 Shaun P. Stice
Shaun P. Stice Spencer D. Stumpf2†
Spencer D. Stumpf2† Brian H. Kvitko
Brian H. Kvitko Bhabesh Dutta
Bhabesh Dutta- 1Department of Plant Pathology, University of Georgia, Athens, GA, United States
- 2Department of Plant Pathology, University of Georgia, Tifton, GA, United States
- 3The Plant Center, University of Georgia, Athens, GA, United States
Pantoea ananatis is a member of the family Enterobacteriaceae and an enigmatic plant pathogen with a broad host range. Although P. ananatis strains can be aggressive on onion causing foliar necrosis and onion center rot, previous genomic analysis has shown that P. ananatis lacks the primary virulence secretion systems associated with other plant pathogens. We assessed a collection of fifty P. ananatis strains collected from Georgia over three decades to determine genetic factors that correlated with onion pathogenic potential. Previous genetic analysis studies have compared strains isolated from different hosts with varying diseases potential and isolation sources. Strains varied greatly in their pathogenic potential and aggressiveness on different cultivated Allium species like onion, leek, shallot, and chive. Using multi-locus sequence analysis (MLSA) and repetitive extragenic palindrome repeat (rep)-PCR techniques, we did not observe any correlation between onion pathogenic potential and genetic diversity among strains. Whole genome sequencing and pan-genomic analysis of a sub-set of 10 strains aided in the identification of a novel series of genetic regions, likely plasmid borne, and correlating with onion pathogenicity observed on single contigs of the genetic assemblies. We named these loci Onion Virulence Regions (OVR) A-D. The OVR loci contain genes involved in redox regulation as well as pectate lyase and rhamnogalacturonase genes. Previous studies have not identified distinct genetic loci or plasmids correlating with onion foliar pathogenicity or pathogenicity on a single host pathosystem. The lack of focus on a single host system for this phytopathgenic disease necessitates the pan-genomic analysis performed in this study.
Introduction
Pantoea ananatis is a member of the Enterobacteriaceae family and is a ubiquitous phytopathogenic bacterium capable of infecting diverse monocotyledonous hosts including maize, rice and pineapple as well as the woody host, Eucalyptus (Serrano, 1928; Tabei et al., 1988; Paccola-Meirelles et al., 2001; Coutinho et al., 2002; Coutinho and Venter, 2009). P. ananatis strains have also been recovered from opportunistic infections in humans (De Baere et al., 2004). In the United States, P. ananatis is of primary concern as a causative agent of center rot of onion (Allium cepa L.). The first outbreak of center rot of onion in the United States occurred in Georgia in 1997 and since then it has been identified in different onion growing regions (Gitaitis and Gay, 1997; Schwartz and Otto, 2000; Carr et al., 2010). P. ananatis outbreaks are a recurring challenge for onion production and outbreaks resulting in considerable crop loss have been documented (Walcott et al., 2002). P. ananatis infection can be transmitted via seed but it is also a common epiphyte on weeds and is known to be transmitted transiently between plants by two different species of thrips (Gitaitis et al., 2002; Walcott et al., 2002; Dutta et al., 2014). Infection of onions is facilitated through wounds or thrips damage. P. ananatis strains are known to express the ice-nucleation active (InaZ) cell surface protein which efficiently nucleates ice crystals and is associated with increased incidence of frost damage (Lindow, 1983; Abe et al., 1989). Upon leaf invasion, P. ananatis reaches high levels in leaf blades and causes foliar necrosis. The pathogen can also progress from leaf blade to the corresponding onion bulb scale where, alongside colonization by secondary pathogens, it causes center rot symptoms in central scales (Carr et al., 2013).
Management and accurate diagnosis of P. ananatis has been complicated due to the phenotypic variation among different strains in onion pathogenicity. P. ananatis strains collected from non-onion sources (thrips, weeds) may either be highly aggressive on onions or completely onion non-pathogenic. Currently no strain features have been described that can predict onion pathogenic potential.
Pantoea ananatis is an enigma among bacterial phytopathogens. Whole genome sequencing has shown that phytopathogenic P. ananatis strains lack Type II, Type III, and Type IV virulence-associated secretion systems as well as known phytotoxin synthesis genes (De Maayer et al., 2014; Sheibani-Tezerji et al., 2015; Weller-Stuart et al., 2017). In the absence of these pathogenicity factors, secretion systems used to deliver Cell Wall Degrading Enzymes (CWDEs) outside of the bacterium (i.e., T2SS), or virulence effector proteins directly into the plant cells (i.e., T3SS/T4SS); however, it remains an open question as to what host interaction mediated pathogenicity factors are associated with P. ananatis infection of onion. Progressing leaf necrosis has been shown to be dependent on pathogen quorum sensing and swimming motility (Morohoshi et al., 2007). In addition, the Type VI Secretion System (T6SS), most commonly associated with inter-bacterial competition, was shown to play a role in foliar necrosis symptoms but with little effect on pathogen populations (Morohoshi et al., 2007; Shyntum et al., 2015; Weller-Stuart et al., 2017).
In this study we assessed a collection of fifty Georgia P. ananatis strains collected over three decades (1997–2015) from diverse sources to determine what genetic factors correlated with onion pathogenic potential. Using multi-locus sequence analysis (MLSA) and repetitive extragenic palindrome (rep)-PCR techniques, we did not observe strong correlations between onion pathogenic potential and genetic diversity among strains. We found that strains varied greatly in their pathogenic potential and aggressiveness on different cultivated Allium species like onion, shallot (Allium cepa var. aggregatum), chives (Allium schoenoprasum), and leeks (Allium porrum). Several strains that were non-pathogenic on onion displayed significant virulence on leek. Pan-genome analysis derived from whole genome sequencing of a sub-set of six onion pathogenic and four onion non-pathogenic Georgia strains identified four loci associated only with the six onion pathogenic strains. These four loci were present on a single contig. We named these loci as the Onion Virulence Regions A through D (OVR-A-D). The OVR loci contains pectate lyase and rhamnogalacturonase genes as well as genes that are involved in redox regulation.
Materials and Methods
Bacterial Strains, Identification, and Culture Preparation
Fifty P. ananatis strains used in this study were isolated from onion and other sources including weeds and thrips throughout the state of Georgia (Table 1). These strains were stored in sterile 15% aqueous glycerol solution at −80°C. The strains which were isolated from either symptomatic onion foliage, bulb, or from contaminated onion seeds were designated as “PNA” in our culture collection. Strains isolated from asymptomatic weeds or from the gut of thrips (Thrips tabaci and Frankliniella fusca) were designated as “PANS.” The source, year of isolation, and county of origin in Georgia for these strains are shown in Table 1. These strains were identified as P. ananatis by their morphological and physiological characteristics such as Gram-negative, facultative anaerobic, positive for indole production, negative for nitrate reductase and phenylalanine deaminase and by PCR amplification of a 398 base pair (bp) fragment using P. ananatis species-specific primers (Gitaitis et al., 2002).
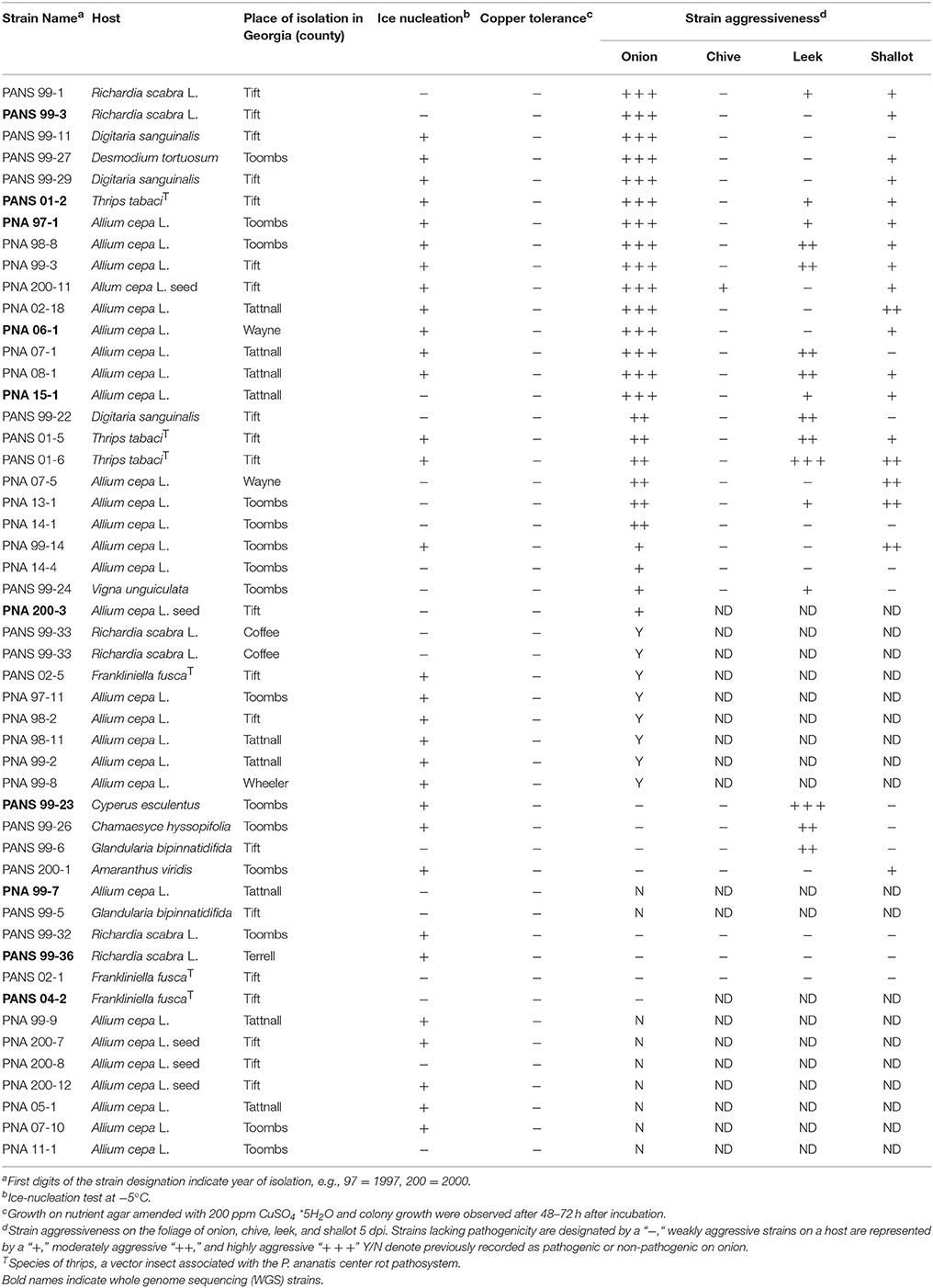
Table 1. Isolates of Pantoea ananatis and their associated phenotypic characteristics.
