 Josephin N. Rashida Gnanaprakasam
Josephin N. Rashida Gnanaprakasam Ruohan Wu
Ruohan Wu Ruoning Wang
Ruoning Wang- Center for Childhood Cancer & Blood Diseases, Hematology/Oncology & BMT, The Research Institute at Nationwide Children’s Hospital, Ohio State University, Columbus, OH, United States
A robust adaptive immune response requires a phase of proliferative burst which is followed by the polarization of T cells into relevant functional subsets. Both processes are associated with dramatically increased bioenergetics demands, biosynthetic demands, and redox demands. T cells meet these demands by rewiring their central metabolic pathways that generate energy and biosynthetic precursors by catabolizing and oxidizing nutrients into carbon dioxide. Simultaneously, oxidative metabolism also produces reactive oxygen species (ROS), which are tightly controlled by antioxidants and plays important role in regulating T cell functions. In this review, we discuss how metabolic rewiring during T cell activation influence ROS production and antioxidant capacity.
Introduction
T cells are central orchestrators of antigen-specific adaptive immunity and tolerance. Upon stimulation of antigen receptors, T cells rapidly transit from naïve to an active state followed by massive clonal expansion. Depending on the nature of pathogens and the surrounding cytokine milieu, proliferating T cells can differentiate into diverse phenotypic and functional subsets to elicit a robust immune response. After the clearance of pathogens, the majority of effector T cells die through apoptosis and the remaining memory T (Tmem) cells are responsible for immunity upon re-exposure to the same pathogen. Accumulating evidence suggests that a coordinated rewiring of cellular metabolism is required for T cell activation and differentiation by fulfilling the bioenergetic, biosynthetic, and redox demands (1–9). Importantly, different phenotypic and functional T cell subsets are characterized by distinct metabolic programs (Table 1), which are largely controlled by immune modulatory signaling cascades (10–17). Naïve T (Tnai) cells, Tmem cells, and immune-suppressive regulatory T (Treg) cells predominantly rely on fatty acid oxidation (FAO) and oxidative phosphorylation (OXPHOS) to meet their relatively low-energy needs (14, 15, 18, 19). However, persistent aerobic glycolysis, the pentose phosphate pathway (PPP), and glutaminolysis are required to drive cell growth, clonal expansion, and effector functions in both CD4+ subsets and CD8+ effector T (Teff) cells (Table 1) (10, 15, 16, 18, 20–31).
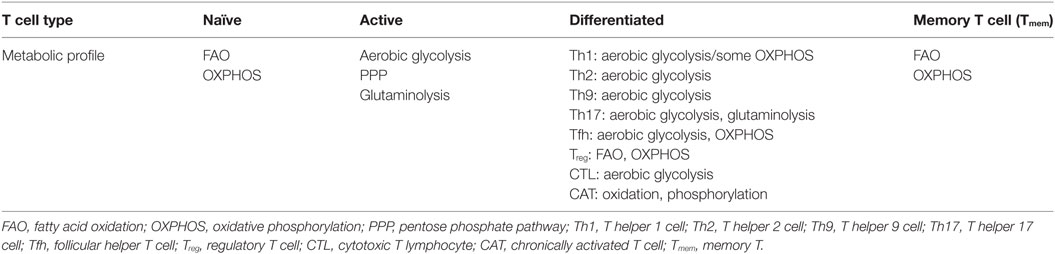
Table 1. The metabolic profiles of T cell subsets.
These metabolic programs actively support ATP production by providing mitochondrial OXPHOS substrates, support biomass accumulation by generating metabolic precursors for the biosynthesis of protein, lipids, and nucleic acids, and maintain redox balance through generation and elimination of reactive oxygen species (ROS).
Mitochondrial OXPHOS and NADPH Oxidases (NOXs) in Generating ROS in T Cells
The mitochondria are the central metabolic hub and powerhouse of all eukaryotic cells. The oxidation of acetyl-CoA to carbon dioxide (CO2) by the tricarboxylic acid (TCA) cycle is the central metabolic process for fueling ATP production. While glycolysis and FAO primarily provide the OXPHOX substrate, acetyl-CoA, for mitochondria in Tnai cells, Tmem cells, and Treg cells (14, 15, 18, 19), heightened mitochondrial biogenesis during T cell activation leads to higher numbers of mitochondria and likely the enhanced mitochondrial dependent metabolic flux in Teff cells compared with Tnai cells (23, 32, 33). In particular, a surplus of 3-, 4-, and 5-carbon metabolites (anaplerotic substrates) including pyruvate, malate, and α-ketoglutarate (α-KG) feed into the TCA cycle during the catabolism of glutamine and other amino acids (5, 13, 15, 34). The electron transport chain (ETC) constantly transfers electrons from NADH and FADH2 to oxygen while allowing protons (H+) to pass through the inner mitochondrial membrane to form an electrochemical proton gradient that drives ATP synthesis. However, both protons and electrons can leak from the ETC due to the uncoupling of ATP synthase from the proton gradient and a premature exit of electron before reaching cytochrome c oxidase, respectively. Electron leak largely occurs at the sites of complex I (NADH–ubiquinone oxidoreductase) and complex III (ubiquinone–cytochrome c oxidoreductase) in the ETC and results in the partial reduction of oxygen, generating superoxide . Subsequently, mitochondrial dismutase acts to convert superoxide to hydrogen peroxide (H2O2), which is free to diffuse into cytosol and act as a redox signaling molecule to elicit different cellular responses (35–37). Thus, increased ROS production in T cells can occur as a result of metabolic reprogramming during T cell activation. Besides mitochondria, cytoplasmic ROS is generated by NOXs, which is also an important source of ROS in T cell. NOX family proteins are transmembrane proteins that transport the electrons from nicotinamide adenine dinucleotide (phosphate), NAD(P)H, to oxygen and generate superoxide anion as the intermediate product of oxidase and subsequently H2O2, as the product of dismutation of the superoxide. There are different isoforms of the NOX enzyme including NOX1, NOX2, NOX3, NOX4, NOX5, dual oxidase 1, and DUOX2, and the expression of these subunits varies among different tissues. NOX-2 is an important source of ROS in T cells (38, 39). The ROS production by NOX is regulated at various levels including the assembly of functional NOX complex, the availability of prosthetic group, flavin adenine dinucleotide, the intracellular concentration of calcium, cell surface receptor signals mediated by G protein-coupled receptors, complement, T cell receptor (TCR), and CD28 (35–37, 40, 41).
