 Nicole B. Crux1,2
Nicole B. Crux1,2 Shokrollah Elahi1,2*
Shokrollah Elahi1,2*
- 1Faculty of Medicine and Dentistry, Department of Dentistry, University of Alberta, Edmonton, AB, Canada
- 2Faculty of Medicine and Dentistry, Department of Medical Microbiology and Immunology, University of Alberta, Edmonton, AB, Canada
The genetic factors associated with susceptibility or resistance to viral infections are likely to involve a sophisticated array of immune response. These genetic elements may modulate other biological factors that account for significant influence on the gene expression and/or protein function in the host. Among them, the role of the major histocompatibility complex in viral pathogenesis in particular human immunodeficiency virus (HIV) and hepatitis C virus (HCV), is very well documented. We, recently, added a novel insight into the field by identifying the molecular mechanism associated with the protective role of human leukocyte antigen (HLA)-B27/B57 CD8+ T cells in the context of HIV-1 infection and why these alleles act as a double-edged sword protecting against viral infections but predisposing the host to autoimmune diseases. The focus of this review will be reexamining the role of classical and non-classical HLA alleles, including class Ia (HLA-A, -B, -C), class Ib (HLA-E, -F, -G, -H), and class II (HLA-DR, -DQ, -DM, and -DP) in immune regulation and viral pathogenesis (e.g., HIV and HCV). To our knowledge, this is the very first review of its kind to comprehensively analyze the role of these molecules in immune regulation associated with chronic viral infections.
Introduction
Chronic viral infection imposes major selective pressure on the host’s immune system. Previously, it has been shown that susceptibility and/or disease progression in a patient with a chronic viral infection, such as hepatitis C virus (HCV) or human immunodeficiency virus (HIV), is influenced by host genetic factors (1, 2). Following their discovery in the 1970s (3), human leukocyte antigen (HLA) loci appeared to be a leading genetic candidate for infectious disease susceptibility. HLAs are classified as the major histocompatibility complexes (MHCs) because of their important role in enabling the immune system to recognize “self” versus “non-self” antigens (4). HLA in human is analogous to the MHC in other animals such as mouse. The classical HLA loci consist of class Ia (HLA-A, -B, -C), class Ib (HLA-E, -F, -G, -H), and class II (HLA-DR, -DQ, -DM, and -DP), which are involved in antigen presentation to CD8+ T cells, natural killer cells (NK cells), and CD4+ T cells, respectively (5, 6). They are encoded in a ~3,500 kb segment on human chromosome 6p21.3, which is the most variable region in the human genome (7) (Figure 1).
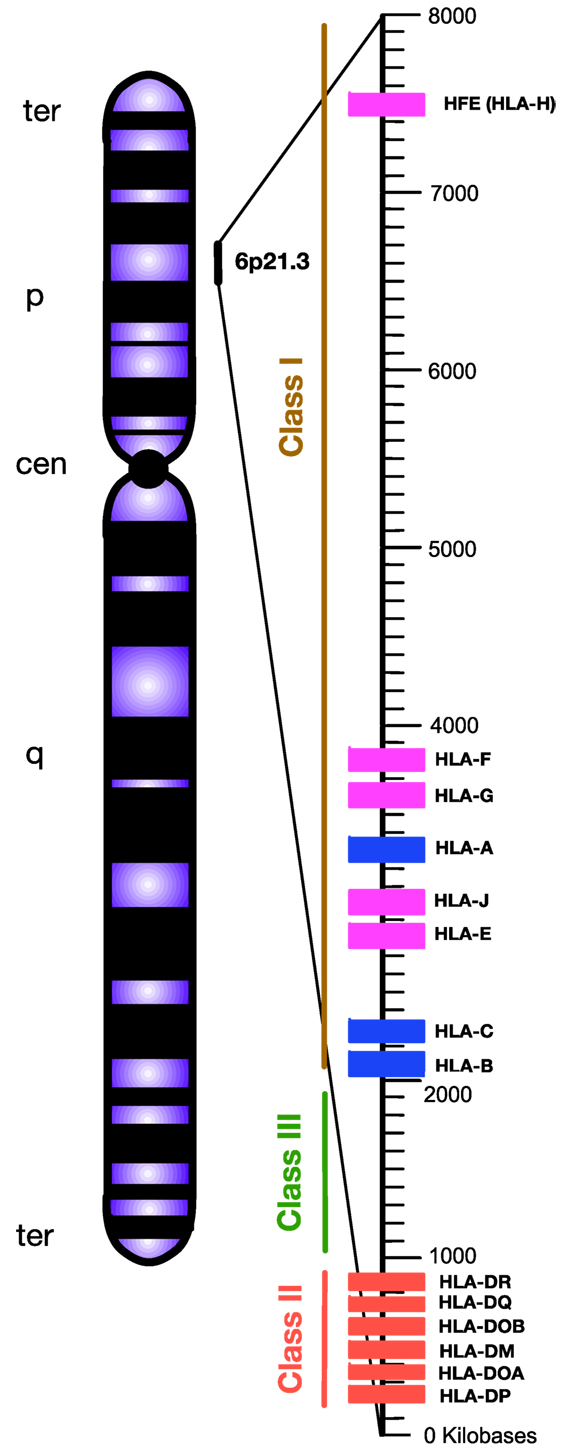
Figure 1. Simplified diagram of the position and organization of human leukocyte antigen (HLA) genes on human chromosome 6. HLA-class I encompasses “classical” HLA-Ia and “non-classical” HLA-Ib loci, which are differentiated by blue and pink, respectively. HLA-class II are encoded by various HLA-II loci labeled by red. Unlike HLA-class I and II, the HLA-class III region does not encode HLAs. Instead, this densely packed region encodes for various inflammatory molecules, complement, and heat shock proteins. Telomere, ter; p-arm, short arm; q-arm, long arm; Centromere, cen.
Human leukocyte antigens are one of the most polymorphic genes in humans, with several thousand alleles encoding for functional polypeptides (8, 9). This high level of polymorphism and heterozygosity enable the immune system with a selective advantage against the diversity of microorganisms and antigens the host encounters. However, extreme polymorphism and possible mutations in the MHC increases the chance of autoimmune diseases (10). Thus, these molecules play an important role in the regulation of host’s immune response as presenters of self-and/or foreign peptides/antigens to T cell receptors (TCRs) for initiating of tolerance and cytotoxic T cell (CTL) or helper T cell response (11) (Figure 2).
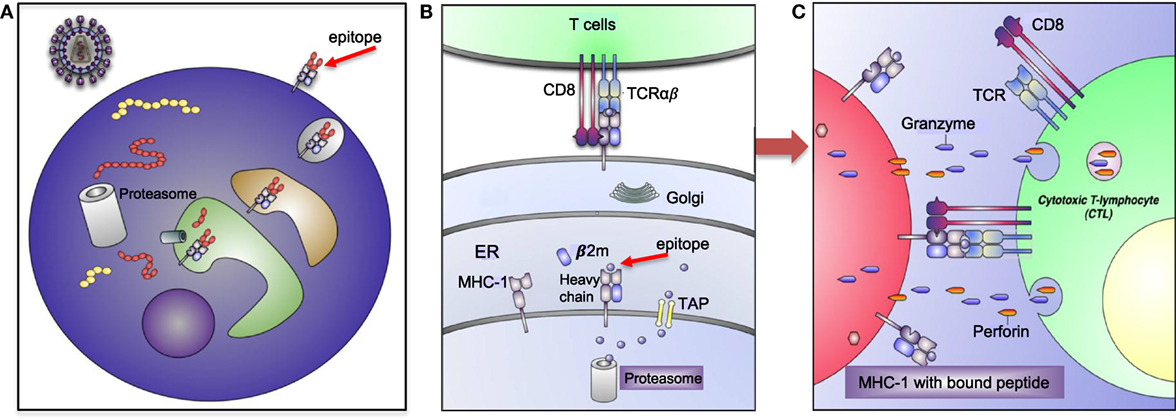
Figure 2. Human leukocyte antigen (HLA)-I antigen-processing and presentation pathway. (A) This model depicting how a pathogen (e.g., human immunodeficiency virus) is taken up and processed into small peptides (8–16aa) before binding to HLA-I molecule. (B) HLA-I is assembled in endoplasmic reticulum (ER) and consists of heavy chain and β-2 microglobulin (β2M). Proteins and antigen are degraded/processed by cytosolic and nuclear proteasomes resulting in short peptides called epitopes. Epitopes are translocated into the ER via transporter associated with antigen presentation (TAP). These peptides may require additional trimming in the ER prior to binding to HLA-I molecules. Eventually, epitope:HLA complex will be transported via the Golgi and presented on the cell surface of CD8+ T cell. (C) Recognition of the epitope:HLA-I complex via T cell receptor (TCR) activates cytotoxic T cell (CTL) that releases perforin and granzyme B (GzmB) to eliminate the infected cell.
Human NK cells mainly use a group of germ-line encoded killer cell immunoglobulin-like receptors (KIRs) to interact with HLA-class I molecules as their major ligands (12). Expression of HLA-I-binding inhibitory receptors (e.g., KIR, NKG2A/CD94, ILT2, and LIR1) results in tolerance of NK cells toward normal cells. The NK cells usually attack abnormal cells that show down regulation of surface HLA-I molecules, termed “missing self recognition” (13). However, “missing self” results in susceptibility to licensed NK cells, which lyse cells lacking HLA-I (14). In rare cases, such as certain tumor cells and viral infected cells, loss of HLA-I/MHC-I expression occurs as a mechanism of avoiding CD8+ T cells response; in these circumstances, NK cells can eliminate HLA-I/MHC-I-deficient cells (15). Therefore, HLAs present host peptides allowing for “self” recognition and evasion of autoimmunity, as well as licensing of NK cells (8). Classical HLA molecules have unique functions in cell–cell interaction and immune response, pending on their expression levels, physical structure, and peptide affinity (16). This has led to the identification of crucial role for HLA alleles in adaptive immune response alongside the innate immune response in viral control (17). Thus, certain combinations of KIR and HLA genes may contribute into the susceptibility of host to viral infections, autoimmune disorders, and even cancers.
Currently, a large body of evidence indicate a genetic association between classical HLA loci and important infectious diseases, such as HIV, hepatitis, malaria, tuberculosis, leprosy, leishmaniasis, and schistosomiasis (18–21). Although there are some inconsistencies in the interpretation of data for bacterial and parasitic infections, the role of HLA alleles in HIV infection and HCV has been widely documented. In the context of viral infections, HLAs have been associated with events, such as drug hypersensitivity, spontaneous clearance, or viral persistence in HCV, as well as better disease outcome in some HIV-infected individuals defined as elite controllers (ECs) and/or long-term non-progressors (LTNPs) (22–24). Different groups have defined LTNPs in different ways but, in our studies, we have defined them as individuals who have been infected with HIV more than 10 years, showing low detectable plasma viremia (<10,000 HIV-RNA copies/ml), CD4 count of ≥500, and remain antiviral therapy naïve (22). ECs or natural controllers have normal CD4 count, plasma viral load persistently below 50 copies/ml, and treatment naïve (25).
Specific HLA alleles and their cognate epitopes have been associated with viral clearance or susceptibility in individuals based on preadaptation in the host and escape mechanisms from HLA recognition (26). For instance, preexisting adaptation of the incoming virus to certain HLA could influence the disease outcome in patients expressing the appropriate HLA-type. In agreement, the preadaptation of the source sequence to HL A-B*08 has been associated with the reduced ability of the hosts with HLA-B*08 to control the virus (26).
Recently, we described how CTLs restricted by HLA-B*57 and HLA-B*27 alleles are responsible for superior control of HIV as being independent of epitope specificity of CD8+ T cells (22). These collective data suggest a model of natural immunity and/or immune robustness in virus-infected individuals with specific HLA-types, or conversely, certain genotypes may result in susceptibility to disease progression. For instance, HLA-B*35Px has been shown to be associated with rapid disease progression in HIV-infected individuals (27). Therefore, HLAs play a complex role in immunomodulation during chronic viral infections and interact in epitope-dependent or independent pathways to mediate immune responses. In addition to viral infections, the role of HLAs in conditions, such as autoimmune diseases, malignancies, and allogeneic responses, has also been widely investigated (28–30).
This review focuses on identifying the contribution of classical and non-classical HLA allele group in immunoregulation and appreciating the diverse impact of HLAs in HIV and HCV pathogenesis.
Specific HLA-A Allele Group Employ Diverse Mechanisms to Either Protect Against Viral Infection or Enhance Viral Persistence
During chronic viral infections, three qualities of CTLs lead to a successful antiviral response. First, if the CTL targets a conserved as opposed to a variable viral region/epitope, it will intuitively be superior in viral control (22, 31–33). Another ability that enhances the efficiency of viral clearance is polyfunctionality. For example, in LTNPs CTLs are more polyfunctional and, thus, more effective in HIV control (22, 32–34). Third the proliferative capacity, presumably proliferation of CTLs, links to their ability to kill infected targets. This is also well exemplified in LTNPs and ECs (32, 35). By contrast, targeting variable regions/epitopes, lack of polyfunctionality, and/or poor proliferative capacity can lead to an unfavorable outcome (31). Therefore, if a HLA allele is able to influence any of these characteristics, this may lead to either a protective or unfavorable immune response.
The role of HLA-A allele group in HIV-1 infection is less appreciated than HLA-B alleles, possibly due to observed differences in their CTLs function (32, 34, 35). The HIV-1 protein Nef downregulates HLA-I and, thus, prevents HLA-I molecule from presenting antigens to the cell surface and evades host immune surveillance (36). However, it appears that HLA-A cytoplasmic domains are more susceptible to HIV-1 Nef-mediated downregulation than HLA-B cytoplasmic domains (37). Thus, relative resistance to Nef-mediated downregulation by the cytoplasmic domains of HLA-B might be a contributing factor for better disease outcome in HLA-B+ individuals. This raises the question of whether higher polyfunctionality, more avidity and greater clonal expansion of HLA-B restricted alleles could be in part due to HIV Nef evasion in these individuals or other factors enabling HLA-B restricted CTLs confer better protection against HIV-1 infection. Although the role of HLA-A alleles in disease protection is debatable, some studies indicate certain HLA-A variants, such as HLA-A*03 and HLA-A*30, are associated with effective antiviral therapy. For instance, HLA-A*03 is associated with spontaneous clearance of HCV and HLA-A*25 with unfavorable treatment outcome in HCV infection (17, 38). In the context of HIV infection, HLA-A*03 has been shown to be dominant in targeting CTL epitopes in both Nef and p24 regions, but HLA-A*02 favors the amino acids in the p24 dense region (39). Therefore, this highlights the role of HLA-A types in selecting the hydrophobic and conserved amino acid positions within Nef and Gag proteins.
Other HLA-A subtypes have also been associated with disease outcome. For instance, among HIV seroconverters in Zambia, HLA-A*36:01 has been associated with high viral load. By contrast, HLA*74:01 has been associated with low viral load (40). Similarly, there has been association of HLA*74:01 with lower viral load, higher CD4+ count, and overall strong protection from HIV-1 acquisition and disease progression in a cohort in Tanzania (41). Although the mechanism of protection is unknown, there is a possibility that HLA-A*74:01 might be in strong linkage equilibrium with another protective polymorphic gene in this population. Alternatively, HLA-A*74:01 may prime the innate immune response against HIV-1 infection. Because HLA-A*74:01 shares common peptide-binding properties with HLA-A*32:01 which has been associated with lower viral load in Caucasians (42). HLA-A*32:01 serves as a ligand for KIR3DL1 and KIR3DS1 on NK cells and, thus, may act prior to the onset of the adaptive immune responses without need for priming (41). A functional supertype, including HLA-A*02:02, HLA-A*02:05, HLA-A*02:14, and HLA-A*68:02, has been reported to be strongly associated with low risk of HIV-1 transmission in female sex workers, suggesting that different mechanisms are involved in disease acquisition versus progression (41, 43). Despite polymorphic nature of HLA molecules and different classifications, HLA-I molecules can be clustered into groups, defined as supertypes which reflects some common structural features of the peptide-binding grooves of these HLA-I molecules (44). By contrast HLA*23:01 has been associated with increased risk of HIV-1 transmission, but HLA*24:02 marginally was correlated with decreased risk of HIV-1 seroconversion (43). In addition, HLA-A*23:01 has been reported to be associated with accelerated disease progression to AIDS in a cohort of white homosexual men (45) and young children (46). Although the underlying mechanism for these observations are unknown, one can reasonably argue that protective alleles may present highly conserved HIV-1 epitopes to elicit stronger protective cellular immunity. In agreement, women in this study exhibited vigorous CTL responses to highly conserved epitopes presented by HLA-A*02:02 and HLA-A*68:02 (43). Alternatively, HLA alleles that tolerate more epitope variation could be associated with protection and those that are sensitive to mutation could be linked to increased risk of disease progression (47). In our studies with a wealth of functional data, no protective role for CTLs restricted by a variety of HLA-A alleles (e.g., HLA-A*03, A*24, A*25, A*02) was implicated (22). More importantly, we discovered that CTLs restricted by HLA-A alleles, in particular HLA-A*03, express significantly higher levels of T-cell immunoglobulin and mucin domain-containing molecule 3 (Tim-3) compared with CTLs restricted by HLA-B alleles and become suppressed by regulatory T cells (Tregs) and, thus, are associated with bad disease outcome in the context of HIV-1 infection (22) (Figure 3). In addition, we found that CTLs restricted by HLA-*A02 group recognizing herpes simplex virus (HSV-2) or Epstein–Barr virus (EBV) epitopes were also susceptible to Tregs-mediated suppression (22). This indicates that suppression of CTL proliferation by Tregs is dependent on the HLA allele restriction, but independent of the epitope specificity of the suppressed cell. With regard to HCV infection, HLA-A*01 and HLA-A*02 have been shown to confer unfavorable outcomes over the course of infection with a poor response to antiviral therapy, HLA-A*24 displays a somewhat favorable response, and HLA-A*25 is indicative of a poor IFN treatment outcome (38). However, no HLA associations to our knowledge have been reported with current direct acting antiviral HCV drugs. Another associated protective factor is viral polymorphism in NS3 and NS5B proteins of HCV as reported in an Australia cohort (26). This suggests that viral epitopes in these regions are important in disease control and escape within these target epitopes could impact disease outcome. Similarly, HLA-A*09 plays a protective role against HCV and correlates with low viral load, whereas HLA-A*11 has been reported to be associated with viral persistence (48).