Inoculum was prepared by transferring single colonies of each bacterial strain from 48-h old cultures on nutrient agar (NA) medium to nutrient broth or lysogeny broth (LB). The broth was shaken overnight on a rotary shaker (Inova; New Brunswick Scientific, Edison, NJ) at 150 rpm. After a 12-h of incubation, 10 ml of each bacterial suspension were centrifuged at 6,000 × g (Allegra 25R, Beckman Coulter, Fullerton, CA) for 5 min. The supernatant was discarded and the pellet was re-suspended in 0.1 M phosphate-buffered saline (PBS). Later, inoculum was adjusted using a spectrophotometer (Spectronic 20; Bausch and Lomb, Rochester, NY) to an optical density of 0.3 at 600 nm [≈1 × 108 colony forming unit (CFU)/ml]. The bacterial suspension was serially diluted in PBS to obtain the desired concentration according to each experiment. PNA 97-1R is a spontaneous rifampicin (Rf) resistant clone of PNA 97-1 recovered after plating a dense suspension of PNA 97-1 onto an LB Rf 60 plate.
Multi-locus Sequence Analysis (MLSA) of P. ananatis Strains
Total microbial genomic DNA was extracted from a 3 ml volume of overnight cultures grown at 28°C in nutrient broth using an UltraClean Microbial DNA Kit (MO BIO, Carlsbad, CA) according to the manufacturer's instructions. Extracted genomic DNA was quantified on a NanoDrop 1000 spectrophotometer (Thermo Scientific, Wilmington, U.S.A.) and 50 ng of genomic DNA were used for each PCR reaction. Six primer pairs of housekeeping genes, fusA, gyrB, leuS, pyrG, rpl, and rpoB, were chosen based on a previous MLSA scheme (Salerno et al., 2007; Deletoile et al., 2009). The concatenation of these six genes represented a total of 2,961 bp. Primer sequences and PCR cycles are outlined in Supplementary Table 3. Each PCR reaction was performed in a 50 μl reaction consisted of 10 μl of 5x PCR buffer, 0.2 mM of dNTPs, 0.2 μM of each primer, 1.25 U HotStart Taq Polymerase (Qiagen, Valencia, Ca) and 50 ng/μl of template DNA. PCR products were subjected to gel electrophoresis in Tris-acetate-EDTA buffer on a 1.5% agarose gel at 100 V for 45 min. Reactions resulting in positive amplifications and expected fragment size were purified with a DNA Clean and Concentrator Kit (Zymo Research Inc., Irvine, Ca), according to the manufacturer's instructions. Fragment sequencing was conducted by Eurofins, (Louisville, KY).
Phylogenetic Analysis of Sequencing Data
Sequences were manually trimmed and verified based on peak quality of forward and reverse chromatograms in Geneious 8.1.8 (Biomatters, Auckland, New Zealand) and were aligned using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/). The nucleotide sites presenting alignment gaps were excluded from analysis and were then concatenated. Consensus sequences were used to generate a maximum-likelihood (ML) tree based on the Tamura-Nei Model of MEGA 6.06 with bootstrapping repetitions of 1,000. Sequences from P. agglomerans 97-1 (accession no. FN434113.1) from GenBank were used as outgroups. MLST sequences generated from this study were deposited in GenBank under accession numbers MF925240–MF925289, MF939901–MF939950, MF939951–MF940000, MF940001–MF940050, MF940051–MF940100, and MF964629–MF964678.
Repetitive Extragenic Palindrome (rep)-PCR Amplification and Analysis
Genomic DNA from strains were isolated as described above and subjected to rep-PCR using REP1R-I (III ICG ICG ICA TCI GGC) and REP 2-I (ICG ICT TAT CIG GCC TAC) primers (Versalovic et al., 1991). The amplification was carried out in a total of 25 μl reactions containing 50 ng of DNA, 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, 200 μM dNTPs, 2.5 U pureTaq DNA polymerase, and 0.5 μM of each primer pair. Amplifications were done using the following conditions: an initial denaturation at 95° C for 4 min, 45 cycles consisting of 94°C for 1 min, 45°C for 1 min, and 65°C for 8 min, followed by cooling for 4 min. Samples were separated by gel electrophoresis in Tris-acetate-EDTA buffer on a 1.5% agarose gel for 2 h at 100 V stained with GelGreen. DNA fingerprinting profile for each P. ananatis strain were compared using Dice's (1945) coefficient of analysis with the aid of BioNumerics software package (Applied Math, Kortrijk, Belgium), and the unweighted pairwise group method with arithmetic mean algorithm was used to generate a dendrogram indicating strain relatedness.
Aggressiveness of P. ananatis Strains on Onion, Chive, Leek, and Shallot
The aggressiveness of 33 selected strains were tested on 6-week old shallots (cv. “Camelot F1”), chives (cv. “Dolores”), leeks (cv. “King Richard”), and onion (cv. “Century”) under greenhouse conditions. Strains from 11 clades (n = 23) and from unresolved portion (n = 10) of the MLSA phylogenetic tree were selected for this assay. These strains represented 66% of the total P. ananatis strains from 11 clades and unresolved portion. Seedlings of each plant species were established in 10 × 8 cm (diameter × height) plastic pots (Hummert International, Earth City, MO) containing commercial potting mix. The seedlings were kept in greenhouse conditions and maintained at 25–28°C and 80–90% RH with a 12L:12D photoperiod. Seedlings were fertilized with osmocote smart-release plant food (14-14-14) (Scotts Miracle-Gro Company, Marysville, OH). Inoculum preparation and standardization were conducted as described above. Seedlings were inoculated by cutting the central leaf 1 cm from the apex with a sterile pair of scissors. Using a micropipette, a 10 μl drop of bacterial suspensions containing 1 × 108 CFU/mL (≈1 × 106 CFU) were placed at the cut end of the seedlings. Seedlings mechanically inoculated with PBS as described above were used as negative controls. Four replications per strain per host were used in a single experiment and this experiment was repeated one more time. The experiment was arranged in a randomized complete block design. Seedlings were checked visually for symptom development every day until 5 days after inoculation (DAI). At 5 DAI, lesion length for each seedling was measured and mean lesion length for each strain on each plant species were compared using Fisher's protected least significant difference test at P < 0.05 level.
To confirm that symptoms observed were caused by P. ananatis, bacteria from symptomatic tissue (n = 5 symptomatic plants per host) were isolated from a region adjoining the necrotic and healthy tissue and streaked onto Tryptic soy broth agar (TSBA) plates and incubated for 48-h at 28°C. After 48-h of incubation, yellow-pigmented colonies were isolated and tested for P. ananatis using physiological tests (as previously described) and species-specific TaqMan based polymerase-chain reaction (PCR) assays (Walcott et al., 2002). Briefly, presumptive P. ananatis colonies were picked using a sterile inoculation loop and suspended separately in a 2 ml micro-centrifuge tubes containing 25 μl of sterile deionized water. The bacterial suspension was heated (Modular Dry Block Heaters, Cole Parmer, IL) for 3 min at 95°C. A suspension (5 μl) was amplified in 20 μl of PCR master-mix containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 0.1% Triton X-100, 1.5 mM MgCl2, and 0.2 mM of each nucleotide (dATP, dCTP, dGTP, and dTTP), 25 μM each of primer PanITS1 (5′-GTCTGATAGAAAGATAAAGAC-3′) and EC5 (5′- CGGTGGATGCCCTGGCA-3′) and 10 μM of TaqMan probe 6-FAM TAGCGGTTAGGACTCCGCCCTTTCA-BHQ. The PCR reaction was conducted in a Cepheid Smart Cycler (Sunnyvale, CA) using the following thermal profile: denaturation at 95°C for 180 s, 35 cycles each of denaturation at 95°C for 15 s, and annealing at 60°C for 40 s. Samples with cycle threshold (Ct) values <35 were considered positive for P. ananatis.
Red Onion Scale Assay
Consumer produce red onions (Allium cepa. L., Red Onion Delana Farms, CA) were purchased, sliced to remove any diseased tissue, cut to approximately 3 cm wide scales, sterilized in a 3% bleach solution for 1 min and promptly removed and rinsed in dH2O. Scales were placed in a potting tray (27.0 × 52.0 cm) containing two layers of paper towels pre-moistened with 90 ml of distilled water. Individual onion scales were wounded on the inner surface with a sterile pipette tip, and 10 μl of a prepared bacterial suspension (previously described) (106 CFU/ml) was inoculated into the wound. The tray was covered with a plastic humidity dome. The onion scales were incubated at room temperature for 72 h in the dark. The size of lesion necrotic zone and pigment-clearing zone was recorded to assess pathogenicity. Pathogenic strains that cleared the red anthocyanin pigment and caused maceration were labeled pathogenic and those that did not were labeled non-pathogenic. Six replicates were carried out for each strain. Sterile deionized water was used as negative control.
Ice Nucleation Test
To test the ice nucleating ability of P. ananatis strains, five separate drops of 10 μl aliquots of bacterial suspension (>1 × 109 CFU/ml) were pipetted onto a sterile weigh boat in an ice-ethanol water bath adjusted to −5°C. Sterile water was used as a negative control and PNA 97-1, an ice-nucleating strain, was used as a positive control. Samples that froze concurrently with the positive control were considered ice nucleation positive and samples that remained in liquid form or with delayed ice-nucleation activity were considered ice nucleation negative. Fifty P. ananatis strains were used and the experiment was repeated twice.
Copper Tolerance
Strains were grown and re-suspended to an optical density of 0.3 at 600 nm, as previously described, and suspensions were serially diluted to a concentration of 106 CFU/ml. Aliquots of 50 μl were spread onto nutrient agar (NA) and NA amended with 200 ppm CuSO4*5H2O. Plates were incubated at 28C for 48–72 h and observed for bacterial growth.