ROS Signaling in Regulating T Cell Activation and Differentiation
T cell activation requires ligation of TCR and the major histocompatibility complex molecules. This interaction will initiate the signaling cascade and activation of transcriptional factors such as nuclear factor of activated T cells (NFAT), activator protein 1 (AP-1), and nuclear factor of kappa light chain enhancer in B cells (42). It has been reported that TCR ligation increases the production of ROS from OXPHOS and cytoplasmic ROS from NADPH oxidases (NOXs), a family of plasma membrane-associated oxidases (36, 40, 41). ROS-mediated signaling events are required for driving T cell activation, proliferation, and differentiation (Figure 1) (36, 41). T cells with reduced production of mitochondrial ROS display impaired production of interleukin-2 and antigen-specific proliferation, indicating an essential signaling role for mitochondrial ROS in driving optimal TCR signaling. The proximal TCR signaling machinery, including zeta chain-associated protein kinase 70, linker of activated T cell, SH2 domain-containing leukocyte protein, phospholipase Cγ1, and protein kinase Cθ, is involved in driving ROS production upon T cell activation (36, 41, 43). Conversely, physiologically relevant levels of ROS facilitate the activation of oxidation-dependent transcription factors, such as NF-κB and AP-1, which are required for driving essential signaling events to support T cell-mediated immune responses (44–46). However, excessive ROS production following ablation of de novo glutathione (GSH) synthesis suppresses the activity of mammalian target of rapamycin and the expression of transcription factors NFAT and c-MYC, the latter of which control metabolic reprogramming following T cell activation (15, 47, 48). Thus, T cells fail to meet their increased energy and biosynthetic needs and display compromised proliferation (48). In addition, uncontrolled ROS production is involved in the activation-induced T-cell death by affecting expression of apoptosis related genes including Bcl-2 and FasL and mitochondrial membrane potential (43, 49–52). NOX-derived ROS modulates the function of GATA-binding protein 3, signal transducer and activator of transcription, and T-box transcription factor to collectively control T cell activation and differentiation. T cells from NOX-deficient animals showed a skewed Th17 phenotype, whereas NOX-intact cells exhibited a preferred Th1 response (39, 53–55). In CD8 T cells, NOX-derived ROS is involved in regulating the production of IFN-γ and CD39 expression through c-Jun N-terminal kinase and NFκB signaling (40, 56). Importantly, the impact of ROS on T cell activation can be extended to the later T cell differentiation stages. Fine tuning of ROS is required for polarizing T cell in part by engaging lineage-specific transcription factors and modulating cytokine profiles, and consequently directs T cell-mediated inflammatory responses (39, 40, 53–55, 57–61).
