 Andrea Morandi
Andrea Morandi Maria Letizia Taddei
Maria Letizia Taddei Paola Chiarugi
Paola Chiarugi Elisa Giannoni
Elisa Giannoni- 1Department of Experimental and Clinical Biomedical Sciences, University of Florence, Florence, Italy
- 2Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy
- 3Excellence Centre for Research, Transfer and High Education DenoTHE, University of Florence, Florence, Italy
The epithelial-to-mesenchymal transition (EMT) process allows the trans-differentiation of a cell with epithelial features into a cell with mesenchymal characteristics. This process has been reported to be a key priming event for tumor development and therefore EMT activation is now considered an established trait of malignancy. The transcriptional and epigenetic reprogramming that governs EMT has been extensively characterized and reviewed in the last decade. However, increasing evidence demonstrates a correlation between metabolic reprogramming and EMT execution. The aim of the current review is to gather the recent findings that illustrate this correlation to help deciphering whether metabolic changes are causative or just a bystander effect of EMT activation. The review is divided accordingly to the catabolic and anabolic pathways that characterize carbohydrate, aminoacid, and lipid metabolism. Moreover, at the end of each part, we have discussed a series of potential metabolic targets involved in EMT promotion and execution for which drugs are either available or that could be further investigated for therapeutic intervention.
Introduction
The epithelial-to-mesenchymal transition (EMT) process allows the trans-differentiation of a cell with epithelial features into a cell with mesenchymal characteristics (Figure 1). This process has an essential role in physiological conditions (e.g., development, wound healing, and stem cell maintenance) and has been extensively reported to contribute pathologically to fibrosis and cancer progression. Interestingly, cells can also undergo the mesenchymal-to-epithelial transition (MET) process. MET is also essential in physiological conditions (e.g., during embryogenesis) but a key role in the formation of secondary metastases was reported (1, 2). The focus of the current review is on EMT in cancer progression although this process shares common characteristic with the physiological EMT program. The molecular mechanisms that an epithelial cell undergoes during EMT are tightly regulated and influenced by the cell-to-cell signaling network and by environmental factors. Independently of the stimuli that induce EMT, loss of E-cadherin is considered the key event and Snail1, Snail2 (also known as Slug), Twist, and ZEB1 are established transcription factors that can regulate E-cadherin expression. These mechanisms have been elegantly reviewed in Ref. (3). Conversely, the metabolic rewiring that sustains an epithelial cell undergoing EMT has been poorly investigated. However, since the metabolic reprogramming is now considered a hallmark of cancer (4), increasing evidence links metabolic deregulation to EMT program. This review gathers the recent findings on EMT and metabolic reprogramming in cancer and discusses how targeting certain metabolic pathways/hubs may impact on EMT and therefore on cancer progression.
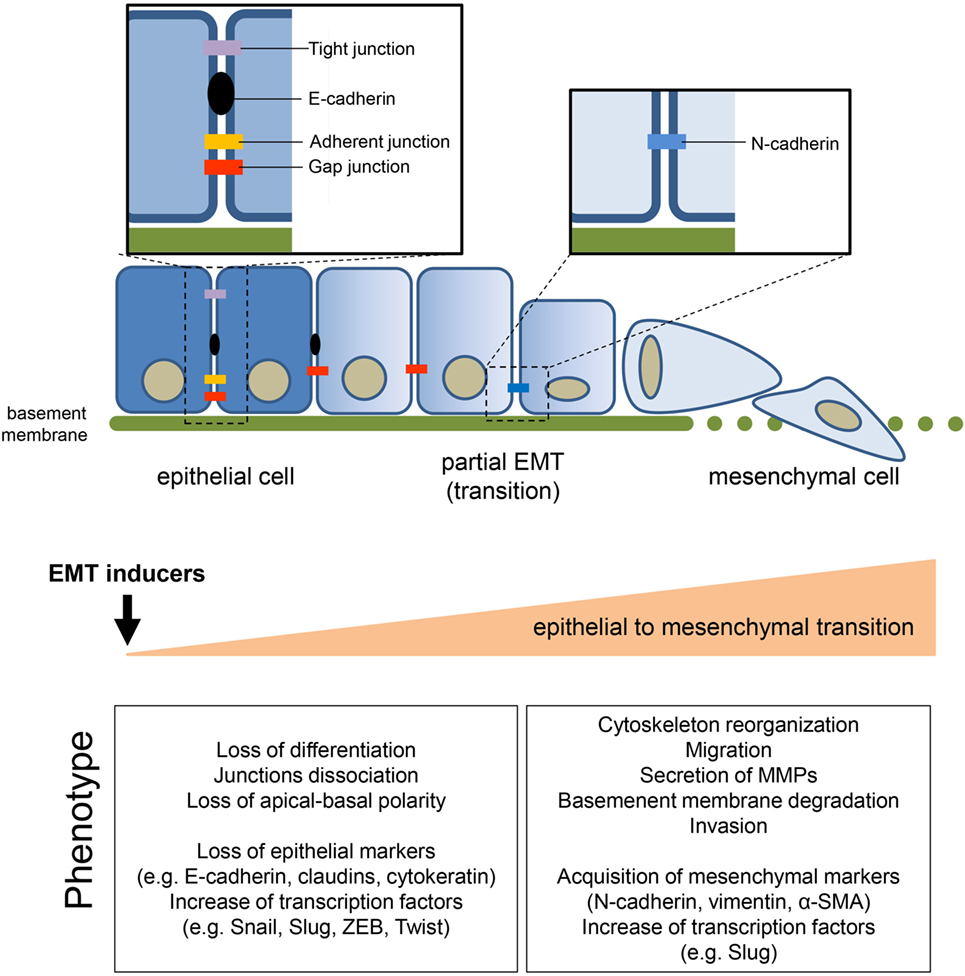
Figure 1. General features of epithelial-to-mesenchymal transition (EMT). The transition of epithelial cells toward a mesenchymal phenotype, induced by several environmental or soluble factors, is characterized by the loss of cell–cell contact and cell polarity, which disrupts the epithelial architecture and endows the mesenchymal cells with migratory and invasive competences. EMT is accompanied by the modulation of well-known markers, among which the loss of epithelial marker E-cadherin, induced by the upregulation of its transcriptional repressors (i.e., Snail1/2, Twist, ZEB1/2), is one of the priming event. The concomitant acquisition of mesenchymal markers sustains and stabilizes the newly acquired phenotype.
Metabolic Pathways that Controls EMT
Targeting Carbohydrates Metabolic Reprogramming in EMT
Non-proliferating differentiated cells preferentially metabolize glucose to pyruvate through glycolysis and then oxidize this pyruvate via the tricarboxylic acid (TCA) cycle and subsequent oxidative phosphorylation (OXPHOS). This maximizes the efficiency of ATP generation from a single molecule of glucose. Otto Warburg first described that most cancer cells show increased glucose conversion into lactate even in oxygen-rich condition (aerobic glycolysis) (5). Aerobic glycolysis allow proliferating cell to satisfy three basic needs of cells in rapid division: (i) fast ATP; (ii) carbohydrates redirection to biosynthetic pathways; and (iii) cellular redox status homeostasis (6). As a direct consequences of a net increase in glucose consumption, many cancers exhibit abnormal lactic acid release and a more acidic extracellular pH (pHe) (7). Moreover, high level of lactate correlates with metastases of several types of human cancers (8). In this scenario, it is not surprising that hypoxia, low pHe, and glucose utilization are established features of many solid tumors and concur to EMT and cancer dissemination. Thus, interfering with the glycolytic pathway could impair EMT and subsequent tumor progression and could be a potential anticancer strategy. However, to our knowledge, none of glycolysis inhibitors that have shown promising results in preclinical models are currently used in the clinic.
Several glycolytic enzymes have been found associated with invadopodia structures, protrusions of the plasma membrane (PM) that have a major role in extracellular matrix (ECM) degradation, and metastasis (9). In addition, the glycolytic-derived ATP is the main source for cell survival during metastatic dissemination (10, 11). Several papers indeed show a strict correlation between transforming growth factor-β (TGF-β)-induced EMT, glycolytic switch, and repression of mitochondrial function (12, 13). Dong et al. have demonstrated that in breast cancer, loss of fructose-1,6-bisphosphatase together with the loss of E-cadherin promote cancer stem cell (CSC)-like properties and cancer cell dissemination by enhancing β-catenin signaling and EMT program. These events are concomitant with the induction of glycolysis, increase of glucose uptake, and inhibition of oxygen consumption (14). Additionally, EMT induction in breast cancer cells is paralleled by the expression of glucose transporters, lactate dehydrogenase (LDH), monocarboxylate transporters (MCTs), and glycogen phosphorylase isoforms, key players in sustaining enhanced aerobic glycolysis (15). Moreover, the acquisition of a malignant and chemo-resistant phenotype is associated with EMT and aerobic glycolysis in gastric cancer (16).
The importance of glycolysis for EMT is also reinforced by a metabolite profiling approach conducted in pancreatic ductal adenocarcinoma (PDAC) cells. This profiling identified three main subpopulations with different phenotypes: interestingly, the clone that is enriched for glycolytic-related metabolites is characterized by a mesenchymal phenotype. Since mesenchymal properties are positive correlated with cancer aggressiveness and diseases progression, the study reinforced the functional relevance of glycolytic metabolism in disease progression (17). In addition, the exposure of PDAC cells to established EMT inducers [i.e., tumor necrosis factor-α (TNF-α) and TGF-β] increases glucose uptake and lactate secretion, without affecting OXPHOS metabolism (18).
Glycolytic metabolism has been also associated to CSC, a phenotype that shares common molecular pathways with EMT, and characterizes the cells that within the tumor tissue are responsible for tumor repopulation, therapy resistance, and relapse in several cancer types (19). However, contradictory results described the CSC metabolic phenotype as glycolytic or OXPHOS addicted not only in various tumor types, but also within individual cancer types. Some studies have postulated that glycolysis supports the self-renewal ability of CSCs by maintaining low ROS levels (19, 20) and in line with this glucose deprivation reduces the number of CSCs in in vitro studies (21). In addition, the enhancement of the glycolytic flux has been shown to be paralleled by a decrease in mitochondrial metabolism (i.e., TCA cycle and OXPHOS) with respect to their differentiated counterparts (22–26). Gammon et al. demonstrated that the CSC fraction of head and neck squamous cell carcinoma that undergoes hypoxia-induced EMT is characterized by glycolysis, a reduction in the oxygen consumption rate and decreased mitochondrial mass and ROS levels (27). Notably, Dong et al. demonstrated the concomitant acquisition of CSC-like properties, EMT, and induction of glycolysis following fructose-bisphosphatase 1 silencing in breast cancer (14).
We now proceed analyzing the principal players involved in sustaining the Warburg metabolism with a particular focus on the transporters and glycolytic enzymes whose deregulation has been linked to the acquisition of EMT traits and subsequent enhanced metastatic potential in cancer cells.
Hypoxia
In fast-growing solid cancers, the inner zone of the tumor becomes progressively hypoxic and acid. These environmental inputs drive the expression and stabilization of the hypoxia-inducible factor-1α (HIF-1α). HIF-1α is responsible for the transcriptional activation of genes involved in the restoration of local oxygen perfusion and in the enhancement of the glycolytic flux (28, 29). In addition to its canonical role, HIF-1α is able to regulate the expression of matrix metalloproteases (MMPs) (30) and several hypoxia-inducible EMT-related genes (31) such as Snail1, Slug, and Twist. In addition, HIF-1α regulates the autocrine motility factor (AMF), the secreted form of the glycolytic enzyme glucose phosphate isomerase (PGI). AMF ectopic expression triggers EMT in breast cancer cells (32). Since hypoxia is a well-known inducer of EMT in epithelial cancers, such as PDAC (33), hepatocellular (34), ovarian (35), and lung cancers (36), we can speculate that this may be partially due to the metabolic reprogramming orchestrated by HIF-1α. More recently, also in glioblastoma, it has been demonstrated that HIF-1α silencing is able to prevent EMT induced by the hypoxic microenvironment (37). Studies from our laboratory have shown that cancer-associated fibroblasts (CAFs) promote EMT of prostate cancer cells through an ROS-dependent HIF-1α stabilization. This transition is paralleled by the acquisition of stem-like and metastatic traits of cancer cell (38, 39). In keeping, hypoxic senescent fibroblasts, which display a CAF-like phenotype with increased lactate production, are also capable of promoting EMT in prostate cancer cells (40). Due to its important roles, HIF-1α is a potential target for anticancer therapy. Among the molecules described to be able to target HIF-1α (41), the compounds BAY87-2243, an inhibitor of its activity and stability, and the antisense oligonucleotide EZN-2968 entered phase I clinical trials (42). Hypoxia induces EMT not only via the transcriptional regulation of HIF-1α-dependent EMT genes but also via epigenetic mechanism of gene regulation (Figure 2). Indeed, HIF-1α is able to increase the expression of ten–eleven translocation 1 (TET1), which catalyzes the conversion of 5-methylcytosine to 5-hydroxymethylcytosine, thus inducing DNA demethylation. Hypoxia-induced expression of TET1 promotes EMT by complexing with HIF-1α and HIF-1β and activating the transcriptional activation of HIF-dependent EMT genes (43, 44).