High Quality Draft Genome of PNA 97-1R
Genomic DNA from the prototype strain PNA 97-1R was obtained from overnight cultures started from single colonies using the Gentra Puregene Yeast/Bact kit (Qiagen Inc), quantified using biospectrometer (Eppendorf), and visually assessed for quality on a 0.75% agarose gel. DNA quality, sequencing and assembly were carried out at the University of Delaware DNA Sequencing & Genotyping Center (Newark, DE). DNA was assessed for quality metrics via Fragment Analyzer (Advanced Analytical Technologies). Sequence reads were generated by a Pacific Biosciences RSII Single-Molecule DNA Sequencer following SMRT Bell library preparation. Basic de-novo assembly was carried out using PacBio HGAP assembler (v3) with seed reads 17 kilo base (kb) and above. Contigs were polished using Quiver and annotation was performed via Rapid Annotation using Subsystem Technology (RAST v2.0) or Prokaryote Genome Annotation Pipeline (PGAP v4.2.) (Zhao et al., 2012; Overbeek et al., 2014).
Draft Genomes of 10 Diverse Strains
Genomic DNA from 10 diverse strains (PNA 97-1R, PNA 06-01, PNA 200-3, PNA 15-1, PNA 99-7, PANS 04-02, PANS 01-02, PANS 99-23, PANS 99-36, and PANS 99-3) was obtained from overnight cultures started from single colonies using the Gentra Puregene Yeast/Bact Kit (Qiagen Inc.), quantified using Biospectrometer (Eppendorf), and visually assessed as above on a 0.75% agarose gel. Sequencing, assembly, annotation, and variant calling were carried out at MicrobesNG (Birmingham, U.K.). Sequence reads were generated by an Illumina HiSeq using 250-bp paired-end libraries, and assembled using SPAdes (version 3.10.1). Reads were mapped to the reference genome PNA 97-1R and back to contigs using BWA-MEM to ensure quality and 30X coverage. Annotation was performed via Prokka or RAST (Overbeek et al., 2014; Seemann, 2014).
Pan-Genomic Analysis
The Prokka annotated fragmented de novo assemblies for the ten draft genomes for strains (PNA 97-1R, PNA 06-01, PNA 200-3, PNA 15-1, PNA 99-7, PANS 04-02, PANS 01-02, PANS99-23, PANS 99-36, and PANS 99-3) were loaded in GFF3 format to the ROARY (v3.6.1) program using default parameters to calculate the pan-genome of the 10 strains (Page et al., 2015). ROARY was run on a windows computer through an emulated Linux machine as described by the authors (https://sanger-pathogens.github.io/Roary/). The resulting newick tree files were edited using Geneious (v10.1.3). The “gene_presence_absence.csv” output file was analyzed using Microsoft excel to identify gene that are present in the pathogenic strains and absent in the non-pathogenic strains (Supplementary File 1).
The average nucleotide identity of the strains (ANI) was calculated using the online ANI calculator server with default parameters to compare WGS strains to the genomic type strain LMG 20103 (http://enve-omics.ce.gatech.edu/ani/). Additional Pan-genomic analysis was conducted using RAST BLASTP pairwise alignment with PNA-97-1 listed as reference genome. The computationally intensive Pan Genomic Analysis Pipeline (PGAP) was run on the UGA Sapelo computer cluster (Zhao et al., 2012). The resulting “5.Orthologs_Cluster_Function” file was opened with Microsoft excel and genes clustering in pathogenic strains and non-pathogenic strains were cross referenced with the results from ROARY analysis. The resulting putative virulence genes were then identified in the contigs of the draft genomes using Geneious. The contigs containing putative virulence associated genes were extracted and aligned using the Geneious Mauve plugin (v2.3.1) (Darling et al., 2004). Putative secretion target signals were assessed by running genes through the Signal P (v4.1) Server and TMHMM Server (v2.0) for prediction of transmembrane helices in proteins (Darling et al., 2004; Nielsen, 2017). Genomes were also investigated for secondary metabolite non-ribosomal peptide synthetase (NRPS) biosynthesis gene clusters using the antiSMASH bacterial server. Whole genome MLST (wgMSLT) was conducted using PGAdb-builder (Liu et al., 2016). The 95% occurrence allele scheme was selected to generate a wgMLST tree with P. agglomerans PC10 and P. stewartii subsp. indologens LMG 2632 selected as outgroups (Accession NZ_LIME00000000.1 and JPKO00000000.1). Genomic islands were identified using the IslandPath DIMOB function on IslandViewer4 server (Bertelli et al., 2017). Partial and complete phage regions were identified in the genome assemblies of the WGS strains using Phaster. Circular genome comparisons of WGS query strains and the OVR A-D locci to the reference strain PNA 97-1R were conducted using BRIG (GPLv3) (Alikhan et al., 2011).
Nucleotide Sequence Accession Numbers
The Whole Genome Shotgun project of P. ananatis strains (PNA 97-1R, PNA 06-1, PNA 200-3, PNA 15-1, PNA 99-7, PANS 04-2, PANS 01-2, PANS 99-23, PANS 99-36, and PANS 99-3) have been deposited at DDBJ/ENA/Genbank under the accessions (CP020943-CP020945), NMZY00000000, NMZX00000000, NMZZ00000000, NMZW00000000, NMZV00000000, NMZU00000000, NMZS00000000, NMZT00000000, and NMZR00000000, respectively. In addition, all bio-projects and accession numbers related to this sequencing project are linked under the NCBI umbrella project PRJNA400632.
Results
Multi-locus Sequence Analysis
Six housekeeping genes (fusA, gyrB, leuS, pyrG, rpl, and rpoB) from fifty P. ananatis strains were sequenced and a concatenated tree was developed. Sequence identity amongst the strains was >99% indicating low diversity. Overall, 11 small clades consisting of P. ananatis strains (n = 35) with bootstrap values >67 were observed (Figure 1). Among 11 clades, three clades contained strains, which were strictly isolated from onion or onion seed (designated PNA) (Figure 1). In addition, there were eight clades that contained strains isolated from diverse sources including onion or onion seed or weeds or thrips. None of the strains isolated from sources other than onion or onion seed (designated PANS) were exclusively resolved in a separate clade. Fifteen strains remain unresolved (Figure 1). There was no trend among strains with respect to their year of isolation, geographic location, source of isolation or ice-nucleation phenotype.

Figure 1. Maximum-likelihood tree based on the Tamura-Nei Model of Pantoea ananatis strains derived from concatenation of six housekeeping genes (fusA, gyrB, leuS, pyrG, rplB, rpoB). Bootstrap values are based on 1,000 repetitions and values >60% are shown in the figure.
Repetitive Extragenic Palindrome (rep)-PCR
A dendrogram was constructed from the rep-PCR profiles of fifty P. ananatis strains. Strains that showed relatedness more than 60% were clustered into four separate clades containing PNA and PANS strains. Fingerprint patterns were not correlated with isolation source or phenotype. Clade I strains (n = 24) were 80% similar while strains in Clade II (n = 11) had similarity of 86% (Supplementary Figure 1). Five strains displayed identical fingerprint profile within Clade III and were unique from the strains in the clades I–IV (Supplementary Figure 1). Clade IV comprised of ten strains that showed fingerprint profiles different from other clades and displayed 61% genetic similarity (Supplementary Figure 1).
Pathogenicity and Aggressiveness of P. ananatis Strains on Onion, Chive, Leek, and Shallot
Thirty-three strains pathogenic or non-pathogenic on onion based on MLSA findings were selected for pathogenicity and aggressiveness study on onion, chive, leek, and shallot under controlled greenhouse conditions. PBS control plants displayed no foliar symptoms in any test. On onion, 100% of the previously determined pathogenic strains displayed typical foliar necrosis symptoms. However, there were significant differences among strains with respect to the aggressiveness (P < 0.0001; Table 1). Fifteen strains representing a mix of PNA and PANS strains were highly aggressive on onion with lesion length >7.8 cm after 5-days of inoculation under greenhouse conditions. Moderate levels of aggressiveness were displayed by ten strains also representing a mix of PNA and PANS strains. All eight previously determined onion non-pathogenic PANS strains were again determined to be non-pathogenic on onions.
On chive, all strains except PNA 200-11 were non-pathogenic. The PNA 200-11 strain was weakly aggressive on chive with lesion length of 0.92 cm after 5-days of inoculation. Out of 25 onion pathogenic strains, 19 strains were also pathogenic on shallot and they varied significantly in aggressiveness (P = 0.0007). These strains were weakly to moderately aggressive and they produced lesions of >0.6 cm after 5 days of inoculation. Six strains, which were pathogenic on onion, were non-pathogenic on shallot; however, one onion nonpathogenic strain, PANS 200-1 was found to be mildly aggressive on shallot. On leek, of the 25 onion pathogenic strains, 12 strains were also pathogenic and again, they varied significantly in aggressiveness (P < 0.0001). These strains were either weakly or moderately aggressive on leek with lesion length >0.8 cm after 5-days of inoculation. Thirteen onion pathogenic strains were non-pathogenic on leek. However, four onion non-pathogenic strains were weakly to moderately aggressive on leek. All symptomatic samples (n = 5 per host type) assayed were determined as positive for P. ananatis by a real-time PCR assay.
Ice Nucleation and Copper Tolerance
Ice nucleating ability was observed in 66% (33/50) of the strains tested. Of the 33 strains that were positive for ice nucleation, 18 were isolated from onion, three from onion seed, and 12 from other hosts. All 50 P. ananatis strains were unable to grow on NA media amended with 200 ppm CuSO4*5H2O. Confluent growth was observed on all non-amended NA plates (Table 1).
Whole Genome Sequencing Summary
The prototype strain PNA 97-1R was sequenced and assembled to near completion via PacBio sequencing. In addition, whole genome Illumina shotgun sequences (WGS) of PNA 97-1R and nine additional P. ananatis strains from diverse primary and secondary sources associated with the center rot pathosystem were assembled and annotated to generate draft genomes for comparative pan-genomic analysis. The genomes of P. ananatis strain ranged from 4.8 to 5.1 Mbp size which is consistent with the reported sizes P. ananatis isolated from other locations (De Maayer et al., 2014). Genome features are reported in Table 3. The pathogenicity of each strain was assessed using both foliar and bulb scale assays (Figure 2). Annotated genomes were assessed for reported virulence elements including: Acyl homoserine lactone quorum sensing system, flagellar elements, and twitching motility type IV pilli.