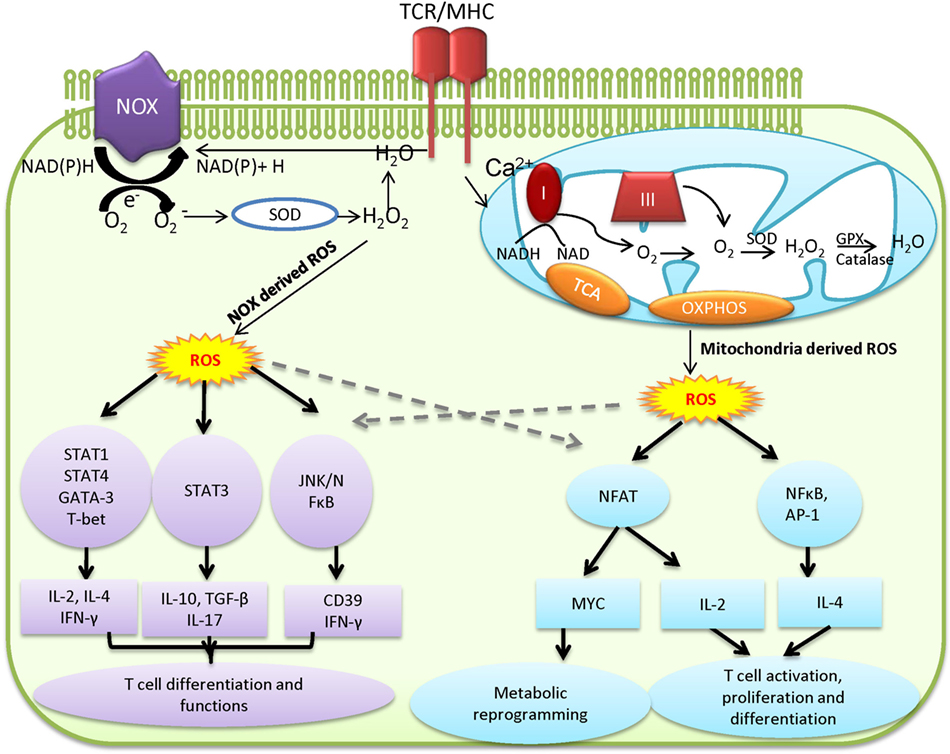
Figure 1. Mitochondria and NADPH oxidases (NOX)-derived reactive oxygen species (ROS) regulates T cell activation, differentiation, and metabolism. Mitochondria and NOX are the two major sources of ROS. The stimulation of T cell receptor (TCR) initiates signaling and metabolic events that drive ROS production in cytoplasm through NOX-dependent reaction and ROS production in mitochondria via mitochondria electron transport chain (ETC). Excess ROS causes damage and cell death. However, physiologically relevant levels of ROS mediate essential redox signaling through nodulation of a wide spectrum of redox-sensitive transcription factors to drive T cell activation and function.
Metabolic Pathways in Modulating Antioxidant Capacities
Excessive ROS production causes collateral damage to macromolecules, cellular organelles, and eventually necrosis, which can lead to uncontrolled hyper-inflammation and tissue damage. Thus, a fine-tuned balance between ROS production and antioxidant capacity ensures appropriate levels of intracellular ROS (Figure 2) (44, 55, 62). GSH, a tripeptide of glutamine, cysteine, and glycine, is the most abundant antioxidant capable of providing reducing equivalents and also serves as a versatile nucleophilic cofactor in a wide spectrum of metabolic reactions in aerobic organisms (63, 64). Thioredoxin (TXN) is a class of small redox proteins that are involved in modulating cell surface receptors and confers tolerance to oxidative stress in T cells (65–69). A reciprocal redox reaction can be coupled between these two antioxidant systems to act as a backup for each other under certain conditions (70–77). Supporting these findings, the inhibition of thioredoxin reductase (TXNRD) conferred an increased susceptibility of cancer cells to GSH depletion (78–80). Glutathione-disulfide reductase (GSR) regenerates GSH from its oxidized form, glutathione disulfide (GSSG), whereas TXNRD is responsible for the regeneration of TXN once it has been oxidized. Importantly, both GSR and TXNRD require NADPH as a reducing agent. Upon antigen stimulation, both PPP and glutaminolysis are significantly upregulated and further enhance T cell antioxidant capacities by generating NADPH through metabolic reactions that are controlled by glucose-6-phosphate dehydrogenase, phosphoglycerate dehydrogenase, malic enzyme 1, and isocitrate dehydrogenase 1. The intracellular GSH concentrations are normally in a range of three orders of magnitude higher than extracellular GSH. Even though some cells are able to recycle extracellular GSH, it may only play a minor role in maintaining intracellular GSH pool (63, 64, 81–86). By contrast, both the regeneration of GSH from GSSG (recycling pathway) and de novo synthesis of GSH, by glutamate-cysteine ligase (GCL) and glutathione synthase (GS), are required to maintain intracellular GSH levels (64, 87). The ligation of glutamate and cysteine to form dipeptide ϒ-glutamylcysteine (ϒ-GC) is the first and also the rate-limiting step of GSH de novo synthesis, which is controlled by ATP-dependent ligase GCL, a heterodimer of a catalytic subunit (GCLC) and modifier subunit (GCLM). Subsequently, GSH is formed by GS-mediated ligation of ϒ-GC and glycine (88, 89). Thus, the supply of intracellular cysteine, glycine, and glutamate must fulfill the need of de novo synthesis of GSH during T cell activation. Supporting this idea, the metabolic processes that are involved in providing three amino acids are tightly regulated upon T cell activation (13, 15, 90–92). Upon T cell activation, heightened glycolysis, PPP, and glutaminolysis intersect with the de novo synthesis of GSH through promoting cysteine uptake and providing glycine, glutamine, and NADPH (93–95). As such, the genetic abrogation of de novo synthesis of GSH, the glucose, or glutamine starvation significantly dampens T cell activation (10, 13, 15, 20, 48).