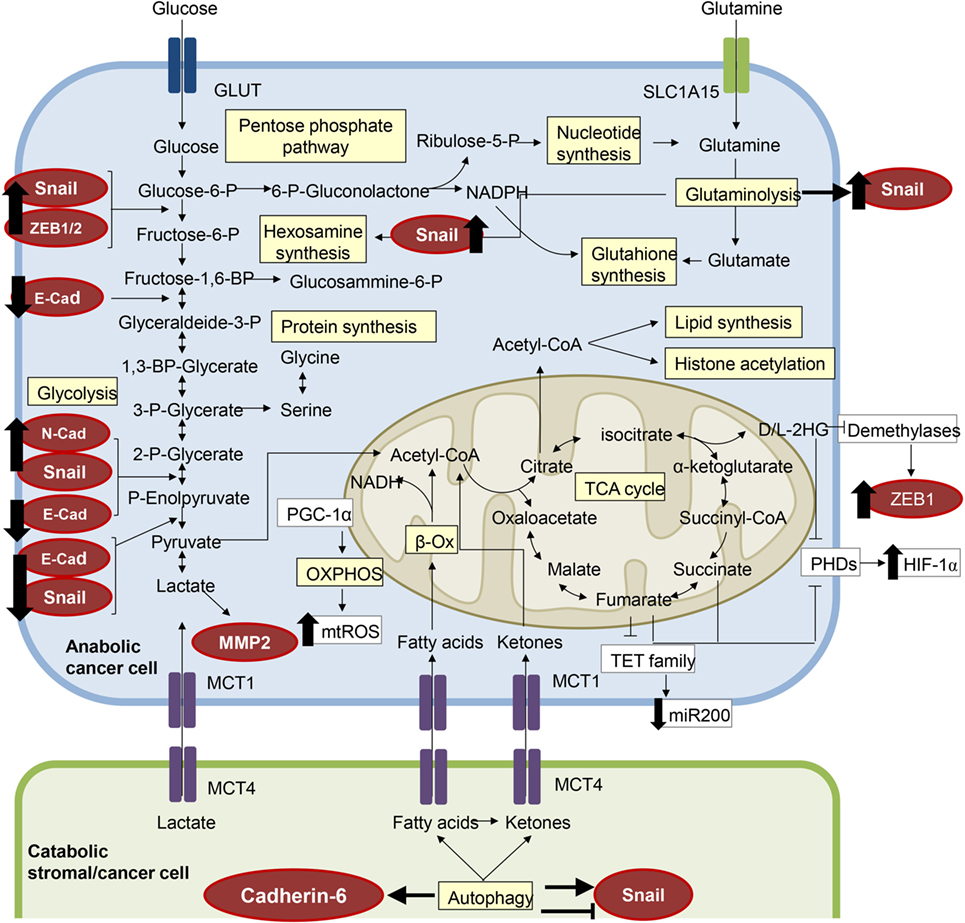
Figure 2. The metabolic features of epithelial-to-mesenchymal transition (EMT). Several components of the molecular pathways driving EMT have a clear impact on cell metabolism and vice versa, resulting in a metabolic rewiring which sustains the EMT transcriptional program. EMT-committed cancer cells could rely on an aerobic glycolytic metabolism or could shift toward the more efficient oxidative phosphorylation (OXPHOS), dependent on tumor type and/or tumor stage. Here, we have highlighted in red circles/ellipses EMT transcriptional factors that are affected by the metabolites reported in different tumor types.
Glucose Transporters
The glucose transporters 1 (GLUT1) and GLUT3 are transcriptionally induced by HIF-1, thus coupling the hypoxia with increased glucose uptake and glycolysis (45). Interestingly, overexpression of GLUT1 increases MMP-2 expression/activity and invasiveness of cancer cell (46). Moreover, GLUT3 expression correlates with both EMT markers (i.e., vimentin, Snail, Slug, ZEB1, ZEB2, Twist1) and glucose uptake in non-small cell lung cancer (NSCLC) cells (47). In addition, enhanced GLUT1 and/or GLUT3 expression is associated with poor prognosis in several type of human tumors (48). Numerous efforts have been made to test the efficacy of GLUT inhibitors as anticancer drugs: WZB117 and the natural flavonoid silibinin are under preclinical studies even if to our knowledge no specific GLUT1 inhibitors have been identified.
Lactate
Since lactate is the by-product of Warburg-dependent metabolism and increase of glycolysis is reported in several carcinomas, high levels of lactate are positively correlate with cancer aggressiveness in several tumors (8, 49). In many types of tumor a significant proportion of pyruvate is reduced into lactate, a reaction catalyzed by LDH5. Elevated LDH5 expression correlates with unfavorable prognosis in many human tumors and silencing of LDH5 impairs tumor initiation and progression (50–53). According to the pivotal role exerted by lactate in the promotion of the metastatic process, lactate can induce the upregulation of TGF-β2 expression (54), which in turn induces a mesenchymal pro-migratory phenotype of glioblastoma cells and promotes MMP-2 activation, ECM remodeling, and metastasis formation (55). It has also been reported that lactate is able to induce the production of TGF-β1 in vivo (56). In keeping, it has been shown that lactate administration promotes in vitro migration of human breast carcinoma cells as well as experimental lung metastases in mice intraperitoneally injected with lactate (57). Lactate is also a sensor of NAD+/NADH ratio, which is crucial for activity of the sirtuins histone deacetylase, a class of NAD+-dependent enzymes that possess either mono-ADP-ribosyltransferase or deacetylase activity (58). High levels of NAD+/NADH ratio correlate with energy stress and promote sirtuins activity. Some studies show that SIRT1 induces both EMT and metastasis by suppressing E-cadherin transcription (59, 60). Furthermore, our group has recently shown that CAF-induced lactate production increases activity of SIRT1 with consequent deacetylation/activation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PPARGC1A, also known as PGC-1α), a master regulator of mitochondrial biogenesis. This activation is crucial for the achievement of EMT. Accordingly, PGC-1α silencing abolishes invasion and EMT of prostate cancer cells. This evidence shows LDH as a possible therapeutic anticancer target. Several different classes of inhibitor have been synthesized, such as the gossypol derivative FX-11, compound GNE-140, galloflavin, and others (61), although none of these molecules are under consideration in the clinic.
Monocarboxylate Transporters
Key elements of the lactate shuttles are the MCTs, being MCT4 mainly involved in export of lactate and MCT1 in the uptake of this catabolite. Several studies demonstrated that silencing or inhibition of MCT1 and MCT4 are able to decrease the migration and invasion of several cancer cells (62–65). In particular, Fiaschi et al. have demonstrated that silencing of MCT1 and inhibition of MCT1-mediate lactate upload by prostate cancer cells impairs tumor growth and lung micrometastasis formation (66). Indeed, the blockade of lactate import and/or export is an interesting target for alternative therapeutic approaches. The α-cyano-4-hydroxycinnamate (CHC), that targets MCTs, has been used successfully in preclinical models without major toxicity in vivo (67–69). The AstraZeneca MCT1 inhibitor AZD3965 is in phase I/II clinical trials in the United Kingdom. Importantly, MCT4 silencing was shown to reduce tumor cell migration in vitro and in vivo (62) and the AstraZeneca MCT4 inhibitor (AZ93) has been reported as an highly efficient and likely selective compound that is currently used in preclinical studies.
Acidity
The regulation of lactate transport directly impacts on extracellular acidity and, together with the increase of the glycolytic flux lead to an unbalance in pH gradient across PM of cancer cells. Indeed, it is well established that tumor tissue are characterized by low pHe and the role of tumor acidity in cancer invasion and metastasis is well recognized (70). Indeed, low pHe leads to apoptosis of normal tissue at the periphery of the tumor while generating a positive selective pressure on cancer cells, selecting subclones that are resistant to acidic environment. Furthermore, acidification promotes angiogenesis through the enhanced expression of the vascular endothelial growth factor (VEGF) (71) and interleukin-8 (72) induces adherens junctions dissociation (73) and ECM degradation and remodeling (70, 74). Moreover, low pHe is crucial for the metastasization process providing secretion/activation of idrolases, such as catepsin (75) and MMPs (76). Acidic pHe was found to induce EMT in human melanoma cells (77) and in Lewis lung carcinoma cells (78). Accordingly, exposure of tumor cells to an acidic environment prior to tail vein injection increases lung colonization in a model of experimental metastasis (79).
Several molecules act as key players of cancer-associated extracellular acidification, such as the sodium-proton exchange 1 (NHE1), carbonic anhydrase IX (CAIX), the sodium bicarbonate transporter 1, and anion exchange 2. These molecules are possible targets for therapy focused on disruption of acid–base balance in cancer cells. In particular, CAIX, a membrane bound isoform of CA transcriptionally regulated by HIF-1α, induces extracellular acidification catalyzing the CO2 hydration and its overexpression is associated with increased metastasis and poor patients survival in several cancers (80). Fiaschi et al. have shown that activation of CAIX in CAFs leads to an extracellular acidification that enhances MMP-2 and MMP-9 secretion, thereby driving the stromal-induced EMT in prostate cancer cells. Moreover, both genetic silencing and pharmacological inhibition of CAIX are sufficient to impede EMT and CAF-induced invasion of prostate cancer cells (81). Furthermore, CAIX silencing in CAFs decreases prostate cancer cell tumor growth and lung micrometastasis formation, indicating the enzyme as an ideal target for anticancer therapy. At the moment, indisulam, an inhibitor of CAIX is tested in clinical trials for the treatment of NSCLC (82).
Glycolytic Enzymes
Glycolysis is finely regulated and articulated in 10 different steps and this could offer an array of potential targets to impair the EMT process. Moreover, the presence of several potential targets allows the possibility to interfere with more steps, either simultaneously or sequentially. Blocking EMT and metabolic reprogramming could therefore be a potential successful therapeutic approach. Hexokinase (HK2) is a HIF-1 target gene that controls the first rate-limiting step of glycolysis and is often overexpressed in cancer (83–86). It has been recently reported that breast CSCs showed increased HK2 expression and glycolytic rate. Notably, the glycolytic inhibitor, 2-deoxyglucose counteracts breast cancer cells undergoing EMT in a dose dependent fashion (87, 88). Furthermore, lonidamine that targets mitochondria-bound HK is now tested in phase II–III clinical trials (89). The HIF-1 dependent PGI/AMF (see above) acts as a pro-metastatic signaling molecule, due to its ability to promote tumor migration, invasion, and metastasis (90–92). Indeed, PGI/AMF overexpression leads to EMT achievement through NF-κB activation and increased expression of Snail1, ZEB1, and ZEB2 (Figure 2), downregulation of miR-200s with concomitant loss of E-cadherin (93). In keeping with these observations, high PGI levels in the serum positively correlate with metastases in colorectal, esophageal squamous cell, and lung tumors (94–96). However, although PGI has an established pro-tumorigenic role, to our knowledge, no selective inhibitors are available for preclinical investigation.
Another potential target of the glycolytic cascade is aldolase that converts the fructose-1,6-bisphosphate to glyceraldehyde-3-phosphate and dihydroxyacetone phosphate. The aldolase A isoenzyme is commonly overexpressed in various cancers (97, 98) and its upregulation has been reported to induce lung carcinoma cell migration and EMT by decreasing the expression of E-cadherin and concomitantly increasing those of fibronectin and vimentin (97).
In addition to the aforementioned glycolytic enzymes, enolase-1 has been recently proposed as a therapeutic target for gene-based therapy. The overexpression of enolase-1 increases glycolysis, proliferation, migration, and invasion in NSCLC cells, a process that is in part mediated by the regulation of EMT genes (99). Indeed, silencing of enolase-1 impaired EMT execution as shown by Snail1 and N-cadherin downregulation and the concomitant increase of E-cadherin expression (99). A role for enolase-1 in promoting migration and invasion has been described also in endometrial carcinoma (100). Small-molecule inhibitors of enolase are available but none of them has entered in the clinical practice (101). Finally, the pyruvate kinase (PK) enzyme has been recently emerged as an important player in tumor progression. PK catalyzes the dephosphorylation of phosphoenolpyruvate into pyruvate, resulting in ATP production. Differentiated cells primarily express the M1 isoform of the PK enzyme, whereas tumor cells often express the embryonic M2 isoform, which can be expressed as a tetramer, in its active form, or as a dimer with lower affinity for phosphoenolpyruvate. Dimeric PKM2 (102) acts as a transcriptional coactivator of HIF-1α in cancer cells, thus promoting glycolysis (103). Recently, our group demonstrated a peculiar role of PKM2 in inducing EMT in prostate cancer cells. Indeed, soluble factors secreted by CAF induce in prostate cancer the oxidation and phosphorylation of PKM2 prompting the nuclear translocation of the enzyme and its association with HIF-1α and the differentially expressed in chondrocyte-1, a transcriptional repressor, which downregulates miR-205. This transcriptional complex induces EMT execution (through the upregulation of ZEB2 and Snail1), as well as the metabolic rewiring of cancer cells toward OXPHOS metabolism (39). To further corroborate the link between PKM2 and EMT, Hamabe et al. recently demonstrated that EMT stimulates nuclear translocation of PKM2 and subsequent interaction with the TGF-β-induced factor homeobox 2, which promotes histone H3K9 deacetylation and the subsequent downregulation of E-cadherin expression (104). The PKM2 inhibitor TLN-232 was assessed for cancer therapy in a phase II clinical trial but despite some initial promising results the recruitment for a second phase II trial has halted for legal reasons in 2010 (105). Other efforts have been made to identify novel small-molecule inhibitors selective for PKM2 (106). However, emerging data suggest that, at least some tumors do not require PKM2 (107, 108), thus lowering the interest about the targeting of PKM2 in cancer.
Moreover, the switch between PKM1 and PKM2 couples glycolysis to pentose phosphate pathway (PPP), a metabolic pathway parallel to glycolysis that partially oxidizes glucose to pentoses and generates NADPH. The PPP has been described to support tumor cell proliferation (109) and to counteract ROS production, due to the generation of NADPH. Indeed, several studies show that PPP is necessary to handle the enhancement of ROS levels in stress condition, such as anchorage independent growth and anoikis (110, 111). Despite the PPP has a role in invasion, little is known about its contribution to EMT. Indeed, scanty data have shown that 6-phophogluconate dehydrogenase downregulation reduces in vitro migration of lung carcinoma cells (112) and high expression of the transketolase-like protein 1 isoform was positively correlated with invasion and metastasis of several carcinoma (113–116).
In addition, the hexosamine biosynthetic pathway (117), that accounts for 2–5% of total glucose metabolism and is intimately interconnected with the glycolytic pathway, shows a correlation with EMT. Indeed, glucose that enters the glycolytic pathway can be diverted to produce O-linked N-acetyl-glucosamine (O-GlcNAc), a molecule that has a signaling and structural role in the cells (117). It has been demonstrated that the addition of an O-GlcNAc motif at serine 112 prevented Snail1 O-phosphorylation-mediated degradation thus promoting its stabilization (118), thereby providing a direct molecular link between glucose metabolism and EMT.