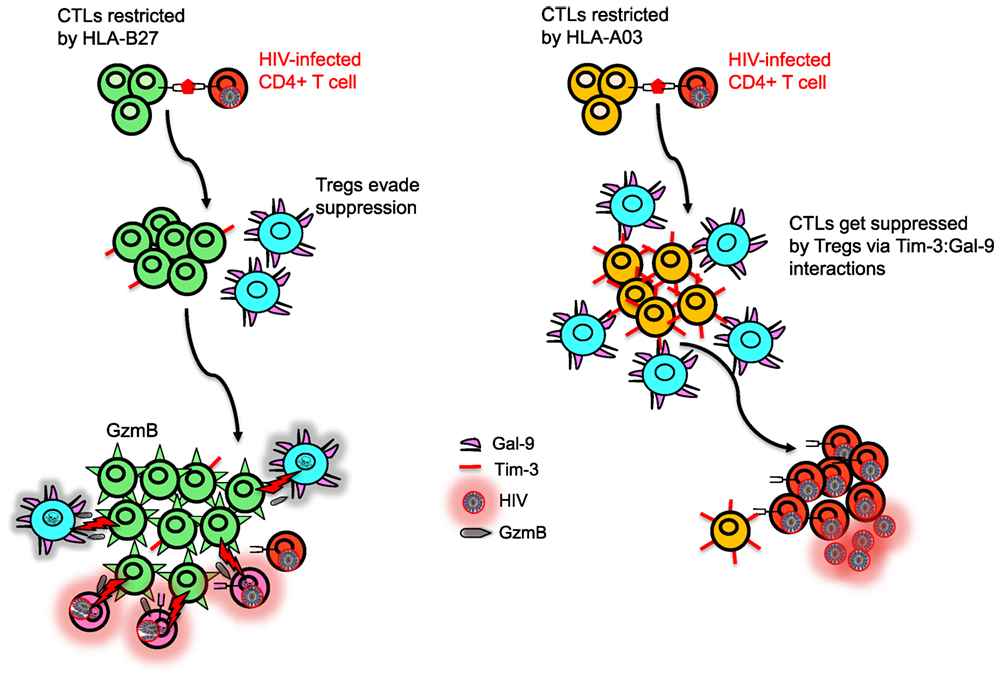
Figure 3. Model depicting differential regulation by CD8+ T cells restricted with different human leukocyte antigen (HLA) alleles. HLA-B27 and/or B57-restricted CD8+ T cell do not upregulate T-cell immunoglobulin and mucin domain-containing molecule 3 (Tim-3) upon recognition of their cognate epitopes on human immunodeficiency virus (HIV)-infected CD4+ T cells, whereas HIV-specific non-B27/B57 such as A03-restricted CD8+ T cell upregulate high levels of Tim-3. Regulatory T cells (Tregs) express Galectin 9 (Gal-9) and suppress and kill A03-restricted CD8+ T cells due to their high expression of Tim-3 but cannot suppress proliferation of B27-restricted CD8+ T cells. Polyfunctional B27-restricted CD8+ T cells upregulate high levels of granzyme B (GzmB) and kill not only infected CD4+ T cells but also Tregs that they encounter. Therefore, B27-restricted CD8+ T cells are capable of keeping HIV replication in control during chronic infection, whereas A03-restricted CD8+ T cells lack this ability.
How Do HLA-B Supertypes Confer Protection Against HIV/HCV Infections?
A significant body of functional and genetic evidence has implicated CTL response as the major determinant of disease outcome following presentation of HIV-1 peptides by HLA-class I molecules (49). These immunogenetic effects have been found through large association studies, with the HLA-B alleles; HLA-B*08, HLA-B*35, HLA-B*53, HLA-B*55, and HLA-B*56 being correlated with worse disease outcome (27, 50, 51), while HLA-B*27 and HLA-B*57 are enriched in HIV controllers (32, 52, 53). The first HIV genome-wide association study (GWAS) demonstrated a major role for HLA-B*57 in maintenance of CD4+ T cells and reduction of viral load (54). Overwhelming evidence in support of this has been reported by different groups (22, 32, 45, 53). Although the protective role of HLA-B*57 has been observed mainly in HLA-targeted studies and in GWAS, it should be noted that the majority of HIV-1-infected patients having HLA-B*57 eventually progress to disease at a rate slower to that of those without HLA-B*57 (55). Although the mechanism(s) of disease progression in these individuals is not fully understood but our longitudinal analyses in HLA-B*57 allele group confirmed that when clinical progression to disease occurs, HLA-B57-restricted HIV-specific CTLs lose their ability to evade Tregs (22). However, it is impossible with these observations to determine whether disease progression in B57+ individuals is a cause or effect of loss of ability to evade Tregs. No single genetic variant yet identified uniformly confers elite control of HIV-1, and it is likely that additive or synergistic effects of multiple genetic variants are required for HIV-1 control, as seems to be the case for HLA-B*57. In agreement, another GWAS implied that diversity at the HLA-B gene locus is the primary host genetic influence and identified key amino acid positions in the peptide-binding groove of HLA-B which were strongly associated with HIV-1 control (56, 57). Furthermore, it has been reported that the magnitude of the cross-reactive CTL repertoire negatively correlates with viral load as individuals with a more cross-reactive CTL repertoire control viral loads better during the acute phase of the infection (57). Thus, enhanced cross-reactivity of the T cell repertoire restricted by HLA-B*57 that binds to a few self-peptides leads to greater immune pressure on the infecting and emergent HIV-1 mutants compared to those with T cells restricted by HLA molecules that bind more types of self-peptides (57). This should also confer protection against other fast-mutating viruses such as HCV (58). By contrast, HLA alleles that have greater binding diversity to self-peptides (e.g., HLA-B*08) or in other words have limited cross-reactive response to immunodominant epitopes are associated with faster disease progression in HIV (59) and HCV (60). Therefore, the correlation between the diversity of peptides presented in the thymus during T cell development may impact disease control or progression. In agreement, T cells restricted by HLA-B*57 that bind to few self-peptides are subject to less rigorous negative selection in the thymus and should be more susceptible to recognizing self-peptides (57). In fact, HLA-B*57 has been associated with autoimmune psoriasis and hypersensitivity reactions (61, 62). Similarly, enhanced cross-reactivity of HLA-B27-restricted CTLs and their unique features (e.g., misfolding, homodimers) may contribute to the enhanced risk of autoimmunity associated [e.g., ankylosing spondylitis (AS)] with this allele (63, 64). As the HLA-B*27 allele group is responsible for about 50% of all genetic susceptibility to AS, other genes may also contribute in the pathogenesis of AS (65).
The immune system has evolved to reduce excessive cytotoxicity by upregulation of inhibitory receptors on activated immune cells (66). For instance, Tim-3 is an inhibitory receptor present on a wide range of immune cells (67) and interacts with Galectin 9 (Gal-9) that is constitutively expressed on Tregs to inhibit CTL response (22). We have shown that CTLs restricted by HLA-B27 and HLA-B57 are able to evade Treg-mediated suppression during chronic infection by killing Tregs they encounter in a granzyme B (GzmB)-dependent manner (22). We have shown that most HIV-specific CTLs restricted by non-B27/B57 HLA alleles upregulate Tim-3 when encounter their cognate epitopes and are, therefore, susceptible to Treg-mediated suppression via Tim-3:Gal-9 interactions. By contrast, CTLs restricted by HLA-B27 and HLA-B*57 highly upregulate GzmB (33) but do not express significant levels of Tim-3 upon epitope recognition and, therefore, they are not suppressed by Tregs via Tim-3:Gal-9 interactions (22) (Figure 3). Interestingly, we have shown that even healthy and HIV negative individuals with HLA-B27/B57+ have lower Tregs frequency (22). As such, evasion of Tregs suppression by HIV-specific CTLs restricted by HLA-B*27 or HLA-B*57 may account for delayed disease progression in individuals restricted by these allele groups (22). Furthermore, we found that resistance to Treg-mediated suppression of HLA-B27 and B57-restricted CTL is irrespective of the epitopes recognized by the T cells, suggesting that resistance to Treg-mediated peripheral tolerance is related to the allele restriction rather than the epitope specificity. In agreement, HSV-2 or EBV B27-restricted CD8+ CTL evade Treg-mediated suppression, whereas HSV-2- and EBV-specific CD8+ CTL restricted by other alleles were susceptible (22). This phenomenon acts as a double-edged sword, as evading Tregs-mediated suppression could be beneficial for the host in chronic viral infections but may mediate autoimmune conditions (68, 69). The mechanisms responsible for reduced upregulation of Tim-3 and increased levels of GzmB upon recognition of their cognate epitopes by B27 and B57-restricted CTLs compared with other HIV-specific CTLs upon recognition of their cognate epitopes are unknown. Understanding the TCR signaling by CTLs restricted by different HLA alleles will be important to understand the mechanism for potential therapeutic interventions that “switch on” virus-specific CTLs that are restricted by other HLA alleles. Similar studies need to be conducted in cancer patients to determine whether individuals with HLA-B*27 and HLA-B*57 alleles are more protective than the others. Thus, our finding may in part explain why the HLA-B*27 and HLA-B*57 allele groups are also associated with resolution of other chronic infections (e.g., HCV) and why both HLA-B*27 and HLA-B*57 allele groups are associated with autoimmunity.
In addition to being protective in HIV-infected LTNPs (53, 70), these protective alleles are associated with spontaneous clearance of HCV infection (71). Studies have shown that HLA-B*57 provides the greatest protection against HIV progression, and HLA-B*57:01 and HLAB*57:03 are the most common HLA-B*57 allele group in Caucasian and African populations, respectively (72). These HLA alleles seem equally protective and the CTL dominant response in HIV infection is to be the Gag epitope KF11 (KAFSPEVIPMF) (72). In addition, HIV restriction factors, such as AOPBEC3C, APOBECC3D, TRIM26, TRIM32, and CTR9, are elevated in elite controls with these protective HLA alleles (73). Increased expression of antiviral genes, including APOBEC3A, APOBEC3B, ISG15, and BST-2/tetherin are also reported in HLA-B*57 positive healthy individuals (73), suggesting another mechanism of protective immunity against HIV-1 that falls outside of classical HLA-mediated functions.
Despite some similar functionality, HLA-*B57 and HLA-B*27 alleles are completely different. HLA-B*27 has a tendency to misfold that was discovered serendipitously during mutagenesis studies (74) and associated with autoimmune diseases, such as AS, psoriasis, reactive arthritis, and inflammatory bowel disease (75). This allele has three unusual features, distinct from that of a typical molecule of HLA-B allele; altered peptide repertoire, tendency to misfold, and often forms heavy chain homodimers during cell surface recycling (76). More mechanistic studies are required to better understand the intrinsic biological consequences of misfolding, the recognition of the abnormal heavy chains by immune effectors, as well as the peptide presentation of an altered repertoire to CTLs.
As we discussed above, CTLs play a major role in the protective effects of HLA-B*57 and HLA-B*27 in early HIV-1 infection and CTLs restricted by these alleles better define disease progression than HLA-genotype alone (77). However, HIV-1 may escape from CTL responses through selection of mutations that inhibit CTL-mediated killing (78). In HLA-B*57, the major escape of HIV-1 occurs from the immunodominant TW10 (TSTLQEQIGW) epitope through the T242N CTL gene mutation which has a negative effect on viral replication (78, 79). Similarly, the importance of CTLs in patients carrying the HLA-B*27 allele is underscored by the observation that viral escape from CTL epitope targeting KK10 (KRWIILGLNK) is associated with increased viral load and disease progression (80–82). In the context of HCV and HIV, it has been shown that HLA-B*27 presents single immunodominant epitopes, which often results in spontaneous clearance of HCV and slow progression of HIV (45, 60, 83). Recently, it has been shown that the protective HLA-B*27 allele specifically presents CTL epitopes from the NS5B, which harbors the most conserved region of the virus (84). Similarly, HLA-B*27 targets NS5B2841–2849 during HCV infections and a conserved p24 Gag epitope in the context of HIV (85, 86). During both infections, escape from the targeted immunodominant epitope is very difficult. Thus, it is thought that the time spent by the virus trying to escape may give the immune response an opportunity to clear it, resulting in a protective phenotype. For HCV escape, two mutations must occur. During the early phase of infection, a single mutation may be selected, but this is not adequate for viral escape. Later in infection, a compensatory mutation outside of the epitope occurs and there is a second mutation at the HLA-B*27 binding anchor of the epitope (82, 87). This epitope is present in the viral RNA-dependent RNA polymerase. The ability to mutate at the main HLA-B*27-binding anchor is dependent on viral fitness (86). Misfolded HLA-B*27 may also be unable to engage KIR3DL1, resulting in increased NK cell activation (88, 89). It has been recently shown that HLA-B*27 only is protective in individuals infected with HCV 1 genotype. This is due to the virus containing the conserved NS5B2841–2849 epitope. This explains why HLA-B*27 only appears to be protective in certain individuals, as HCV genotypes other than genotype 1 lack this epitope (90). In addition, HLA-B*27 exhibits a strong “HLA footprint” in a novel HCV NS3 epitope (TVYHGAGTK), which is predominantly targeted in women (23), highlighting sex-specific differences involving the mechanism of viral control and clearance.
As previously mentioned, HLA variants can also cause unfavorable outcomes in chronic infection. A more recent study has suggested that HLA-B*15:02 may play a protective role against HCV but B*15:01 acts as a risk allele. Similar association was reported between the HLA-B*07:01 and HLA-B*07:05 alleles in this Chinese cohort (91). It appears that the differences in the amino acids encoded by these polymorphic alleles may impact the HLA properties at antigen binding sites and this could contribute to the divergent immune response to the virus and subsequently different disease outcomes.
Human leukocyte antigen-B*35 is usually found in haplotypic association with HLA-C*04 and is the most HLA allele associated with rapid HIV disease progression. Two variants of HLA-B*35 have been identified: HLA-B*35-Py (HLA-B*35:01, HLA-B*35:08) and HLA-B*35-Px (HLA-B*35:02, HLA-B*35:03, HLA-B*35:04, HLA-B*53:01). HLA-B*35-Py binds peptides with a proline (P) anchor residue at position 2 of the peptide-binding region and tyrosine (Y) at position 7 (92). HLA-B*35-Px also bind P at position 2, however, they have much more flexibility in peptide binding at position 9. In addition, both can bind non-conventional peptides (Los Alamos database, http://www.hiv.lanl.gov/). Individuals expressing HLA-B*35-Px, but not HLA-B*35-Py subtype, progress rapidly to AIDS (27). The HLA-B*35-Px molecule B*3503 binds with higher affinity to the inhibitory MHC I receptor immunoglobulin-like transcript 4 (ILT4) on dendritic cells (DCs) that does HLA-B*35-Py molecule B*3501 (27). This preferential binding of B*3503 with DCs is associated with impaired DC functionality in vitro and ex vivo compared with carriers of the B*3501 allele (27). Thus, inhibitory impulses due to preferential interactions of HLA-B*3503 (Px) subtypes with ILT4 could lead to DC dysfunction and, subsequently, accelerated disease progression in these individuals. Interestingly, rapid disease progression associated with HLA-B*3503 (Px) begins during primary HIV-1 and remains noticeable during the course of infection (77). This might be due to lack of CTL response to HIV infection in HLA-B*35-Px compared to those with HLA-B*35-Py (93). Another possible explanation for accelerated HIV disease progression among individuals with B*35-Px is that these alleles do not bind HIV epitopes and fail to mediate a protective immune response in infected individuals (49). The HLA-B*53 allele is included with B*35Px because of its close phylogenic connection with HLA-B35. HLA-B*5301 is more prevalent than HLA-B*35 in African-Americans and associated with rapid disease progression in this population (92, 94). A more recent study has reconfirmed the role of HLA-B35 allele in HIV disease progression and also indicated that individuals with HLA-B*51 allele exhibit rapid CD4 T cells decline and are more prone to opportunistic infections associated with HIV progression (95).
Abacavir Hypersensitivity and Long-Term Non-Progression in Individuals with HLA-B*57 Serve as a Paradigm for the Epitope-Independent and Epitope-Dependent Model of Immune Regulation
Although pathogens have exerted selective pressure on HLAs to diversify as a mechanism of immune adaptation, this has resulted in the high potential for drug hypersensitivity reactions (96). These reactions seem to be due to HLA-allotype-specific interactions, stimulating T cells to respond to drug-exposed tissues (97). Abacavir is a widely used reverse transcriptase nucleoside analog widely used to treat HIV and HCV infections. In a fraction of HLA-B*57:01 individuals, abacavir hypersensitivity syndrome (AHS) can occur (98, 99), resulting in systemic disease (62). Asp114Asn and Ser116116Tyr polymorphisms in the HLA-B*57:01 binding region, as well as abacavir-responsive T cells, are sufficient to alter peptide recognition, and (100) shifting of peptide binding to the unique C-F binding site and recognition of mostly smaller, aliphatic residues (96, 101). This shift in peptide repertoire results in a diverse naïve and memory T-cell response (98, 102). Fully understanding the mechanism of AHS might allow for alteration of abacavir’s chemical structure so that non-immunogenic analogs may be generated with retained antiviral potency (103). This molecular model of AHS suggests that HLA-peptide recognition is highly epitope-independent and that differences in the peptide-binding site result in an altered peptide repertoire. In turn, this causes downstream effects different from that of regular HLAs.