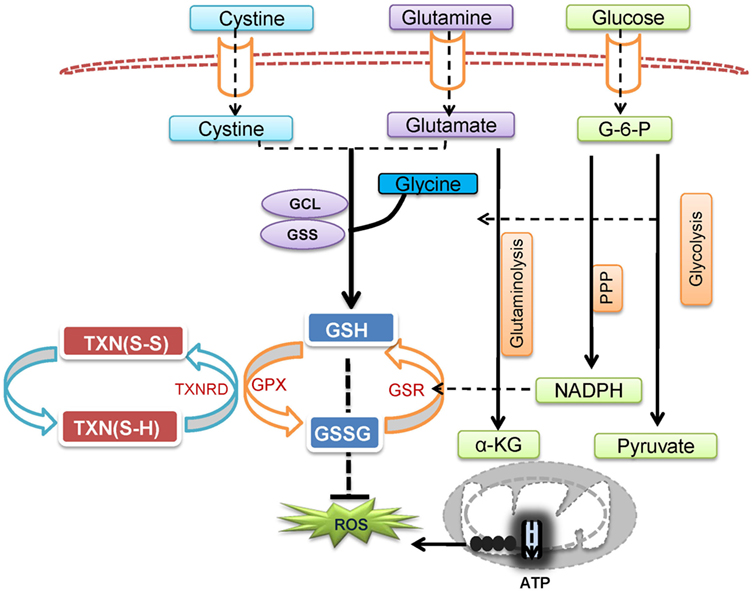
Figure 2. T cell metabolic programs that link to reactive oxygen species (ROS) production and the de novo synthesis of GSH. Pyruvate that is derived from glucose via glycolysis is shuttled to the mitochondria and drives the tricarboxylic acid (TCA) cycle and fuels oxidative phosphorylation (OXPHOS). Glucose-derived glucose-6-phosphate feeds into the pentose phosphate pathway (PPP) and produces NADPH in the cytoplasm. In addition, glutamate feeds the TCA cycle through α-ketoglutarate (α-KG) to fuel OXPHOS and generate ROS. Excessive ROS production is regulated by glutathione (GSH), a tripeptide of glutamine, cysteine, and glycine, which is synthesized de novo by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS). In addition, NADPH, glutathione-disulfide reductase (GSR), and glutathione peroxidase (GPX) are involved in regenerating GSH from glutathione disulfide (GSSG), whereas thioredoxin reductase (TXNRD) is responsible for the regeneration of thioredoxin (TXN) to control oxidative stress in T cell.
Glutamine Catabolism in Coordinating the Production of ROS and GSH
Glutamine has been known as a key nutrient, which supports a diverse range of cellular functions (93–102). Glutamine provides high proportions of the energy from OXPHOS, provides precursors for various biosynthetic pathways, as a key nitrogen and carbon donor, and also is catabolized to various intermediate metabolites that have signaling roles in modulating cellular processes. In specialized cells, such as the cells of the nervous system, glutamine catabolism intersects with signaling networks to support the production of central neurotransmitters including glutamate, GABA, and aspartate (103–106). To meet bioenergetic and biosynthetic demand during T cell growth and proliferation, glutaminolysis replenishes the anapleurotic substrate α-KG that fuels OXPHOS via the TCA cycle and also provides sources of nitrogen and carbon to support the biosynthesis of nonessential amino acids, lipids, nucleotides, and polyamines (13, 15, 102, 107). Similar to cancer cells, de novo synthesis of GSH in T cells, which relies on glutamine to provide precursors, plays an essential role in suppressing oxidative stress. Accordingly, glutaminolysis is a branched pathway that consists of several paths, enabling energy production through oxidation and biomolecule production, including GSH through biosynthesis (93–95). While the ATP generating capacity of glutaminolysis is considered to be redundant with glucose oxidation and/or FAO, the oxidation of glutamine is indispensable for driving T cell proliferation and differentiation (13, 15, 102). However, enhanced glutamine oxidation in the mitochondria also increases the production of its by-product, mitochondrial ROS, the main source of cellular ROS in T cells (35, 37). Therefore, glutamate represents a key branch point in glutaminolysis that can be committed toward mitochondrial oxidation to produce ATP and ROS, or toward de novo synthesis of GSH to modulate redox balance and suppress oxidative stress. In addition, the high rate of glutaminolysis ensures that the capacity to supply glutamate, the most abundant intracellular metabolite in cells, exceeds the demand for glutamate from each of the downstream metabolic branches. The branched pathways in glutaminolysis enable the production of counteracting metabolites, i.e., ROS and GSH, from a common metabolic precursor, and permit a fine-tuned coordination between the metabolic flux shunted toward GSH synthesis and the metabolic flux shunted toward OXPHOS. Consistent with this idea, the overall high consumption rate of glutamine in proliferative cells is suggested to provide a sensitive and precise regulation on intermediate metabolites that can be committed toward several metabolic branches, hence permitting rapid responses to meet the demands for energy production or antioxidant production (99, 108). In addition to increasing antioxidant capacity, T cells may adapt by shifting glucose catabolism from OXPHOS toward aerobic glycolysis, which could provide biosynthetic precursors and rapidly produce ATP by the substrate level of phosphorylation.
Conclusion and Perspective
Reactive oxygen species is not only a by-product of cellular metabolic programs but also a key signaling molecule involved in directing T cell activation and differentiation. However, uncontrolled ROS production causes collateral damage to biomolecules and cellular organelles. Under pathophysiological conditions, ROS generation from mitochondria can contribute to the initiation and progression of inflammatory and autoimmune diseases. However, oxidative stress caused by elevated ROS may also render key immune effector cells vulnerable to agents that can either modulate stress response or modulate metabolic pathways for ROS and GSH production (Figure 3). Redox signaling is essential to regulate T cell metabolism. Technological advancement in genetic models and metabolomics will allow us to understand the key metabolic processes that dictate T cell fate through modulation ROS and GSH production. Thus, further research is expected to illustrate the complex interplay between cellular metabolism and redox signaling in T cells, thereby offering novel therapies for treating inflammatory and autoimmune diseases.