Impact of Amino Acids Metabolism on EMT and Targeting Approaches
Glucose dependency of fast-growing tumor is paralleled by the higher amino acids requirements of these aggressive cancers. Indeed, targeting amino acid metabolism is now considered a potential therapeutic approach in many cancer types (119). Particularly, the role of glutamine in cancer progression has been extensively investigated due to the fact that glutamine is an essential amino acid for proliferating tumor cells. Glutamine can enter the oxidative TCA cycle, contribute to reductive carboxylation, and alter the NADPH production and redox balance. Glutaminase (GLS) catabolized glutamine conversion into glutamate, which is then oxidized by glutamate dehydrogenase (GDH) into α-ketoglutarate (α-KG), an intermediate of the TCA cycle. Silencing of GLS1 or impairing glutamine metabolism were able to counteract the induction of EMT mediated by growth factors (e.g., TGF-β) in breast and colon cancers by increasing Snail1-targeting miRNA expression and hence impacting on Snail stability (Figure 2). Importantly, GLS1 silencing impaired tumor growth and metastases formation (120). Moreover, since GDH was reported to control tumor aggressiveness and therapy response (121), Liu and colleagues hypothesized a link between GDH expression/activity and EMT. Indeed, they reported an association of GDH overexpression with metastasis formation and poor prognosis in colorectal cancer patients. These effects were demonstrated to be dependent, at least in vitro, on the EMT induction mediated by GDH/STAT3 axis (122). However, Aguilar and colleagues reported a differential contribution of amino acid metabolism to EMT. Indeed, prostate cancer cells that were selected to display a stable EMT phenotype uncoupled to CSC behaviors were shown to (i) reduce the consumption rate of glutamine and other ketogenic amino acids (i.e., leucine, isoleucine, lysine, threonine, tyrosine, tryptophan, and phenylalanine) and (ii) be less sensitive to glutamine metabolism inhibition (123). The differences observed may be due to the fact that the model used was peculiar and CSC and EMT programs were uncoupled. Indeed, it is usually difficult to distinguish whether the metabolic alterations observed in CSC are dependent of EMT or CSC states, since recent studies have found that the acquisition of CSC properties may occur in cancer cells independently of EMT (124).
Drugs that target glutamine transport into the cell or the conversion to α-KG have been designed and tested. GLS inhibitors have shown positive results in preclinical models; bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide impairs cancer cells growth in vitro and in vivo (125, 126), the GLS inhibitor CB-839 is effective against triple-negative breast cancer (TNBC) and hematological tumors in preclinical studies (127, 128) and is currently moved on to clinical trials (129). Therefore, targeting GLS could be an effective strategy to block the EMT process and therefore impair invasive abilities of aggressive cancer cells (130).
An alternative source of amino acids in cancer cell is due to degradation and recycling of cellular components (e.g., autophagy). A role for protein degradation in controlling EMT has been postulated recently by investigating how interfering with lysosomal cathepsin proteases (i.e., lysosomal-dependent protein turnover) affects TGF-β-induced EMT. Indeed, Kern and colleagues found increased lysosome activity during EMT of mammary epithelial cells derived from the MMTV-PyMT mouse model and showed that cathepsin inhibitor E64d was able to impair TGF-β-induced EMT and invasion (131). Moreover, Akalay and coworkers reported that tumor cells undergoing EMT-induced autophagy regulate target recognition and lysis of cytotoxic T lymphocytes and may be exploited for immunotherapeutic strategies to block immune escape (132). However, conflicting results revealed that liver-specific autophagy-deficient mice showed reduced expression levels of epithelial-related genes and increased of the mesenchymal related suggesting a direct link between autophagy and EMT. Indeed, induction of autophagy degrades Snail1 thus inhibiting the TGF-β-mediated EMT (133). This inverse correlation was also reported for glioblastoma cells (134). The diverging results may be ascribed to the different models used; however the link between autophagy and EMT seems to be conceivable. Recently, this connection has been further reinforced by the demonstration that cadherin-6 (Figure 2), a type 2 cadherin that marks cells that undergo EMT and correlates with metastatic ability in papillary thyroid cancers, restrains autophagy, and promotes reorganization of mitochondrial network (135). Autophagy targeting via hydroxychloroquine has now moved into clinical trials and a recent review (136) discusses how inducing or inhibiting autophagy can be achieved in preclinical models.
Finally, amino acid-related metabolic pathways control the EMT process via epigenetic modifications. Indeed, the methionine synthesis pathway plays a pivotal role in controlling promoters’ methylation and subsequent gene expression regulation. DNA methyltransferases and histone methyltransferases transfer a methyl group from the S-adenosyl methionine (SAM) to DNA or to positive charged amino acidic residues of histones with the formation of the by-product S-adenosyl homocysteine (SAH) (137). Since SAH inhibits DNA methyltransferases and histone methyltransferases, the ratio between SAM and SAH is important for regulating DNA and histone methylation (138). Threonine, glycine, and serine are the amino acids that sustain SAM generation through the folate metabolism (139). Threonine dehydrogenase converts threonine into glycine and acetyl-CoA and together with methionine adenosyl transferase are the key enzymes involved in SAM biosynthesis. Since methionine adenosyl transferase can be localized into the nucleus, it provides an effective system for chromatin-localized biosynthesis of metabolites to allow an adequate and well-organized regulation of the histone and DNA methylation processes. This mechanism can be exploited for targeting the epigenetic regulation of EMT-related genes. Indeed, SAM-competitive inhibitors and SAH hydrolase inhibitors have been shown to inhibit the methyltransferase EZH2 thus impairing the overall methylation histone status leading to transcription reactivation of silenced genes, such as the EMT-related gene E-cadherin (140, 141).
Impact of Lipid Metabolism on EMT and Targeting Approaches
Despite glucose and glutamine alterations have been extensively investigated in the context of cancer metabolism, increasing evidence are now pointing to a pivotal role of alterations to lipid-associated pathways (142, 143). Indeed, proliferating cancer cells are characterized by increased lipids and cholesterol dependency, which can be fulfilled by either increasing the upload of exogenous lipids or by over-activating their endogenous synthesis. The excess of lipids in cancer cells are stored in lipid droplets (LDs) and high LDs and stored-cholesteryl ester content in tumors are now considered as hallmarks of cancer aggressiveness (144).
Lipids encompass a vast class of biomolecules of unique chemical structure. Among their multiple biological functions, they contribute to cell compartmentalization, cell signaling, protein trafficking, membrane organization, and energy storage and production. Despite their established role in regulating a variety of processes during cancer development (145–147), little is known about the impact of lipid composition and metabolism during EMT.
A lipidomic approach performed on prostate cancer cells that underwent EMT following TNFα treatment revealed a significant increase in the synthesis of triacylglicerols (TAGs), sustained by a concomitant upregulation of fatty acid synthase (FASN) (148). It is conceivable that the newly synthesized TAGs, stored into LDs, may act not only as a reservoir of energy, but also of fatty acids (FAs), which could serve for protein modification, cell membrane remodeling, and the generation of pro-tumorigenic signals which support EMT-induced cell motility. In keeping with the increase of FASN during EMT, administration of the FASN inhibitor osthole to the MCF7 breast cancer cells abolishes cell scattering, EMT execution, migration, and invasion induced by the hepatocyte growth factor (149). In addition, in breast cancer cells undergoing EMT an increased expression of FASN resulted in saturated FAs accumulation, which are then relocated to the cell membrane and regulate lipid rafts organization, resulting in the activation of the EMT-inducer VEGF/VEGFR2 signaling (150).
Although the above-described reports deal with a supporting role of FASN-driven lipogenesis for EMT and cell migration, opposing observations are also described in the literature. The exposure of NSCLC cells to the EMT-inducer TGF-β suppresses the transcription of enzymes involved in lipogenesis, including FASN and acetyl-CoA carboxylase (ACC) (151). These enzymes are controlled by the carbohydrate-responsive element-binding protein (ChREBP) and sterol regulatory element-binding proteins (SREBPs). Snail1 mediated the suppression of the lipogenic program by repressing SREBPs and ChREBP levels. In addition to a decreased lipogenesis, TGF-β-induced EMT also promoted higher oxygen consumption and elevated intracellular ATP. Indeed, decreased FAs synthesis could divert acetyl-CoA into the catabolic pathways, e.g., TCA cycle and OXPHOS, which efficiently yield high amount of energy to support the elevated cell migration required for metastatic spreading. In addition, an increased availability of acetyl-CoA could foster histone acetylation/activation of genes responsible for EMT engagement, i.e., the transcription factors ZEB1, ZEB2, and Slug; the mesenchymal markers vimentin, N-cadherin, and fibronectin; and the CSC markers Sox2 and Nanog (152). Notably, the expression of the lipogenic enzymes FASN and ACC are downregulated in circulating cancer cells, when compared to their primary cancer counterpart (153). Interestingly, this metabolic switch toward a catabolic signature is reversible upon withdrawal of EMT-inducing stimuli. The functional role of this metabolic reversion is that, when metastatic cells finally localize to secondary sites, they have the potential to re-activate FASN expression, switching their metabolism back to lipogenic pathways, in order to sustain rapid cell proliferation and metastatic colonies formation. The observation that decreased lipogenesis might stimulate cell invasion and metastases raises significant attention in considering FASN targeting and/or other lipogenic enzymes as potential therapeutic target for cancer treatment. Indeed, this therapeutic intervention, besides inducing a temporary cancer growth inhibition could, in the long-term, increase the risk of metastasis.
Another strategy used by cancer cells to improve FAs availability for anabolic and catabolic purposes, is to increase the expression of acyl-CoA synthetases (ACSLs), which converts the long-chain FAs into acyl-CoA, critical for phospholipids and TAGs synthesis, lipid modification of proteins as well as for FAs β-oxidation (154). In colorectal cancer cells, the simultaneous overexpression of ACSL1, ACSL4, and stearoyl-CoA desaturase-1 (SCD), the rate-limiting enzyme converting saturated FAs into monounsaturated FAs, induced EMT and increased cellular migration and invasion (155). Furthermore, the combination of low doses of pharmacological inhibitors of ACSL and SCD selectively reduces cell viability of chemotherapy resistant colorectal cells, with no effects on normal colonocytes. The clinical relevance of these findings is emphasized in colorectal patients with tumors displaying ACSL1/ACSL4/SCD simultaneous overexpression, that show a poorer outcome and a higher risk of relapse compared to other patients within the same clinico-pathological stage.
In addition to the above discussed increase in TAGs, a reduction of C16:0 ceramide levels was also associated with EMT (148, 156). A recent study documented that TGF-β-induced EMT in human breast epithelial cells downregulates the expression of ceramide synthase 6 (CerS6), which produces C16:0 ceramide, further metabolized to C16:0 sphingolipids, such as sphingomyelin and glycosphingolipids, both enriched in the PM microdomains (156). CerS6 mRNA and protein levels are also reduced in TNBC mesenchymal cells as compared with non-TNBC epithelial cells. Downregulation of CerS6 in TNBC patients correlates with increased PM fluidity, a required feature to foster cell motility and metastatic spreading. Hence, CerS6 is emerging as a novel EMT-regulated gene, whose reduction in cancer correlates with a mesenchymal phenotype and, in turn, with tumor aggressiveness and poor prognosis. Accordingly, it is predictable that in patients with aggressive TNBC characterized by CerS6 downregulation, increasing the levels of C16:0 ceramide could impair the metastatic potential of this aggressive breast cancer subtype.
Further evidence indicates that EMT is associated with changes in sphingolipids metabolism. Guan and colleagues reported that EMT is associated with the alteration of ganglioside (i.e., sialic acid-containing glycosphingolipids) metabolism in normal mouse mammary gland and bladder cancer (157). Gg4 is one of the most relevant ganglioside involved in EMT execution, whose content is reduced upon TGF-β-induced EMT. Gg4 was reported to physically interact with key epithelial cell molecules, such as E-cadherin and β-catenin, likely facilitating epithelial intercellular adhesion via stabilization of the E-cadherin/β-catenin complex at the cell surface (157). The modulation of ganglioside pattern upon EMT is related, at least in part, to the altered expression of genes encoding ganglioside metabolizing enzymes, which are under the control of different transcription factors involved in EMT (i.e., Snail1, Twist, ZEB1) (158–160). Alongside the role of gangliosides, an involvement of sphingosine-1-phosphate (S1P) in EMT has also been reported. In hepatocellular carcinoma, where high levels of S1P in the serum correlate with poor prognosis (161), S1P activates the PI3K/AKT signaling pathway, resulting in MMP-7 upregulation. MMP-7 mediates the shedding of syndecan-1, a transmembrane heparan sulfate proteoglycan that regulate cell–matrix interaction, with a consequent increase in TGF-β1 production, a well-established inducer of EMT (162). A role of S1P in the modulation of other MMPs which promote cell invasion, such as MMP-2 and MMP-9, has also been proposed (163, 164).
In parallel with the acquisition of an aggressive phenotype, the EMT is marked by a profound rewiring of cell signaling pathways, most of them originating from PM located mediators (165, 166). It is therefore conceivable that alterations in the properties of the PM may impact on the overall signaling network rearrangements associated with EMT. Recent evidence showed that EMT is associated with a reorganization of the PM and in particular with a destabilization of lipid raft domains (Figure 3) (167). Alterations of the PM biophysical properties are required to maintain the mesenchymal state, associated with a CSC phenotype, and an increase in metastatic potential. Interestingly, stabilization of lipid rafts is emerging as a target for therapeutic strategies aimed to reduce the metastatic potential driven by EMT in cancer. The stabilization of lipid raft domains is a therapeutically attractive approach, since a number of pharmacological and nutritional factors have been shown to positively affect raft stability and function (168, 169). Among them, the essential ω-3 polyunsaturated FA docosahexaenoic acid (DHA) has been recently discovered to stabilize lipid raft and to antagonize both EMT and the acquisition of stem-like features (170). It suggests that the PM properties are subject to dietary inputs and that long-term perturbations in lipid metabolism (e.g., hypercholesterolemia, DHA supplementation) may profoundly affect tumor progression. In addition, alkylphospholipid drugs (i.e., miltefosine and edelfosine) that have been described as “dis-rafters” for their ability to affect raft organization have potent anti-neoplastic activity (169).