Interaction of Diverse Host KIRs with HLA-C Variants is a Determinant of Viral Disease Progression
Human leukocyte antigen-A, HLA-B, and HLA-C are highly polymorphic and are expressed on the surface of nearly all nucleated cells (26, 104). Engagement of HLA-I is critical for CD8+ T cell activation, as well as NK cell licensing, but is not necessary for NK cell activation (105).
Human leukocyte antigen-C has homology with HLA-A and HLA-B, with its α-helix being particularly similar to HLA-B, but it differs on several levels: (1) HLA-C has a highly conserved α1 domain containing the KYRV motif of residues 66, 67, 69, and 76, which is only present in HLA-B*46, and a conserved glycine (Gly) at amino acid 45 (106), (2) four conserved regions in the α2 domain, (3) reduced diversity in its binding region, correlated with a restricted set of self-peptides, unlike HLA-A and HLA-B (107, 108), and (4) HLA-C is expressed at only about 10% on the surface of cells, which is 15–35% lower than its counterparts (109). All HLA-C alleles are divided into two groups, HLA-C1 and C2, depending on the amino acid presented at position 80 of the molecule (110). Due to reduced diversity in the peptide-binding region, it seems HLA-C is more specific for NK cell regulation; whereas HLA-A and HLA–B seem to be more specific for CTL regulation (111).
Human leukocyte antigen-B and HLA-C are in direct linkage disequilibrium and, therefore, it has been suggested that HLA-C may not directly play a role in disease progression itself but may only influence HLA-B expression (112). However, other studies have demonstrated that incorporation of HLA-C into HIV-1 envelope enhances viral infectivity and reduces susceptibility to neutralizing antibodies (113, 114). This effect appears to be linked with changes in viral envelope proteins conformation such as GP120 as this association between HLA-C molecules and viral envelope enhances the infectivity of both HIV R5 and X4 isolates (114). A most recent study has mechanistically confirmed the role of HLA-C in enhanced HIV infectivity (115). This study suggests that although HIV-1 infection does not upregulate HLA-C expression, HIV-1 envelope may facilitate dissociation of β2M from HLA-C, leading to higher levels of HLA-C free chain molecules at the cell surface influencing HIV-1 infectivity (115). In agreement, another study has demonstrated an important role for HLA-C in HIV pathogenesis. In this study, higher surface expression levels of HLA-C has been associated with reduced viral load and maintained CD4+ T cells count in European Americans and African cohorts (116). In particular, a single nucleotide polymorphism (SNP) rs9264942, which is 35kb upstream from HLA-C and is associated with expression of HLA-C mRNA, suggested that HLA-C contributes to viral load set point in HIV-1 infection (117). However, this may not be the direct effect of HLA-C or SNP-rs9264942 on HIV-1 control but polymorphic miRNA-148a may be responsible for differential HLA-C expression (118). This −35 SNP shows a functional effect (e.g., insertion or deletion) in the 3′ untranslated region (UTR) of HLA-C that directly impacts expression of different HLA-C allotypes through miRNA-148a recognition (119). Thus, disease-associated haplotypes might enforce their action via multiple mechanisms such as the characteristics of peptides they interact with and their expression level. Despite a recent study has NK cells play a crucial role in early defense against viruses and malignancies, as they are able to respond rapidly to pathogens without prior sensitization (120, 121). Although CTLs are the main players in viral control, even a strong CTL response fails during chronic HIV infection (e.g., mutation) and impaired CTL response by HCV leads to a persistent infection (122). In HIV-infected patients with a poor CTL response, NK cells play a significant role in viral control (123). NK cells are able to shape the immune response through production of cytokines and chemokines, as well as through direct cell-to-cell interactions with their stimulatory/inhibitory receptors (124–127). NK cells such as T cells need to discriminate self from altered-self. This education, termed “licensing, arming or tuning” results in maturation of NK cell functional repertoire and mediated via the engagement of the MHC-I inhibitory receptors (105). NK cell becomes more responsive when licensed and requires a regulatory mechanism, thus preventing excessive cytotoxicity. The “rheostat model” suggests that cytotoxicity of NK cell, which is acquired through licensing, is dependent on the HLA/receptor combinations and the strength of inhibitory signaling always overrides activating signals (128, 129).
It has been widely reported that HIV Nef downregulates HLA-A and HLA-B (130). However, unlike previously thought, HIV-Vpu downregulates HLA-C (131, 132). Primary HIV-1 clones vary in their ability to downregulate HLA-C, possibly due to whether the hosts CTL or NK cell dominate immune pressure. Therefore, in the context of HIV-1 infection NK cells may not be able to maintain licensure. This could result in a high population of unlicensed NK cells, which are normally superior at undergoing antibody-dependent cytotoxicity (ADCC) to kill virally infected cells (133). However, Vpu antagonizes type I IFN-inducible factor tetherin that enhances the presentation of viral antigens on the surface of infected CD4+ T cells, thereby disabling NK-cell-mediated killing via ADCC (134–136). In support of this, HLA-C on HIV-infected cells diminishes response by NK cells, but this suppressed responsiveness of NK cells depends on host’s HLA-C genotype but independent of viral strain (137). Interestingly, in rhesus macaques infected with SIV lacking functional Nef, viremia was controlled. However, over time SIV become virulent due to mutation and regained Nef function (138). This suggests that Nef is crucial for virulence possibly due to its ability to downregulate HLA-class I.
Epidemiological and functional studies demonstrated that NK cells play a crucial role in HIV control via recognition of infected cells by both activating or inhibitory KIRs (139–141). The expression levels of HLA and KIR molecules on NK cells and also their strength of interactions have crucial impact on NK function (141). These KIR genes are located on chromosome 19q13.4 in the leukocyte receptor complex, expressed on NK cells, some T cells, and interact with HLA-class I molecules (142, 143). They have been widely implicated to influence viral disease progression due to their heritability and high polymorphism (144–146), along with their frequent and diverse interactions with HLAs (147, 148). Cross talk between self-specific inhibitory KIRs and their cognate HLA-I ligands is essential for “licensing” as absence of KIRs for self HLA make NK cells “unlicensed” and hyporesponsive (149). These observations indicate that NK cell ability to secrete cytokine and cytolysis is dependent on MHC-I molecule. Thus, KIRs play an important role in HCV and HIV pathogenesis, as different combinations of KIRs have been widely shown to influence different disease outcomes (150, 151). KIRs can be stimulatory or inhibitory and the presented peptide can further modulate the outcome (152). The nomenclature of KIRs is based on the presence of either two or three C-type-immunoglobulin domains (2D or 3D), as well as a long or short tail (L or S). Long-tailed KIRs possess immunoreceptor tyrosine-based inhibitory motifs, which recruit tyrosine phosphates that are critical to inhibitory activity. Conversely, short-tailed KIRs associate with the transmembrane signaling protein DAP12 that causes NK cell activation. The single exception to this short/long-tailed rule is KIR2DL4, which is expressed on CD56bright NK cells and induces cytokine production rather than cytotoxicity (15). Although HLA-C more extensively interacts with KIRs, some subsets of HLA-A and HLA-B present the HLA-Bw4 epitope, which also interacts with certain KIRs, suggesting that NK cells may principally be regulated by HLA-C (153). For example, HLA-C allotypes recognize KIR2DL2 and KIR2DL3 defined by Asn80 (HLA-C1); KIR2DL1, and to a lesser degree some KIR2DL2/3 allotypes, recognize HLA-C allotypes defined by Lys80 (HLA-C2) (154). Recently, it has been reported that the avidity of the ligand and receptor (the strength of the interaction) as well as receptor density are functionally important during HIV infection (155). In addition, KIR–KIR ligand combinations impact NK cell education and functional responsiveness (156, 157). Most recent studies have demonstrated a differentiating role for HLA-C in ECs versus LTNPs. These studies have shown that KIR2DS receptors and their HLA-C2 ligands escape miRNA-148a regulation in ECs but not LTNPs (158). Therefore, escaping miRNA-148a regulation in ECs supports a dominant role for NK cell-mediated HIV-1 control.
Viremic HIV-infected individuals display reduced expression of NK cell activating receptors (3, 159–161) but increased expression of NK cell inhibitory receptors (159, 162). As blockade of interaction between HLAs (e.g., HLA-C and HLA-E) and their corresponding inhibitory receptors enhances killing of HIV-infected cells with viral strains that reduce MHC-I expression (163). This indicates that the killing ability of NK cells depends on the capacity of HIV isolate to alter MHC-I expression. KIRs or combinations of KIR and HLA-I have different effects on infectious diseases (e.g., HIV and HCV) outcome. For instance, KIR2DL5 facilitates HIV-1 infection in both sexes but KIR2DL2 only in females, whereas KIR2DS1 facilitates the disease course in intravenous drug users only. By contrast, KIR3DS1 is protective in intravenous drug users and KIR2DL3 in all individuals (164). As such, KIR3DS1 and 2DS1 are associated with resistance to HIV infection, while 2DS1, 2DL5, and 2DL2 genes are correlated with enhanced HIV acquisition. Despite the role of KIRs in HIV-1 disease progression, their role in disease acquisition has not been fully supported (165). However, KIR2DS4*001, the only allele of KIR2DS4, has been associated with higher viral load and accelerated HIV-1 transmission in a heterosexual cohort in Zambia (166). Different mechanisms might be associated with differential roles for KIRs in viral infections. For example, KIR2DL2 binds virus-derived peptides on HLA-C resulting in reduced NKC function and a selective advantage for the virus to escape the protective role of this KIR (139, 167). KIR2DL3 binds HLA-C with lower affinity resulting in weaker NK cell inhibition and, subsequently, a protective role for KIR2DL3:HLA-C (168), which explains how subtle differences result in the differing effects of these KIRs (169). KIR2DL2 promotes HLA-I restricted CTL adaptive immunity as expression of KIRs on both NK cells and CTLs have been associated with shaping the adaptive immune system (170). In agreement, KIR2DL2 expressing CTLs survive longer and, therefore, exhibit prolonged immune protection, as KIR2DL2 homozygotes with HLA-B*57 are almost 5 times more likely to clear HCV infection (171). Likewise, CTLs restricted by HLA alleles associated with bad disease outcome survive longer once they carry KIR2DL2 and, therefore, can be detrimental in chronic viral infection (171). A recent study suggests an alternative mechanism for the observed effects of KIR2DL2 in chronic viral infection (172). In HCV infection, KIR2DL2 enhances the detrimental effects of HLA-C*08 and increases the protective effects of HLA-B*54 and HLA-B*57. As the copy number of KIR2DL2 increases, so does the enhancing effects (171). Interestingly, cells with iKIRs have been shown to survive for a longer period in the presence of chronic stimulation (173). Thus, these NK cells with alleles either promoting or protecting against disease progression will survive for a longer time and their effects will be enhanced (172).
Interestingly, in the majority of HIV-infected individuals, there is an increased frequency in HLA-C-specific inhibitory natural killer receptors (iNKRs) in viremic subjects versus aviremic subjects and this upregulation in iNKRs is associated with impaired NK cell cytolytic function (160). In addition, the synergistic effect of KIR3DS1 with HLA-Bw4 to delay HIV progression shows a model of genetic epistasis (174). By contrast, KIR3DS1 alone in the absence of HLA-Bw4 was liked to accelerated HIV disease progression (174). Thus, this lack of protective effects for KIR3DS1 (in the absence of HLA-B Bw4-80Ile) could be associated with other genes in linkage disequilibrium with KIR3DS1. Taken together, the pieces of evidence from different viral infections consistently demonstrate a role for NK cells in viral control. However, viruses can evade NK cell-mediated immune pressure by selecting for variants that prevent the recognition of infected cells by KIRs (e.g., MHC-I alteration).
HLA-E Signaling Dysfunction as a Viral Strategy to Alter NK Cell Activity and Skewing of Populations
Human leukocyte antigen-E is a non-classical HLA-Ib with broad tissue distribution but least polymorphic of all the MHC-class I molecules. It is upregulated by microenvironmental stresses such as hypoxia and glucose deprivation in tumors (175) but transcribed at lower rates than HLA-Ia molecules (176). Seventeen alleles are known to date with two functional variants, HLA-E*01:01 (HLA-E107R) and HLA-E*01:03 (HLA-E107G) (177). These variants differ in a single amino acid at position 107 when an arginine (Arg) is replaced by a Gly in HLA-E*0103 (16), resulting in different thermal stabilities and lengths of interaction with cognate receptors (16). HLA-E is mainly occupied by conserved epitopes derived from leader sequences of HLA-class I molecules. The frequencies of HLA-E variants are balanced between populations, though the HLA-E*01:03 variant is usually expressed at higher levels than HLA-E*01:01 (16) This Arg–Gly mismatch may also alter either the conformation of HLA-E or its association with β2M resulting in the presentation of different peptide repertoires (178). In the absence of Tapasin (MHC assembly factor), both variants of HLA-E present drastically different peptide repertories (HLA-E*01:01 presents non-canonical peptides, while under normal physiological conditions and in the absence of Tapasin, HLA-E*01:03 binds non-americ peptides) (179). So, in a transporter associated with antigen presentation/Tapasin impaired environment (e.g., viral infection and tumor), changes in peptide repertoire especially in peptide-binding motif for HLA-E mirrors that of HLA-A*0201(179).
Human leukocyte antigen-E interacts with activating and inhibitory natural killer receptors (NKRs) to regulate NK cell activity. Endothelial cells constitutively express HLA-E and are ubiquitously present along the endothelium of the circulatory system, thus being accessible to NK cells (180). NKRs are made up of CD94, which dictates specificity of NKR interactions with peptides presented on HLA-E, and an isoform of NKG2 that dictates the affinity of the NKR (181). These interactions can either be activating responses (i.e., NKG2C/CD94) or inhibitory responses (i.e., NKG2A/CD94). However, in viral infections, the balance between inhibitory and stimulatory signals could be altered. In murine models, HLA-E also interacts with the TCR (e.g., αβTCR), directly inhibiting CTLs by Qa-1-restricted interaction (182). Thus, HLA-E has a dual role and bridges the innate and adaptive immune response (182).
During HCV infection, natural cytotoxicity receptors, such as NKp46 and NKp30, are downregulated compared to healthy controls (183). The HCV core protein YLLPRRGPRL stabilizes HLA-E expressing cells resulting in impaired NK cell-mediated cytotoxicity. In addition, NKG2A is upregulated on CD8+ T cells and NK cells in chronic HCV infection (183). Therefore, enhanced expression of NKG2A on NK cell from HCV patients is associated with decreased cytotoxicity against HLA-E expressing cells and altered NK cell-induced modulation of DC functions in chronic HCV (184). Of note, the two identified HLA-E alleles are HLA-ER and HLA-EG, but the HLA-ER/HLA-ER homozygous genotype appears to be associated with increased resistance to infection or spontaneous resolution of HCV infection, resulting in an abundance of the permissive HLA-EG in HCV patients (185). Although the exact mechanism of this association remains to be determined, HCV-induces upregulation of the inhibitory receptors NKG2A receptor on both CD8+ T cells and NK cell rendering these cells less resistant to inhibition via HLA-E/NKG2A interactions as discussed above. Moreover, HLA-ER/HLA-ER homozygous genotype is associated with low surface expression of HLA-E, therefore, HLA-E-mediated inhibition might be less efficient in carriers of a homozygous HLA-ER genotype in chronic HCV patients. Taken together, it seems that HCV core amino acids 35–44 will be recognized by CD8+ T cells in the context of non-classical HLA-E allele and HLA-E allelic variants impact the course of HCV infection (185). Treatment-induced clearance has also been associated with the HLA-E*01:01 genotype since these patients have a high frequency of antiviral HLA-E-restricted CD8+ T cells with increased IFN-γ production, which is correlated with sustained antiviral response in HIV/HCV coinfected patients (186). In this context, Zimbabwean women with the HLA-EG genetic variant (the HLA-E*01:03 allele) are four times less likely to be infected with HIV and interestingly women carrying HLA-EG homozygous and HLA-G*01:05N heterozygous genotypes exhibit 12.5-fold lower risk of HIV infection than those carrying neither genotype (187). It is possible to speculate that individuals restricted by HLA-EG have high levels of the HLA-E molecule with great affinity for viral antigens, which may increase NK cell and CD8+ T cell lysis of HIV-infected cells and subsequent protection from infection.