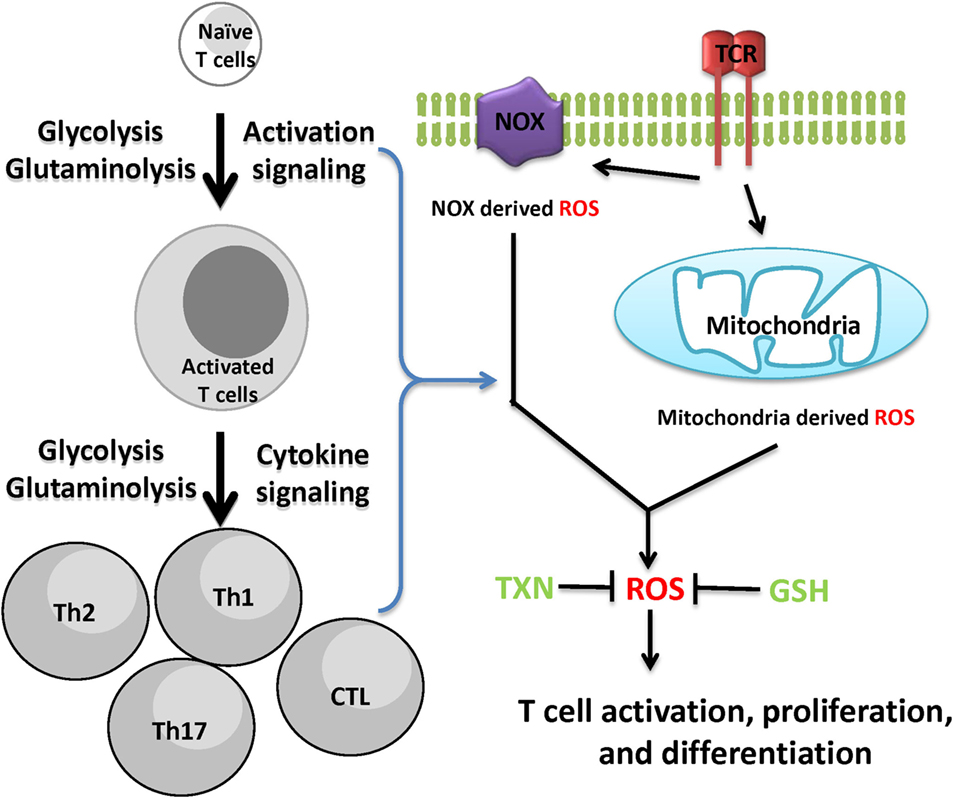
Figure 3. Cellular redox homeostasis is essential for mounting an effective T cell-mediated immune response. In addition to generate ATP and provide biosynthetic precursors, T cell activation-induced metabolic reprogramming actively regulates redox homeostasis. The coordination of de novo synthesis of glutathione (GSH) and the production of reactive oxygen species (ROS) ensures T cell redox balance and a fine-tuned T cell response.
Author Contributions
JG and RHW wrote the manuscript. RW wrote and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by R21AI117547 and 1R01AI114581 from National Institutes of Health, V2014-001 from the V-Foundation and 128436-RSG-15-180-01-LIB from the American Cancer Society (RW).
References
1. Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol (2011) 11:109–17. doi:10.1038/nri2888
2. Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15. doi:10.1038/ni.2386
3. Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43. doi:10.1016/j.immuni.2013.04.005
4. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity (2015) 42:406–17. doi:10.1016/j.immuni.2015.02.002
5. O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol (2016) 16:553–65. doi:10.1038/nri.2016.70
6. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell (2017) 169:570–86. doi:10.1016/j.cell.2017.04.004
7. Ma EH, Poffenberger MC, Wong AH, Jones RG. The role of AMPK in T cell metabolism and function. Curr Opin Immunol (2017) 46:45–52. doi:10.1016/j.coi.2017.04.004
8. Patel CH, Powell JD. Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr Opin Immunol (2017) 46:82–8. doi:10.1016/j.coi.2017.04.006
9. Zeng H, Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol (2017) 46:103–11. doi:10.1016/j.coi.2017.04.005
10. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77. doi:10.1016/S1074-7613(02)00323-0
11. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell (2008) 134:97–111. doi:10.1016/j.cell.2008.04.052
12. Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood (2008) 111:2101–11. doi:10.1182/blood-2007-06-096297
13. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol (2011) 185:1037–44. doi:10.4049/jimmunol.0903586
14. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, Maciver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186:3299–303. doi:10.4049/jimmunol.1003613
15. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi:10.1016/j.immuni.2011.09.021
16. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med (2012) 209:2441–53. doi:10.1084/jem.20112607
17. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14:489–99. doi:10.1038/ni.2570
18. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi:10.1038/nature08097
19. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
20. Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol (2001) 167:6869–76. doi:10.4049/jimmunol.167.12.6869
21. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol (2008) 180:4476–86. doi:10.4049/jimmunol.180.7.4476
22. Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33:168–73. doi:10.1016/j.it.2012.01.010
23. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8(+) T cell memory development. Immunity (2012) 36(1):68–78. doi:10.1016/j.immuni.2011.12.007
24. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity (2015) 42:1033–47. doi:10.1016/j.immuni.2015.05.024
25. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun (2015) 6:6692. doi:10.1038/ncomms7692
26. Ray JP, Staron MM, Shyer JA, Ho P-C, Marshall HD, Gray SM, et al. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity (2015) 43:690–702. doi:10.1016/j.immuni.2015.08.017
27. Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, et al. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun (2016) 7:13683. doi:10.1038/ncomms13683
28. Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell (2016) 166:63–76. doi:10.1016/j.cell.2016.05.035
29. Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4+ T cells. Immunity (2016) 44:1337–49. doi:10.1016/j.immuni.2016.05.009
30. Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev (2017) 35:7–14. doi:10.1016/j.cytogfr.2017.04.003
31. Binger KJ, Côrte-Real BF, Kleinewietfeld M. Immunometabolic regulation of interleukin-17-producing T helper cells: uncoupling new targets for autoimmunity. Front Immunol (2017) 8:311. doi:10.3389/fimmu.2017.00311