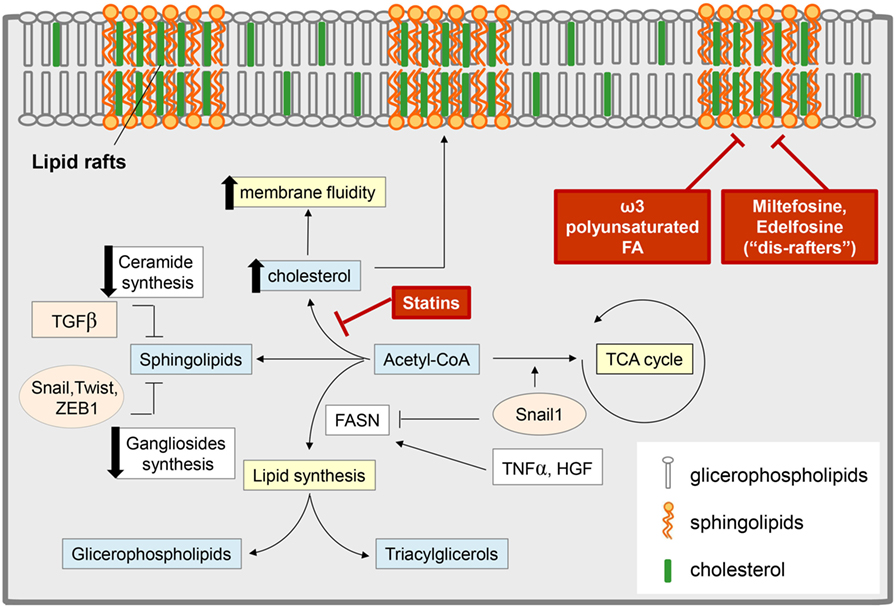
Figure 3. Epithelial-to-mesenchymal transition (EMT) and lipid metabolism: interconnection and targeting. Several EMT-induced stimuli modulate the content of different classes of lipids, with an impact on both cell metabolism and membrane composition. Some EMT inducers [i.e., tumor necrosis factor-α (TNF-α), hepatocyte growth factor (HGF)] promote lipogenesis by stimulating fatty acid synthase (FASN) and leading to an increase in glicerophospholipids (which are transferred to the cell membrane and regulate lipid rafts organization) and in triacylglycerol [stored in lipid drops as a reservoir of fatty acids (FAs) for catabolic or anabolic purpose]. On the other hand, other reports support a role for transforming growth factor-β (TGF-β)-derived Snail1 in the suppression of the lipogenic program by repressing FASN expression, thus diverting acetyl-CoA toward catabolic pathways, such as the tricarboxylic acid (TCA) cycle. TGF-β-induced EMT also correlates with a reduction in the pool of sphingolipids by (i) the downregulation of the expression of ceramide synthase, which results in the reduction of ceramide levels; (ii) the lowering of ganglioside content due to the control of EMT-related transcription factors (i.e., SNAIL1, Twist, Zeb1) on the expression of genes encoding ganglioside metabolizing enzymes. These events result in an increase of plasma membrane fluidity and destabilization of lipid rafts, to which significantly concurs also the upregulation of cholesterol content. Pharmacological inhibitors of cholesterol synthesis (i.e., statins), the administration of alkyl phospholipid drugs (the so-called “dis-rafters”), and nutritional factors, such as the ω-3 polyunsaturated FA docosahexaenoic acid, result in the stabilization of lipid raft and in the reduction of membrane fluidity, thereby counteracting EMT, invasion, and the acquisition of stem-like features.
To reinforce the link between PM composition and EMT, alterations in membrane fluidity, e.g., by modulating cholesterol content (Figure 3), has been shown to induce or inhibit the conversion toward a mesenchymal phenotype (167). Indeed, the lipid compositions of epithelial or mesenchymal cells are distinct (171) and when a given cell undergo the EMT process it exhibits a significant increase in cholesterol content, which strongly contributes, together with the reduction in ceramide pool, to the enhancement in membrane fluidity. Accordingly, cancer cells undergoing EMT are more sensitive to cholesterol lowering drugs, such as statins, that have been reported to deplete cholesterol content in mesenchymal cells, thereby reducing PM fluidity and impairing cell motility and metastatic potential (172). Increased membrane fluidity is an emerging necessary feature of metastatic cancers that can be controlled by many currently available drugs, offering a feasible therapeutic opportunity to prevent cancer metastasis. A fascinating study has recently identified, using an in silico drug screening, a series of pharmacological compounds that can repress the metastatic phenotype of cancer cells by inhibiting a gene expression signature associated with EMT. The compounds discovered using this analysis, including previously acknowledged anti-metastatic drugs, appeared to restrict the metastatic capacity through a common mechanism, i.e., the ability to modulate the fluidity of cell membranes (173). In keeping, the treatment of breast cancer cell lines with some of these anti-metastatic agents (i.e., alprostadil, amitriptyline, haloperidol, and maprotiline, as well as, salinomycin and thioridazine) reduced membrane fluidity, resulting in EMT impairment, decreased cell motility, and stem cell-like properties, culminating in the impairment of spontaneous metastasis in animal models, thus validating the in silico analysis. The impact of fluidity on the metastatic behavior and the strict correlation between membrane fluidity and cholesterol content was further supported by the finding that in breast cancer patients, the overexpression of the cholesterol efflux channel ABCA1 was associated with increased metastatic success and was revealed in 41% of metastatic tumors (173).
Role and Targeting of TCA Cycle Intermediates and OXPHOS in EMT
Although the contribution of mitochondria to the pathogenesis of cancer was underestimated for a long time, it is now established that mitochondria have an essential role during tumorigenesis. Indeed, mitochondrial alterations are essential for the metabolic rewiring that characterized a given cancer cells and play a role in a series of additional cellular processes during the development and progression of cancer.
We have discussed in details in the first paragraph how aerobic glycolysis is extensively exploited by rapidly proliferating cancer cells to meet their energetic demands and to accumulate biosynthetic precursors. However, several reports support the opposite notion, i.e., that the metabolic requirements of invasive and metastatic cancer cells are characterized by enhanced mitochondrial respiration and ATP generation. This review does not discuss whether and what are the circumstances in which glycolysis or OXPHOS are exploited by cancer cells for promoting their survival and distant colonization. However, we would like to emphasize that contradictory results described the metabolic phenotype of invasive EMT-committed cancer cells as glycolytic or OXPHOS addicted, not only in various tumor types, but also within individual cancer types. As confirmed by clinical analysis of human invasive breast cancers, the transcription coactivator PGC-1α is overexpressed in invasive cancer cells and stimulates mitochondrial biogenesis and OXPHOS during their transit to metastatic sites (153). PGC-1α silencing in cancer cells significantly impaired their invasion ability and decreased the frequency of metastasis without affecting cell proliferation and primary tumor growth. Notably, the PGC-1α-induced metabolic conversion toward OXPHOS is synergistically coupled to a functional EMT program and although PGC-1α-induced pathways are not essential for cancer cells during the acquisition of a mesenchymal phenotype, both pathways concur and correlate with the achievement of invasive and metastatic properties (153).
Accordingly, in both mouse melanoma and human breast cancer models, an overloading of the mitochondrial electron transport chain (ETC) was reported to promote an invasive tumor phenotype associated with increased mitochondrial reactive oxygen species (mtROS) generation (174, 175). However, this study underlined that a dysfunctional mitochondrial activity and a partial lowering of ETC activity strictly resembled ETC overloading, generating a similar pro-oxidant mitochondrial milieu. In both cases, superoxide generation induced mitochondrial Src activation, which enhanced the expression of Pyk2, a FAK family member protein tyrosine kinase that was previously reported to promote EMT and migratory abilities (174, 176). Targeting mtROS generation by treating with inhibitors of the ETC Complex I activity (Ebselen) or using specific mitochondria-targeted antioxidants [MitoTEMPO or MitoQ, a mitochondria-targeted form of coenzyme Q10 currently tested in clinical trials (177)] was sufficient to abolish metastasis formation in vivo (174).
Although mitochondrial superoxide acts as an inducer/sustainer of EMT, an uncontrolled production of this pro-oxidant can shift cell fate toward senescence or cell death (178, 179). To maintain ROS generation to levels that allow EMT execution, mitochondrial superoxide dismutase 2 (SOD2) have been found upregulated upon TGF-β-mediated EMT, prompting the conversion toward a CSC-like phenotype during the early stage of EMT (180). ZEB2, but not ZEB1, together with NF-κB seems to control the transcriptional regulation of SOD2. In addition, a positive feedback loop has been described between SOD2 and NF-κB during EMT in lung adenocarcinoma cells: NF-κB can promote SOD2 transcriptional activation and concomitantly SOD2 induces EMT activating the axis NF-κB/IκB kinase β (IKKβ) (181).
Since an overloading of the respiratory mitochondrial metabolism is often associated with EMT and with the acquisition of pro-invasive skills by cancer cells, drugs that inhibit OXPHOS may be proposed as effective strategies to cope with the acquisition of a motile mesenchymal phenotype endowed with CSC-like features and metastatic competency. Arsenic trioxide, which interfere with OXPHOS by inhibiting the complex III of ETC and metformin, acting on complex I are FDA-approved treatments already used in the clinical practice (182, 183). Crucially, arsenic trioxide has been proposed for the clinical management of relapsed or refractory acute promyelocytic leukemia (183), while metformin particularly exerts anticancer activity in patients with breast, endometrial, or prostate cancer (184, 185).
However, a recent study reported data that partially disagree with the preeminent role of OXPHOS for metastatic potential of cancer cells. In this study, the authors compared mRNA expression of metabolic genes in 20 different solid cancers to investigate the link between metabolic transformation of cancer cells and patient prognosis (186). Data revealed that the inhibition of mitochondrial metabolism is a common signature for all the cancer samples. The downregulation of OXPHOS-related genes correlates with metastatic potential and poor clinical outcome across several cancer types and it is associated with the presence of EMT (186). These results support previous finding that partial mitochondrial dysfunction increases metastatic potential of cancer cells (174) and encourage future investigations on the role of dysregulated metabolism during tumor progression, in order to suggest more suitable targets for clinical intervention.
One of the main metabolic pathways active within mitochondria is the TCA cycle. Numerous enzymes of the TCA cycle show mutation or deregulated expression pattern in both sporadic and hereditary cancers. These alterations identify a subset of patients characterized by poor prognosis (187, 188) and sometimes specific alterations of these mitochondrial enzymes are directly linked to EMT induction.
Consistent with the frequent metabolic rewiring toward OXPHOS observed in invasive cancer cells, epithelial cells that have activated the EMT program may increase the amount of glucose diverted into the TCA cycle by regulating the pyruvate dehydrogenase (PDH) complex, the gatekeeper of the cycle. In particular, in lung cancer cells the execution of the EMT program is accompanied by a downregulation of pyruvate dehydrogenase kinase 4 (PDK4) expression levels, which results in an increase of PDH activity (189). Downregulation of PDK4 is sufficient to drive EMT and promotes erlotinib resistance in EGFR mutant lung cancer cells. In keeping, analysis of human lung adenoma tumor samples reveals PDK4 suppression as a predictor of poor prognosis, consistent with its role during EMT (189).
Besides the deregulation of the expression, specific cancer-associated mutations of some enzymes of the TCA cycle, associated with an alteration of their catalytic function, have been also recognized. Among the mutated enzymes, the most characterized are those involved in the generation of the so-called oncometabolites, including isocitrate dehydrogenase (IDH) (190), succinate dehydrogenase (SDH) (191), and fumarate hydratase (FH) (191).
Isocitrate dehydrogenase catalyzes the reversible conversion of isocitrate into α-KG. Three isoforms of the enzymes have been identified: the NADPH-dependent IDH1 and IDH2 and the NADH-dependent IDH3. Mutations in the cytoplasmic IDH1 and in the mitochondrial IDH2 mutations occur in various human cancers, including gliomas, glioblastoma, and acute myelogenous leukemias (AMLs) (192–194). The mutated forms of the enzymes reduce α-KG into the d-2-hydroxyglutarate (d-2HG). d-2HG levels are low in normal tissues but can reach higher concentration levels (up to millimolar) in IDH1 or IDH2-mutated tumors. High levels of d-2HG interfere with the function of α-KG-dependent dioxygenases, including prolyl hydroxylases (PHDs), essential for the regulation of the stability of HIFs, Jumonji family of histone demethylases, and TET family of DNA demethylases, whose inhibition inevitably impacts on the epigenetic control of gene expression (195, 196). High d-2HG levels paralleled by an altered epigenetic fingerprint was also described in breast tumors characterized by c-Myc overexpression and subsequent metabolic reprogramming, independently of IDH mutations (197). The enantiomer l-2HG (L2HG) can be generated by the non-canonical activity of malate dehydrogenase and LDH and not by the IDH1-/IDH2-mutated enzymes (198, 199). Recently, in colorectal cancer cells an elevation of both d-2HG and its enantiomer l-2HG has been observed in the absence of IDH mutations and it is promoted by glutamine anaplerosis. It has been reported that only d-2HG is able to increase the histone H3 methylation pattern of the ZEB1 promoter region, resulting in a direct upregulation of ZEB1 and downregulation of miR-200, two key drivers of the EMT process (200, 201). Clinical specimens with higher levels of d-2HG associate with colonization of distant organs, supporting the significant contribution of this metabolite in cancer metastasis. Treatment strategies designed to reduce the levels of d-2HG or inhibiting its downstream effects could be effective in colorectal cancer. Interestingly, 2HG has been reported as one of the few metabolites whose levels are reduced upon aspirin administration, currently the most effective drug available for chemoprevention of colorectal cancer (202), suggesting d-2HG as a potential target for therapeutic intervention, with low risk of side effects, since the physiological role of this metabolite is not known. IDH-mutated tumors represent the perfect scenario to test the specific targeting of tumor metabolism with minimal interference with that of normal cells. Inhibitors of the mutant form of IDH have been tested glioma and AML patients harboring IDH mutations (202).