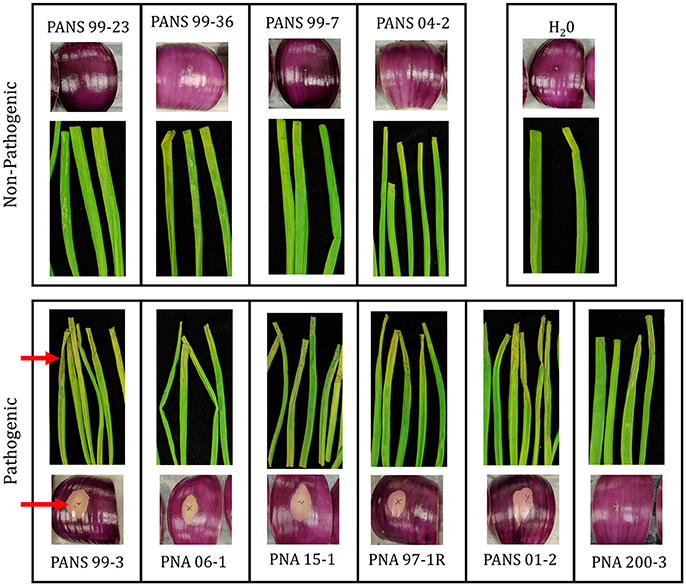
Figure 2. Pathogenicity of Pantoea anantis strains on red onion scale and onion leaves. Pathogenic strains cause clearing of pigmentation and associated weakening of onion scale. Pathogenic strains in foliar assay cause severe wilting and chlorosis along the length of the leaf blade. Blade and onion scales were inoculated with 20 uL suspension containing 1 × 106 CFU/ml for each isolate. Scales were imaged 3 days post inoculation. Leaves were imaged 5 days post inoculation (Table 1). Red arrows highlight foliar wilting and scale clearing among pathogenic strains.
Universally, P. ananatis strains lacked complete virulence-type T3SS and T4SS secretion systems as well as the T2SS commonly used by soft rot causing Enterobacteriacea pathogens to delivery CWDEs and other virulence factors across the outer membrane. A scan of the genomes to identify NRPS/ Polyketide Synthase (PKS) clusters associated with secondary metabolites resulted in identification of clusters that encode synthetic genes for siderophores and bacteriocins. In addition, a biosynthesis cluster for the exopolysaccharide, stewartan, associated with virulence in P. stewartii subsp. stewartii was also identified (Coplin et al., 1992). Gene clusters associated with known phytotoxins were not identified. Pan-genomic analysis conducted using two pipelines with different computational strategies could differentiate onion pathogenic strains from onion non-pathogenic strains (Figure 3). Comparative analysis identified a subset of genes specific to pathogenic strains that were localized to approximately four virulence-associated regions. The largest being 31 kbp in length. We named these loci OVR-A-D- (Onion Virulence Region) due to their correlation with the onion pathogenicity (Figures 4, 5, Table 4).
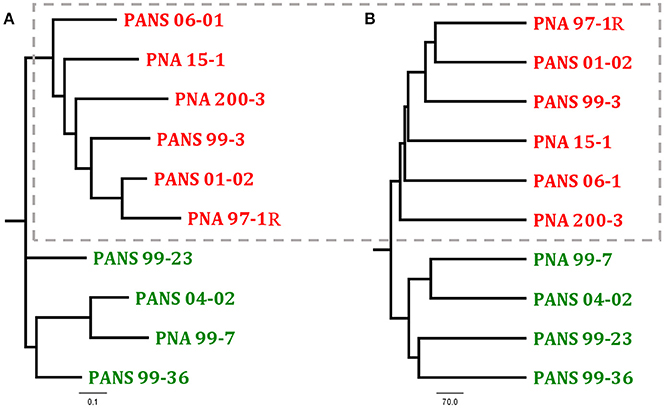
Figure 3. Comparison of two dendrograms of Pantoea ananatis strains generated through different pan-genome analysis programs (A) ROARY presence-absence based tree generated uses the first 4,000 genes in accessory genomes and Fast Tree to calculate distances (Page et al., 2015) (B) PGAP pan-genomic based UPGMA dendrogram is calculated with phylip (Zhao et al., 2012). Dotted box highlights foliar pathogen isolates. See NCBI accession numbers (Table 2).
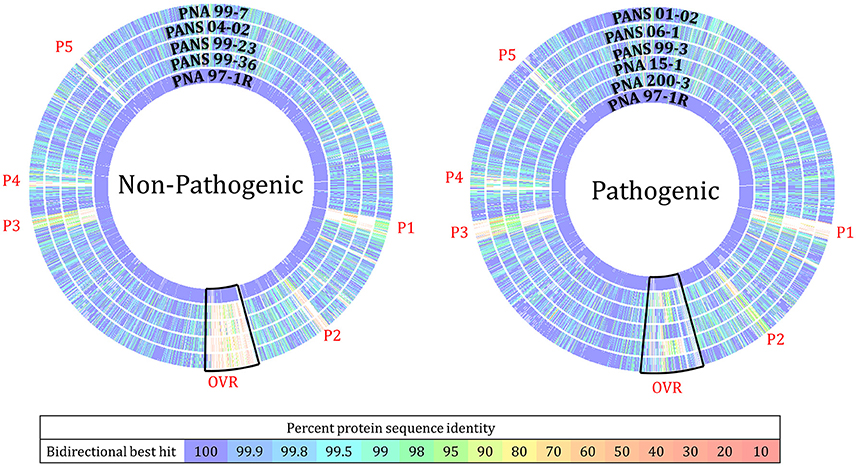
Figure 4. RAST Seed Viewer protein sequence-based comparison analysis of Pantoea ananatis strains to pathogenic proto-type strain PNA 97-1R. BLASTP analysis compares every protein in the concatenation of the reference genome (PNA 97-1R PacBio) to every protein in the comparison genome (9 other strains, Illumina HiSeq). Overlapping regions and heat map indicate level of homology between proteins. (P1) Phage related proteins including: hypothetical proteins, phage lytic proteins, phage tail proteins, phage replication proteins, phage DNA binding proteins, phage lysine and lysozymes (P2) Phage associated genes (OVR) onion virulence region loci including: cell wall degrading enzymes, metabolic related proteins, amino acid transporters, and sugar transporters. (P3) Hypothetical proteins, phage proteins, transporters, and transposable DNA elements. (P4) hypothetical proteins, and putative transcriptional regulators. This plot should not be used to determine the location of genes in the comparison genome but highlights regions that may require further analysis.
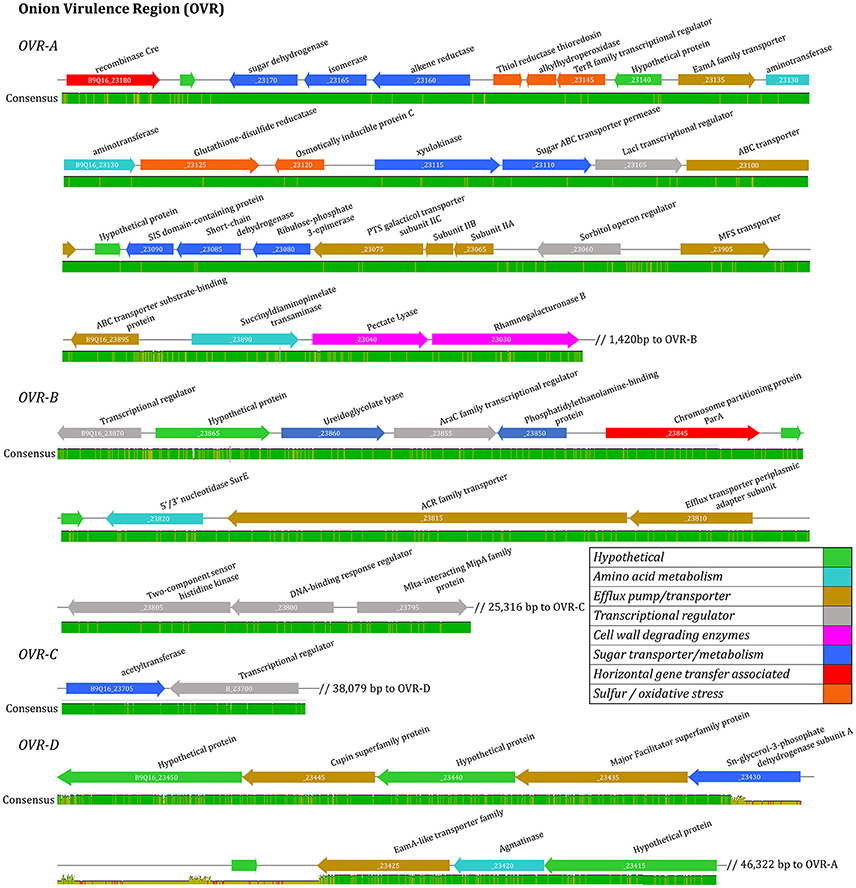
Figure 5. Virulence associated gene alignment for six foliar pathogenic isolates of Pantoea ananatis. Four clusters of virulence associated genes were identified including OVR-A, OVR-B, OVR-C, and OVR-D. The linear open-reading-frame maps of the virulence regions were identified after pan-genomic analysis and aligned using MAUVE. Genes are represented by arrows oriented in direction of transcription. Colored arrows are annotated to indicate predicated function based on manual annotation. Consensus level is illustrated with bar below ORF maps.
Red Onion Scale Assay
The ability of P. ananatis strains to clear red onion scales, and cause scale weakening was observed in five of the six onion pathogenic WGS strain (Figure 2, Table 2). The weakly aggressive PNA 200-3 did not exhibit scale clearing, but did cause traditional foliar necrotic symptoms (Figure 2, Table 2). The negative control did not exhibit clearing in the onion scale assay or blighting in the foliar assay. Thus, all six strains containing the OVR-A-D region exhibited blight symptoms in foliar assay and five of six strains containing the OVR-A-D region exhibited pigment clearing in the onion scale assay (Table 2).
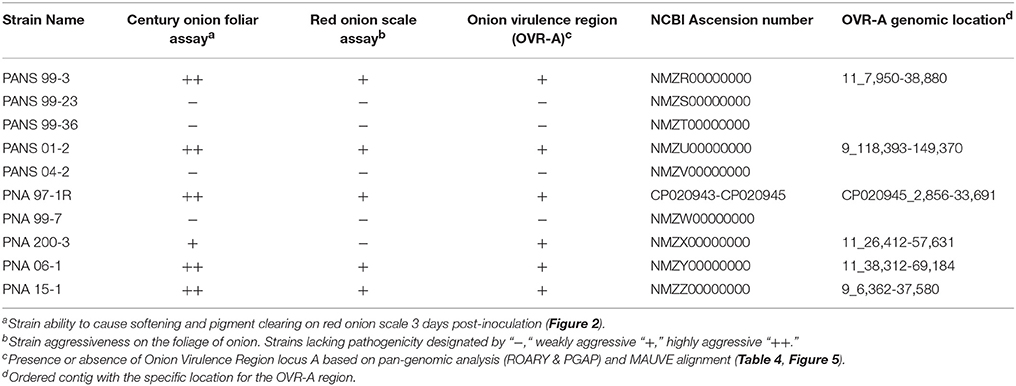
Table 2. Pantoea ananatis WGS strains and associated phenotypes.