32. D’Souza AD, Parikh N, Kaech SM, Shadel GS. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion (2007) 7:374–85. doi:10.1016/j.mito.2007.08.001
33. Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab (2016) 24:104–17. doi:10.1016/j.cmet.2016.06.007
34. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–842.e13. doi:10.1016/j.cell.2016.09.031
35. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J (2009) 417:1–13. doi:10.1042/BJ20081386
36. Kaminski MM, Sauer SW, Kaminski M, Opp S, Ruppert T, Grigaravicius P, et al. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep (2012) 2:1300–15. doi:10.1016/j.celrep.2012.10.009
37. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol (2014) 24:R453–62. doi:10.1016/j.cub.2014.03.034
38. Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev (2007) 87:245–313. doi:10.1152/physrev.00044.2005
39. Hubert MT, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, et al. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol (2010) 185:5247–58. doi:10.4049/jimmunol.1001472
40. Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol (2004) 5:818–27. doi:10.1038/ni1096
41. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity (2013) 38:225–36. doi:10.1016/j.immuni.2012.10.020
42. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol (2009) 27:591–619. doi:10.1146/annurev.immunol.021908.132706
43. Kamiński M, Kießling M, Süss D, Krammer PH, Gülow K. Novel role for mitochondria: protein kinase Cθ-dependent oxidative signaling organelles in activation-induced T-cell death. Mol Cell Biol (2007) 27:3625–39. doi:10.1128/MCB.02295-06
44. Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation. J Exp Med (2002) 195:59–70. doi:10.1084/jem.20010659
45. Kaminski MM, Roth D, Krammer PH, Gulow K. Mitochondria as oxidative signaling organelles in T-cell activation: physiological role and pathological implications. Arch Immunol Ther Exp (Warsz) (2013) 61:367–84. doi:10.1007/s00005-013-0235-0
46. Murphy MP, Siegel RM. Mitochondrial ROS fire up T cell activation. Immunity (2013) 38:201–2. doi:10.1016/j.immuni.2013.02.005
47. Klein Geltink RI, O’Sullivan D, Pearce EL. Caught in the cROSsfire: GSH controls T cell metabolic reprogramming. Immunity (2017) 46:525–7. doi:10.1016/j.immuni.2017.03.022
48. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity (2017) 46:675–89. doi:10.1016/j.immuni.2017.03.019
49. Hildeman DA, Mitchell T, Teague TK, Henson P, Day BJ, Kappler J, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity (1999) 10:735–44. doi:10.1016/S1074-7613(00)80072-2
50. Li-Weber M, Weigand MA, Giaisi M, Süss D, Treiber MK, Baumann S, et al. Vitamin E inhibits CD95 ligand expression and protects T cells from activation-induced cell death. J Clin Invest (2002) 110:681. doi:10.1172/JCI0215073
51. Hildeman DA. Regulation of T-cell apoptosis by reactive oxygen species. Free Radic Biol Med (2004) 36:1496–504. doi:10.1016/j.freeradbiomed.2004.03.023
52. Takahashi A, Hanson MG, Norell HR, Havelka AM, Kono K, Malmberg K-J, et al. Preferential cell death of CD8+ effector memory (CCR7− CD45RA−) T cells by hydrogen peroxide-induced oxidative stress. J Immunol (2005) 174:6080–7. doi:10.4049/jimmunol.174.10.6080
53. Purushothaman D, Sarin A. Cytokine-dependent regulation of NADPH oxidase activity and the consequences for activated T cell homeostasis. J Exp Med (2009) 206:1515–23. doi:10.1084/jem.20082851
54. Shatynski KE, Chen H, Kwon J, Williams MS. Decreased STAT5 phosphorylation and GATA-3 expression in NOX2-deficient T cells: role in T helper development. Eur J Immunol (2012) 42:3202–11. doi:10.1002/eji.201242659
55. Belikov AV, Schraven B, Simeoni L. T cells and reactive oxygen species. J Biomed Sci (2015) 22:85. doi:10.1186/s12929-015-0194-3
56. Bai A, Moss A, Rothweiler S, Longhi MS, Wu Y, Junger WG, et al. NADH oxidase-dependent CD39 expression by CD8+ T cells modulates interferon gamma responses via generation of adenosine. Nat Commun (2015) 6:8819. doi:10.1038/ncomms9819
57. Zhi L, Ustyugova IV, Chen X, Zhang Q, Wu MX. Enhanced Th17 differentiation and aggravated arthritis in IEX-1-deficient mice by mitochondrial reactive oxygen species-mediated signaling. J Immunol (2012) 189:1639–47. doi:10.4049/jimmunol.1200528
58. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125:194–207. doi:10.1172/JCI76012