The other two oncometabolites that exert their effects outside of the conventional metabolic network are succinate and fumarate, two TCA cycle intermediates which reach elevated concentrations in some tumors as a consequence of loss-of-function mutations in the SDH complex or the FH, respectively (203–205). Like d-2HG, succinate and fumarate have been found to interfere with dioxygenase activity, underlining the oncogenic role of both these metabolites in the inhibition of PHDs and the subsequent stabilization of HIF-1 (206) and supporting the notion that a general property of oncometabolites is the ability to regulate epigenetics (191, 207).
Interestingly, fumarate possesses another unique property linked to its chemical structure. Indeed, fumarate can covalently bind to cysteine residues of proteins in a process called succination (208, 209). Several proteins are succinated in FH-deficient cells, including aconitase (210), Kelch-like ECH-associated protein 1 (208, 209), and glutathione (211). Notably, in a subset of FH-deficient human renal cell carcinomas, fumarate at high concentration directly bounds the antioxidant glutathione both in vitro and in vivo to produce the metabolite succinated glutathione (211). Succinated glutathione acts as an alternative substrate to glutathione reductase to decrease NADPH levels and enhance mtROS and HIF-1 activation, two mandatory events fostering EMT execution.
More recently, Sciacovelli and colleagues demonstrate a fascinating role of fumarate, which accumulates in FH-deficient renal cancers, in the epigenetic suppression of miR-200 through the inhibition of Tet-mediated demethylation of a regulatory region of the anti-metastatic miRNA cluster mir-200ba429 (212). Fumarate-dependent miR-200 downregulation leads to the expression of EMT-related transcription factors and enhanced aggressive features and it is associated with a poor clinical outcome (212). Although it has been extensively reported that FH deficiency is associated to fumarate-dependent epigenetic deregulation, supporting the notion of FH as a tumor suppressor, the molecular mechanisms by which FH gene expression is controlled is not well clarified. It has been recently reported that in nasopharyngeal carcinoma, the chromatin remodeling factor lymphoid-specific helicase binds the FH promoter and recruits the epigenetic silencer factor G9a to inhibit the transcription of FH (213). The FH reduction promoted an unbalance in the TCA intermediates, with a decrease in malate levels and a concomitant increase in αKG amount. Deregulation of TCA metabolites in nasopharyngeal carcinoma cells induces the recruitment of IKKα to the promoter regions of genes involved in the EMT program, causing the downregulation of E-cadherin and ZO-1 and the parallel upregulation of the mesenchymal marker vimentin, leading to EMT, migration and invasion in vitro, and increase metastasis in vivo (213).
Deregulation of different components of the SDH complex, enzyme involved in both the ETC and the TCA cycle, have been reported in several cancer types (214, 215). SDH is composed of four essential subunits: the flavoprotein SDHA, the iron-sulfur protein SDHB, and two units anchored to the mitochondrial membrane SDHC and SDHD. Mutations in SDHx genes (encoding SDH subunits) lead to succinate accumulation that inhibits DNA demethylases (TET enzymes), leading to a global hypermethylation of DNA (191, 216, 217). Crucially, SDHx mutations have been associated with EMT in hereditary pheochromocytoma and paraganglioma, as a result of epigenetic alterations (218, 219). In particular, Aspuria et al. demonstrated that Sdhb knockdown in mouse ovarian cancer cells resulted in a global hypermethylation pattern that promotes EMT and induces a metabolic reprogramming of the central carbon metabolism, together with additional mitochondrial dysfunction, ultimately leading to altered glucose and glutamine utilization (220). More recently, Loriot and colleagues clarified how the succinate-mediated epigenetic modulation impacts on EMT. First, using transcriptome profiling of a large cohort of metastatic pheochromocytomas and paragangliomas, the authors reported that SDHB-malignant samples displayed a change in the expression pattern of Twist1, Twist2, Snail1, and N-cadherin, all indications of EMT program activation (221). Moreover, cytokeratin 19 (KRT19), a component of the intermediate filaments, already implicated in conferring enhanced migratory and invasive properties in squamous cell carcinomas, neuroblastomas, renal, and breast cancers (222–225), has been identified as one of the most significantly hypermethylated and downregulated gene in SDHB-deficient mouse chromaffin cells, supporting the EMT-dependent invasive properties and the metastatic behavior of these neuroendocrine cancer cells (218).
Besides the four subunits that constitute the SDH complex, SDH5 has been identified as a mitochondrial protein necessary for the flavination of SDHA and for the assembly of the SDH complex. Additionally, several independent reports pointed to a role of SDH5 as a tumor-suppressor gene and SDH5 deregulated expression correlate with higher tumor incidence (226–228). An interesting study reported that SDH5 loss in lung cancers promotes tumor aggressiveness and metastasis, hence providing an additional functional support on the link between SDH5 and EMT. In particular, SDH5 acts as a modulator of the glycogen synthase kinase 3β (GSK-3β)/β-catenin signaling, since the physical interaction between SDH5 and GSK-3β induces GSK-3β dephosphorylation/activation, the subsequent decrease of β-catenin nuclear accumulation and activation, thereby ultimately inhibiting Wnt-β-catenin signaling (229). The downregulation of SDH5 observed in lung cancers accounts for the SDH5-mediated β-catenin stabilization and transcriptional activity, which in turn induces Slug and Twist1 gene expression, thereby repressing E-cadherin and contributing to EMT (229).
Besides SDH5 repression, other mitochondrial signaling pathways concur to regulate the GSK-3β/β-catenin pathway, impacting on EMT. In lung cancer cells, the downregulation of the TU translation elongation factor mitochondrial (TUFM), a key factor in the translational expression of mitochondrial DNA, is associated to the maintenance of the mesenchymal phenotype of cancer cells and to the acquisition of more aggressive features. TUFM downregulation results in a reduced expression of mtDNA, leading to mitochondrial dysfunction and cellular stresses such as ATP deficiency and ROS production, which trigger AMPK activation. It has been reported that activated AMPK induces the phosphorylation of GSK-3β and increases the nuclear accumulation of β-catenin, leading to the induction of EMT and increased migration and metastatic competency of lung cancer cells (230). These results provide a molecular link between mitochondrial dysfunction and EMT, which is implicated in lung cancer progression.
Malic enzyme 1 (ME1) is reported to be a major metabolic enzyme, both localized in cytosol and mitochondria, catalyzing the oxidative decarboxylation of malate into pyruvate, thus fueling the TCA cycle, and concomitantly reducing NADP+ into NADPH and contributing to macromolecular biosynthesis and protection from excessive oxidative stress. For its essential metabolic roles, ME1 is ubiquitously expressed in different human tissues. However, up to now, there are no evidence about its oncological functions and clinic significance. A significant upregulation of ME1 has been associated to aggressive hepatocellular carcinoma and to reduced overall survival and progression-free survival in this tumor type (231). Silencing of ME1 in hepatocellular carcinoma cells inhibits migration and invasion by blocking the EMT program (i.e., restoring E-cadherin while downregulating N-cadherin and vimentin expression) in a ROS-dependent way, suggesting ME1 as a poor prognostic predictor for hepatocellular carcinoma-bearing patients (231).
Concluding Remarks
Epithelial-to-mesenchymal transition is a specific program that confers the cancer cell a series of stem-like properties and migratory abilities thus increasing cancer aggressiveness. The hostile microenvironment (i.e., hypoxia, low pH, and low glucose) and the metabolic requirements of a fast-growing tumor sustain EMT execution allowing cancer cells to bypass the nutrients and oxygen supply limitation caused by the rapid primary cancer growth and colonize secondary sites to secure the adequate support of energy and nutrients. However, this is not simply a process to acquire migration and invasion abilities but a more complex rewiring of signaling, metabolic, genetic, and epigenetic networks that allow differentiated epithelial cancer cells to revert into a more undifferentiated state and stem cell functions. Additionally, it is plausible to speculate that aggressive cancer cells are characterized by the ability to undergoing EMT, acquire stem cell-like traits, and rewire their metabolism to gain a plethora of strategies to survive in different environmental conditions and/or in presence of anticancer drugs. This plasticity reinforces the conclusion that “it is not the fittest of the species (in this case of the tumor cell bulk) that survives but the most adaptable.” The challenges we now face are (i) the identification of distinct metabolic hallmarks exclusive to cancer-associated EMT and (ii) the identification of therapeutic window that allows EMT targeting in cancer patients. These are key questions that need to be addressed to reduce the off target effects caused by general metabolic therapies and importantly to avoid the administration of anti-EMT compounds to cause a MET in cells that have already left the primary site and may give rise to metastases. However, none of the clinical trials using anti metabolic drugs, extensively reviewed in Ref. (6), have investigated whether the effects exerted by these compounds have an impact on the EMT process and therefore further confirmation in preclinical studies and the design of large prospective clinical trials should be planned.
Author Contributions
AM, MT, PC, and EG were responsible for the conception, design and drafting of the article, and final approval; agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MB and handling editor declared their shared affiliation, and the handling editor states that the process nevertheless met the standards of a fair and objective review.
Funding
The work was funded by Fondazione Umberto Veronesi to AM, Associazione Italiana Ricerca sul Cancro (AIRC) (grant 8797 to PC), AIRC and Fondazione Cassa di Risparmio di Firenze (grant 19515 to PC and AM), Istituto Toscano Tumori (grant 0203607 to PC), and Programma operativo regionale Obiettivo “Competitività regionale e occupazione” della Regione Toscana cofinanziato dal Fondo europeo di sviluppo regionale 2007-2013 (POR CReO FESR 2007-2013, grant to PC).
Abbreviations
ACC, acetyl-CoA carboxylase; ACSLs, acyl-CoA synthetases; AMF, autocrine motility factor; CAFs, cancer-associated fibroblasts; CAIX, carbonic anhydrase IX; CerS6, ceramide synthase 6; CHC, α-cyano-4-hydroxycinnamate; ChREBP, carbohydrate-responsive element-binding protein; CSC, cancer stem cell; DHA, docosahexaenoic acid; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; ETC, electron transport chain; FAs, fatty acids; FASN, fatty acid synthase; FBP1, fructose-1,6-bisphosphatase; FH, fumarate hydratase; GDH, glutamate dehydrogenase; GLS, glutaminase; GLUT, glucose transporter; GSK-3β, glycogen synthase kinase 3β; d-2HG, d-2-hydroxyglutarate; HGF, hepatocyte growth factor; HIF-1α, hypoxia-inducible factor-1α; HK2, hexokinase; IDH, isocitrate dehydrogenase; IL-8, interleukin-8; α-KG, α-ketoglutarate; LDs, lipid droplets; LDH, lactate dehydrogenase; MCTs, monocarboxylate transporters; ME1, malic enzyme 1; MET, mesenchymal-to-epithelial transition; MMPs, matrix metalloproteases; mtROS, mitochondrial reactive oxygen species; NHE1, sodium-proton exchange 1; NSCLC, non-small cell lung cancer; O-GlcNAc, O-linked N-acetyl-glucosamine; OXPHOS, oxidative phosphorylation; PDAC, pancreatic ductal adenocarcinoma; PDH, pyruvate dehydrogenase; PDK4, pyruvate dehydrogenase kinase 4; PGI, glucose phosphate isomerase; PM, plasma membrane; PK, pyruvate kinase; PPARGC1A/PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; PPP, pentose phosphate pathway; SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine; SCD, stearoyl-CoA desaturase-1; SDH, succinate dehydrogenase; SIRT, sirtuin; SOD2, superoxide dismutase 2; SREBPs, sterol regulatory element-binding proteins; TAGs, triacylglicerols; TCA, tricarboxylic acid; TDH, threonine dehydrogenase; TET1, ten–eleven translocation 1; TGF-β, transforming growth factor-β; TGIF2, TGF-β-induced factor homeobox 2; TKTL1, transketolase-like protein 1 isoform; TNBC, triple-negative breast cancer; TNF-α, tumor necrosis factor-α; TUFM, TU translation elongation factor mitochondrial; VEGF, vascular endothelial growth factor.