Draft Genomes of 10 P. ananatis Strains from Georgia
Quality assessment of extracted PNA 97-1R prototype strain DNA yielded fragments >50 kb. PacBio reads were assembled resulting in a high quality draft genome of prototype strain PNA 97-1R. The assembly consists of three contigs with sizes 4,571,292 bp; 295,591 bp; 180,232 bp, GC content 53.6, 52.4, and 49.6%, respectively; with an estimated 200X genome coverage (GenBank accession numbers CP020943-CP020945 respectively). The second largest contig 295,591 bp falls in the range of the large universal Pantoea plasmid LPP-1 280–789 kb (De Maayer et al., 2012). The presence of core factors such as thiamine biosynthesis proteins (thiaOSF), and MAUVE alignment with the AJ3355 LPP-1 pEA320 (NC_017533.1) aided in characterizing this contig as homologous to the widely reported universal plasmid LPP-1 (Supplementary Figure 2; Weller-Stuart et al., 2017). MAUVE alignment yielded a single block with homology between the proposed PNA 97-1R LPP-1 plasmid (CP020944) and the AJ3355 LPP-1 plasmid (Supplementary Figure 2).
MicrobesNG Illumina draft genome sequencing yielded 354,367–2,036,787 bp range in reads for assemblies. The average insert size ranged from 344 to 649 bp (Supplementary Table 1). Average coverage for draft-genome sequences ranges from 28.9 to 167.46%. The GC content ranged from 53.29 to 53.52%. The RAST annotation resulted in prediction of 4,681–4,992 protein coding sequences (CDS) among WGS strains, roughly 1,165–1,330, were predicted as hypothetical coding sequences (Supplementary Table 2). The number of predicted phages, prophages, and transposable elements varied widely among strains from 2 to 53 CDS (Supplementary Table 2). Most proteins coding sequences are devoted to carbohydrates, amino acids and derivatives; 455–630 CDS on average. No traditional complete virulence associated plant pathogen secretion systems were identified. Two proteins associated with the type II secretion system including gspE (cytosolic ATPase) and gspF (inner membrane protein) were identified in WGS genomes of all strains.
Known Virulence Factors
Virulence factors previously recorded as having importance in the pathogenicity of P. ananatis including: acyl-homoserine lactone mediated quorum sensing system, twitching motility, flagellar motility, two clusters of type VI secretion system (C1, C3) associated genes and effectors were present in WGS of all 10 P. ananatis strains. Among these, PANS 99-3 contained all three previously described type VI clusters (C1, C2, C3) (De Maayer et al., 2011).
The Ice Nucleation Gene (inaZ) Is Present in All Strains
Although only five of the 10 WGS strains have an ice nucleation positive phenotype, the ice-nucleation protein gene inaZ was found to be present in all strains sequenced in this study. The inaZ gene was observed to vary in lengths from 3,474 bp (PANS 04-2) to 4,209 bp (PANS 06-1). PNA 200-3 has three ORFs with homology to inaZs, which are severely truncated being, 750, 450, and 1,287 bp. The severely truncated nature of the inaZ loci in PNA 200-3 likely explains the ice nucleation minus phenotype in this strain. PANS 99-3, PNA 15-1, PNA 99-7, and PANS 04-2 encoded the inaZ gene but lacked an ice nucleation phenotype in vitro.
Pan-Genomic Comparative Analysis of P. ananatis Strains from Georgia
The ROARY pan genomic analysis identified a core genome consisting of 3,750 protein coding sequences (CDS) and an accessory genome consisting of 1,395 CDS. Dendrograms generated through pan-genomic pipelines based on presence or absence of genes, differentiated onion pathogenic strains from onion non-pathogenic strains (Figure 3). ROARY BLASTP comparisons clearly identified CDS with homology to the prototype strain PNA 97-1R while also indicating phage, and virulence associated regions absent from onion non-pathogenic strains, but present in onion pathogenic strains (Figure 4, Supplementary Figure 6). Comparative analysis identified 57 CDS on four contiguous gene clusters on one contig in all pathogenic strains, but absent in onion non-pathogenic strains (Supplementary File 1). wgMLST analysis resulted in several different clades with the outgroups, P. agglomerans and P. stewartii subsp. indologens clustering from the P. ananatis strains (Supplementary Figure 4). Strains PANS 99-36 and PANS 99-23 clustered from other strains and had similar years of isolation. The pathogenic strain PANS 99-3 and non-pathogenic strain PNA 99-7 clustered together. Genomic islands were identified in all P. ananatis strains. Strains had between 9 and 15 predicted genomic islands. The size of the islands varied from 5,430 to 150,125 bp (Supplementary Table 4). The described OVR A-D regions were predicted as genomic islands in pathogenic strains. Islands unique to non-pathogenic strains were not identified. Partial and complete phage regions were identified in all WGS strains. The relative orientation and location of 5 phage regions can be observed in the type strain PNA 97-1R (Supplementary Figure 5).
Identification of Genomic Regions Associated with Onion Pathogenic Strains
The 57 CDS shared between onion foliar pathogen strains were present on four contiguous regions, and were localized on a single contig among pathogenic strains (Table 2). The contiguous regions contained 30, 16, 2, and 9 CDS and were ordered based on orientation in the prototype strain PNA 97-1R PacBio genome (CP020945). Of the 57 CDS, 17 CDS are predicted, hypothetical proteins based on NCBI PGAP annotations (Table 4). The onion virulence associated regions were termed onion virulence regions A-D (OVR-A through OVR-D) (Table 4, Figure 5). All four regions are located on the smallest 180,232 bp contig from PNA 97-1R PacBio assembly (CP020945). PGAP pan-genomic analysis, which utilizes computationally intensive strategy to BLAST every gene to each genome confirmed, 20 of the pathogen associated genes were present only once in the onion pathogenic strains and entirely absent in the non-pathogenic strains (Table 4, Figure 5). MAUVE alignment of pathogenic strains along these regions indicated high levels of homology (Figure 5).
The OVR-A locus begins with a gene for a Cre-like phage recombinase, which may indicate integration of the 31-kb OVR-A locus as a mobilized genomic island. A gene closely following Cre is predicted to be a TetR family transcriptional regulator. Closely following this regulator are sugar transport proteins including a sugar ABC transporter permease and ribulose-phosphate-3-epimerase. The OVR-A contains three genes associated with sulfur, redox and oxidative stress, such as a thiol reductase and alkylhydroperoxidase. Several efflux pumps and transporters are present including an ABC transporter and PTS galacticol transporter subunit IIA-C. Amino acid metabolism-transport and plant cell wall degrading enzymes (CWDE) including a pectate lyase, and rhamnogalacturoase B are also present in the OVR-A region (Table 4, Figure 5).
OVR-B consists of 16 CDS. Three proteins are predicted to have DNA-binding regulatory functions such as the AraC family transcriptional regulator. A ureidoglycolate lyase is associated with amino acid degradation. Two outer membrane transport proteins are present including an ACR family transporter. Two protein kinases are present including a predicted two-component sensor histidine kinase and a phosphatidylethanolamine-binding protein with a predicted secretion target signal. In addition, three proteins are present related to transposable DNA elements including an IS110 family transposase. An Mlta-interacting family protein lytic transglycosylases, which is related to protein binding membrane, is also present (Table 4).
OVR-C contains a transcriptional repressor with homology to PuuR, and an acetyl-transferase YjaB (Table 4). OVR-D contains nine predicted CDS. The PGAP annotation pipeline characterized all as hypothetical proteins. Porkka annotation predicted two transport associated proteins including a Major Facilitator Superfamily CDS (Table 4). In addition, two proteins involved in carbon and amino acid metabolism, including agmatinase are also present.
Several of the CDS in the regions specific to onion pathogenic strains have predicted transmembrane or Tat-secreted target signal. CDS of interest with Tat-secreted target signals include: CWDE pectate lyase, and rhamnogalacuroase in the OVR-A and two protein signal regulators in OVR-B. Most proteins predicted to be transport-related contained transmembrane domains (Table 4).
Discussion
Phenotypic Variation amongst P. ananatis Strains
A set of fifty P. ananatis strains from Georgia was assembled to maximize diversity of host and geographic sources of isolation as well as diversity of previously determined onion pathogenicity and ice nucleation phenotypes. We observed extensive phenotypic variation among a sub-set of 33 strains tested for pathogenicity and aggressiveness on multiple Allium hosts. In addition to the 15 primary onion and onion seed strains, 10 of 25 PANS strains collected from weed and thrips species were re-confirmed to be pathogenic on onion. However, aggressiveness of strains on Allium sp. varied considerably. Among the 33 strains, we observed similar levels of variation in pathogenic potential and aggressiveness on shallot and leek although the strains were generally less aggressive on these hosts than on onion. Pathogenic potential and aggressiveness of a strain on onion or leek or shallot did not generally correspond with its potential to be pathogenic on the other Allium hosts. In addition, there were multiple examples of onion non-pathogenic strains that were pathogenic on leek or shallot and vice versa such as PANS 99-23. This could indicate some degree of host specialization among P. ananatis strains tested. Unlike other Allium hosts, the chive cv. Dolores was infected by only a single P. ananatis strain with mild symptoms. This may highlight the potential of Allium schoenoprasum to serve as a source of genetic resistance against P. ananatis.
We used MLSA and rep-PCR based genetic diversity analysis to determine whether phenotypic diversity correlated with underlying phylogeny. Contrary to the high phenotypic variation, both analyses indicated little genetic diversity amongst the fifty P. ananatis strains. By MLSA all strains strain were found to be >99% identical. The majority of branches in the phylogeny had poor bootstrap value support and were composed of a mixture of PNA and PANS strains. We observed no underlying correlation between the isolate phylogeny and pathogenic phenotypes or isolation sources using either method of diversity analysis.