59. Abimannan T, Peroumal D, Parida JR, Barik PK, Padhan P, Devadas S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic Biol Med (2016) 99:352–63. doi:10.1016/j.freeradbiomed.2016.08.026
60. Padgett LE, Tse HM. NADPH oxidase-derived superoxide provides a third signal for CD4 T cell effector responses. J Immunol (2016) 197:1733–42. doi:10.4049/jimmunol.1502581
61. Xu T, Stewart KM, Wang X, Liu K, Xie M, Kyu Ryu J, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature (2017) 548:228–33. doi:10.1038/nature23475
62. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell (2012) 48:158–67. doi:10.1016/j.molcel.2012.09.025
63. Kosower NS, Kosower EM. The glutathione status of cells. Int Rev Cytol (1978) 54:109–60. doi:10.1016/S0074-7696(08)60166-7
64. Meister A. Metabolism and function of glutathione: an overview. Biochem Soc Trans (1982) 10:78–9. doi:10.1042/bst0100078
65. Tagaya Y, Wakasugi H, Masutani H, Nakamura H, Iwata S, Mitsui A, et al. Role of ATL-derived factor (ADF) in the normal and abnormal cellular activation: involvement of dithiol related reduction. Mol Immunol (1990) 27:1279–89. doi:10.1016/0161-5890(90)90032-U
66. Wakasugi N, Tagaya Y, Wakasugi H, Mitsui A, Maeda M, Yodoi J, et al. Adult T-cell leukemia-derived factor/thioredoxin, produced by both human T-lymphotropic virus type I- and Epstein-Barr virus-transformed lymphocytes, acts as an autocrine growth factor and synergizes with interleukin 1 and interleukin 2. Proc Natl Acad Sci U S A (1990) 87:8282–6. doi:10.1073/pnas.87.21.8282
67. Rosen A, Lundman P, Carlsson M, Bhavani K, Srinivasa BR, Kjellstrom G, et al. A CD4+ T cell line-secreted factor, growth promoting for normal and leukemic B cells, identified as thioredoxin. Int Immunol (1995) 7:625–33. doi:10.1093/intimm/7.4.625
68. Matthias LJ, Yam PT, Jiang XM, Vandegraaff N, Li P, Poumbourios P, et al. Disulfide exchange in domain 2 of CD4 is required for entry of HIV-1. Nat Immunol (2002) 3:727–32. doi:10.1038/ni815
69. Mougiakakos D, Johansson CC, Jitschin R, Bottcher M, Kiessling R. Increased thioredoxin-1 production in human naturally occurring regulatory T cells confers enhanced tolerance to oxidative stress. Blood (2011) 117:857–61. doi:10.1182/blood-2010-09-307041
70. Mustacich D, Powis G. Thioredoxin reductase. Biochem J (2000) 346(Pt 1):1–8. doi:10.1042/0264-6021:3460001
71. Johansson C, Lillig CH, Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J Biol Chem (2004) 279:7537–43. doi:10.1074/jbc.M312719200
72. Hanschmann EM, Lonn ME, Schutte LD, Funke M, Godoy JR, Eitner S, et al. Both thioredoxin 2 and glutaredoxin 2 contribute to the reduction of the mitochondrial 2-Cys peroxiredoxin Prx3. J Biol Chem (2010) 285:40699–705. doi:10.1074/jbc.M110.185827
73. Tan SX, Greetham D, Raeth S, Grant CM, Dawes IW, Perrone GG. The thioredoxin-thioredoxin reductase system can function in vivo as an alternative system to reduce oxidized glutathione in Saccharomyces cerevisiae. J Biol Chem (2010) 285:6118–26. doi:10.1074/jbc.M109.062844
74. Du Y, Zhang H, Lu J, Holmgren A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J Biol Chem (2012) 287:38210–9. doi:10.1074/jbc.M112.392225
75. Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med (2014) 66:75–87. doi:10.1016/j.freeradbiomed.2013.07.036
76. Couto N, Wood J, Barber J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic Biol Med (2016) 95:27–42. doi:10.1016/j.freeradbiomed.2016.02.028
77. Lei XG, Zhu JH, Cheng WH, Bao Y, Ho YS, Reddi AR, et al. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiol Rev (2016) 96:307–64. doi:10.1152/physrev.00010.2014
78. Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A (2007) 104:12288–93. doi:10.1073/pnas.0701549104
79. Mandal PK, Schneider M, Kolle P, Kuhlencordt P, Forster H, Beck H, et al. Loss of thioredoxin reductase 1 renders tumors highly susceptible to pharmacologic glutathione deprivation. Cancer Res (2010) 70:9505–14. doi:10.1158/0008-5472.CAN-10-1509
80. Wang Y, Lu H, Wang D, Li S, Sun K, Wan X, et al. Inhibition of glutathione synthesis eliminates the adaptive response of ascitic hepatoma 22 cells to nedaplatin that targets thioredoxin reductase. Toxicol Appl Pharmacol (2012) 265:342–50. doi:10.1016/j.taap.2012.09.001
81. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science (1992) 257:1496–502. doi:10.1126/science.1523409