References
1. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest (2009) 119(6):1420–8. doi: 10.1172/JCI39104
2. Moreno-Bueno G, Peinado H, Molina P, Olmeda D, Cubillo E, Santos V, et al. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat Protoc (2009) 4(11):1591–613. doi:10.1038/nprot.2009.152
3. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi:10.1038/nrm3758
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
5. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi:10.1085/jgp.8.6.519
6. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol (2017) 14(1):11–31. doi:10.1038/nrclinonc.2016.60
7. Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci (2016) 73(7):1333–48. doi:10.1007/s00018-015-2098-5
8. Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol (2004) 14(3):267–74. doi:10.1016/j.semradonc.2004.04.004
9. Attanasio F, Caldieri G, Giacchetti G, van Horssen R, Wieringa B, Buccione R. Novel invadopodia components revealed by differential proteomic analysis. Eur J Cell Biol (2011) 90(2–3):115–27. doi:10.1016/j.ejcb.2010.05.004
10. Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature (2009) 461(7260):109–13. doi:10.1038/nature08268
11. Danhier P, Copetti T, De Preter G, Leveque P, Feron O, Jordan BF, et al. Influence of cell detachment on the respiration rate of tumor and endothelial cells. PLoS One (2013) 8(1):e53324. doi:10.1371/journal.pone.0053324
12. Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH, Yoo MA, et al. Dlx-2 is implicated in TGF-β- and Wnt-induced epithelial-mesenchymal, glycolytic switch, and mitochondrial repression by Snail activation. Int J Oncol (2015) 46(4):1768–80. doi:10.3892/ijo.2015.2874
13. Yang L, Hou Y, Yuan J, Tang S, Zhang H, Zhu Q, et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget (2015) 6(28):25755–69. doi:10.18632/oncotarget.4697
14. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell (2013) 23(3):316–31. doi:10.1016/j.ccr.2013.01.022
15. Kondaveeti Y, Guttilla Reed IK, White BA. Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett (2015) 364(1):44–58. doi:10.1016/j.canlet.2015.04.025
16. Qin W, Li C, Zheng W, Guo Q, Zhang Y, Kang M, et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget (2015) 6(37):39839–54. doi:10.18632/oncotarget.5674
17. Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci U S A (2015) 112(32):E4410–7. doi:10.1073/pnas.1501605112
18. Liu M, Quek LE, Sultani G, Turner N. Epithelial-mesenchymal transition induction is associated with augmented glucose uptake and lactate production in pancreatic ductal adenocarcinoma. Cancer Metab (2016) 4:19. doi:10.1186/s40170-016-0160-x
19. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res (2016) 18(1):55. doi:10.1186/s13058-016-0712-6
20. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer (2016) 114(12):1305–12. doi:10.1038/bjc.2016.152
21. Liu PP, Liao J, Tang ZJ, Wu WJ, Yang J, Zeng ZL, et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ (2014) 21(1):124–35. doi:10.1038/cdd.2013.131
22. Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis (2014) 5:e1336. doi:10.1038/cddis.2014.285
23. Emmink BL, Verheem A, Van Houdt WJ, Steller EJ, Govaert KM, Pham TV, et al. The secretome of colon cancer stem cells contains drug-metabolizing enzymes. J Proteomics (2013) 91:84–96. doi:10.1016/j.jprot.2013.06.027
24. Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One (2014) 9(1):e84941. doi:10.1371/journal.pone.0084941
25. Palorini R, Votta G, Balestrieri C, Monestiroli A, Olivieri S, Vento R, et al. Energy metabolism characterization of a novel cancer stem cell-like line 3AB-OS. J Cell Biochem (2014) 115(2):368–79. doi:10.1002/jcb.24671
26. Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, Keating MJ, et al. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem (2011) 286(37):32843–53. doi:10.1074/jbc.M111.260935
27. Gammon L, Mackenzie IC. Roles of hypoxia, stem cells and epithelial-mesenchymal transition in the spread and treatment resistance of head and neck cancer. J Oral Pathol Med (2016) 45(2):77–82. doi:10.1111/jop.12327
28. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev (2010) 20(1):51–6. doi:10.1016/j.gde.2009.10.009
29. Taddei ML, Giannoni E, Comito G, Chiarugi P. Microenvironment and tumor cell plasticity: an easy way out. Cancer Lett (2013) 341(1):80–96. doi:10.1016/j.canlet.2013.01.042
30. Lin JL, Wang MJ, Lee D, Liang CC, Lin S. Hypoxia-inducible factor-1alpha regulates matrix metalloproteinase-1 activity in human bone marrow-derived mesenchymal stem cells. FEBS Lett (2008) 582(17):2615–9. doi:10.1016/j.febslet.2008.06.033
31. Jiang J, Tang YL, Liang XH. EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther (2011) 11(8):714–23. doi:10.4161/cbt.11.8.15274
32. Funasaka T, Hogan V, Raz A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial and mesenchymal phenotype conversions in breast cancer. Cancer Res (2009) 69(13):5349–56. doi:10.1158/0008-5472.CAN-09-0488
33. Liu L, Salnikov AV, Bauer N, Aleksandrowicz E, Labsch S, Nwaeburu C, et al. Triptolide reverses hypoxia-induced epithelial-mesenchymal transition and stem-like features in pancreatic cancer by NF-κB downregulation. Int J Cancer (2014) 134(10):2489–503. doi:10.1002/ijc.28583
34. Liu Y, Yan X, Xu Y, Luo F, Ye J, Yan H, et al. HIFs enhance the migratory and neoplastic capacities of hepatocellular carcinoma cells by promoting EMT. Tumour Biol (2014) 35(8):8103–14. doi:10.1007/s13277-014-2056-0
35. Du J, Sun B, Zhao X, Gu Q, Dong X, Mo J, et al. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol Oncol (2014) 133(3):575–83. doi:10.1016/j.ygyno.2014.02.034
36. Shaikh D, Zhou Q, Chen T, Ibe JC, Raj JU, Zhou G. cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial-mesenchymal transition, migration, and invasion in lung cancer cells. Cell Signal (2012) 24(12):2396–406. doi:10.1016/j.cellsig.2012.08.007
37. Joseph JV, Conroy S, Pavlov K, Sontakke P, Tomar T, Eggens-Meijer E, et al. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett (2015) 359(1):107–16. doi:10.1016/j.canlet.2015.01.010
38. Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid Redox Signal (2011) 14(12):2361–71. doi:10.1089/ars.2010.3727
39. Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs OXPHOS and prostate cancer metastatic spread. Oncotarget (2015) 6(27):24061–74. doi:10.18632/oncotarget.4448
40. Taddei ML, Cavallini L, Comito G, Giannoni E, Folini M, Marini A, et al. Senescent stroma promotes prostate cancer progression: the role of miR-210. Mol Oncol (2014) 8(8):1729–46. doi:10.1016/j.molonc.2014.07.009
41. Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med (2009) 13(9A):2780–6. doi:10.1111/j.1582-4934.2009.00876.x
42. Jeong W, Rapisarda A, Park SR, Kinders RJ, Chen A, Melillo G, et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother Pharmacol (2014) 73(2):343–8. doi:10.1007/s00280-013-2362-z
43. Kao SH, Wu KJ, Lee WH. Hypoxia, epithelial-mesenchymal transition, and TET-mediated epigenetic changes. J Clin Med (2016) 5(2). doi:10.3390/jcm5020024
44. Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, et al. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol (2014) 15(12):513. doi:10.1186/s13059-014-0513-0
45. Zhao M, Zhang Z. Glucose transporter regulation in cancer: a profile and the loops. Crit Rev Eukaryot Gene Expr (2016) 26(3):223–38. doi:10.1615/CritRevEukaryotGeneExpr.2016016531
46. Ito H, Duxbury M, Benoit E, Farivar RS, Gardner-Thorpe J, Zinner MJ, et al. Fibronectin-induced COX-2 mediates MMP-2 expression and invasiveness of rhabdomyosarcoma. Biochem Biophys Res Commun (2004) 318(2):594–600. doi:10.1016/j.bbrc.2004.04.070
47. Masin M, Vazquez J, Rossi S, Groeneveld S, Samson N, Schwalie PC, et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab (2014) 2:11. doi:10.1186/2049-3002-2-11
48. Ito S, Fukusato T, Nemoto T, Sekihara H, Seyama Y, Kubota S. Coexpression of glucose transporter 1 and matrix metalloproteinase-2 in human cancers. J Natl Cancer Inst (2002) 94(14):1080–91. doi:10.1093/jnci/94.14.1080
49. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, et al. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys (2001) 51(2):349–53. doi:10.1016/S0360-3016(01)01630-3
50. Koukourakis MI, Giatromanolaki A, Winter S, Leek R, Sivridis E, Harris AL. Lactate dehydrogenase 5 expression in squamous cell head and neck cancer relates to prognosis following radical or postoperative radiotherapy. Oncology (2009) 77(5):285–92. doi:10.1159/000259260
51. Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Trarbach T, Folprecht G, et al. Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin Cancer Res (2011) 17(14):4892–900. doi:10.1158/1078-0432.CCR-10-2918
52. Koukourakis MI, Giatromanolaki A, Panteliadou M, Pouliliou SE, Chondrou PS, Mavropoulou S, et al. Lactate dehydrogenase 5 isoenzyme overexpression defines resistance of prostate cancer to radiotherapy. Br J Cancer (2014) 110(9):2217–23. doi:10.1038/bjc.2014.158
53. Koukourakis MI, Kakouratos C, Kalamida D, Bampali Z, Mavropoulou S, Sivridis E, et al. Hypoxia-inducible proteins HIF1α and lactate dehydrogenase LDH5, key markers of anaerobic metabolism, relate with stem cell markers and poor post-radiotherapy outcome in bladder cancer. Int J Radiat Biol (2016) 92(7):353–63. doi:10.3109/09553002.2016.1162921
54. Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol (2009) 11(4):368–80. doi:10.1215/15228517-2008-106
55. Wick W, Naumann U, Weller M. Transforming growth factor-beta: a molecular target for the future therapy of glioblastoma. Curr Pharm Des (2006) 12(3):341–9. doi:10.2174/138161206775201901
56. Ahmed S, Tsuchiya T. Novel mechanism of tumorigenesis: increased transforming growth factor-beta 1 suppresses the expression of connexin 43 in BALB/cJ mice after implantation of poly-l-lactic acid. J Biomed Mater Res A (2004) 70(2):335–40. doi:10.1002/jbm.a.30090
57. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle (2010) 9(17):3506–14. doi:10.4161/cc.9.17.12731
58. Palmirotta R, Cives M, Della-Morte D, Capuani B, Lauro D, Guadagni F, et al. Sirtuins and cancer: role in the epithelial-mesenchymal transition. Oxid Med Cell Longev (2016) 2016:3031459. doi:10.1155/2016/3031459
59. Eades G, Yao Y, Yang M, Zhang Y, Chumsri S, Zhou Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J Biol Chem (2011) 286(29):25992–6002. doi:10.1074/jbc.M111.229401
60. Byles V, Zhu L, Lovaas JD, Chmilewski LK, Wang J, Faller DV, et al. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene (2012) 31(43):4619–29. doi:10.1038/onc.2011.612
61. Rani R, Kumar V. Recent update on human lactate dehydrogenase enzyme 5 (hLDH5) inhibitors: a promising approach for cancer chemotherapy. J Med Chem (2016) 59(2):487–96. doi:10.1021/acs.jmedchem.5b00168
62. Gallagher SM, Castorino JJ, Wang D, Philp NJ. Monocarboxylate transporter 4 regulates maturation and trafficking of CD147 to the plasma membrane in the metastatic breast cancer cell line MDA-MB-231. Cancer Res (2007) 67(9):4182–9. doi:10.1158/0008-5472.CAN-06-3184
63. Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang KY, et al. Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells. Cancer Sci (2011) 102(5):1007–13. doi:10.1111/j.1349-7006.2011.01908.x
64. De Saedeleer CJ, Porporato PE, Copetti T, Pérez-Escuredo J, Payen VL, Brisson L, et al. Glucose deprivation increases monocarboxylate transporter 1 (MCT1) expression and MCT1-dependent tumor cell migration. Oncogene (2014) 33(31):4060–8. doi:10.1038/onc.2013.454
65. Zhao Z, Wu MS, Zou C, Tang Q, Lu J, Liu D, et al. Downregulation of MCT1 inhibits tumor growth, metastasis and enhances chemotherapeutic efficacy in osteosarcoma through regulation of the NF-κB pathway. Cancer Lett (2014) 342(1):150–8. doi:10.1016/j.canlet.2013.08.042
66. Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res (2012) 72(19):5130–40. doi:10.1158/0008-5472.CAN-12-1949
67. Colen CB, Shen Y, Ghoddoussi F, Yu P, Francis TB, Koch BJ, et al. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: an in vivo study. Neoplasia (2011) 13(7):620–32. doi:10.1593/neo.11134
68. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest (2008) 118(12):3930–42. doi:10.1172/JCI36843
69. Marchiq I, Pouysségur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl) (2016) 94(2):155–71. doi:10.1007/s00109-015-1307-x
70. Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res (2006) 66(10):5216–23. doi:10.1158/0008-5472.CAN-05-4193
71. Shi Q, Le X, Wang B, Abbruzzese JL, Xiong Q, He Y, et al. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene (2001) 20(28):3751–6. doi:10.1038/sj.onc.1204500
72. Shi Q, Abbruzzese JL, Huang S, Fidler IJ, Xiong Q, Xie K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin Cancer Res (1999) 5(11):3711–21.
73. Chen KH, Tung PY, Wu JC, Chen Y, Chen PC, Huang SH, et al. An acidic extracellular pH induces Src kinase-dependent loss of beta-catenin from the adherens junction. Cancer Lett (2008) 267(1):37–48. doi:10.1016/j.canlet.2008.03.005
74. Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol (2007) 244(4):703–13. doi:10.1016/j.jtbi.2006.09.010
75. Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res (1994) 54(24):6517–25.
76. Kato Y, Ozawa S, Tsukuda M, Kubota E, Miyazaki K, St-Pierre Y, et al. Acidic extracellular pH increases calcium influx-triggered phospholipase D activity along with acidic sphingomyelinase activation to induce matrix metalloproteinase-9 expression in mouse metastatic melanoma. FEBS J (2007) 274(12):3171–83. doi:10.1111/j.1742-4658.2007.05848.x
77. Peppicelli S, Bianchini F, Calorini L. Extracellular acidity, a “reappreciated” trait of tumor environment driving malignancy: perspectives in diagnosis and therapy. Cancer Metastasis Rev (2014) 33(2–3):823–32. doi:10.1007/s10555-014-9506-4
78. Suzuki A, Maeda T, Baba Y, Shimamura K, Kato Y. Acidic extracellular pH promotes epithelial mesenchymal transition in Lewis lung carcinoma model. Cancer Cell Int (2014) 14(1):129. doi:10.1186/s12935-014-0129-1
79. Riemann A, Schneider B, Gündel D, Stock C, Thews O, Gekle M. Acidic priming enhances metastatic potential of cancer cells. Pflugers Arch (2014) 466(11):2127–38. doi:10.1007/s00424-014-1458-6
80. Hussain SA, Ganesan R, Reynolds G, Gross L, Stevens A, Pastorek J, et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br J Cancer (2007) 96(1):104–9. doi:10.1038/sj.bjc.6603530
81. Fiaschi T, Giannoni E, Taddei ML, Cirri P, Marini A, Pintus G, et al. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle (2013) 12(11):1791–801. doi:10.4161/cc.24902
82. Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ. Targeting the metabolic microenvironment of tumors. Adv Pharmacol (2012) 65:63–107. doi:10.1016/B978-0-12-397927-8.00004-X
83. Lyshchik A, Higashi T, Hara T, Nakamoto Y, Fujimoto K, Doi R, et al. Expression of glucose transporter-1, hexokinase-II, proliferating cell nuclear antigen and survival of patients with pancreatic cancer. Cancer Invest (2007) 25(3):154–62. doi:10.1080/07357900701208931
84. Rho M, Kim J, Jee CD, Lee YM, Lee HE, Kim MA, et al. Expression of type 2 hexokinase and mitochondria-related genes in gastric carcinoma tissues and cell lines. Anticancer Res (2007) 27(1A):251–8.