Recently, it was shown that HLA-E presents a highly conserved Gag-derived peptide (AISPRTLNA or AA9) that is unable to interact with NKG2A/CD94, resulting in NK cell-mediated lysis of HIV-infected cells by NKG2A/CD94 NKCs (137). Based on these reports that NKG2A/CD94+ NK cells respond more robustly to HIV-infected T-cells than those lacking this inhibitory receptor, which can be speculated that HLA-E on HIV-infected cells is not recognized by NKG2A/CD94. Consistent with this hypothesis, NKG2A/CD94+ NK cells degranulate more extensively than NK cells lacking NKG2A when exposed to K562 cells lacking surface HLA-E (137). Of note, NKG2A/CD94+ CD56bright NK cells, contrary to the popular belief, are more successful at lysing HIV-infected cells than CD56dim NK cells as they possess considerable levels of perforin, granzymes, and granulysin (137). CD56bright NK cells express the high-affinity IL-2 receptor and are, therefore, more active than CD56dim NK cells in the presence of IL-2 (137). CD56bright NKCs commonly express the FcγIIIa receptor and undergo ADCC early during infection. The observation that, overall, CD56bright NK cells are better at lysing HIV-infected cells is consistent with their predominant expression of NKG2A/CD94 and relative lack of KIR expression. By contrast, CD56dim NK cells are less likely to express NKG2A/CD94, but express higher frequencies of KIR2DL receptors and are negatively affected by HLA-C on the HIV-infected cell surface (137). During HIV infection, CD94-NKG2A expressing NK cells decrease dramatically which corresponds with the significant expansion of a distinct population of cells expressing a functional activating CD94–NKG2C receptor and HLA-C-specific inhibitory KIRs (188). This NK cell skewing does not appear to be associated with the viral load but resembles those associated with immune activation and concomitant infections (188). Early in HIV infection, the NKG2A:NKG2C ratio is very low, whereas in the later stages of disease, the ratio becomes much higher and NKG2A gets upregulated in the cytotoxic NK cell subset of HIV-1-infected patients with advanced clinical status (189). Thus, elevated levels of NKG2A might be associated with escape of HIV-infected CD4+ T cells from NK cells in late stage of disease since NKG2A-CD94 and HLA-E interactions result in an inhibitory signal. In support, high NK cell diversity results in susceptibility to HIV infection and faster disease progression as repertoire diversity may decrease potency of NK cell-mediated antiviral response (190). Of interest, NKG2C is upregulated in response to HIV infection and it is a major triggering receptor in the response of Vδ1 T-cells to HIV-infected CD4+ T cells (191). Early in HIV infection, NKG2C is protective independent of viral strain, as it allows for immediate detection and destruction of HIV via ADCC (192). Therefore, upregulation of NKG2C in HIV patients, in combination with genetic-associated NKG2C deletion results in increased susceptibility to HIV infection, higher HIV viral set point, and a faster disease progression, demonstrating a significant role for this activation receptor in HIV infection and progression (192).
HLA-F Preferentially Interacts with KIR3DS1 to Elicit an Antiviral Response
Human leukocyte antigen-F is the most enigmatic and understudied HLA with tightly regulated tissue distribution (193–195). Tissue expression of HLA-F is controversial, it mainly presents in reticulum endothelium, however, can be upregulated in most lymphocyte subsets upon activation except Tregs (196, 197). Interestingly, other studies were unable to detect HLA-F transcripts in T cell lines but instead reported expression of HLA-F protein only in B cells and tissues containing B cells, such as tonsils and fetal liver (198). It is highly conserved, with 22 reported alleles encoding for four functional variants (199), suggesting a critical role in the immune response depicted by its expression on activated T-cells (200). HLA-F is expressed on monocytes and macrophages, as well on activated CD4+ T-cells as an open conformation that does not require β2M or peptide-binding (194, 197, 200). It has been suggested that due to its co-expression with other MHCs, it may have overlapping or interdependent functions with HLA-Ia, or participate in antigen-cross presentation (201).
Recently, HLA-F was found to be a high-affinity ligand for activating NK cell receptor KIR3DS1, leading to degranulation of NK cells and production of antiviral cytokines upon interaction. It has also been liked to delayed disease progression in HIV-1 infection (196). Of note, HIV-1 infection increases the transcription of HLA-F mRNA but decreases its binding to KIR3DS1, indicative of a mechanism for evading recognition by KIR3DS1+ NK cells (196). HLA-F also interacts with the leukocyte immunoglobulin-like receptors, such as LILBR1, LILBR2, and lineage II KIRs (195, 202). Although expression of HLA-F is upregulated in healthy activated CD4+ T-cells, HLA-F mRNA is also upregulated in HIV-infected CD4+ T-cells, but the binding of KIR3DS1 is significantly reduced due to downregulation of HLA-F over time through an unknown mechanism (196). It’s possible to speculate that HIV-1 infection downregulates HLA-F expression on the surface of infected cells but this hasn’t been investigated due to lack of antibody to HLA-F. Interactions of HLA-F with KIR3DS1 inhibit HIV-1 replication in vitro; however, HIV control only occurs in HLA-Bw4+ individuals. As patients with KIR3DS1, KIR3DL1, and HLA-Bw4 progress slowly to AIDS and have reduced viremia (203, 204). This can be attributed to the synergistic effects of HLA-F:KIR3DS1 and HLA-Bw4:KIR3DL1, the latter of which delays HIV progression (140). KIR3DL1 plays a role in sustained virologic response (SVR) in HCV and HCV/HIV coinfected patients (205), suggesting that during chronic viral infection, a stronger HLA-F:KIR3DS1 interaction is beneficial. Therefore, any host polymorphisms resulting in higher HLA-F or KIR3DS1 expression or increased interaction between two molecules would be beneficial in viral control.
The Complex Role of HLA-G in Immunomodulation
Human leukocyte antigen-G belongs to the HLA-class I heavy chain paralogs. It was first characterized by its expression at the fetal–maternal interface, where it was later reported to shield the fetus from the maternal immune system (206, 207). It is distinct from other HLAs in that (1) its coding region is less polymorphic (208); (2) it can be expressed in either an insoluble membrane-bound form (HLA-G1, -G2, -G3, -G4) or a soluble form (HLA-G5, -G6, -G7) (209, 210); (3) it has a highly restricted tissue distribution (211); and (4) it exerts immunosuppressive properties on different immune cells (Figure 4) (212). Soluble HLA-G1 (sHLA-G1) is produced by the proteolytic cleavage of HLA-G1 and is dependent on metalloproteinase activity (213), which is regulated by nitric oxide and the TNF-α/NF-κB pathway (214). sHLA-G1, HLA-G1, and HLA-G5 are the most common forms of HLA-G expressed in healthy tissues (210). HLA-G expression is involved in tumor escape from immunesurveillance (215), tolerogenic responses in transplantation (216), and importantly, blood HLA-G expression increases in viral infections (217–223).
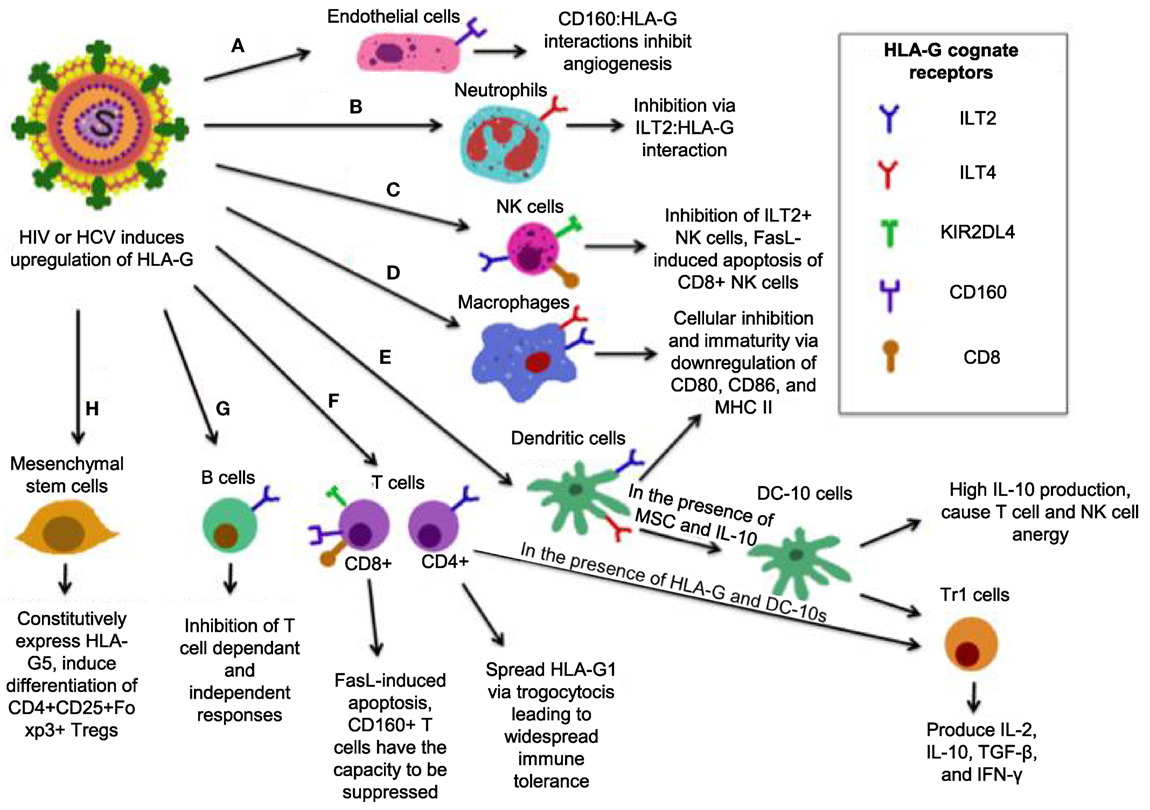
Figure 4. Simplified model of how human leukocyte antigen (HLA)-G mediates its inhibitory functions. (A) Interactions between CD160 on endothelial cells with HLA-G inhibit angiogenesis. (B) ILT2 expression on neutrophils renders them susceptible to inhibition by HLA-G, resulting in a tolerogenic phenotype. (C) The interaction between HLA-G and CD8 on some natural killer (NK) cells triggers FasL-mediated apoptosis, while ILT2:HLA-G impairs NK cell function. (D) ILT2 and immunoglobulin-like transcript 4 (ILT4) present on macrophages interacts with HLA-G, resulting a tolerogenic, immature phenotype with reduced activation markers CD80/86, and major histocompatibility complex (MHC) II. (E) In the presence of HLA-G, mesenchymal stem cells (MSC), and HLA-G, dendritic cells (DCs) that express ILT2 and ILT4 differentiate into DC-10s, which produce high IL-10 and mediate differentiation of Tr1 cells and immaturity of macrophages, and induce widespread immune tolerance. (F) HLA-G:CD8 triggers FasL-mediated apoptosis. KIR2DL4:HLA-G inhibits cytotoxicity, while CD160:HLA-G results in inhibition of proliferation. Upon induction of HLA-G1 on ILT4+ CD4+ T-cells, DC-10 are able to cause their differentiation into Tr1 regulatory cells, which secrete high IL-10 and TGF-β and kill myeloid cells in a Grzm-dependent manner. (G) B-cells express ILT2, it is likely that B-cells are susceptible to HLA-G-mediated suppression. (H) MSC constitutively express HLA-G5 and via HLA-G5 suppress other cells. MSCs also induce CD4+ CD25+ Foxp3+ regulatory T cells (Tregs) via cell-to-cell interaction.
The immunomodulatory activities of HLA-G antigens occur via direct interaction with adaptive and other immune cell inhibitory receptors: ILT2/CD85j (expressed on lymphoid cells) (224), ILT4/CD85d (expressed on monocytes, macrophages, and DCs) (225), KIR2DL4 (CD158d) (expressed on NKCs) (226), and CD160 (expressed on endothelial cells, NK cells, and T cells) (227). Though ILT2 and ILT4 may bind other ligands, HLA-G seems to be their receptor of preference than classical HLA-I molecules (228). Therefore, HLA-G through these receptors directly interacts with antigen-presenting cells (APCs), NK cells, and small populations of T cells (229, 230), and exerts its immunoregulatory functions. Of note, HLA-G may utilize its function indirectly by upregulating the expression of HLA-E, which via the inhibitory receptor CD94/NKG2A to inhibit both NK cell and T cell functions (231). Although HLA-G can modulate cytokine production by human peripheral mononuclear cells toward a Th2 phenotype, its expression could be upregulated by anti-inflammatory cytokines, such as IL-4, IL-5, and pro-inflammatory cytokines, such as IFN-γ, TNF-α, IL-1β and IL-6 (232), IL-10 (233), and growth factors TGF-β, GM-CSF, and G-CSF (221, 234). Indolemine 2,3-dioxygense (IDO) is another important molecule that plays a role in HLA-G mediated immunoregulation. It has been shown that upon tryptophan depletion, IDO induces HLA-G expression in DCs, induces Tregs, and inhibits HLA-G surface expression in monocytes (235), suggesting a cross talk between IDO and HLA-G expression in immunoregulation.
Human leukocyte antigen-G expression levels are positively correlated with the acceptance of human allograft, suggesting that HLA-G may regulate the function of both CD4+ and CD8+ T cells (236). For instance, HLA-G interaction with CD4+ T cells causes a cell cycle arrest at G1 phase, resulting in inhibition of CD4+ T cell proliferation and limiting the induction of Th1 cytokines (237). HLA-G inhibits CTL proliferation and lysis of virally infected cells by interacting with ILT2 (238, 239). The ILT4:HLA-G interaction inhibits monocyte-derived DC maturation. This leads to downregulation of MHC-II and maturation markers CD80 and CD86, and differentiation of DCs into regulatory IL-10 producing DC-10s that cause T cell and NK cell anergy and generation of Tr1 cells (240, 241). Tr1 cells produce high levels of IL-10 and TGF-β, low IL-2 and IFN-γ, and may kill APC via perforin and GzmB-dependent manner (242). HLA-G:CD8 interactions induce TCR-independent FasL upregulation and secretion (243), resulting in apoptosis of CD8+ T cells and NK CD8+ cells by Fas/sFasL interaction (244). Interestingly, mesenchymal stem cells, which express only very low MHC-I level and do not induce T cell activation are able to induce differentiation of naïve T cells into Tregs in the presence of HLA-G (245). B-cell antibody production is also inhibited by HLA-G (246), which may be due to direct effects on B-cells or indirectly by inhibition of helper T cells function (247). HLA-G:CD160 interactions were recently shown to control angiogenesis, which is consistent with vascularization that occurs during pregnancy (248). Neutrophils also have the capacity to be suppressed by HLA-G as they express ILT4 (249). CD160 is also expressed on the majority of NK cells and inhibits them upon HLA-G interaction (249). Similarly, this occurs on CD8+ T cells, which may result in CTL exhaustion (250).
Despite the coding region displaying low allelic polymorphism, the HLA-G 5′ regulatory region and 3′ UTR are highly polymorphic. Seven polymorphisms at the 3′ UTR are reported to play a role in HLA-G expression: a 14 bp insertion/deletion (INS/DEL) rs1704 polymorphism (251), and the SNPs +3003 T/C, +3010 G/C, +3035 C/T, +3142 C/G, +3187 A/G, +3196 C/G (252). The 14 bp INS/DEL polymorphism plays a role in alternative splicing, which removes 92 bp, including the region where the 14 bp region is resulting in a higher yield of stable HLA-G mRNA transcripts in vitro (253). However, in vivo this allele is associated with decreased HLA-G production, deemed the “14 bp paradox” (254, 255). The presence or absence of this 14 bp region alters the set of miRNAs that bind to this locus, thus repressing translation and negatively influencing HLA-G mRNA turnover (252). The only SNP polymorphism reported to alter miRNA binding is the +3142 C/G (256). In this polymorphic site, the G allele favors the targeting of three miRNAs (miR-148a, miR-148b, and miR-152), resulting in reduced HLA-G mRNA production. These polymorphisms exist in linkage disequilibrium (256, 257). The 14 bp (D/D) genotype has been associated with higher levels of vertical transmission of HCV independent from the other non-immunogenetic parameters (258) and HIV-1 (259). In addition, HLA-G*0105N polymorphisms are associated with augmented risk for HIV infection in heterosexual individuals (260). Therefore, maternal HLA-G alleles and/or SNPs that modulate maternal HLA-G expression at the maternal–fetal interface could influence vertical HCV and HIV-1 transmission. Although the exact mechanism of increased vertical transmission is unknown, alter HLA-G expression may influence the function of NK cells, CD4+, and CD8+ T cells at the fetal–maternal interface and, ultimately, influence the susceptibility to HIV transmission. In addition, the homozygous HLA-G −14-bp (D/D) genotype has been associated with higher viral load, lower CD4+ T cell count, and lower survival rate compared with HLA-G +14-bp (D/D) carriers (261). In patients with sickle cell anemia, HLA-G +3142 C/G increases susceptibility to HCV infection (262). In addition, HLA-G genotypes have been associated with susceptibility to HIV infection. HLA-G*01:05N increases in HIV infection, which may be due to lack of HLA-G1 and HLA-G5 expression resulting in compensation by other HLA-G isoforms, which then may inhibit NK cell activation (263). It has also been shown that the HLA-G*01:01:01 genotype is significantly enriched in HIV-resistant women, while HLA-G*01:04:04 is associated with increased rates of seroconversion (264).
Accumulating evidence suggests that during HCV infection, the inactivation of immune effector cells by HLA-G may lead to inability to clear the virus. As CTLs are crucial for restricting viral replication, yet the magnitude of CTL response in chronic HCV decreases dramatically compared with acute HCV (265–267). The tolerizing functions of HLA-G seem to be playing a significant role in viral clearance in non-responders to IFN-α2α and ribavirin therapy. Patients who are able to clear HCV infection express only low levels of HLA-G, while non-responders express drastically higher levels of HLA-G (268). Many patients with high levels of HLA-G who failed to respond to antiviral treatment also had a −1082 G/G genotype, resulting in overproduction of IL-10, higher levels of HLA-G, and inadequate viral clearance (267). In addition, high levels of HLA-G in HCV and hepatocellular carcinoma are associated with shortened overall survival and increased tumor recurrence rate (267, 269). Therefore, it is likely that HLA-G plays a role in virus-associated malignant cell transformation. In agreement, the levels of HLA-G activation positively correlate with the amount of mast cell activation and liver fibrosis (270). For instance, IFN-α upregulates HLA-G, as well as various chemokines and cytokines that are produced around the fibrotic tissue. It is possible that the interaction of HLA-G with IL-T2 on mast cells creates an autocrine loop, and the immune response against HCV-induces liver fibrosis via hepatic stellate cells (270, 271). Significant elevation of sHLA-G (sHLA-G1 and HLA-G5) in chronic HCV patients, given its immunotolerant nature, may facilitate persistence of HCV infection (267).
