, Vojtesek B, et al. Autophagic degradation of the inhibitory p53 isoform Δ133p53α as a regulatory mechanism for p53-mediated senescence. Nat Commun (2014) 5:4706. doi:10.1038/ncomms5706
96. Bourdon J-C, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev (2005) 19:2122–37. doi:10.1101/gad.1339905
97. Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, et al. p53 isoforms Δ133p53 and p53β are endogenous regulators of replicative cellular senescence. Nat Cell Biol (2009) 11:1135–42. doi:10.1038/ncb1928
98. Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis, via glycolysis and ketone production. Cell Cycle (2012) 11:2285–302. doi:10.4161/cc.20718
99. Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene (2012) 31:1949–62. doi:10.1038/onc.2011.394
100. Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell (2009) 5:279–89. doi:10.1016/j.stem.2009.06.017
101. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle (2009) 8:1888–95. doi:10.4161/cc.8.12.8606
102. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) (2010) 2:344–52. doi:10.18632/aging.100160
103. Leontieva OV, Demidenko ZN, Gudkov AV, Blagosklonny MV. Elimination of proliferating cells unmasks the shift from senescence to quiescence caused by rapamycin. PLoS One (2011) 6:e26126. doi:10.1371/journal.pone.0026126
104. Young ARJ, Narita M, Narita M. Cell senescence as both a dynamic and a static phenotype. Methods Mol Biol (2013) 965:1–13. doi:10.1007/978-1-62703-239-1_1
105. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle (2008) 7:3355–61. doi:10.4161/cc.7.21.6919
106. Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells (2012) 33:597–604. doi:10.1007/s10059-012-0042-1
107. Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science (2011) 332:966–70. doi:10.1126/science.1205407
108. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell (2010) 141:290–303. doi:10.1016/j.cell.2010.02.024
109. Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans (2013) 41:1103–30. doi:10.1042/BST20130134
110. García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, et al. Autophagy maintains stemness by preventing senescence. Nature (2016) 529:37–42. doi:10.1038/nature16187
111. Kang HT, Lee KB, Kim SY, Choi HR, Park SC. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One (2011) 6:e23367. doi:10.1371/journal.pone.0023367
112. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science (2015) 349:aaa5612–5612. doi:10.1126/science.aaa5612
113. Wang Y, Wang XD, Lapi E, Sullivan A, Jia W, He Y-W, et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc Natl Acad Sci U S A (2012) 109:13325–30. doi:10.1073/pnas.1120193109
114. Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J (1999) 18:3888–96. doi:10.1093/emboj/18.14.3888
115. Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer (2005) 5:773–85. doi:10.1038/nrc1714
116. Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol (2010) 20:R285–95. doi:10.1016/j.cub.2010.01.034
117. Fededa JP, Gerlich DW. Molecular control of animal cell cytokinesis. Nat Cell Biol (2012) 14:440–7. doi:10.1038/ncb2482
118. Sagona AP, Nezis IP, Pedersen NM, Liestøl K, Poulton J, Rusten TE, et al. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol (2010) 12:362–71. doi:10.1038/ncb2036
119. Thoresen SB, Pedersen NM, Liestøl K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res (2010) 316:3368–78. doi:10.1016/j.yexcr.2010.07.008
120. You SY, Park YS, Jeon H-J, Cho D-H, Jeon HB, Kim SH, et al. Beclin-1 knockdown shows abscission failure but not autophagy defect during oocyte meiotic maturation. Cell Cycle (2016) 15:1611–9. doi:10.1080/15384101.2016.1181235
121. Sagona AP, Nezis IP, Bache KG, Haglund K, Bakken AC, Skotheim RI, et al. A tumor-associated mutation of FYVE-CENT prevents its interaction with Beclin 1 and interferes with cytokinesis. PLoS One (2011) 6:e17086. doi:10.1371/journal.pone.0017086
122. Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol (2011) 12:469–82. doi:10.1038/nrm3149
123. Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev (2007) 17:157–62. doi:10.1016/j.gde.2007.02.011
124. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature (2005) 437:1043–7. doi:10.1038/nature04217
125. Steigemann P, Wurzenberger C, Schmitz MHA, Held M, Guizetti J, Maar S, et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell (2009) 136:473–84. doi:10.1016/j.cell.2008.12.020
126. Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J (2008) 410:1–17. doi:10.1042/BJ20071427
127. Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res (2013) 73:4311–22. doi:10.1158/0008-5472.CAN-12-4142
128. Piekny A, Werner M, Glotzer M. Cytokinesis: welcome to the Rho zone. Trends Cell Biol (2005) 15:651–8. doi:10.1016/j.tcb.2005.10.006
129. Yüce Ö, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol (2005) 170:571–82. doi:10.1083/jcb.200501097
130. Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadéa G, et al. A novel function for Cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol (2012) 196:147–62. doi:10.1083/jcb.201102085
131. den Elzen N, Pines J. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol (2001) 153:121–35. doi:10.1083/jcb.153.1.121
132. Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, Hunt T. Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol (2001) 153:137–47. doi:10.1083/jcb.153.1.137
133. Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, et al. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol Biol Cell (1995) 6:185–97. doi:10.1091/mbc.6.2.185
134. Caballe A, Wenzel DM, Agromayor M, Alam SL, Skalicky JJ, Kloc M, et al. ULK3 regulates cytokinetic abscission by phosphorylating ESCRT-III proteins. Elife (2015) 4:1–70. doi:10.7554/eLife.06547
135. Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol (2010) 11:556–66. doi:10.1038/nrm2937
136. Nähse V, Christ L, Stenmark H, Campsteijn C. The abscission checkpoint: making it to the final cut. Trends Cell Biol (2017) 27:1–11. doi:10.1016/j.tcb.2016.10.001
137. Dubreuil V, Marzesco AM, Corbeil D, Huttner WB, Wilsch-Bräuninger M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J Cell Biol (2007) 176:483–95. doi:10.1083/jcb.200608137
138. Ettinger AW, Wilsch-Bräuninger M, Marzesco A-M, Bickle M, Lohmann A, Maliga Z, et al. Proliferating versus differentiating stem and cancer cells exhibit distinct midbody-release behaviour. Nat Commun (2011) 2:503. doi:10.1038/ncomms1511
139. Kieserman EK, Glotzer M, Wallingford JB. Developmental regulation of central spindle assembly and cytokinesis during vertebrate embryogenesis. Curr Biol (2008) 18:116–23. doi:10.1016/j.cub.2007.12.028
140. Isakson P, Lystad AH, Breen K, Koster G, Stenmark H, Simonsen A. TRAF6 mediates ubiquitination of KIF23/MKLP1 and is required for midbody ring degradation by selective autophagy. Autophagy (2013) 9:1955–64. doi:10.4161/auto.26085
141. Kuo T-C, Chen C-T, Baron D, Onder TT, Loewer S, Almeida S, et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol (2011) 13:1467–1467. doi:10.1038/ncb2405
142. Pohl C, Jentsch S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat Cell Biol (2009) 11:65–70. doi:10.1038/ncb1813
143. Kisurina-Evgenieva OP, Sutiagina OI, Onishchenko GE. Biogenesis of micronuclei. Biochem (2016) 81:453–64. doi:10.1134/S0006297916050035
144. Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Mariño G, et al. Autophagic removal of micronuclei. Cell Cycle (2012) 11:170–6. doi:10.4161/cc.11.1.18564
145. Sagona AP, Nezis IP, Stenmark H. Association of CHMP4B and autophagy with micronuclei: implications for cataract formation. Biomed Res Int (2014) 2014:974393. doi:10.1155/2014/974393
146. An Z, Tassa A, Thomas C, Zhong R, Xiao G, Fotedar R, et al. Autophagy is required for G1/G0 quiescence in response to nitrogen starvation in Saccharomyces cerevisiae. Autophagy (2014) 10:1702–11. doi:10.4161/auto.32122
147. Matsui A, Kamada Y, Matsuura A. The role of autophagy in genome stability through suppression of abnormal mitosis under starvation. PLoS Genet (2013) 9:e1003245. doi:10.1371/journal.pgen.1003245
148. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol (2011) 12:385–92. doi:10.1038/nrm3115