Motivated by the high degree of P. ananatis phenotypic variation that is unexplained by the low levels of core genome diversity, we sought to conduct a pan-genome analysis to determine whether fine scale variations in gene presence-absence might correlate with phenotypic variation. A sub-set of 10 strains from the original fifty strains were selected for whole genome Illumina shotgun sequencing with the goal of maximizing diversity of phenotypes and sources of isolation. Additionally, the Georgia prototype onion pathogenic strain PNA 97-1R was sequenced using PacBio to create a high quality near-closed genome assembly. Among the 10 strains selected for WGS, six were able to cause foliar necrosis in onion blades and five of those also were also able to clear red onion scales (Figure 2). The phenotypic variation seen in red-onion scale assays may potentially be a useful tool for identifying isolates with onion pathogenic potential as it is easy to replicate and rapid with minimal space requirements. It would be beneficial to utilize more strains to determine if correlations between red onion scale clearing and onion foliar blight potential occur.
Pan-Genome Analysis of Sequenced Isolates
Dendrograms generated from pan-genomic gene presence-absence analysis clearly indicate a clustering of onion pathogenic strains from non-pathogenic strains. This suggests an underlying genetic difference between the two groups that was not detected using MLSA or rep-PCR approaches. The majority of the gene differences between onion pathogenic and non-pathogenic strains resides in four contiguous gene clusters the largest being 31 kb that we termed as the Onion Virulence Region (OVR-A). RAST BlastP further illustrated this point by identifying genes present in onion pathogenic strains, but absent in non-pathogenic strains when compared with the pathogenic prototype strain PNA 97-1R. The level of core genome homology amongst all the strains is notable. There is a high degree of CDS similarity. Most gene variation is the result of phage, transposase, and integrase proteins. In addition, homologs to the Lux1, type VI, V, I s, and Tat secretion systems previously noted as candidate virulence factors were identified in both onion pathogenic and non-pathogenic strains. Phytotoxin synthetic clusters were not identified (Weller-Stuart et al., 2017).
The de-novo genome assembly of the Georgia prototype strain PNA 97-1R resulted in three contigs, of 4.9 Mbp, 295 kbp, and 180 kbp (CP020943-CP020945). The 4.9 Mbp contig corresponds to the Pantoea circular chromosome. The 295 kbp contig was identified as the Large Pantoea Plasmid LPP-1 based on previously described genes and MAUVE alignment. The genomes of the sequenced strains contain LPP-1 with a high level of conservation (Supplementary Figure 3). The third 180 kbp PNA 97-1R contig encompasses the four OVR loci which correlate with onion pathogenicity based on pangenomic analysis. This contig aligned with single contigs of varying sizes (74–167 kbp) from onion pathogenic WGS strains. The 180 kbp contig shares complete homology in 6 MAUVE alignment blocks with all genes in a 60 kbp contig (CAEI01000108.1) of a single sequenced strain B1-9 of P. ananatis in the NCBI database (NZ_CAEI00000000.1). Strain B1-9 is an isolate from green onion described by researchers in South Korea associated with growth promotion of pepper (Kim et al., 2012). It is intriguing that a growth promoting strain from South Korea share all genes associated with onion virulence among our Georgia strains. We know that the pathogenicity of P. ananatis strains can vary widely between different species of Allium (Table 1). It would be interesting to determine if B1-9 is pathogenic on onion and on other Allium sp. In addition to sharing homology with this contig from B1-9, the 180 kbp contig shares some genes with those found on the pEcWSU1 plasmid of the onion pathogen, Enterobacter cloacae associated with onion bulb-rot (Schroeder et al., 2009). Highest homology is shared between a histidine kinase protein in OVR-B, and the PTS galacticol transporter system A-C in the OVR-A loci. Interestingly, plasmid partitioning genes and repA plasmid replication genes were present on the WGS contigs that contained the OVR-A loci in each of the onion pathogenic strains indicating that these contigs may, in fact, represent an accessory plasmid.
Pantoea ananatis is a species that is frequently associated with diverse eukaryotic hosts. In the onion-center rot system, it is associated epiphytically with weed species, and pathogenically and endophytically with onions, and persists temporarily in the thrips vector commonly associated with center-rot disease outbreaks (Gitaitis et al., 2002; Walcott et al., 2002; Dutta et al., 2014). Our strains were isolated from various sources such as thrips, weeds, seeds, and symptomatic onions. Despite these strains inhabiting different niches, there existed a high degree of genetic similarity among them. Furthermore, a high average nucleotide identity was observed among Georgia isolates and other strains isolated in different regions around the world (Table 3). This suggests that the core genome is highly conserved among P. ananatis strains (Supplementary Figure 3). The core genome of 3,750 CDS identified through ROARY pan-genomic analysis is consistent with the core genome identified through previous pan-genomic analysis methods (De Maayer et al., 2014; Sheibani-Tezerji et al., 2015). The accessory genome, where most differences were identified, revealed the Onion Virulence regions (OVR-AD), as well as differences among poorly characterized hypothetical coding sequences, phages, transposases, integrases, and mobile genetic elements. The presence of the Cre recombinase in the OVR-A loci could be indicative of a possible horizontal gene transfer mechanism.
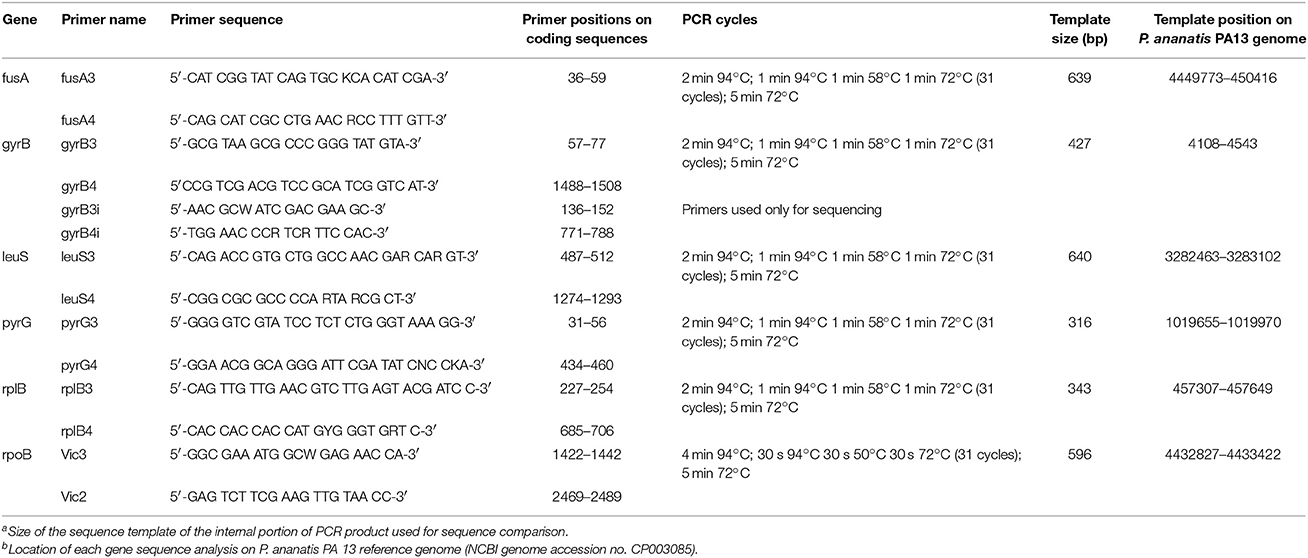
Table 3. Primers and conditions used for amplification and sequencing.
Genetic Content of the OVR-A Loci
The OVR-A loci has not been previously described and may offer insights into the ability of P. ananatis to cause bacterial disease in onions. This region was present in all Georgia strains that caused the foliar symptoms on onion (Figure 2). Except for strain PNA 200-3, this region was present in all P. ananatis strains that caused scale clearing in the red onion scale assay. The region begins with a Cre recombinase followed by amino acid transporters, metabolic enzymes, transcriptional regulators and CWDEs (Table 4).
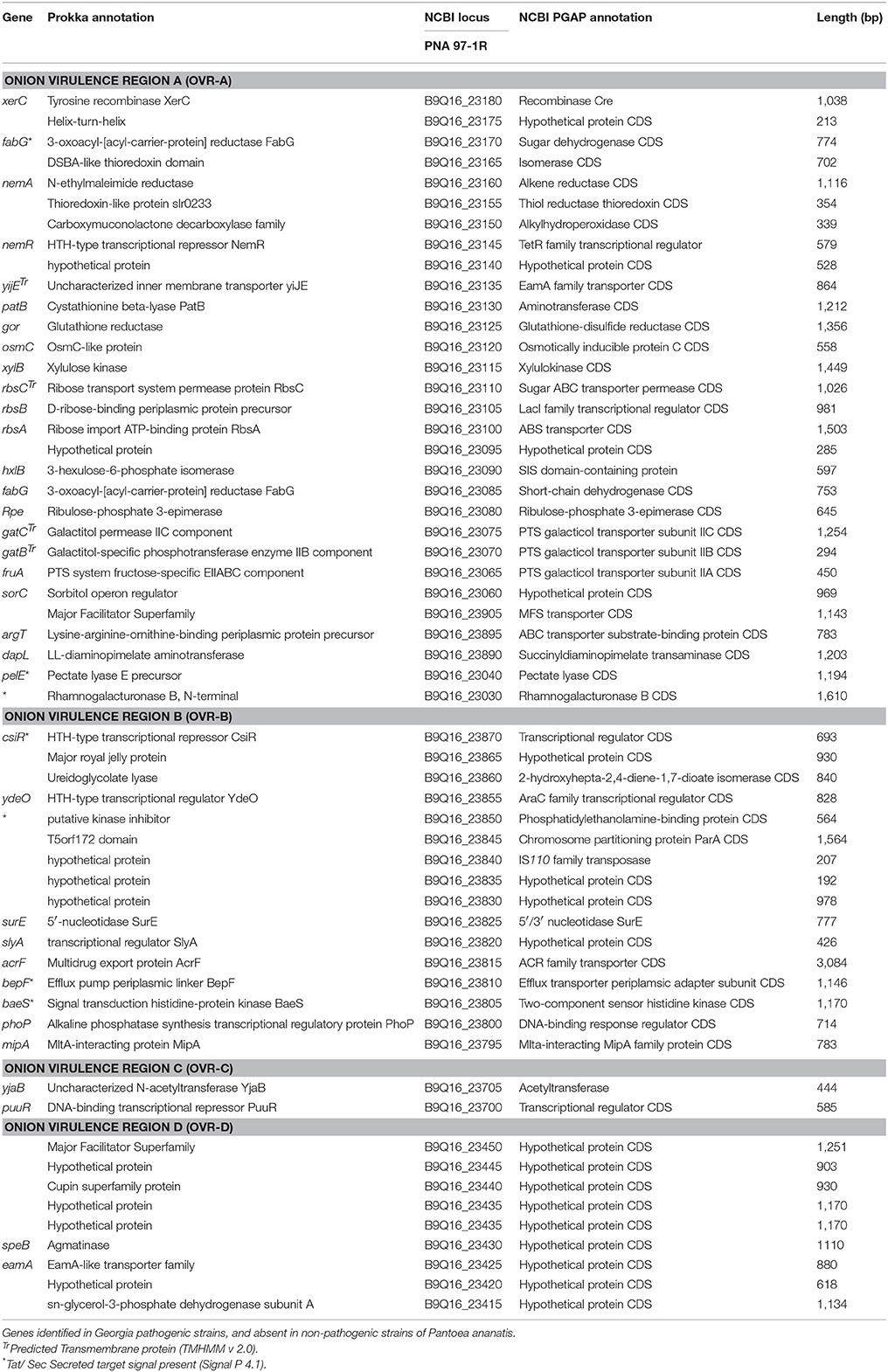
Table 4. ROARY presence-absence based comparison.