Differential splicing and translation of HLA-G may also alter viral disease progression by various mechanisms. For instance, HLA-G5 is reported to induce TNF-α, IFN-γ, and IL-10 but downregulates expression and function of CCR2, CXCR3, and CXCR4 in different subsets of T cells, while HLA-G1 promotes Th2 polarization of naïve T cells (272–274). There are some important basic questions in biology of HLA-G that merits further investigation. Among them is better understanding how its expression levels can be modulated in different pathological conditions. This knowledge could assist us to induce HLA-G when immune suppression is needed (e.g., pregnancy, organ transplant, and autoimmunity) or to inhibit its expression when can be potentially harmful (e.g., tumor or chronic viral infections).
Nef-Mediated Downregulation of HLA-H Expression Contributes to Disruption of Iron Homeostasis
Human leukocyte antigen-H, also known as the High Fe (HFE) gene was discovered in patients with hereditary hemochromatosis (HH) who often have mutated HFE variants (275). HFE is structurally homologous to HLA-A*2 with a wide tissue distribution (276). It is in strong linkage disequilibrium with MHC-Ia, which may be due to competition between HFE and MHC-Ia to bind intracellular components resulting in differential cellular surface expression (277, 278). HFE directly inhibits CD8+ T cells via their TCR in a peptide-independent manner and interacts with transferrin receptors (TfR) (279). Transferrin receptor 1 (TfR1) and transferrin receptor 2 (TfR2) compete for HFE binding. HFE is expressed strongly with the TfR2 on Kupffer cells, circulating monocytes and macrophages, as well as duodenal epithelial cells (280). At the basal state, HFE:TfR1 interactions inhibit iron overload by lowering the affinity of TfR1 for iron-loaded transferrin and reducing cellular iron uptake (Figure 5A) (280, 281). However, in the presence of high ironbound transferrin, HFE binds TfR2 allowing for cellular iron uptake and hepcidin production (Figure 5B). Hepcidin is a crucial iron-regulatory peptide, which is normally released in the liver in response to iron loading and inflammatory stimuli, such as TNF-α and IL-6 via the JAK/STAT3 pathway (282). Once released, hepcidin degrades ferroportin present on TfR1 molecules, preventing further cellular uptake of iron. In monocytes and intestinal cells, HFE inhibits gut iron absorption and macrophage iron efflux via a TfR1-independent mechanism, leading to intracellular accumulation of iron, low serum iron, and anemia due to chronic inflammation (283). This occurs via TfR2 signaling in which HFE binds TfR2 on the bone morphogenic protein (BMP) receptor type 1 (Alk3) inhibiting its ubiquitination and proteosomal degradation (284). This allows for BMP/SMAD signaling and hepcidin expression (Figure 5B). However, TfR1 signaling occurs through pro-inflammatory-mediated NF-κB signaling (285) and plays an essential role in lymphocyte development (286). Expression of TfR1 is downregulated in iron overload state due to coordination of mRNA-encoding proteins with iron-regulatory proteins and inhibition of TfR1 expression in T cells prevents cell cycle transition from G1 to S phase (287, 288).
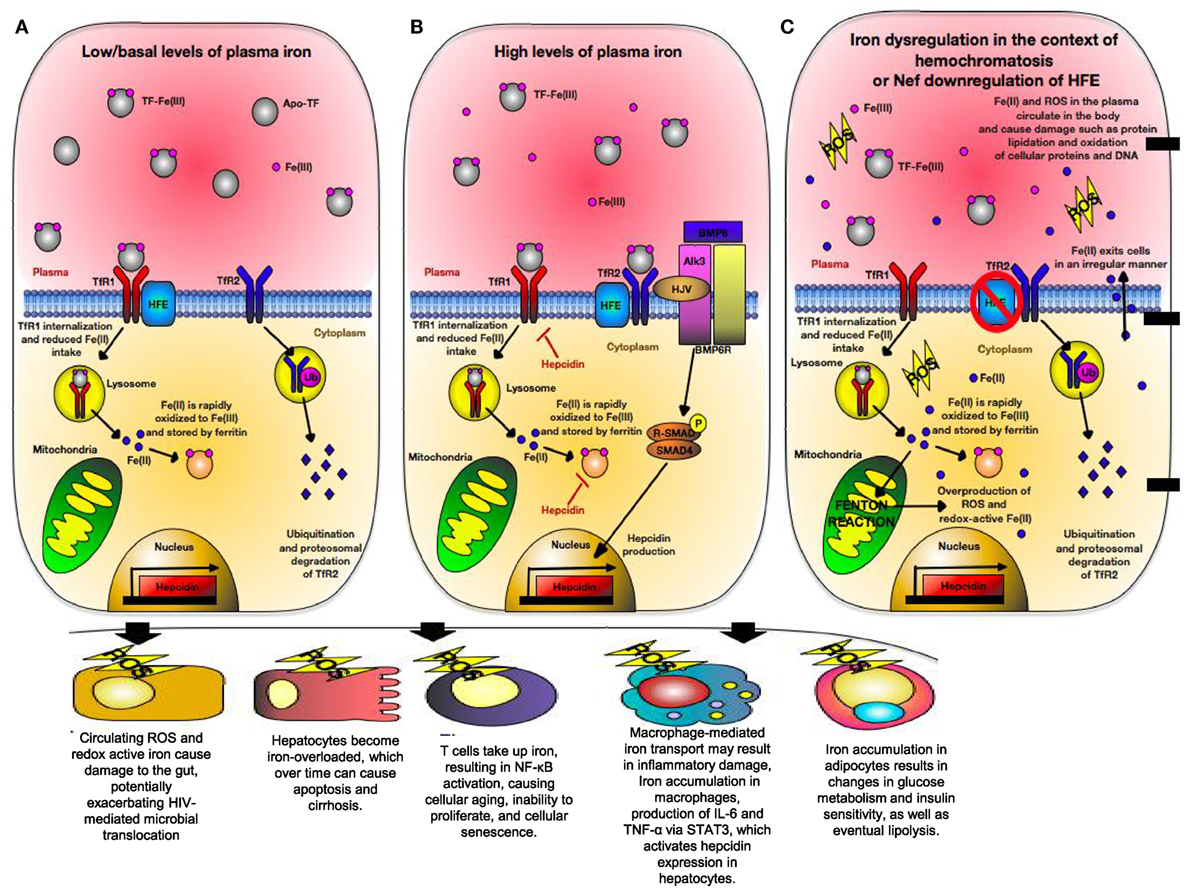
Figure 5. Iron regulation of hepatocytes during normal physiological condition versus conditions of hereditary hemochromatosis (HH) or human immunodeficiency virus (HIV) infection. (A) Under normal physiological iron concentration, transferrin-bound iron is taken up upon binding of saturated transferrin to transferrin receptor 1 (TfR1), which is endocytosed and recycled. Fe(III) (indicated in pink) is oxidized to Fe(II) (indicated in blue) in the endosome and is reduced back to Fe(III). Fe(III) then is stored intracellularly by ferritin. TfR1 and transferrin receptor 2 (TfR2) are in competition for High Fe (HFE). When not associated with HFE, TfR2 is endocytosed, ubiquitinated, and thus degraded by the proteasome. This maintains uptake of iron into the hepatocyte. (B) When too much iron is present in the plasma, HFE interacts with TfR2, preventing its ubiquitination. A multiprotein complex consisting of HFE, GPI-anchor HJV, and TfR2 form on the hepatocyte membrane. HJV is the bone morphogenic protein (BMP) co-receptor, which assists in activating hepcidin gene expression via BMP/SMAD signaling. Hepcidin then limits cellular iron intake by promoting degradation of cellular ferritin, as well as downregulating TfR1 expression. (C) In situations where HFE is absent (i.e., HIV or certain mutations in HFE), TfR2 is unable to interact with HFE and is thus degraded. In states of high physiological iron, TfR2 is unable to mediate hepcidin gene expression. Due to this, cells become iron overloaded labile cell iron (LCI) and non-transferrin-bound labile plasma iron (LPI). These forms of redox-active iron are able to move across cell membranes in a non-regular manner. LCI participates in a Fenton reaction, resulting in overproduction of reactive oxygen species (ROS) that cause lipid peroxidation, protein nitration, and DNA damage. LPI is able to enter other cells irregularly, thus causing even more production of ROS. Eventually, hepcidin production will be triggered due to STAT3 signaling caused by ROS-induced inflammation.
One of the first lines of defense against pathogens is the withholding of nutrients deemed “nutritional immunity” with the most significant form being the sequestration of iron (289). Excess iron storage increases the virulence of numerous pathogens such as HIV and inversely associated with survival of HIV-infected individuals (290). During HIV and HCV infections, patients experience iron overload that exceeds the binding capacity of transferrin, resulting in creation of non-transferrable unbound iron (NTBI) that cannot be internalized by TfR1 (291, 292). NTBI is toxic and moves into the cytosol of cells in an irregular manner. It then deposits on the cell surface leading to creation of labile cell iron, which is redox-active and generates reactive oxygen species (ROS) (286). As a result, iron overload alters the balance between ROS production and removal, causing oxidative stress that results in DNA damage, mitochondrial dysfunction, lipid peroxidation, and protein nitration (Figure 5C) (287). Iron also impacts T cells development, for example, iron overload induces a shift in CD4+ T cells to a glycolytic metabolism via upregulation of the Glut1 transmembrane protein (293). CD4+ T cells have a low spare respiratory capacity and, therefore, have insufficient mitochondrial energy to handle stress and viral infection (294). Because energy metabolism is tightly coupled to intracellular redox state, a functional link between glucose metabolism and iron metabolism exists (295). As a result, CD4+ T cells exhibit low stress tolerance and HIV intracellular protein Tat101 and Vpr also contribute to additional oxidative stress during HIV infection (296, 297). Therefore, iron level could impact the functionality of CD4+ T cells as the prime target for HIV infection.
Unfortunately, the role of HLA-H or HFE has been understudied in the context of viral infections. However, various HFE mutations as seen in HH patients suggest that HFE dysregulation contributes strongly to the particular phenotype seen in viral infections. The most common HFE mutation causing hemochromatosis is C282Y, which results in the inability of β-microglobulin to bind HFE intracellularly and, thus, is not transported to the cell membrane (284). Patients with knockout mutations in HFE have impaired and low CD8+ T cell counts (298, 299), modified CD4+ T cell polarization (300), and are susceptible to autoimmune conditions such as multiple sclerosis, further showcasing the importance of HLA-H in immune regulation (301).
Like HLA-A and HLA-B; HLA-H contains a conserved motif (342YxxA, in which x is any amino acid), allowing its expression to be downregulated by HIV Nef by 90% (302). Of note, Nef protein of HIV-1 plays an important role in HIV pathogenesis. As we discussed in the HLA-A section, Nef is a multifunctional protein and one of its roles is to downregulate surface MHC-I molecules, assisting infected cells to evade CTL response (302). Interactions of HLA-H with HIV-1 regulates cellular iron metabolism, possibly benefiting viral growth (302). In HIV infection, macrophages, microglia, endothelial cells, bone marrow, muscle, and liver are often iron loaded, and degree of iron loading inversely correlates with patients’ survival rate (303). HIV-1 replication exploits the iron-dependent host enzyme ribonuclease reductase (304), which is activated in macrophages via altered cellular redox state activating the NF-κB pathway (305). During iron overload oxidative stress highly upregulates NF-κB that acts in synergy with the HIV-1 tat-III gene to enhance viral replication in CD4+ T cells (304). Monocytes and macrophages are critical in iron homeostasis and they mediate inflammatory damage to target tissues in lipoatrophy and peripheral neuropathy in iron overload conditions (283, 306). Adipocytes also take up high levels of iron during iron overload (307), altering the expression of various pro-inflammatory cytokines and adipokines, which over time can result in lipolysis (308). This may contribute to the lipoatrophy observed in some HIV-infected individuals and may precede other metabolic complications (309–311). High-fat diets are associated with insulin resistance, modified hepatic lipid and iron metabolism, increased mitochondrial dysfunction, and oxidative stress (312) that are comorbidities associated with both HIV and HCV. Interestingly, the 187C>G HFE SNP in A5005 mice is protective from lipoatrophy (313).
In HIV infection, hepcidin levels are elevated (314, 315). However, hepcidin levels are not associated with severity of anemia, except during coinfection with Mycobacterium tuberculosis (316). Degree of surface expression of HFE influences hepatic hepcidin, as well as iron transport (313), consistent with the role of HFE in inflammation (317). In chronic HCV, hepcidin mRNA expression positively correlates with alanine aminotransferase activity and serum iron concentration. Although low expression of hepcidin has no correlation with tissue iron overload in those with chronic HCV, in univariate analysis, HCV viral load and efficacy of antiviral treatment are not significantly associated with hepcidin mRNA expression, suggesting that hepcidin expression is not due to viral load (292).
Macrophages expressing wildtype HFE once infected with HIV accumulated iron, ferritin, and increased cellular HIV-1 Gag expression. However, when this experiment was attempted with Nef deleted HIV-1 or with macrophages from HH patients expressing mutated HFE, iron was unable to accumulate (302). Besides the critical involvement of Nef and HFE in dysregulation of iron homeostasis, individuals with genetic mutation in their HFE gene lack functional HFE molecules on their cells, resulting in impaired iron uptake (Figure 5C).
Human leukocyte antigen-H impacts HCV disease severity and treatment outcome as chronic HCV patients with HFE mutations often have more transferrin saturation and advanced liver fibrosis. For instance, only 27.5% of HFE-mutated carriers achieved response at the end of treatment with pegylated-interferon and ribavirin, as compared to 60% of non-carriers (318), suggesting that individuals with HFE mutations may be more susceptible to chronic HCV due to iron accumulation. In agreement, patients heterozygous for HH infected with HCV displayed higher liver fibrosis than those who were homozygous wildtype, providing additional support that iron dysregulation is a critical component of liver fibrosis (319, 320). However, another study reported no correlation between HFE polymorphisms and HCV in Majorcan patients treated with pegylated-interferon plus ribavirin (321). By contrast, heterozygosity for H63D and/or C282Y HFE gene mutation has been shown to be associated with the lack of SVR to pegylated-IFN and ribavirin combination therapy in patients with chronic HCV (322). Taken together, the MHC-Ib protein HLA-H/HFE is strongly expressed by a variety of cells such as Kupffer cells, circulating monocytes/macrophages and intestinal crypt cells. It plays an essential role in iron homeostasis and C282Y mutation of HLA-H results in protein misfolding, which is associated with the iron overload and as we discussed impacts pathogenesis and treatment outcome of viral infections.
Diversity of HLA-Class II Genes and Their Influence on Viremia and Viral Dissemination
Major histocompatibility complex-II molecules specifically present antigen to CD4+ T cells and the genes encoding them are extremely polymorphic, especially in the peptide-binding groove (323). This renders them highly specific to the epitopes they bind. Genetic correlations between HIV-1 and HLA-II loci have not received as much attention as those observed for HLA-I, indicating more effectiveness of CTL response than humoral immunity in these studies. Although CD4+ T cells are preferentially infected by HIV-1, they provide help for the generation of high-affinity antibodies and the function of CD8+ T cells and B cells (324), HLA-II genes, specifically functional variants of HLA-DRβ, have been shown to play a fundamental role in the formation of HIV CD4+ T cell syncytia (324). However, HIV-infected individuals fail to mount an immune response to this region, possibly due to lack of recognition by the immune system (325). During HIV infection, CTLA-4, CD28, and CD3 are upregulated and assist in syncytia formation with APCs (112). Thus, we may assume that the formation of syncytia with APCs and T cells results in dissemination of HIV from infected to uninfected cells. In addition to CTLA-4, LAG-3 is another inhibitory receptor associated with immune suppression that may function in syncytia to suppress APCs and maintain their immaturity (326, 327). In a study of seroconverted HIV-1 patients, HLA DRB1*13-DQB1*06 was shown to be the only class II haplotype associated with a trend toward increased duration of AIDS-free period (328). Furthermore, inheritance of DRB1*13 alleles has been associated with long-term survival among children with vertically transmitted HIV-1 infection (46). Therefore, the properties of class II molecules, and particularly HLA DRB1*13-DQB1*06, influence HIV-1 pathogenesis and disease outcome. Interestingly, inheritance of the DRB1*13-DQB1*06 haplotype is associated with durable virus suppression and HIV-specific T helper responses (e.g., higher proliferative capacity and IFN-γ production) among early-treated patients (329). Such studies, in which HLA associations are correlated with functional assays, provide strong evidence for the crucial role of certain HLA-II alleles in disease protection. This haplotype may exert its protective effects by presenting crucial epitopes with avid class II binding in highly conserved regions of HIV-1 that may be potentially important targets for HIV-1 vaccines (330). In agreement, patients expressing DRB1*1303 allele exhibit significantly lower mean plasma viral loads and increased CD4+ T cell counts, indicating a protective role for this allele in HIV-1 infection (331). The protective role of DRB1*13 has also been shown in other infections, in particular hepatitis B virus infection, where this allele is associated with a higher rate of clearance and/or better clinical disease outcome (332, 333). In addition, studies in human papillomavirus (HPV) infection have associated DRB1*13 with protection against the HPV-16 serotype, and negative correlation of DRB1*13 with the occurrence of cervical carcinoma (334). By contrast, DRB1*13 enrichment has been reported in subjects with autoimmune diseases (335, 336). DRB1*1303 might, therefore, share some similarities with the HLA-Ia allele B*2705, which is associated with protection in HIV and HCV infection but is linked to autoimmune disorders (337). Therefore, it can be speculated that the effect of DRB*13 on disease pathogenesis may be related to particularly effective antigen presentation capacity, which confers protection against viral infections, but may trigger autoimmunity through molecular mimicry with self-antigens (338). It remains uncertain, however, whether the observed associations can be attributed to a stronger and more effective T cell response or whether other mechanisms mediate the DRB1*13 effects. Most recently it has been shown that alleles HLA-DRB1*15:02 and HLA-DRB1*03:01 are significantly associated with HIV control and progression, respectively, and the strength of these HLA-DRB1-mediated effects is independent of HLA-B*57, HLA-B*27, and HLA-B*35Px (330). The protective effects seem to be mediated primarily by the protein specificity of CD4+ T cell responses to HIV Gag and Nef proteins (330). The relevance of the genetic background with HCV vertical transmission demonstrate a protective role for HLA-DRB1*04, while HLA-DRB1*10 is reported as a risk factor (339). Interestingly, it was revealed that having a mismatch in the HLA-DRB1 locus between mother and child is a protective factor, indicating that alloreactive immune responses are involved in preventing HCV vertical transmission (339). In addition, HLA-DRB1*04:01 and DRB1*01:01 are shown to be present in patients with spontaneous clearance of HCV, while HLA-DQB1*02:01 is associated with chronic infection (17). In support of these observations, association of other genotypes, such as DRB1*11:01, DQB1*03:01, HLA-DRB1*01:01, and HLA-DQB1*03:01 alleles, with viral clearance in HCV is documented (340). This suggests that MHC-II alleles could present particular viral epitopes to CD4+ T cells, resulting in an effective immune response against the virus. However, there have been some contradicting observations regarding the associations of DRB1*01:01and DQB1*03:01 with protection against HCV. For instance, in a study the protective role for DQB1*03:01 was only observed in African-Americans but not in white Americans (341). Interestingly, another study reported a protective role for DRB1*01:01/HLA-DQB1*03:01 against HCV in white Americans but not in African-Americans (342). However, DQB1*07 is associated with viral persistence (343, 344).