149. Inoue T, Nakayama Y, Li Y, Matsumori H, Takahashi H, Kojima H, et al. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J (2014) 281:2623–37. doi:10.1111/febs.12810
150. Maskey D, Yousefi S, Schmid I, Zlobec I, Perren A, Friis R, et al. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun (2013) 4:2130. doi:10.1038/ncomms3130
151. Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol Cell (2011) 44:878–92. doi:10.1016/j.molcel.2011.11.005
152. Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, et al. Genome-wide survey of protein kinases required for cell cycle progression. Nature (2004) 432:980–7. doi:10.1038/nature03160
153. Thaiparambil JT, Eggers CM, Marcus AI. AMPK regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol (2012) 32:3203–17. doi:10.1128/MCB.00418-12
154. Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature (2007) 447:1017–20. doi:10.1038/nature05828
155. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor AMP-activated protein kinase α (AMPKα) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle (2009) 8:2385–98. doi:10.4161/cc.8.15.9082
156. Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol (2005) 15:371–7. doi:10.1016/j.tcb.2005.05.004
157. Rosenblatt J, Cramer LP, Baum B, McGee KM. Myosin II-dependent cortical movement is required for centrosome separation and positioning during mitotic spindle assembly. Cell (2004) 117:361–72. doi:10.1016/S0092-8674(04)00341-1
158. Sellers JR. Myosins: a diverse superfamily. Biochim Biophys Acta (2000) 1496:3–22. doi:10.1016/S0167-4889(00)00005-7
159. Vicente-manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol (2009) 10:778–90. doi:10.1038/nrm2786
160. Chew T-L, Masaracchia RA, Goeckeler ZM, Wysolmerski RB. Phosphorylation of non-muscle myosin II regulatory light chain by p21-activated kinase (γ-PAK). J Muscle Res Cell Motil (1998) 19:839–54. doi:10.1023/A:1005417926585
161. Ito M, Nakano T, Erdodi F, Hartshorne DJ. Myosin phosphatase: structure, regulation and function. Mol Cell Biochem (2004) 259:197–209. doi:10.1023/B:MCBI.0000021373.14288.00
162. Matsumura F, Hartshorne DJ. Myosin phosphatase target subunit: many roles in cell function. Biochem Biophys Res Commun (2008) 369:149–56. doi:10.1016/j.bbrc.2007.12.090
163. Tuazon PT, Traugh JA. Activation of actin-activated ATPase in smooth muscle by phosphorylation of myosin light chain with protease-activated kinase I. J Biol Chem (1984) 259:541–6.
164. Tang H-W, Wang Y-B, Wang S-L, Wu M-H, Lin S-Y, Chen G-C. Atg1-mediated myosin II activation regulates autophagosome formation during starvation-induced autophagy. EMBO J (2011) 30:636–51. doi:10.1038/emboj.2010.338
165. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol (2012) 13:251–62. doi:10.1038/nrm3311
166. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, et al. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol (2003) 2:28. doi:10.1186/1475-4924-2-28
167. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A (2004) 101:3329–35. doi:10.1073/pnas.0308061100
168. Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LGD, Neumann D, et al. LKB1 Is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol (2003) 13:2004–8. doi:10.1016/j.cub.2003.10.031
169. Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, et al. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab (2005) 2:9–19. doi:10.1016/j.cmet.2005.05.009
170. Woods A, Dickerson K, Heath R, Hong S, Momcilovic M, Johnstone SR, et al. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab (2005) 2:21–33. doi:10.1016/j.cmet.2005.06.005
Keywords: autophagy, cancer, cell cycle, cell stress, senescence, mitosis, cytokinesis, p53
Citation: Mathiassen SG, De Zio D and Cecconi F (2017) Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 7:51. doi: 10.3389/fonc.2017.00051
Received: 01 February 2017; Accepted: 10 March 2017;
Published: 31 March 2017
Edited by:
Jon Lane, University of Bristol, UKReviewed by:
Viktor Korolchuk, Newcastle University, UKSimon Wilkinson, University of Edinburgh, UK
Copyright: © 2017 Mathiassen, De Zio and Cecconi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela De Zio, dzio@cancer.dk;
Francesco Cecconi, cecconi@cancer.dk