The presence of redox-related/sulfur reducing enzymes could play a role in withstanding host sulfur containing antimicrobials, or interrupting ROS signaling that is important during plant immunity (Torres et al., 2006). In addition, amino acid transporters may play a role in the absorption of host nutrients. The specific localization of P. ananatis in host tissue has not been well characterized and may offer insights into nutrient acquisition for this bacterium (Fatima and Senthil-Kumar, 2015). Sugar transporters and major super facilitator proteins have been shown to play an important role in the pathogenicity of bacterial pathogens such as Dickeya dadantii where they can play a role in the anti-microbial resistance, and chemotaxis (De Maayer et al., 2011).
Plant cell wall degrading enzymes such as pelE and rhamnogalacturonase, have been found to contribute to pathogenicity in a number of other bacterial pathogens, notably Dickeya dadantii (Hassan et al., 2013). A protein in OVR-D was computationally annotated as a member of the EamA transport family shares homology with the PecM, a transporter in D. dadantii, which plays a major role in regulating virulence factor synthesis such as, PelE and other CWDEs (Praillet et al., 1996). However, as P. ananatis lacks a T2SS it is unclear by what mechanism these proteins would be secreted out of the cell. The protein kinase and kinase inhibitor found in OVR-B are intriguing as they both have predicted secretion signals (putative kinase inhibitor, two-component sensor histidine kinase) (Table 4). They could play a role in signal transduction in the bacteria or perhaps could be involved in interrupting host immunity by some as-of-yet uncharacterized host cell delivery mechanism. Transporter and transmembrane proteins located in onion virulence correlated regions could be involved in secretion of proteins across the outer membrane, or surface interactions between the bacteria and host cells.
The localization of OVR A-D to single contigs from the genome constructs offers some supportive evidence that the region is plasmid localized. Symbiotic and pathogenic plasmids are important for pathogenicity in a number of phytopathogenic bacterial species. In plant pathogens mega plasmids have been shown to contribute to virulence in bacteria such as Pseudomonas syringae pv. phaseolicola (Jackson et al., 1999). The contigs constructs containing OVR locci contain parA plasmid partitioning proteins. In our future work work we will seek to confirm whether the OVR loci are, in fact, plasmid-borne.
Few pan-genomic analyses of P. ananatis strains have been conducted. The first pan-genomic study of P. ananatis utilized NCBI draft and complete genomes of strains isolated from various hosts. It is difficult to infer the genomic diversity of strains isolated from a single geographic location or host-pathosysem from such, studies, as hosts, isolation sources, and locations varied dramatically (De Maayer et al., 2014). Currently, only one study elucidated the genomic variation among P. ananatis strains, isolated from the same host, maize seed. The researchers observed a high degree of genetic similarity among P. ananatis strains with diverse phenotypes; epiphytic, plant growth-promoting, and pathogenic (Sheibani-Tezerji et al., 2015). Despite high levels of genetic similarity, minute differences in genes encoding protein secretion systems, putative effectors, and transpose/integrases/phage related genes were noted. For example, the pathogenic P. ananatis genome contained genes relating to cell cycle control, cell division, chromosome partitioning, and amino acid transport that differentiated it from the growth promoting, and endophytic strain. This study found subtle variations between the pathogenic and non-pathogenic strains. Other protein coding sequences that differed between the strains included transposase related proteins, chemotaxis proteins, and T6SS loci (Sheibani-Tezerji et al., 2015). Conversely, our results identified a distinct subset of genes in contiguous regions that correlated with onion pathogenicity.
The presence of genes within the OVR-A loci associated with virulence and pathogenicity in other onion pathogens such as Enterobacter cloacae illustrates a need to further characterize genes in this region and other onion virulence correlated regions. Future work will include the deletion of the OVR-A loci from PNA 97-1R to determine what role this region plays in onion pathogenicity.
Strain PNA 200-3 has the OVR-A region but lacks the scale clearing phenotype. The negative-scale clearing phenotype may be explained by SNP missense mutations in genes encoding CWDEs of PNA 200-3's OVR-A or could indicate variation in expression of genes associated with this loci. The OVR-A loci may simply represent a vertically inherited locus or plasmid common to Georgia onion pathogenic strains through a common clonal origin of onion pathogenic strains in Georgia, and possible relation to strains in South Korea. Thus, while the OVR-A loci is common among Georgia onion pathogenic strains, it may not in itself, carry pathogenicity determinants that are critical to infection. Many other currently sequenced strains have been reported as pathogenic on onion, while apparently lacking the OVR loci (Morohoshi et al., 2007; Shyntum et al., 2015).
Conclusions
Overall, our study illustrates the importance of WGS in differentiating closely related strains. MLSA and rep-PCR analysis indicated little diversity among the P. ananatis strains in Georgia, although the strains were phenotypically diverse for Allium infection phenotypes. The WGS revealed that although these strains share similar core genomes, the universal plasmid LPP-1, and previously identified candidate virulence factors, differences in the mobile genetic elements, as well as the OVR-A loci differentiated the sequenced strains into two groups that shared common scale-clearing and foliar pathogenicity phenotypes. Thus, the differences between pathogenic and non-pathogenic strains in P. ananatis may not be as subtle as previously suggested (Sheibani-Tezerji et al., 2015). In any case, the contribution of the OVR loci to P. ananatis onion virulence remains to be determined. Subtle differences in loci gene expression and host defense reactions may further differentiate host range and aggressiveness of P. ananatis strains. The transcriptional analysis of mutants with and without these loci may provide insights into the nature of this regions contributions to virulence. Future studies will involve the use of mutational analysis to assess the role of the OVR-A loci in onion pathogenicity and determine if this genetic locus is chromosomal or plasmid-borne.
Author Contributions
SPS and SDS contributed equally to this work. BK and BD are co-senior on this work. SPS, SDS, RG, BK, and BD conceived and designed the experiments. SDS performed MLSA, rep-PCR and phenotypic testing. SPS conducted experiments and analysis related to whole genome sequencing and Pan-genome analysis. SPS, SDS, BK, and BD wrote and edited the manuscript.
Funding
We would like to acknowledge the Vidalia Onion Commitee for partial funding to BD as well as NIFA HATCH (Project: GEO00791) and the State of Georgia for funding to BK.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00184/full#supplementary-material
Supplementary Figure 1. Dendrogram derived by cluster analysis of similarities between Pantoea ananatis strains based on repetitive extragenic palindromic polymerase chain reaction (REP-PCR) DNA genomic profiles. Analysis was performed using Dice's (1945) coefficient in BioNumerics software package (Applied Math, Kortrijk, Belgium).
Supplementary Figure 2. Large Pantoea Plasmid LPP-1 alignment of proto-type strain PNA 97-1 with AJ13355 pEA320 (Accession CP020943; NC_017533.1) Alignment generated with Geneious MAUVE plugin.
Supplementary Figure 3. Venn diagram of genes shared among the pathogenic isolate PNA 97-1R; nonpathogenic isolates PANS 99-36, and genomic type strain LMG 20103 (Accession CP001875.2). The number of coding sequences are derived from the gene_presence_absense.csv output of the ROARY pan-genome analysis using default parameters (Supplementary File 2).
Supplementary Figure 4. wgMLST cladogram of Pantoea strains generated through PGAdb-builder platform. Outgroups P. agglomerans P10C and P. stewartii subsp. indologens LMG 2632 (Accession NZ_LIME00000000.1 and JPKO00000000.1).
Supplementary Figure 5. Phaster predicted phage regions in PNA 97-1R. Green indicated intact phage regions whereas red indicates incomplete phage regions.
Supplementary Figure 6. BRIG analysis of P. ananatis strains and the OVR A-D loci to the reference strain PNA 97-1R. BRIG is a locally run analysis that generates figures based on blastn comparisons of query genomes to a reference strain.
Supplementary Table 1. Sequencing and assembly statistics for Illumina draft-genomes of Pantoea ananatis strains.
Supplementary Table 2. RAST functional categories for predicted protein coding sequences of Pantoea ananatis strains.
Supplementary Table 3. Primers and conditions used for amplification and sequencing.
Supplementary Table 4. IslandPath DIMOB genomic islands identified in WGS P. ananatis strains.
Supplementary File 1. ROARY 10 strain WGS Analysis. The presence of each CDS in the respective genome is listed below each labeled strain column. If a cell is blank the respective strain does not have the gene. Logic equations were used to highlight cells, resulting in genes present in pathogenic strains and absent in non-pathogenic strains.
Supplementary File 2. ROARY 3 strain WGS Analysis. The presence of each CDS in the respective genome is listed below each labeled strain column. If a cell is blank the respective strain does not have the gene. Summation equations were used to calculate the numeric values for the Venn Diagram in Supplementary Figure 3.