How Does HLA Impact Mother-to-Child HIV Transmission and Progression?
The sharing of HLA alleles between the mother and the offspring occurs routinely in HIV vertical transmission. Mother-to-child transmission occurs in utero, intrapartum, or postpartum (breastfeeding) (345). The risk of vertical transmission is associated with the viral load in the mother as higher viral load is the indicative of a non-protective HLA allele (e.g., HLA-B*18, HLA-B*5802, and HLA-B35Px) (27, 346). Thus, HLA alleles may contribute in HIV pathogenesis in early life as defined with rapid CD4+ T cells decline, higher plasma viral load, and accelerated disease progression in two different ways (347–349). First, infected children share ≥50% of their mother HLA-type and, therefore, less likely to have protective HLA alleles compared with adults (350). So, this population carries less protective and more non-protective HLA alleles. Second, even if the child possesses a protective allele (e.g., HLA-B*27 and HLA-B*57), this allele cannot protect the child if the vertically transmitted virus has escape mutations within the conserved Gag-epitopes (351, 352). However, a study conducted in a cohort of 61-HIV-infected children in South Africa suggests that HIV-infected children may benefit from protective HLA-B alleles similar to adults, as slow disease progression was associated with the mother or child having one of the protective HLA alleles but more pronounced once they carried non-shared protective HLA-B alleles (346). In addition, they have found that mothers expressing these protective alleles more frequently transmitted escape variants within the Gag-epitopes presented by these HLA-B protective alleles compared with mothers lacking these protective alleles (346). Finally, this study reported that infected children are able to generate Gag-specific CTL response if they carry protective HLA-B alleles without preadaptation of the transmitted virus (346).
Furthermore, the role of HLA-G has also been investigated in a cohort in Zambia and presence of HLA-G 14bp insertion polymorphism has been associated with protection against HIV vertical transmission (259). These pieces of evidence indicate that HLA alleles influence HIV vertical transmission and disease progression in the newborn. Thus, determining the HLA-type of the mother and the child could help to identify children who may experience rapid disease progression.
Conclusion Remarks
Although the role of HLA alleles in HIV and HCV is well documented. However, some findings have not been confirmed or have been conducted in a very small patient cohort, therefore, these data need to be evaluated with caution. Some differences in the methodology and clinical endpoints might be accountable for some discrepancies reported for some HLA alleles by different groups. To solve these issues, there is an immediate need to conduct similar studies in much larger cohorts especially from different ethnicities, such as African, African-American, Middle-Eastern, and South East Asian populations, to reexamine these observations in these populations. Since the importance role of sex and gender in medical and biologicals studies have become more noticeable, these factors in association with HLA alleles should be taken in consideration. In addition, rigorous functional and mechanistic studies, longitudinal planning, larger sample size and power, reliable statistical analysis, better defining of clinical phenotypes, and reproducibility by different groups are all important elements that will contribute to the confidence of HLA associations with HIV and HCV infection and disease outcome. Another major issue associated with the interpretation of some data is that genes encoding the MHC-I/MHC-II molecules occur within a cluster of other MHC genes with a broad spectrum of functions linked to the innate and adaptive immunity.
Although the role of classical HLAs in HIV/HCV pathogenesis has been widely studied, the role of non-classical HLAs, such as HLA-G, HLA-F, and HFE, still remains enigmatic, despite their potential role in immune regulation and viral pathogenesis. It is critical to have a depth understanding of the role of these HLA alleles in viral infections (e.g., HIV and HCV). For instance, iron metabolism plays a complex but important role in immune regulation in the host. It appears that iron dysregulation, potentially through HFE, plays a larger role in the shift from oxidative phosphorylation to a glycolytic metabolism. It also seems to be highly involved in production of ROS leading to cell senescence with speculative effects on progression to AIDS and inflammaging. Therefore, any host genetic factors affecting iron regulation may impact viral pathogenesis.
Taken together, with the advancement in the available tools, larger GWAS and more in-depth analysis, SNP chip analysis of a larger number of SNPs across the region, and sequence-based genotyping of the HLAs would be essential to better delineate the role of HLA alleles in viral infections in terms of infection susceptibility or disease progression.
Finally, when designing vaccines or drugs from an epidemiological perspective, the major variants of certain genes such as HLA alleles and how these variants affect viral escape mutations, viral load heritability, and disease outcome should be considered.
Author Contributions
NC wrote the first version of manuscript and designed Figures 1, 4 and 5. SE proposed the idea, designed the article, designed Figures 2 and 3, rewrote some sections, and revised and edited the whole manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by an Open Operating grant, a Foundation Scheme grant by the Canadian Institutes of Health Research (CIHR) and a CIHR New Investigator award.
References
1. Lama J, Planelles V. Host factors influencing susceptibility to HIV infection and AIDS progression. Retrovirology (2007) 4:52. doi:10.1186/1742-4690-4-52
2. Matsuura K, Tanaka Y. Host genetic variants influencing the clinical course of hepatitis C virus infection. J Med Virol (2016) 88(2):185–95. doi:10.1002/jmv.24334
3. Silvestr D, Kourilsk FM, Niccolai MG, Levy JP. Presence of HLA antigens on human reticulocytes as demonstrated by electron microscopy. Nature (1970) 228(5266):67–8. doi:10.1038/228067a0
4. Hudson LE, Allen RL. Leukocyte Ig-like receptors – a model for MHC class I disease associations. Front Immunol (2016) 7:281. doi:10.3389/fimmu.2016.00281
5. Allard M, Oger R, Benlalam H, Florenceau L, Echasserieau K, Bernardeau K, et al. Soluble HLA-I/peptide monomers mediate antigen-specific CD8 T cell activation through passive peptide exchange with cell-bound HLA-I molecules. J Immunol (2014) 192(11):5090–7. doi:10.4049/jimmunol.1303226
6. Leddon SA, Sant AJ. Generation of MHC class II-peptide ligands for CD4 T-cell allorecognition of MHC class II molecules. Curr Opin Organ Transplant (2010) 15(4):505–11. doi:10.1097/MOT.0b013e32833bfc5c
7. Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet (2009) 54(1):15–39. doi:10.1038/jhg.2008.5
8. Little AM, Parham P. Polymorphism and evolution of HLA class I and II genes and molecules. Rev Immunogenet (1999) 1(1):105–23.
9. Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh SG. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res (2015) 43(Database issue):D423–31. doi:10.1093/nar/gku1161
10. Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet (2013) 14:301–23. doi:10.1146/annurev-genom-091212-153455
11. Bjorkman PJ, Parham P. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu Rev Biochem (1990) 59:253–88. doi:10.1146/annurev.bi.59.070190.001345
12. Hamerman JA, Ogasawara K, Lanier LL. NK cells in innate immunity. Curr Opin Immunol (2005) 17(1):29–35. doi:10.1016/j.coi.2004.11.001
13. Ljunggren HG, Karre K. In search of the missing self – MHC molecules and NK cell recognition. Immunol Today (1990) 11(7):237–44. doi:10.1016/0167-5699(90)90097-S
14. Orr MT, Lanier LL. Natural killer cell licensing during viral infection. Crossroads Innate Adapt Immun III (2011) 780:37–44. doi:10.1007/978-1-4419-5632-3_4
15. Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology (2011) 132(3):315–25. doi:10.1111/j.1365-2567.2010.03398.x
16. Strong RK, Holmes MA, Li PW, Braun L, Lee N, Geraghty DE. HLA-E allelic variants – correlating differential expression, peptide affinities, crystal structures, and thermal stabilities. J Biol Chem (2003) 278(7):5082–90. doi:10.1074/jbc.M208268200
17. Fitzmaurice K, Hurst J, Dring M, Rauch A, McLaren PJ, Gunthard HF, et al. Additive effects of HLA alleles and innate immune genes determine viral outcome in HCV infection. Gut (2015) 64(5):813–9. doi:10.1136/gutjnl-2013-306287
18. Huy NT, Hamada M, Kikuchi M, Nguyen TPL, Yasunami M, Zamora J, et al. Association of HLA and post-schistosomal hepatic disorder: a systematic review and meta-analysis. Parasitol Int (2011) 60(4):347–56. doi:10.1016/j.parint.2011.05.008
19. Ribas-Silva RC, Ribas AD, dos Santos MCG, da Silva WV, Lonardoni MVC, Borelli SD, et al. Association between HLA genes and American cutaneous leishmaniasis in endemic regions of Southern Brazil. BMC Infect Dis (2013) 13:198. doi:10.1186/1471-2334-13-198
20. Sakhno LV, Shevela EY, Tikhonova MA, Nikonov SD, Ostanin AA, Chernykh ER. Impairments of antigen-presenting cells in pulmonary tuberculosis. J Immunol Res (2015) 2015:793292. doi:10.1155/2015/793292
21. Sauer ME, Salomao H, Ramos GB, D’Espindula HR, Rodrigues RS, Macedo WC, et al. Genetics of leprosy: expected-and unexpected-developments and perspectives. Clin Dermatol (2016) 34(1):96–104. doi:10.1016/j.clindermatol.2015.10.005
22. Elahi S, Dinges WL, Lejarcegui N, Laing KJ, Collier AC, Koelle DM, et al. Protective HIV-specific CD8(+) T cells evade T-reg cell suppression. Nat Med (2011) 17(8):989–95. doi:10.1038/nm.2422
23. Fitzmaurice K, Petrovic D, Ramamurthy N, Simmons R, Merani S, Gaudieri S, et al. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut (2011) 60(11):1563–71. doi:10.1136/gut.2010.228403
24. Zaunders J, van Bockel D. Innate and adaptive immunity in long-term non-progression in HIV disease. Front Immunol (2013) 4:95. doi:10.3389/fimmu.2013.00095
25. Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity (2007) 27(3):406–16. doi:10.1016/j.immuni.2007.08.010
26. Merani S, Petrovic D, James I, Chopra A, Cooper D, Freitas E, et al. Effect of immune pressure on hepatitis C virus evolution: insights from a single-source outbreak. Hepatology (2011) 53(2):396–405. doi:10.1002/hep.24076
27. Huang J, Goedert JJ, Sundberg EJ, Cung TD, Burke PS, Martin MP, et al. HLA-B*35-Px-mediated acceleration of HIV-1 infection by increased inhibitory immunoregulatory impulses. J Exp Med (2009) 206(13):2959–66. doi:10.1084/jem.20091386
28. Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene (2008) 27(45):5869–85. doi:10.1038/onc.2008.273
29. Fleischhauer K, Beelen DW. HLA mismatching as a strategy to reduce relapse after alternative donor transplantation. Semin Hematol (2016) 53(2):57–64. doi:10.1053/j.seminhematol.2016.01.010
30. Thorsby E, Lie BA. HLA associated genetic predisposition to autoimmune diseases: genes involved and possible mechanisms. Transpl Immunol (2005) 14(3–4):175–82. doi:10.1016/j.trim.2005.03.021
31. Kunwar P, Hawkins N, Dinges WL, Liu Y, Gabriel EE, Swan DA, et al. Superior control of HIV-1 replication by CD8+ T cells targeting conserved epitopes: implications for HIV vaccine design. PLoS One (2013) 8(5):e64405. doi:10.1371/journal.pone.0064405
32. Horton H, Frank I, Baydo R, Jalbert E, Penn J, Wilson S, et al. Preservation of T cell proliferation restricted by protective HLA alleles is critical for immune control of HIV-1 infection. J Immunol (2006) 177(10):7406–15. doi:10.4049/jimmunol.177.10.7406
33. Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, et al. Lytic granule loading of CD8(+) T cells is required for HIV-infected cell elimination associated with immune control. Immunity (2008) 29(6):1009–21. doi:10.1016/j.immuni.2008.10.010
34. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood (2006) 107(12):4781–9. doi:10.1182/blood-2005-12-4818
35. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, et al. HIV-specific CD8(+) T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol (2002) 3(11):1061–8. doi:10.1038/ni845
36. Tokarev A, Guatelli J. Misdirection of membrane trafficking by HIV-1 Vpu and Nef: keys to viral virulence and persistence. Cell Logist (2011) 1(3):90–102. doi:10.4161/cl.1.3.16708
37. Rajapaksa US, Li DM, Peng YC, McMichael AJ, Dong T, Xu XN. HLA-B may be more protective against HIV-1 than HLA-A because it resists negative regulatory factor (Nef) mediated down-regulation. Proc Natl Acad Sci U S A (2012) 109(33):13353–8. doi:10.1073/pnas.1204199109
38. Czerwionka-Szaflarska M, Los-Rycharska E, Szaflarska-Poplawska A, Gronkowska A, Wozniakowska-Gesicka T, Kups J, et al. [The analysis of the hepatitis C virus infection course and efficacy of treatment dependence on HLA A antigens in children and youth]. Pol Merkur Lekarski (2008) 25(146):124–31.
39. Chakraborty S, Rahman T, Chakravorty R, Kuchta A, Rabby A, Sahiuzzaman M. HLA supertypes contribute in HIV type 1 cytotoxic T lymphocyte epitope clustering in Nef and Gag proteins. AIDS Res Hum Retroviruses (2013) 29(2):270–8. doi:10.1089/AID.2012.0160
40. Tang J, Malhotra R, Song W, Brill I, Hu L, Farmer PK, et al. Human leukocyte antigens and HIV type 1 viral load in early and chronic infection: predominance of evolving relationships. PLoS One (2010) 5(3):e9629. doi:10.1371/journal.pone.0009629
41. Koehler RN, Walsh AM, Saathoff E, Tovanabutra S, Arroyo MA, Currier JR, et al. Class I HLA-A*7401 is associated with protection from HIV-1 acquisition and disease progression in Mbeya, Tanzania. J Infect Dis (2010) 202(10):1562–6. doi:10.1086/656913
42. Fellay J, Ge D, Shianna KV, Colombo S, Ledergerber B, Cirulli ET, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet (2009) 5(12):e1000791. doi:10.1371/journal.pgen.1000791
43. MacDonald KS, Fowke KR, Kimani J, Dunand VA, Nagelkerke NJ, Ball TB, et al. Influence of HLA supertypes on susceptibility and resistance to human immunodeficiency virus type 1 infection. J Infect Dis (2000) 181(5):1581–9. doi:10.1086/315472
44. Sidney J, Peters B, Frahm N, Brander C, Sette A. HLA class I supertypes: a revised and updated classification. BMC Immunol (2008) 9:1. doi:10.1186/1471-2172-9-1
45. Kaslow RA, Carrington M, Apple R, Park L, Munoz A, Saah AJ, et al. Influence of combinations of human major histocompatibility complex genes on the course of HIV-1 infection. Nat Med (1996) 2(4):405–11. doi:10.1038/nm0496-405
46. Chen Y, Winchester R, Korber B, Gagliano J, Bryson Y, Hutto C, et al. Influence of HLA alleles on the rate of progression of vertically transmitted HIV infection in children: association of several HLA-DR13 alleles with long-term survivorship and the potential association of HLA-A*2301 with rapid progression to AIDS. Hum Immunol (1997) 55(2):154–62. doi:10.1016/S0198-8859(97)00092-X
47. Hendel H, Caillat-Zucman S, Lebuanec H, Carrington M, O’Brien S, Andrieu JM, et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J Immunol (1999) 162(11):6942–6.