85. Peng SY, Lai PL, Pan HW, Hsiao LP, Hsu HC. Aberrant expression of the glycolytic enzymes aldolase B and type II hexokinase in hepatocellular carcinoma are predictive markers for advanced stage, early recurrence and poor prognosis. Oncol Rep (2008) 19(4):1045–53.
86. Palmieri D, Fitzgerald D, Shreeve SM, Hua E, Bronder JL, Weil RJ, et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol Cancer Res (2009) 7(9):1438–45. doi:10.1158/1541-7786.MCR-09-0234
87. Luo M, Davis A, McDermott S, Jiagge E, Brooks M, Gheordunescu E, et al. Abstract 2311: targeting EMT and MET breast cancer stem cell states through simultaneous inhibition of glycolytic and antioxidant pathways. Proceedings: AACR 106th Annual Meeting 2015. Vol. 75. Philadelphia, PA: Cancer Research (2015). doi:10.1158/1538-7445.AM2015-2311
88. Bacci M, Giannoni E, Fearns A, Ribas R, Gao Q, Taddei ML, et al. miR-155 drives metabolic reprogramming of ER+ breast cancer cells following long-term estrogen deprivation and predicts clinical response to aromatase inhibitors. Cancer Res (2016) 76(6):1615–26. doi:10.1158/0008-5472.CAN-15-2038
89. Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene (2006) 25(34):4633–46. doi:10.1038/sj.onc.1209597
90. Niizeki H, Kobayashi M, Horiuchi I, Akakura N, Chen J, Wang J, et al. Hypoxia enhances the expression of autocrine motility factor and the motility of human pancreatic cancer cells. Br J Cancer (2002) 86(12):1914–9. doi:10.1038/sj.bjc.6600331
91. Funasaka T, Yanagawa T, Hogan V, Raz A. Regulation of phosphoglucose isomerase/autocrine motility factor expression by hypoxia. FASEB J (2005) 19(11):1422–30. doi:10.1096/fj.05-3699com
92. Tsutsumi S, Fukasawa T, Yamauchi H, Kato T, Kigure W, Morita H, et al. Phosphoglucose isomerase enhances colorectal cancer metastasis. Int J Oncol (2009) 35(5):1117–21. doi:10.3892/ijo_00000427
93. Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, et al. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res (2011) 71(9):3400–9. doi:10.1158/0008-5472.CAN-10-0965
94. Nakamori S, Watanabe H, Kameyama M, Imaoka S, Furukawa H, Ishikawa O, et al. Expression of autocrine motility factor receptor in colorectal cancer as a predictor for disease recurrence. Cancer (1994) 74(7):1855–62. doi:10.1002/1097-0142(19941001)74:7<1855::AID-CNCR2820740705>3.0.CO;2-1
95. Maruyama K, Watanabe H, Shiozaki H, Takayama T, Gofuku J, Yano H, et al. Expression of autocrine motility factor receptor in human esophageal squamous cell carcinoma. Int J Cancer (1995) 64(5):316–21. doi:10.1002/ijc.2910640506
96. Takanami I, Takeuchi K, Naruke M, Kodaira S, Tanaka F, Watanabe H, et al. Autocrine motility factor in pulmonary adenocarcinomas: results of an immunohistochemical study. Tumour Biol (1998) 19(5):384–9. doi:10.1159/000030031
97. Du S, Guan Z, Hao L, Song Y, Wang L, Gong L, et al. Fructose-bisphosphate aldolase a is a potential metastasis-associated marker of lung squamous cell carcinoma and promotes lung cell tumorigenesis and migration. PLoS One (2014) 9(1):e85804. doi:10.1371/journal.pone.0085804
98. Chaerkady R, Harsha HC, Nalli A, Gucek M, Vivekanandan P, Akhtar J, et al. A quantitative proteomic approach for identification of potential biomarkers in hepatocellular carcinoma. J Proteome Res (2008) 7(10):4289–98. doi:10.1021/pr800197z
99. Fu QF, Liu Y, Fan Y, Hua SN, Qu HY, Dong SW, et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J Hematol Oncol (2015) 8:22. doi:10.1186/s13045-015-0117-5
100. Zhao M, Fang W, Wang Y, Guo S, Shu L, Wang L, et al. Enolase-1 is a therapeutic target in endometrial carcinoma. Oncotarget (2015) 6(17):15610–27. doi:10.18632/oncotarget.3639
101. Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature (2012) 488(7411):337–42. doi:10.1038/nature11331
102. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature (2008) 452(7184):230–3. doi:10.1038/nature06734
103. Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab (2012) 23(11):560–6. doi:10.1016/j.tem.2012.06.010
104. Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci U S A (2014) 111(43):15526–31. doi:10.1073/pnas.1407717111
105. Sborov DW, Haverkos BM, Harris PJ. Investigational cancer drugs targeting cell metabolism in clinical development. Expert Opin Investig Drugs (2015) 24(1):79–94. doi:10.1517/13543784.2015.960077
106. Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, et al. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem Pharmacol (2010) 79(8):1118–24. doi:10.1016/j.bcp.2009.12.003
107. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science (2011) 334(6060):1278–83. doi:10.1126/science.1211485
108. Cortés-Cros M, Hemmerlin C, Ferretti S, Zhang J, Gounarides JS, Yin H, et al. M2 isoform of pyruvate kinase is dispensable for tumor maintenance and growth. Proc Natl Acad Sci U S A (2013) 110(2):489–94. doi:10.1073/pnas.1212780110
109. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
110. Sun W, Liu Y, Glazer CA, Shao C, Bhan S, Demokan S, et al. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res (2010) 16(3):857–66. doi:10.1158/1078-0432.CCR-09-2604
111. Ciou SC, Chou YT, Liu YL, Nieh YC, Lu JW, Huang SF, et al. Ribose-5-phosphate isomerase A regulates hepatocarcinogenesis via PP2A and ERK signaling. Int J Cancer (2015) 137(1):104–15. doi:10.1002/ijc.29361
112. Chan B, VanderLaan PA, Sukhatme VP. 6-Phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem Biophys Res Commun (2013) 439(2):247–51. doi:10.1016/j.bbrc.2013.08.048
113. Langbein S, Zerilli M, Zur Hausen A, Staiger W, Rensch-Boschert K, Lukan N, et al. Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br J Cancer (2006) 94(4):578–85. doi:10.1038/sj.bjc.6602962
114. Langbein S, Frederiks WM, zur Hausen A, Popa J, Lehmann J, Weiss C, et al. Metastasis is promoted by a bioenergetic switch: new targets for progressive renal cell cancer. Int J Cancer (2008) 122(11):2422–8. doi:10.1002/ijc.23403
115. Krockenberger M, Honig A, Rieger L, Coy JF, Sutterlin M, Kapp M, et al. Transketolase-like 1 expression correlates with subtypes of ovarian cancer and the presence of distant metastases. Int J Gynecol Cancer (2007) 17(1):101–6. doi:10.1111/j.1525-1438.2007.00799.x
116. Zerilli M, Amato MC, Martorana A, Cabibi D, Coy JF, Cappello F, et al. Increased expression of transketolase-like-1 in papillary thyroid carcinomas smaller than 1.5 cm in diameter is associated with lymph-node metastases. Cancer (2008) 113(5):936–44. doi:10.1002/cncr.23683
117. Hanover JA, Krause MW, Love DC. Bittersweet memories: linking metabolism to epigenetics through O-GlcNAcylation. Nat Rev Mol Cell Biol (2012) 13(5):312–21. doi:10.1038/nrm3334
118. Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J (2010) 29(22):3787–96. doi:10.1038/emboj.2010.254
119. Ananieva E. Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World J Biol Chem (2015) 6(4):281–9. doi:10.4331/wjbc.v6.i4.281
120. Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH, Park HG, et al. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget (2016) 7(7):7925–39. doi:10.18632/oncotarget.6879
121. Yang C, Sudderth J, Dang T, Bachoo RM, Bachoo RG, McDonald JG, et al. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res (2009) 69(20):7986–93. doi:10.1158/0008-5472.CAN-09-2266
122. Liu G, Zhu J, Yu M, Cai C, Zhou Y, Fu Z, et al. Glutamate dehydrogenase is a novel prognostic marker and predicts metastases in colorectal cancer patients. J Transl Med (2015) 13:144. doi:10.1186/s12967-015-0500-6
123. Aguilar E, Marin de Mas I, Zodda E, Marin S, Morrish F, Selivanov V, et al. Metabolic reprogramming and dependencies associated with epithelial cancer stem cells independent of the epithelial-mesenchymal transition program. Stem Cells (2016) 34(5):1163–76. doi:10.1002/stem.2286
124. Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell (2015) 16(1):67–79. doi:10.1016/j.stem.2014.12.002
125. Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab (2012) 15(1):110–21. doi:10.1016/j.cmet.2011.12.009
126. Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest (2015) 125(6):2293–306. doi:10.1172/JCI75836
127. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood (2015) 126(11):1346–56. doi:10.1182/blood-2015-01-621870
128. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi:10.1158/1535-7163.MCT-13-0870
129. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16(10):619–34. doi:10.1038/nrc.2016.71
130. Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget (2016) 7(38):61890–904. doi:10.18632/oncotarget.11301
131. Kern U, Wischnewski V, Biniossek ML, Schilling O, Reinheckel T. Lysosomal protein turnover contributes to the acquisition of TGFβ-1 induced invasive properties of mammary cancer cells. Mol Cancer (2015) 14:39. doi:10.1186/s12943-015-0313-5
132. Akalay I, Janji B, Hasmim M, Noman MZ, André F, De Cremoux P, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res (2013) 73(8):2418–27. doi:10.1158/0008-5472.CAN-12-2432
133. Grassi G, Di Caprio G, Santangelo L, Fimia GM, Cozzolino AM, Komatsu M, et al. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis (2015) 6:e1880. doi:10.1038/cddis.2015.249
134. Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol (2015) 9(8):1612–25. doi:10.1016/j.molonc.2015.04.016
135. Gugnoni M, Sancisi V, Gandolfi G, Manzotti G, Ragazzi M, Giordano D, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene (2017) 36(5):667–77. doi:10.1038/onc.2016.237
136. Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene (2016) 35(1):1–11. doi:10.1038/onc.2015.99
137. Selhub J, Miller JW. The pathogenesis of homocysteinemia: interruption of the coordinate regulation by S-adenosylmethionine of the remethylation and transsulfuration of homocysteine. Am J Clin Nutr (1992) 55(1):131–8.
138. Luka Z, Mudd SH, Wagner C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J Biol Chem (2009) 284(34):22507–11. doi:10.1074/jbc.R109.019273
139. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer (2013) 13(8):572–83. doi:10.1038/nrc3557
140. Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE, et al. Pharmacologic disruption of polycomb repressive complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer (2011) 10:40. doi:10.1186/1476-4598-10-40
141. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature (2012) 492(7427):108–12. doi:10.1038/nature11606
142. Cruz PM, Mo H, McConathy WJ, Sabnis N, Lacko AG. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: a review of scientific findings, relevant to future cancer therapeutics. Front Pharmacol (2013) 4:119. doi:10.3389/fphar.2013.00119
143. Ackerman D, Simon MC. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol (2014) 24(8):472–8. doi:10.1016/j.tcb.2014.06.001
144. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis (2016) 5:e189. doi:10.1038/oncsis.2015.49
145. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov (2013) 12(9):688–702. doi:10.1038/nrd4099
146. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer (2010) 10(3):181–93. doi:10.1038/nrc2809
147. Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol (2013) 35(2):123–37. doi:10.1007/s00281-012-0342-8
148. Dalmau N, Jaumot J, Tauler R, Bedia C. Epithelial-to-mesenchymal transition involves triacylglycerol accumulation in DU145 prostate cancer cells. Mol Biosyst (2015) 11(12):3397–406. doi:10.1039/c5mb00413f
149. Hung CM, Kuo DH, Chou CH, Su YC, Ho CT, Way TD. Osthole suppresses hepatocyte growth factor (HGF)-induced epithelial-mesenchymal transition via repression of the c-Met/Akt/mTOR pathway in human breast cancer cells. J Agric Food Chem (2011) 59(17):9683–90. doi:10.1021/jf2021489
150. Li J, Dong L, Wei D, Wang X, Zhang S, Li H. Fatty acid synthase mediates the epithelial-mesenchymal transition of breast cancer cells. Int J Biol Sci (2014) 10(2):171–80. doi:10.7150/ijbs.7357
151. Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro-o M, et al. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene (2015) 34(30):3908–16. doi:10.1038/onc.2014.321
152. Kong D, Ahmad A, Bao B, Li Y, Banerjee S, Sarkar FH. Histone deacetylase inhibitors induce epithelial-to-mesenchymal transition in prostate cancer cells. PLoS One (2012) 7(9):e45045. doi:10.1371/journal.pone.0045045
153. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(10):992–1003,1–15. doi:10.1038/ncb3039
154. Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baró MR. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr (2002) 132(8):2123–6.