References
Abe, K., Watabe, S., Emori, Y., Watanabe, M., and Arai, S. (1989). An ice nucleation active gene of erwinia-ananas-sequence similarity to those of pseudomonas species and regions required for ice nucleation activity. FEBS Lett. 258, 297–300. doi: 10.1016/0014-5793(89)81678-3
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Carr, E. A., Bonasera, J. M., Zaid, A. M., Lorbeer, J. W., and Beer, S. V. (2010). First report of bulb disease of onion caused by Pantoea ananatis in New York. Plant Dis. 94, 916–916. doi: 10.1094/PDIS-94-7-0916B
Carr, E. A., Zaid, A. M., Bonasera, J. M., Lorbeer, J. W., and Beer, S. V. (2013). Infection of onion leaves by Pantoea ananatis leads to bulb infection. Plant Dis. 97, 1524–1528. doi: 10.1094/PDIS-06-12-0597-RE
Coplin, D. L., Frederick, R. D., Majerczak, D. R., and Tuttle, L. D. (1992). Characterization of a gene-cluster that specifies pathogenicity in erwinia-stewartii. Mol. Plant Microbe Interact. 5, 81–88. doi: 10.1094/MPMI-5-081
Coutinho, T. A., Preisig, O., Mergaert, J., Cnockaert, M. C., Riedel, K. H., Swings, J., et al. (2002). Bacterial blight and dieback of Eucalyptus species, hybrids, and clones in South Africa. Plant Dis. 86, 20–25. doi: 10.1094/PDIS.2002.86.1.20
Coutinho, T. A., and Venter, S. N. (2009). Pantoea ananatis: an unconventional plant pathogen. Mol. Plant Pathol. 10, 325–335. doi: 10.1111/j.1364-3703.2009.00542.x
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De Baere, T., Verhelst, R., Labit, C., Verschraegen, G., Wauters, G., Claeys, G., et al. (2004). Bacteremic infection with Pantoea ananatis. J. Clin. Microbiol. 42, 4393–4395. doi: 10.1128/JCM.42.9.4393-4395.2004
Delétoile, A., Decré, D., Courant, S., Passet, V., Audo, J., Grimont, P., et al. (2009). Phylogeny and identification of Pantoea species and typing of Pantoea agglomerans strains by multilocus gene sequencing. J. Clin. Microbiol. 47, 300–310. doi: 10.1128/JCM.01916-08
De Maayer, P., Chan, W. Y., Blom, J., Venter, S. N., Duffy, B., Smits, T. H. M., et al. (2012). The large universal Pantoea plasmid LPP-1 plays a major role in biological and ecological diversification. BMC Genomics 13:625. doi: 10.1186/1471-2164-13-625
De Maayer, P., Chan, W. Y., Rubagotti, E., Venter, S. N., Toth, I. K., Birch, P. R. J., et al. (2014). Analysis of the Pantoea ananatis pan-genome reveals factors underlying its ability to colonize and interact with plant, insect and vertebrate hosts. BMC Genomics 15:404. doi: 10.1186/1471-2164-15-404
De Maayer, P., Venter, S. N., Kamber, T., Duffy, B., Coutinho, T. A., and Smits, T. H. M. (2011). Comparative genomics of the type VI secretion systems of Pantoea and Erwinia species reveals the presence of putative effector islands that may be translocated by the VgrG and Hcp proteins. BMC Genomics 12:576. doi: 10.1186/1471-2164-12-576
Dutta, B., Barman, A. K., Srinivasan, R., Avci, U., Ullman, D. E., Langston, D. B., et al. (2014). Transmission of Pantoea Ananatis and P-agglomerans, causal agents of center rot of onion (Allium cepa), by Onion Thrips (Thrips tabaci) through feces. Phytopathology 104, 812–819. doi: 10.1094/PHYTO-07-13-0199-R
Fatima, U., and Senthil-Kumar, M. (2015). Plant and pathogen nutrient acquisition strategies. Front. Plant Sci. 6:750. doi: 10.3389/fpls.2015.00750
Gitaitis, R. D., and Gay, J. D. (1997). First report of a leaf blight, seed stalk rot, and bulb decay of onion by Pantoea ananas in Georgia. Plant Dis. 81, 1096–1096. doi: 10.1094/P.D.I.S.1997.81.9.1096C
Gitaitis, R., Walcott, R., Culpepper, S., Sanders, H., Zolobowska, L., and Langston, D. (2002). Recovery of Pantoea ananatis, causal agent of center rot of onion, from weeds and crops in Georgia, USA. Crop Protect. 21, 983–989. doi: 10.1016/S0261-2194(02)00078-9
Hassan, S., Shevchik, V. E., Robert, X., and Hugouvieux-Cotte-Pattat, N. (2013). PelN is a new pectate lyase of dickeya dadantii with unusual characteristics. J. Bacteriol. 195, 2197–2206. doi: 10.1128/JB.02118-12
Jackson, R. W., Athanassopoulos, E., Tsiamis, G., Mansfield, J. W., Sesma, A., Arnold, D. L., et al. (1999). Identification of a pathogenicity island, which contains genes for virulence and avirulence, on a large native plasmid in the bean pathogen Pseudomonas syringae pathovar phaseolicola. Proc. Natl. Acad. Sci. U.S.A. 96, 10875–10880. doi: 10.1073/pnas.96.19.10875
Kim, S. N., Cho, W. K., Kim, W. I., Jee, H. J., and Parke, C. S. (2012). Growth promotion of pepper plants by Pantoea ananatis B1-9 and its efficient endophytic colonization capacity in plant tissues. Plant Pathol. J. 28, 270–281. doi: 10.5423/PPJ.OA.02.2012.0026
Lindow, S. E. (1983). The role of bacterial ice nucleation in frost injury to plants. Annu. Rev. Phytopathol. 21, 363–384. doi: 10.1146/annurev.py.21.090183.002051
Liu, Y. Y., Chiou, C. S., and Chen, C. C. (2016). PGAdb-builder: A web service tool for creating pan-genome allele database for molecular fine typing. Sci. Rep. 6:36213. doi: 10.1038/srep36213
Morohoshi, T., Nakamura, Y., Yamazaki, G., Ishida, A., Kato, N., and Ikeda, T. (2007). The plant pathogen Pantoea ananatis produces N-acylhomoserine lactone and causes center rot disease of onion by quorum sensing. J. Bacteriol. 189, 8333–8338. doi: 10.1128/JB.01054-07
Nielsen, H. (2017). Predicting secretory proteins with signalP. Methods Mol. Biol. 1611, 59–73. doi: 10.1007/978-1-4939-7015-5_6
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Paccola-Meirelles, L. D., Ferreira, A. S., Meirelles, W. F., Marriel, I. E., and Casela, C. R. (2001). Detection of a bacterium associated with a leaf spot disease of maize in Brazil. J. Phytopathol. Phytopathol. Zeitschrift 149, 275–279. doi: 10.1046/j.1439-0434.2001.00614.x
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Praillet, T., Nasser, W., RobertBaudouy, J., and Reverchon, S. (1996). Purification and functional characterization of PecS, a regulator of virulence-factor synthesis in Erwinia chrysanthemi. Mol. Microbiol. 20, 391–402. doi: 10.1111/j.1365-2958.1996.tb02626.x
Salerno, A., Deletolle, A., Lefevre, M., Ciznar, I., Krovacek, K., Grimont, P., et al. (2007). Recombining population structure of Plesiomonas shigelloides (Enterobacteriaceae) revealed by multilocus sequence typing. J. Bacteriol. 189, 7808–7818. doi: 10.1128/JB.00796-07
Schwartz, H. F., and Otto, K. (2000). First report of a leaf blight and bulb decay of onion by Pantoea ananitas in colorado. Plant Dis. 84:808. doi: 10.1094/P.D.I.S.2000.84.7.808A
Schroeder, B. K., du Toit, L. J., and Schwartz, H. F. (2009). First report of enterobacter cloacae causing onion bulb rot in the columbia basin of Washington State. Plant Dis. 93, 323–323. doi: 10.1094/PDIS-93-3-0323A
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Serrano, F. B. (1928). Bacterial fruitlet brown-rot of pineapple in the Philippines. Philipp. J. Sci. 36, 271–305.
Sheibani-Tezerji, R., Naveed, M., Jehl, M. A., Sessitsch, A., Rattei, T., and Mitter, B. (2015). The genomes of closely related Pantoea ananatis maize seed endophytes having different effects on the host plant differ in secretion system genes and mobile genetic elements. Front. Microbiol. 6:440. doi: 10.3389/fmicb.2015.00440
Shyntum, D. Y., Theron, J., Venter, S. N., Moleleki, L. N., Toth, I. K., and Coutinho, T. A. (2015). Pantoea ananatis utilizes a Type VI secretion system for pathogenesis and bacterial competition. Mol. Plant Microbe Interact. 28, 420–431. doi: 10.1094/MPMI-07-14-0219-R
Tabei, H., Azegami, K., and Fukuda, T. (1988). Infection site of rice grain with Erwinia herbicola, the causal agent of bacterial palea browning of rice. Ann. Phytopathol. Soc. Japan 54, 637–639. doi: 10.3186/jjphytopath.54.637
Torres, M. A., Jones, J. D. G., and Dangl, J. L. (2006). Reactive oxygen species signaling in response to pathogens. Plant Physiol. 141, 373–378. doi: 10.1104/pp.106.079467
Versalovic, J., Koeuth, T., and Lupski, J. R. (1991). Distribution of repetitive DNA-sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res. 19, 6823–6831. doi: 10.1093/nar/19.24.6823
Walcott, R. R., Gitaitis, R. D., Castro, A. C., Sanders, F. H., and Diaz-Perez, J. C. (2002). Natural infestation of onion seed by Pantoea ananatis, causal agent of center rot. Plant Dis. 86, 106–111. doi: 10.1094/PDIS.2002.86.2.106
Weller-Stuart, T., De Maayer, P., and Coutinho, T. (2017). Pantoea ananatis: genomic insights into a versatile pathogen. Mol. Plant Pathol. 18, 1191–1198. doi: 10.1111/mpp.12517
Keywords: Pantoea ananatis, pangenome, onion, CWDE, MLSA, bacterial secretion systems
Citation: Stice SP, Stumpf SD, Gitaitis RD, Kvitko BH and Dutta B (2018) Pantoea ananatis Genetic Diversity Analysis Reveals Limited Genomic Diversity as Well as Accessory Genes Correlated with Onion Pathogenicity. Front. Microbiol. 9:184. doi: 10.3389/fmicb.2018.00184
Received: 05 October 2017; Accepted: 26 January 2018;
Published: 13 February 2018.
Edited by:
Péter Poór, University of Szeged, HungaryReviewed by:
Stephanus Nicolaas Venter, University of Pretoria, South AfricaSiomar De Castro Soares, Universidade Federal do Triângulo Mineiro, Brazil
Copyright © 2018 Stice, Stumpf, Gitaitis, Kvitko and Dutta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian H. Kvitko, bkvitko@uga.edu
†These authors have contributed equally to this work.
‡Co-Senior Authors.