82. Ganguly D, Srikanth CV, Kumar C, Vats P, Bachhawat AK. Why is glutathione (a tripeptide) synthesized by specific enzymes while TSH releasing hormone (TRH or thyroliberin), also a tripeptide, is produced as part of a prohormone protein? IUBMB Life (2003) 55:553–4. doi:10.1080/15216540310001623064
83. Perrone GG, Grant CM, Dawes IW. Genetic and environmental factors influencing glutathione homeostasis in Saccharomyces cerevisiae. Mol Biol Cell (2005) 16:218–30. doi:10.1091/mbc.E04-07-0560
84. Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol (2009) 5:593–9. doi:10.1038/nchembio.186
85. Morgan B, Sobotta MC, Dick TP. Measuring E(GSH) and H2O2 with roGFP2-based redox probes. Free Radic Biol Med (2011) 51:1943–51. doi:10.1016/j.freeradbiomed.2011.08.035
86. Park JO, Rubin SA, Xu YF, Amador-Noguez D, Fan J, Shlomi T, et al. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol (2016) 12:482–9. doi:10.1038/nchembio.2077
87. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med (2009) 30:42–59. doi:10.1016/j.mam.2008.05.005
88. Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem (2005) 280:33766–74. doi:10.1074/jbc.M504604200
89. Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med (2009) 30:86–98. doi:10.1016/j.mam.2008.08.009
90. Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A (2002) 99:1491–6. doi:10.1073/pnas.022630299
91. Levring TB, Hansen AK, Nielsen BL, Kongsbak M, Von Essen MR, Woetmann A, et al. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci Rep (2012) 2:266. doi:10.1038/srep00266
92. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab (2017) 25:345–57. doi:10.1016/j.cmet.2017.01.014
93. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov (2013) 12:931–47. doi:10.1038/nrd4002
94. Hensley CT, Wasti AT, Deberardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123:3678–84. doi:10.1172/JCI69600
95. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16:619–34. doi:10.1038/nrc.2016.71
96. Kvamme E, Svenneby G. Effect of anaerobiosis and addition of keto acids on glutamine utilization by Ehrlich ascites-tumor cells. Biochim Biophys Acta (1960) 42:187–8. doi:10.1016/0006-3002(60)90779-4
97. Lund P. Glutamine metabolism in the rat. FEBS Lett (1980) 117:K86–92. doi:10.1016/0014-5793(80)80573-4
98. Kovacevic Z, McGivan JD. Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev (1983) 63:547–605. doi:10.1152/physrev.1983.63.2.547
99. Newsholme EA, Crabtree B, Ardawi MS. Glutamine metabolism in lymphocytes: its biochemical, physiological and clinical importance. Q J Exp Physiol (1985) 70:473–89. doi:10.1113/expphysiol.1985.sp002935
100. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104:19345–50. doi:10.1073/pnas.0709747104
101. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, Mcgettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi:10.1038/nature11986
102. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal (2015) 8:ra97. doi:10.1126/scisignal.aab2610
103. Daikhin Y, Yudkoff M. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr (2000) 130:1026S–31S. doi:10.1093/jn/130.4.1026S
104. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem (2006) 98:641–53. doi:10.1111/j.1471-4159.2006.03913.x
105. Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab (2011) 14:724–38. doi:10.1016/j.cmet.2011.08.016
106. Hertz L. The glutamate-glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol (2013) 4:59. doi:10.3389/fendo.2013.00059
107. Wang R, Green DR. Metabolic reprogramming and metabolic dependency in T cells. Immunol Rev (2012) 249:14–26. doi:10.1111/j.1600-065X.2012.01155.x
Keywords: reactive oxygen species, oxidative stress, metabolism, T cell, antioxidant
Citation: Rashida Gnanaprakasam JN, Wu R and Wang R (2018) Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front. Immunol. 9:1075. doi: 10.3389/fimmu.2018.01075
Received: 16 December 2017; Accepted: 30 April 2018;
Published: 16 May 2018
Edited by:
Heitor Affonso Paula Neto, Universidade Federal do Rio de Janeiro, BrazilReviewed by:
Martin Rottenberg, Karolinska Institutet (KI), SwedenPedro Barcellos-de-Souza, Instituto Nacional de Câncer (INCA), Brazil
Copyright: © 2018 Rashida Gnanaprakasam, Wu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruoning Wang, ruoning.wang@nationwidechildrens.org