155. Sánchez-Martínez R, Cruz-Gil S, Gómez de Cedrón M, Álvarez-Fernández M, Vargas T, Molina S, et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget (2015) 6(36):38719–36. doi:10.18632/oncotarget.5340
156. Edmond V, Dufour F, Poiroux G, Shoji K, Malleter M, Fouqué A, et al. Downregulation of ceramide synthase-6 during epithelial-to-mesenchymal transition reduces plasma membrane fluidity and cancer cell motility. Oncogene (2015) 34(8):996–1005. doi:10.1038/onc.2014.55
157. Guan F, Handa K, Hakomori SI. Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc Natl Acad Sci U S A (2009) 106(18):7461–6. doi:10.1073/pnas.0902368106
158. Mathow D, Chessa F, Rabionet M, Kaden S, Jennemann R, Sandhoff R, et al. Zeb1 affects epithelial cell adhesion by diverting glycosphingolipid metabolism. EMBO Rep (2015) 16(3):321–31. doi:10.15252/embr.201439333
159. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest (2012) 122(6):2066–78. doi:10.1172/JCI59735
160. Sarkar TR, Battula VL, Werden SJ, Vijay GV, Ramirez-Peña EQ, Taube JH, et al. GD3 synthase regulates epithelial-mesenchymal transition and metastasis in breast cancer. Oncogene (2015) 34(23):2958–67. doi:10.1038/onc.2014.245
161. Grammatikos G, Schoell N, Ferreirós N, Bon D, Herrmann E, Farnik H, et al. Serum sphingolipidomic analyses reveal an upregulation of C16-ceramide and sphingosine-1-phosphate in hepatocellular carcinoma. Oncotarget (2016) 7(14):18095–105. doi:10.18632/oncotarget.7741
162. Zeng Y, Yao X, Chen L, Yan Z, Liu J, Zhang Y, et al. Sphingosine-1-phosphate induced epithelial-mesenchymal transition of hepatocellular carcinoma via an MMP-7/ syndecan-1/TGF-β autocrine loop. Oncotarget (2016) 7(39):63324–37. doi:10.18632/oncotarget.11450
163. Kim ES, Kim JS, Kim SG, Hwang S, Lee CH, Moon A. Sphingosine 1-phosphate regulates matrix metalloproteinase-9 expression and breast cell invasion through S1P3-Gαq coupling. J Cell Sci (2011) 124(Pt 13):2220–30. doi:10.1242/jcs.076794
164. Xin C, Ren S, Kleuser B, Shabahang S, Eberhardt W, Radeke H, et al. Sphingosine 1-phosphate cross-activates the Smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J Biol Chem (2004) 279(34):35255–62. doi:10.1074/jbc.M312091200
165. Mathias RA, Simpson RJ. Towards understanding epithelial-mesenchymal transition: a proteomics perspective. Biochim Biophys Acta (2009) 1794(9):1325–31. doi:10.1016/j.bbapap.2009.05.001
166. Thomson S, Petti F, Sujka-Kwok I, Mercado P, Bean J, Monaghan M, et al. A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis (2011) 28(2):137–55. doi:10.1007/s10585-010-9367-3
167. Tisza MJ, Zhao W, Fuentes JS, Prijic S, Chen X, Levental I, et al. Motility and stem cell properties induced by the epithelial-mesenchymal transition require destabilization of lipid rafts. Oncotarget (2016) 7(32):51553–68. doi:10.18632/oncotarget.9928
168. Wu H, Mahmood A, Lu D, Jiang H, Xiong Y, Zhou D, et al. Attenuation of astrogliosis and modulation of endothelial growth factor receptor in lipid rafts by simvastatin after traumatic brain injury. J Neurosurg (2010) 113(3):591–7. doi:10.3171/2009.9.JNS09859
169. van Blitterswijk WJ, Verheij M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim Biophys Acta (2013) 1831(3):663–74. doi:10.1016/j.bbalip.2012.10.008
170. Levental KR, Lorent JH, Lin X, Skinkle AD, Surma MA, Stockenbojer EA, et al. Polyunsaturated lipids regulate membrane domain stability by tuning membrane order. Biophys J (2016) 110(8):1800–10. doi:10.1016/j.bpj.2016.03.012
171. Sampaio JL, Gerl MJ, Klose C, Ejsing CS, Beug H, Simons K, et al. Membrane lipidome of an epithelial cell line. Proc Natl Acad Sci U S A (2011) 108(5):1903–7. doi:10.1073/pnas.1019267108
172. Warita K, Warita T, Beckwitt CH, Schurdak ME, Vazquez A, Wells A, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep (2014) 4:7593. doi:10.1038/srep07593
173. Zhao W, Prijic S, Urban BC, Tisza MJ, Zuo Y, Li L, et al. Candidate antimetastasis drugs suppress the metastatic capacity of breast cancer cells by reducing membrane fluidity. Cancer Res (2016) 76(7):2037–49. doi:10.1158/0008-5472.CAN-15-1970
174. Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8(3):754–66. doi:10.1016/j.celrep.2014.06.043
175. Porporato PE, Sonveaux P. Paving the way for therapeutic prevention of tumor metastasis with agents targeting mitochondrial superoxide. Mol Cell Oncol (2015) 2(3):e968043. doi:10.4161/23723548.2014.968043
176. Wendt MK, Schiemann BJ, Parvani JG, Lee YH, Kang Y, Schiemann WP. TGF-β stimulates Pyk2 expression as part of an epithelial-mesenchymal transition program required for metastatic outgrowth of breast cancer. Oncogene (2013) 32(16):2005–15. doi:10.1038/onc.2012.230
177. Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta (2014) 1842(8):1282–94. doi:10.1016/j.bbadis.2013.09.007
178. Kagawa S, Natsuizaka M, Whelan KA, Facompre N, Naganuma S, Ohashi S, et al. Cellular senescence checkpoint function determines differential Notch1-dependent oncogenic and tumor-suppressor activities. Oncogene (2015) 34(18):2347–59. doi:10.1038/onc.2014.169
179. Yoon YS, Lee JH, Hwang SC, Choi KS, Yoon G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene (2005) 24(11):1895–903. doi:10.1038/sj.onc.1208262
180. Kinugasa H, Whelan KA, Tanaka K, Natsuizaka M, Long A, Guo A, et al. Mitochondrial SOD2 regulates epithelial-mesenchymal transition and cell populations defined by differential CD44 expression. Oncogene (2015) 34(41):5229–39. doi:10.1038/onc.2014.449
181. Chen PM, Wu TC, Wang YC, Cheng YW, Sheu GT, Chen CY, et al. Activation of NF-κB by SOD2 promotes the aggressiveness of lung adenocarcinoma by modulating NKX2-1-mediated IKKβ expression. Carcinogenesis (2013) 34(11):2655–63. doi:10.1093/carcin/bgt220
182. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J (2000) 348(Pt 3):607–14. doi:10.1042/bj3480607
183. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med (2013) 369(2):111–21. doi:10.1056/NEJMoa1300874
184. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab (2014) 20(6):953–66. doi:10.1016/j.cmet.2014.09.018
185. Kordes S, Pollak MN, Zwinderman AH, Mathôt RA, Weterman MJ, Beeker A, et al. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol (2015) 16(7):839–47. doi:10.1016/S1470-2045(15)00027-3
186. Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun (2016) 7:13041. doi:10.1038/ncomms13041
187. Gaude E, Frezza C. Defects in mitochondrial metabolism and cancer. Cancer Metab (2014) 2:10. doi:10.1186/2049-3002-2-10
188. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi:10.1126/sciadv.1600200
189. Sun Y, Daemen A, Hatzivassiliou G, Arnott D, Wilson C, Zhuang G, et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab (2014) 2(1):20. doi:10.1186/2049-3002-2-20
190. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462(7274):739–44. doi:10.1038/nature08617
191. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26(12):1326–38. doi:10.1101/gad.191056.112
192. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360(8):765–73. doi:10.1056/NEJMoa0808710
193. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med (2009) 361(11):1058–66. doi:10.1056/NEJMoa0903840
194. Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer (2009) 125(2):353–5. doi:10.1002/ijc.24379
195. Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep (2011) 12(5):463–9. doi:10.1038/embor.2011.43
196. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19(1):17–30. doi:10.1016/j.ccr.2010.12.014
197. Terunuma A, Putluri N, Mishra P, Mathé EA, Dorsey TH, Yi M, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest (2014) 124(1):398–412. doi:10.1172/JCI71180
198. Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia induces production of l-2-hydroxyglutarate. Cell Metab (2015) 22(2):304–11. doi:10.1016/j.cmet.2015.06.023
199. Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab (2015) 22(2):291–303. doi:10.1016/j.cmet.2015.06.021
200. Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, et al. Oncometabolite d-2-hydroxyglutarate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci Rep (2016) 6:36289. doi:10.1038/srep36289
201. Grassian AR, Lin F, Barrett R, Liu Y, Jiang W, Korpal M, et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J Biol Chem (2012) 287(50):42180–94. doi:10.1074/jbc.M112.417832
202. Liesenfeld DB, Botma A, Habermann N, Toth R, Weigel C, Popanda O, et al. Aspirin reduces plasma concentrations of the oncometabolite 2-hydroxyglutarate: results of a randomized, double-blind, crossover trial. Cancer Epidemiol Biomarkers Prev (2016) 25(1):180–7. doi:10.1158/1055-9965.EPI-15-0697
203. Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer (2005) 5(11):857–66. doi:10.1038/nrc1737
204. Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet (2002) 30(4):406–10. doi:10.1038/ng849
205. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science (2000) 287(5454):848–51. doi:10.1126/science.287.5454.848
206. Selak MA, Armour SM, MacKenzie, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7(1):77–85. doi:10.1016/j.ccr.2004.11.022
207. Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG, et al. Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J Biol Chem (2016) 291(8):4256–65. doi:10.1074/jbc.M115.688762
208. Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell (2011) 20(4):511–23. doi:10.1016/j.ccr.2011.08.024
209. Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell (2011) 20(4):524–37. doi:10.1016/j.ccr.2011.09.006
210. Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep (2013) 3(3):689–700. doi:10.1016/j.celrep.2013.02.013
211. Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell (2013) 51(2):236–48. doi:10.1016/j.molcel.2013.05.003
212. Sciacovelli M, Gonçalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature (2016) 537(7621):544–7. doi:10.1038/nature19353
213. He X, Yan B, Liu S, Jia J, Lai W, Xin X, et al. Chromatin remodeling factor LSH drives cancer progression by suppressing the activity of fumarate hydratase. Cancer Res (2016) 76(19):5743–55. doi:10.1158/0008-5472.CAN-16-0268
214. Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta (2011) 1807(11):1432–43. doi:10.1016/j.bbabio.2011.07.003
215. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene (2006) 25(34):4675–82. doi:10.1038/sj.onc.1209594
216. Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov (2013) 3(6):648–57. doi:10.1158/2159-8290.CD-13-0092
217. Morin A, Letouzé E, Gimenez-Roqueplo AP, Favier J. Oncometabolites-driven tumorigenesis: from genetics to targeted therapy. Int J Cancer (2014) 135(10):2237–48. doi:10.1002/ijc.29080
218. Loriot C, Domingues M, Berger A, Menara M, Ruel M, Morin A, et al. Deciphering the molecular basis of invasiveness in Sdhb-deficient cells. Oncotarget (2015) 6(32):32955–65. doi:10.18632/oncotarget.5106
219. Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell (2013) 23(6):739–52. doi:10.1016/j.ccr.2013.04.018
220. Aspuria PJ, Lunt SY, Väremo L, Vergnes L, Gozo M, Beach JA, et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab (2014) 2:21. doi:10.1186/2049-3002-2-21
221. Loriot C, Burnichon N, Gadessaud N, Vescovo L, Amar L, Libé R, et al. Epithelial to mesenchymal transition is activated in metastatic pheochromocytomas and paragangliomas caused by SDHB gene mutations. J Clin Endocrinol Metab (2012) 97(6):E954–62. doi:10.1210/jc.2011-3437
222. Carén H, Djos A, Nethander M, Sjöberg RM, Kogner P, Enström C, et al. Identification of epigenetically regulated genes that predict patient outcome in neuroblastoma. BMC Cancer (2011) 11:66. doi:10.1186/1471-2407-11-66
223. Morris MR, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, et al. Functional epigenomics approach to identify methylated candidate tumour suppressor genes in renal cell carcinoma. Br J Cancer (2008) 98(2):496–501. doi:10.1038/sj.bjc.6604180
224. Ju JH, Yang W, Lee KM, Oh S, Nam K, Shim S, et al. Regulation of cell proliferation and migration by keratin19-induced nuclear import of early growth response-1 in breast cancer cells. Clin Cancer Res (2013) 19(16):4335–46. doi:10.1158/1078-0432.CCR-12-3295
225. Tripathi MK, Misra S, Chaudhuri G. Negative regulation of the expressions of cytokeratins 8 and 19 by SLUG repressor protein in human breast cells. Biochem Biophys Res Commun (2005) 329(2):508–15. doi:10.1016/j.bbrc.2005.02.006
226. Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science (2009) 325(5944):1139–42. doi:10.1126/science.1175689
227. Kaelin WG. SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell (2009) 16(3):180–2. doi:10.1016/j.ccr.2009.08.013
228. Kunst HP, Rutten MH, de Mönnink JP, Hoefsloot LH, Timmers HJ, Marres HA, et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res (2011) 17(2):247–54. doi:10.1158/1078-0432.CCR-10-0420
229. Liu J, Gao L, Zhang H, Wang D, Wang M, Zhu J, et al. Succinate dehydrogenase 5 (SDH5) regulates glycogen synthase kinase 3β-β-catenin-mediated lung cancer metastasis. J Biol Chem (2013) 288(41):29965–73. doi:10.1074/jbc.M113.450106
230. He K, Guo X, Liu Y, Li J, Hu Y, Wang D, et al. TUFM downregulation induces epithelial-mesenchymal transition and invasion in lung cancer cells via a mechanism involving AMPK-GSK3β signaling. Cell Mol Life Sci (2016) 73(10):2105–21. doi:10.1007/s00018-015-2122-9
Keywords: epithelial-to-mesenchymal transition, metabolic reprogramming, Warburg metabolism, OXPHOS, TCA cycle, oncometabolites, amino acid, lipids
Citation: Morandi A, Taddei ML, Chiarugi P and Giannoni E (2017) Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 7:40. doi: 10.3389/fonc.2017.00040
Received: 22 December 2016; Accepted: 28 February 2017;
Published: 14 March 2017
Edited by:
Silvia Giordano, University of Turin, ItalyReviewed by:
K. B. Harikumar, Rajiv Gandhi Centre for Biotechnology, IndiaMaria Felice Brizzi, University of Turin, Italy
Copyright: © 2017 Morandi, Taddei, Chiarugi and Giannoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Morandi, andrea.morandi@unifi.it;
Paola Chiarugi, paola.chiarugi@unifi.it