48. Mosaad YM, Farag RE, Arafa MM, Eletreby S, El-Alfy HA, Eldeek BS, et al. Association of human leucocyte antigen class I (HLA-A and HLA-B) with chronic hepatitis C virus infection in Egyptian patients. Scand J Immunol (2010) 72(6):548–53. doi:10.1111/j.1365-3083.2010.02468.x
49. Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med (2003) 54:535–51. doi:10.1146/annurev.med.54.101601.152346
50. Koga M, Kawana-Tachikawa A, Heckerman D, Odawara T, Nakamura H, Koibuchi T, et al. Changes in impact of HLA class I allele expression on HIV-1 plasma virus loads at a population level over time. Microbiol Immunol (2010) 54(4):196–205. doi:10.1111/j.1348-0421.2010.00206.x
51. Munkanta M, Terunuma H, Takahashi M, Hanabusa H, Miura T, Ikeda S, et al. HLA-B polymorphism in Japanese HIV-1-infected long-term surviving hemophiliacs. Viral Immunol (2005) 18(3):500–5. doi:10.1089/vim.2005.18.500
52. Carrington M, Walker BD. Immunogenetics of spontaneous control of HIV. Annu Rev Med (2012) 63:131–45. doi:10.1146/annurev-med-062909-130018
53. Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, et al. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A (2000) 97(6):2709–14. doi:10.1073/pnas.050567397
54. Fellay J, Shianna KV, Ge DL, Colombo S, Ledergerber B, Weale M, et al. A whole-genome association study of major determinants for host control of HIV-1. Science (2007) 317(5840):944–7. doi:10.1126/science.1143767
55. McLaren PJ, Carrington M. The impact of host genetic variation on infection with HIV-1. Nat Immunol (2015) 16(6):577–83. doi:10.1038/ni.3147
56. International HIV Controllers StudyPereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PI, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science (2010) 330(6010):1551–7. doi:10.1126/science.1195271
57. Kosmrlj A, Read EL, Qi Y, Allen TM, Altfeld M, Deeks SG, et al. Effects of thymic selection of the T-cell repertoire on HLA class I-associated control of HIV infection. Nature (2010) 465(7296):350–4. doi:10.1038/nature08997
58. Thio CL, Gao X, Goedert JJ, Vlahov D, Nelson KE, Hilgartner MW, et al. HLA-Cw*04 and hepatitis C virus persistence. J Virol (2002) 76(10):4792–7. doi:10.1128/JVI.76.10.4792-4797.2002
59. Turnbull EL, Lopes AR, Jones NA, Cornforth D, Newton P, Aldam D, et al. HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently. J Immunol (2006) 176(10):6130–46. doi:10.4049/jimmunol.176.10.6130
60. McKiernan SM, Hagan R, Curry M, McDonald GS, Kelly A, Nolan N, et al. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology (2004) 40(1):108–14. doi:10.1002/hep.20261
61. Bhalerao J, Bowcock AM. The genetics of psoriasis: a complex disorder of the skin and immune system. Hum Mol Genet (1998) 7(10):1537–45. doi:10.1093/hmg/7.10.1537
62. Chessman D, Kostenko L, Lethborg T, Purcell AW, Williamson NA, Chen Z, et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity (2008) 28(6):822–32. doi:10.1016/j.immuni.2008.04.020
63. Bowness P. The 2000 Michael Mason Prize Essay – HLA B27 in health and disease: a double-edged sword? Rheumatology (2002) 41(8):857–68. doi:10.1093/rheumatology/41.8.857
64. Lopez de Castro JA. HLA-B27 and the pathogenesis of spondyloarthropathies. Immunol Lett (2007) 108(1):27–33. doi:10.1016/j.imlet.2006.10.004
65. Seregin SS, Rastall DP, Evnouchidou I, Aylsworth CF, Quiroga D, Kamal RP, et al. Endoplasmic reticulum aminopeptidase-1 alleles associated with increased risk of ankylosing spondylitis reduce HLA-B27 mediated presentation of multiple antigens. Autoimmunity (2013) 46(8):497–508. doi:10.3109/08916934.2013.819855
66. Wiersma VR, de Bruyn M, Helfrich W, Bremer E. Therapeutic potential of Galectin-9 in human disease. Med Res Rev (2013) 33:E102–26. doi:10.1002/med.20249
67. Wang F, Wan LG, Zhang CC, Zheng XF, Li JM, Chen ZK. Tim-3-Galectin-9 pathway involves the suppression induced by CD4(+)CD25(+) regulatory T cells. Immunobiology (2009) 214(5):342–9. doi:10.1016/j.imbio.2008.10.007
68. Mathieu A, Paladini F, Vacca A, Cauli A, Fiorillo MT, Sorrentino R. The interplay between the geographic distribution of HLA-B27 alleles and their role in infectious and autoimmune diseases: a unifying hypothesis. Autoimmun Rev (2009) 8(5):420–5. doi:10.1016/j.autrev.2009.01.003
69. Sorrentino R, Bockmann RA, Fiorillo MT. HLA-B27 and antigen presentation: at the crossroads between immune defense and autoimmunity. Mol Immunol (2014) 57(1):22–7. doi:10.1016/j.molimm.2013.06.017
70. Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, et al. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J Infect Dis (2008) 197(4):563–71. doi:10.1086/526786
71. Kim AY, Kuntzen T, Timm J, Nolan BE, Baca MA, Reyor LL, et al. Spontaneous control of HCV is associated with expression of HLA-B*57 and preservation of targeted epitopes. Gastroenterology (2011) 140(2):686.e–96.e. doi:10.1053/j.gastro.2010.09.042
72. Payne RP, Branch S, Kloverpris H, Matthews PC, Koofhethile CK, Strong T, et al. Differential escape patterns within the dominant HLA-B*57:03-restricted HIV gag epitope reflect distinct clade-specific functional constraints. J Virol (2014) 88(9):4668–78. doi:10.1128/JVI.03303-13
73. Abdel-Mohsen M, Raposo RAS, Deng XT, Li MQ, Liegler T, Sinclair E, et al. Expression profile of host restriction factors in HIV-1 elite controllers. Retrovirology (2013) 10:106. doi:10.1186/1742-4690-10-106
74. Mear JP, Schreiber KL, Munz C, Zhu XM, Stevanovic S, Rammensee HG, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol (1999) 163(12):6665–70.
75. Sheehan NJ. The ramifications of HLA-B27. J R Soc Med (2004) 97(1):10–4. doi:10.1258/jrsm.97.1.10
76. Bird LA, Peh CA, Kollnberger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur J Immunol (2003) 33(3):748–59. doi:10.1002/eji.200323678
77. Gao X, Bashirova A, Iversen AK, Phair J, Goedert JJ, Buchbinder S, et al. AIDS restriction HLA allotypes target distinct intervals of HIV-1 pathogenesis. Nat Med (2005) 11(12):1290–2. doi:10.1038/nm1333
78. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med (2004) 10(3):282–9. doi:10.1038/nm992
79. Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature (2009) 458(7238):641–5. doi:10.1038/nature07746
80. Goulder PJR, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med (1997) 3(2):212–7. doi:10.1038/nm0297-212
81. Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, et al. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med (2001) 193(3):375–85. doi:10.1084/jem.193.3.375
82. Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, et al. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol (2007) 81(22):12382–93. doi:10.1128/JVI.01543-07
83. Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg HC, Killinger T, et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology (2006) 43(3):563–72. doi:10.1002/hep.21049
84. Rao X, Hoof I, van Baarle D, Kesmir C, Textor J. HLA preferences for conserved epitopes: a potential mechanism for hepatitis C clearance. Front Immunol (2015) 6:552. doi:10.3389/fimmu.2015.00552
85. Ammaranond P, van Bockel DJ, Petoumenos K, McMurchie M, Finlayson R, Middleton MG, et al. HIV immune escape at an immunodominant epitope in HLA-B*27-positive individuals predicts viral load outcome. J Immunol (2011) 186(1):479–88. doi:10.4049/jimmunol.0903227
86. Nitschke K, Barriga A, Schmidt J, Timm J, Viazov S, Kuntzen T, et al. HLA-B*27 subtype specificity determines targeting and viral evolution of a hepatitis C virus-specific CD8+ T cell epitope. J Hepatol (2014) 60(1):22–9. doi:10.1016/j.jhep.2013.08.009
87. Goulder PJR, Brander C, Tang YH, Tremblay C, Colbert RA, Addo MM, et al. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature (2001) 412(6844):334–8. doi:10.1038/35085576
88. Marcilla M, de Castro JAL. Peptides: the cornerstone of HLA-B27 biology and pathogenetic role in spondyloarthritis. Tissue Antigens (2008) 71(6):495–506. doi:10.1111/j.1399-0039.2008.01051.x
89. Stewart-Jones GB, di Gleria K, Kollnberger S, McMichael AJ, Jones EY, Bowness P. Crystal structures and KIR3DL1 recognition of three immunodominant viral peptides complexed to HLA-B*2705. Eur J Immunol (2005) 35(2):341–51. doi:10.1002/eji.200425724
90. Neumann-Haefelin C, Oniangue-Ndza C, Kuntzen T, Schmidt J, Nitschke K, Sidney J, et al. Human leukocyte antigen B27 selects for rare escape mutations that significantly impair hepatitis C virus replication and require compensatory mutations. Hepatology (2011) 54(4):1157–66. doi:10.1002/hep.24541
91. Xiong H, Huang J, Rong X, Zhang M, Huang K, Xu R, et al. HLA-B alleles B*15:01 and B*15:02: opposite association with hepatitis C virus infection in Chinese voluntary blood donors. Intervirology (2015) 58(2):80–7. doi:10.1159/000369209
92. Gao X, Nelson GW, Karacki P, Martin MP, Phair J, Kaslow R, et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med (2001) 344(22):1668–75. doi:10.1056/NEJM200105313442203
93. Jin X, Gao X, Ramanathan M Jr, Deschenes GR, Nelson GW, O’Brien SJ, et al. Human immunodeficiency virus type 1 (HIV-1)-specific CD8+-T-cell responses for groups of HIV-1-infected individuals with different HLA-B*35 genotypes. J Virol (2002) 76(24):12603–10. doi:10.1128/JVI.76.24.12603-12610.2002
94. Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science (1999) 283(5408):1748–52. doi:10.1126/science.283.5408.1748
95. Keynan Y, Rueda ZV, Bresler K, Becker M, Kasper K. HLA B51 is associated with faster AIDS progression among newly diagnosed HIV-infected individuals in Manitoba, Canada. Int J Immunogenet (2015) 42(5):336–40. doi:10.1111/iji.12219
96. Illing PT, Mifsud NA, Purcell AW. Allotype specific interactions of drugs and HLA molecules in hypersensitivity reactions. Curr Opin Immunol (2016) 42:31–40. doi:10.1016/j.coi.2016.05.003
97. Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med (2008) 358(6):568–79. doi:10.1056/NEJMoa0706135
98. Hewitt RG. Abacavir hypersensitivity reaction. Clin Infect Dis (2002) 34(8):1137–42. doi:10.1086/339751
99. Lucas A, Lucas M, Strhyn A, Keane NM, McKinnon E, Pavlos R, et al. Abacavir-reactive memory T cells are present in drug naive individuals. PLoS One (2015) 10(2):e0117160. doi:10.1371/journal.pone.0117160
100. Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A (2012) 109(25):9959–64. doi:10.1073/pnas.1207934109
101. Adam J, Wuillemin N, Watkins S, Jamin H, Eriksson KK, Villiger P, et al. Abacavir induced T cell reactivity from drug naive individuals shares features of allo-immune responses. PLoS One (2014) 9(4):e95339. doi:10.1371/journal.pone.0095339
102. Naisbitt DJ, Yang EL, Alhaidari M, Berry NG, Lawrenson AS, Farrell J, et al. Towards depersonalized abacavir therapy: chemical modification eliminates HLA-B*57:01-restricted CD8(+) T-cell activation. AIDS (2015) 29(18):2385–95. doi:10.1097/QAD.0000000000000867
103. Elahi S, Horton H. Association of HLA-alleles with the immune regulation of chronic viral infections. Int J Biochem Cell Biol (2012) 44(8):1361–5. doi:10.1016/j.biocel.2012.05.003
104. Daar AS, Fuggle SV, Fabre JW, Ting A, Morris PJ. The detailed distribution of MHC class-II antigens in normal human organs. Transplantation (1984) 38(3):293–8. doi:10.1097/00007890-198409000-00019
105. Tu MM, Mahmoud AB, Makrigiannis AP. Licensed and unlicensed NK cells: differential roles in cancer and viral control. Front Immunol (2016) 7:166. doi:10.3389/fimmu.2016.00166
106. Zemmour J, Parham P. Distinctive polymorphism at the HLA-C locus – implications for the expression of HLA-C. J Exp Med (1992) 176(4):937–50. doi:10.1084/jem.176.4.937
107. Neisig A, Melief CJM, Neefjes J. Reduced cell surface expression of HLA-C molecules correlates with restricted peptide binding and stable TAP interaction. J Immunol (1998) 160(1):171–9.
108. Turner S, Ellexson ME, Hickman HD, Sidebottom DA, Fernandez-Vina A, Confer DL, et al. Sequence-based typing provides a new look at HLA-C diversity. J Immunol (1998) 161(3):1406–13.
109. Mccutcheon JA, Gumperz J, Smith KD, Lutz CT, Parham P. Low HLA-C expression at cell-surfaces correlates with increased turnover of heavy-chain messenger-RNA. J Exp Med (1995) 181(6):2085–95. doi:10.1084/jem.181.6.2085
110. Biassoni R, Falco M, Cambiaggi A, Costa P, Verdiani S, Pende D, et al. Amino-acid substitutions can influence the natural-killer (NK)-mediated recognition of HLA-C molecules – pole of serine-77 and lysine-80 in the target-cell protection from lysis mediated by group-2 or group-1 NK clones. J Exp Med (1995) 182(2):605–9. doi:10.1084/jem.182.2.605
111. Blais ME, Dong T, Rowland-Jones S. HLA-C as a mediator of natural killer and T-cell activation: spectator or key player? Immunology (2011) 133(1):1–7. doi:10.1111/j.1365-2567.2011.03422.x
112. Nunnari G, Fagone P, Condorelli F, Nicoletti F, Malaguarnera L, Di Rosa M. CD4+T-cell gene expression of healthy donors, HIV-1 and elite controllers: immunological chaos. Cytokine (2016) 83:127–35. doi:10.1016/j.cyto.2016.04.007
113. Cosma A, Blanc D, Braun J, Quillent C, Barassi C, Moog C, et al. Enhanced HIV infectivity and changes in GP120 conformation associated with viral incorporation of human leucocyte antigen class I molecules. AIDS (1999) 13(15):2033–42. doi:10.1097/00002030-199910220-00005
114. Matucci A, Rossolillo P, Baroni M, Siccardi AG, Beretta A, Zipeto D. HLA-C increases HIV-1 infectivity and is associated with gp120. Retrovirology (2008) 5:68. doi:10.1186/1742-4690-5-68
115. Serena M, Parolini F, Biswas P, Sironi F, Miranda AB, Zoratti E, et al. HIV-1 Env associates with HLA-C free-chains at the cell membrane modulating viral infectivity. Sci Rep (2017) 7:40037. doi:10.1038/srep40037
116. Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, et al. Influence of HLA-C expression level on HIV control. Science (2013) 340(6128):87–91. doi:10.1126/science.1232685
117. Majorczyk E, Matusiak L, Nowak I, Pietkiewicz-Sworowska A, Luszczek W, Szepietowski JC, et al. A single nucleotide polymorphism-35 kb T > C (rs9264942) is strongly associated with psoriasis vulgaris depending on HLA-Cw*06. Hum Immunol (2014) 75(6):504–7. doi:10.1016/j.humimm.2014.04.003
118. Martin MP, Carrington M. Immunogenetics of HIV disease. Immunol Rev (2013) 254:245–64. doi:10.1111/imr.12071
119. Kulkarni S, Savan R, Qi Y, Gao XJ, Yuki Y, Bass SE, et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature (2011) 472(7344):495–8. doi:10.1038/nature09914
120. Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol (2013) 31:163–94. doi:10.1146/annurev-immunol-032712-100001
121. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi:10.1038/ni1582
122. Hiroishi K, Eguchi J, Ishii S, Hiraide A, Sakaki M, Doi H, et al. Immune response of cytotoxic T lymphocytes and possibility of vaccine development for hepatitis C virus infection. J Biomed Biotechnol (2010) 2010:263810. doi:10.1155/2010/263810
123. Ravet S, Scott-Algara D, Bonnet E, Tran HK, Tran T, Nguyen N, et al. Distinctive NK-cell receptor repertoires sustain high-level constitutive NK-cell activation in HIV-exposed uninfected individuals. Blood (2007) 109(10):4296–305. doi:10.1182/blood-2006-08-040238
124. Crome SQ, Lang PA, Lang KS, Ohashi PS. Natural killer cells regulate diverse T cell responses. Trends Immunol (2013) 34(7):342–9. doi:10.1016/j.it.2013.03.002
125. Long EO, Kim HS, Liu DF, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol (2013) 31:227–58. doi:10.1146/annurev-immunol-020711-075005
126. Mandal A, Viswanathan C. Natural killer cells: in health and disease. Hematol Oncol Stem Cell Ther (2015) 8(2):47–55. doi:10.1016/j.hemonc.2014.11.006
127. Moretta A, Marcenaro E, Sivori S, Chiesa MD, Vitale M, Moretta L. Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends Immunol (2005) 26(12):668–74. doi:10.1016/j.it.2005.09.008
128. Brodin P, Karre K, Hoglund P. NK cell education: not an on-off switch but a tunable rheostat. Trends Immunol (2009) 30(4):143–9. doi:10.1016/j.it.2009.01.006
129. Joncker NT, Fernandez NC, Treiner E, Vivier E, Raulet DH. NK cell responsiveness is tuned commensurate with the number of inhibitory receptors for self-MHC class I: the rheostat model. J Immunol (2009) 182(8):4572–80. doi:10.4049/jimmunol.0803900
130. Specht A, DeGottardi MQ, Schindler M, Hahn B, Evans DT, Kirchhoff F. Selective downmodulation of HLA-A and -B by Nef alleles from different groups of primate lentiviruses. Virology (2008) 373(1):229–37. doi:10.1016/j.virol.2007.11.019
131. Apps R, Del Prete GQ, Chatterjee P, Lara A, Brumme ZL, Brockman MA, et al. HIV-1 Vpu mediates HLA-C downregulation. Cell Host Microbe (2016) 19(5):686–95. doi:10.1016/j.chom.2016.04.005
132. Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta(2)-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst (1996) 88(2):100–8. doi:10.1093/jnci/88.2.100
133. Tarek N, Le Luduec JB, Gallagher MM, Zheng JT, Venstrom JM, Chamberlain E, et al. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest (2012) 122(9):3260–70. doi:10.1172/JCI62749
134. Alvarez RA, Hamlin RE, Monroe A, Moldt B, Hotta MT, Caprio GR, et al. HIV-1 Vpu antagonism of tetherin inhibits antibody-dependent cellular cytotoxic responses by natural killer cells. J Virol (2014) 88(11):6031–46. doi:10.1128/JVI.00449-14
135. Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, et al. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A (2014) 111(17):6425–30. doi:10.1073/pnas.1321507111
136. Kramski M, Stratov I, Kent SJ. The role of HIV-specific antibody-dependent cellular cytotoxicity in HIV prevention and the influence of the HIV-1 Vpu protein. AIDS (2015) 29(2):137–44. doi:10.1097/QAD.0000000000000523
137. Davis ZB, Cogswell A, Scott H, Mertsching A, Boucau J, Wambua D, et al. A conserved HIV-1-derived peptide presented by HLA-E renders infected T-cells highly susceptible to attack by NKG2A/CD94-bearing natural killer cells. PLoS Pathog (2016) 12(2):e1005421. doi:10.1371/journal.ppat.1005421
138. Parham P, Abi-Rached L, Matevosyan L, Moesta AK, Norman PJ, Older Aguilar AM, et al. Primate-specific regulation of natural killer cells. J Med Primatol (2010) 39(4):194–212. doi:10.1111/j.1600-0684.2010.00432.x
139. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature (2011) 476(7358):96–100. doi:10.1038/nature10237
140. Martin MP, Qi Y, Gao XJ, Yamada E, Martin JN, Pereyra F, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet (2007) 39(6):733–40. doi:10.1038/ng2035
141. Guerini FR, Clerici M. NK cells in human disease: an evolving story. Clin Immunol (2012) 143(3):203–6. doi:10.1016/j.clim.2012.03.003
142. Lanier LL. Activating and inhibitory NK cell receptors. Adv Exp Med Biol (1998) 452:13–8. doi:10.1007/978-1-4615-5355-7_2
143. Lanier LL. NK cell receptors. Annu Rev Immunol (1998) 16:359–93. doi:10.1146/annurev.immunol.16.1.359
144. Middleton D, Gonzelez F. The extensive polymorphism of KIR genes. Immunology (2010) 129(1):8–19. doi:10.1111/j.1365-2567.2009.03208.x
145. Parham P. Influence of KIR diversity on human immunity. Mechanisms of lymphocyte activation and immune regulation X. Innate Immun (2005) 560:47–50. doi:10.1007/0-387-24180-9_6
146. Uhrberg M, Valiante NM, Shum BP, Shilling HG, Lienert-Weidenbach K, Corliss B, et al. Human diversity in killer cell inhibitory receptor genes. Immunity (1997) 7(6):753–63. doi:10.1016/S1074-7613(00)80394-5
147. Jamil KM, Khakoo SI. KIR/HLA interactions and pathogen immunity. J Biomed Biotechnol (2011) 2011:298348. doi:10.1155/2011/298348
148. Parham P, Moffett A. Variable NK cell receptors and their MHC class I ligands in immunity, reproduction and human evolution. Nat Rev Immunol (2013) 13(2):133–44. doi:10.1038/nri3370
149. Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song YJ, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature (2005) 436(7051):709–13. doi:10.1038/nature03847
150. Hens J, Jennes W, Kestens L. The role of NK cells in HIV-1 protection: autologous, allogeneic or both? AIDS Res Ther (2016) 13:15. doi:10.1186/s12981-016-0099-6
151. Plauzolles A, Lucas M, Gaudieri S. Influence of host resistance on viral adaptation: hepatitis C virus as a case study. Infect Drug Resist (2015) 8:63–74. doi:10.2147/IDR.S49891
152. Rajagopalan S, Long EO. Understanding how combinations of HLA and KIR genes influence disease. J Exp Med (2005) 201(7):1025–9. doi:10.1084/jem.20050499
153. Hilton HG, Vago L, Aguilar AMO, Moesta AK, Graef T, Abi-Rached L, et al. Mutation at positively selected positions in the binding site for HLA-C shows that KIR2DL1 is a more refined but less adaptable NK cell receptor than KIR2DL3. J Immunol (2012) 189(3):1418–30. doi:10.4049/jimmunol.1100431
154. Moesta AK, Norman PJ, Yawata M, Yawata N, Gleimer M, Parham P. Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J Immunol (2008) 180(6):3969–79. doi:10.4049/jimmunol.180.6.3969
155. Boudreau JE, Mulrooney TJ, Le Luduec JB, Barker E, Hsu KC. KIR3DL1 and HLA-B density and binding calibrate NK education and response to HIV. J Immunol (2016) 196(8):3398–410. doi:10.4049/jimmunol.1502469
156. Schonberg K, Fischer JC, Kogler G, Uhrberg M. Neonatal NK-cell repertoires are functionally, but not structurally, biased toward recognition of self HLA class I. Blood (2011) 117(19):5152–6. doi:10.1182/blood-2011-02-334441
157. Yawata M, Yawata N, Draghi M, Partheniou F, Little AM, Parham P. MHC class I-specific inhibitory receptors and their ligands structure diverse human NK-cell repertoires toward a balance of missing self-response. Blood (2008) 112(6):2369–80. doi:10.1182/blood-2008-03-143727
158. Malnati MS, Ugolotti E, Monti MC, Battista D, Vanni I, Bordo D, et al. Activating killer immunoglobulin receptors and HLA-C: a successful combination providing HIV-1 control. Sci Rep (2017) 7:42470. doi:10.1038/srep42470
159. De Maria A, Fogli M, Costa P, Murdaca G, Puppo F, Mavilio D, et al. The impaired NK cell cytolytic function in viremic HIV-1 infection is associated with a reduced surface expression of natural cytotoxicity receptors (NKp46, NKp30 and NKp44). Eur J Immunol (2003) 33(9):2410–8. doi:10.1002/eji.200324141
160. Mavilio D, Benjamin J, Daucher M, Lombardo G, Kottilil S, Planta MA, et al. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci U S A (2004) 101(16):6326. doi:10.1073/pnas.0401560101
161. Wong AHW, Williams K, Reddy S, Wilson D, Giddy J, Alter G, et al. Alterations in natural killer cell receptor profiles during HIV type 1 disease progression among chronically infected South African adults. AIDS Res Hum Retroviruses (2010) 26(4):459–69. doi:10.1089/aid.2009.0176
162. Sirianni MC, Ensoli F, Alario C, Fiorelli V, Sacco G, Topino S, et al. Distribution of the natural killer-related receptor for the HLA-C during highly active antiretroviral therapy for human immunodeficiency virus infection. Hum Immunol (2001) 62(12):1328–34.
163. Bonaparte MI, Barker E. Killing of human immunodeficiency virus-infected primary T-cell blasts by autologous natural killer cells is dependent on the ability of the virus to alter the expression of major histocompatibility complex class I molecules. Blood (2004) 104(7):2087–94. doi:10.1182/blood-2004-02-0696
164. Zwolinska K, Blachowicz O, Tomczyk T, Knysz B, Gasiorowski J, Zalewska M, et al. The effects of killer cell immunoglobulin-like receptor (KIR) genes on susceptibility to HIV-1 infection in the Polish population. Immunogenetics (2016) 68(5):327–37. doi:10.1007/s00251-016-0906-1
165. Naranbhai V, Rosa DD, Werner L, Moodley R, Hong H, Kharsany A, et al. Killer-cell Immunoglobulin-like receptor (KIR) gene profiles modify HIV disease course, not HIV acquisition in South African women. BMC Infect Dis (2016) 16:27. doi:10.1186/s12879-016-1361-1
166. Merino A, Malhotra R, Morton M, Mulenga J, Allen S, Hunter E, et al. Impact of a functional KIR2DS4 allele on heterosexual HIV-1 transmission among discordant Zambian couples. J Infect Dis (2011) 203(4):487–95. doi:10.1093/infdis/jiq075
167. Fadda L, Korner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw*0102-restricted HIV-1 p24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS Pathog (2012) 8(7):e1002805. doi:10.1371/journal.ppat.1002805
168. Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science (2004) 305(5685):872–4. doi:10.1126/science.1097670
169. Cassidy S, Mukherjee S, Myint TM, Mbiribindi B, North H, Traherne J, et al. Peptide selectivity discriminates NK cells from KIR2DL2- and KIR2DL3-positive individuals. Eur J Immunol (2015) 45(2):492–500. doi:10.1002/eji.201444613
170. Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol (2004) 5(10):996–1002. doi:10.1038/ni1114
171. al Basatena NKS, MacNamara A, Vine AM, Thio CL, Astemborski J, Usuku K, et al. KIR2DL2 enhances protective and detrimental HLA class I-mediated immunity in chronic viral infection. PLoS Pathog (2011) 7(10):e1002270. doi:10.1371/journal.ppat.1002270
172. Seich Al Basatena NK, Macnamara A, Vine AM, Thio CL, Astemborski J, Usuku K, et al. KIR2DL2 enhances protective and detrimental HLA class I-mediated immunity in chronic viral infection. PLoS Pathog (2011) 7(10):e1002270. doi:10.1371/journal.ppat.1002270
173. Ugolini S, Arpin C, Anfossi N, Walzer T, Cambiaggi A, Forster R, et al. Involvement of inhibitory NKRs in the survival of a subset of memory-phenotype CD8+ T cells. Nat Immunol (2001) 2(5):430–5. doi:10.1038/85246
174. Martin MP, Gao XJ, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet (2002) 31(4):429–34. doi:10.1038/ng934
175. Sasaki T, Kanaseki T, Shionoya Y, Tokita S, Miyamoto S, Saka E, et al. Microenvironmental stresses induce HLA-E/Qa-1 surface expression and thereby reduce CD8(+) T-cell recognition of stressed cells. Eur J Immunol (2016) 46(4):929–40. doi:10.1002/eji.201545835
176. Wei XH, Orr HT. Differential expression of HLA-E, HLA-F, and HLA-G transcripts in human tissue. Hum Immunol (1990) 29(2):131–42. doi:10.1016/0198-8859(90)90076-2
177. Felicio LP, Porto IO, Mendes-Junior CT, Veiga-Castelli LC, Santos KE, Vianello-Brondani RP, et al. Worldwide HLA-E nucleotide and haplotype variability reveals a conserved gene for coding and 3’ untranslated regions. Tissue Antigens (2014) 83(2):82–93. doi:10.1111/tan.12283
178. Kraemer T, Celik AA, Huyton T, Kunze-Schumacher H, Blasczyk R, Bade-Doding C. HLA-E: presentation of a broader peptide repertoire impacts the cellular immune response-implications on HSCT outcome. Stem Cells Int (2015) 2015:346714. doi:10.1155/2015/346714
179. Lampen MH, Hassan C, Sluijter M, Geluk A, Dijkman K, Tjon JM, et al. Alternative peptide repertoire of HLA-E reveals a binding motif that is strikingly similar to HLA-A2. Mol Immunol (2013) 53(1–2):126–31. doi:10.1016/j.molimm.2012.07.009
180. Djaoud Z, Riou R, Gavlovsky PJ, Mehlal S, Bressollette C, Gerard N, et al. Cytomegalovirus-infected primary endothelial cells trigger NKG2C(+) natural killer cells. J Innate Immun (2016) 8(4):374–85. doi:10.1159/000445320
181. Petrie EJ, Clements CS, Lin J, Sullivan LC, Johnson D, Huyton T, et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J Exp Med (2008) 205(3):725–35. doi:10.1084/jem.20072525
182. Sullivan LC, Clements CS, Rossjohn J, Brooks AG. The major histocompatibility complex class Ib molecule HLA-E at the interface between innate and adaptive immunity. Tissue Antigens (2008) 72(5):415–24. doi:10.1111/j.1399-0039.2008.01138.x
183. Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut (2006) 55(6):869–77. doi:10.1136/gut.2005.076463
184. Jinushi M, Takehara T, Tatsumi T, Kanto T, Miyagi T, Suzuki T, et al. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J Immunol (2004) 173(10):6072–81. doi:10.4049/jimmunol.173.10.6072
185. Schulte D, Vogel M, Langhans B, Kramer B, Korner C, Nischalke HD, et al. The HLA-E-R/HLA-E-R genotype affects the natural course of hepatitis C virus (HCV) infection and is associated with HLA-E-restricted recognition of an HCV-derived peptide by interferon-gamma-secreting human CD8(+) T cells. J Infect Dis (2009) 200(9):1397–401. doi:10.1086/605889
186. Guzman-Fulgencio M, Berenguer J, Rallon N, Fernandez-Rodriguez A, Miralles P, Soriano V, et al. HLA-E variants are associated with sustained virological response in HIV/hepatitis C virus-coinfected patients on hepatitis C virus therapy. AIDS (2013) 27(8):1231–8. doi:10.1097/QAD.0b013e32835f5b9c
187. Lajoie J, Hargrove J, Zijenah LS, Humphrey JH, Ward BJ, Roger M. Genetic variants in nonclassical major histocompatibility complex class I human leukocyte antigen (HLA)-E and HLA-G molecules are associated with susceptibility to heterosexual acquisition of HIV-1. J Infect Dis (2006) 193(2):298–301. doi:10.1086/498877
188. Mela CM, Burton CT, Imami N, Nelson M, Steel A, Gazzard BG, et al. Switch from inhibitory to activating NKG2 receptor expression in HIV-1 infection: lack of reversion with highly active antiretroviral therapy. AIDS (2005) 19(16):1761–9. doi:10.1097/01.aids.0000183632.12418.33
189. Zhang RJ, Xu JQ, Hong KX, Yuan L, Peng H, Tang HL, et al. Increased NKG2A found in cytotoxic natural killer subset in HIV-1 patients with advanced clinical status. AIDS (2007) 21:S9–17. doi:10.1097/01.aids.0000304691.32014.19
190. Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med (2015) 7(297):297ra115. doi:10.1126/scitranslmed.aac5722
191. Fausther-Bovendoa H, Wauquier N, Cherfils-Vicini J, Cremer I, Debre P, Vieillard V. NKG2C is a major triggering receptor involved in the V delta 1 T cell-mediated cytotoxicity against HIV-infected CD4 T cells. AIDS (2008) 22(2):217–26. doi:10.1097/QAD.0b013e3282f46e7c
192. Thomas R, Low HZ, Kniesch K, Jacobs R, Schmidt RE, Witte T. NKG2C deletion is a risk factor of HIV infection. AIDS Res Hum Retroviruses (2012) 28(8):844–51. doi:10.1089/AID.2011.0253
193. Boyle LH, Gillingham AK, Munro S, Trowsdale J. Selective export of HLA-F by its cytoplasmic tail. J Immunol (2006) 176(11):6464–72. doi:10.4049/jimmunol.176.11.6464
194. Lee N, Geraghty DE. HLA-F surface expression on B cell and monocyte cell lines is partially independent from tapasin and completely independent from TAP. J Immunol (2003) 171(10):5264–71. doi:10.4049/jimmunol.171.10.5264
195. Lepin EJ, Bastin JM, Allan DS, Roncador G, Braud VM, Mason DY, et al. Functional characterization of HLA-F and binding of HLA-F tetramers to ILT2 and ILT4 receptors. Eur J Immunol (2000) 30(12):3552–61. doi:10.1002/1521-4141(200012)30:12<3552::AID-IMMU3552>3.0.CO;2-L
196. Garcia-Beltran WF, Holzemer A, Martrus G, Chung AW, Pacheco Y, Simoneau CR, et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat Immunol (2016) 17(9):1067–74. doi:10.1038/ni.3513
197. Lee N, Ishitani A, Geraghty DE. HLA-F is a surface marker on activated lymphocytes. Eur J Immunol (2010) 40(8):2308–18. doi:10.1002/eji.201040348
198. Wainwright SD, Biro PA, Holmes CH. HLA-F is a predominantly empty, intracellular, TAP-associated MHC class Ib protein with a restricted expression pattern. J Immunol (2000) 164(1):319–28. doi:10.4049/jimmunol.164.1.319
199. Lima TH, Buttura RV, Donadi EA, Veiga-Castelli LC, Mendes-Junior CT, Castelli EC. HLA-F coding and regulatory segments variability determined by massively parallel sequencing procedures in a Brazilian population sample. Hum Immunol (2016) 77(10):841–53. doi:10.1016/j.humimm.2016.07.231
200. Goodridge JP, Burian A, Lee N, Geraghty DE. HLA-F complex without peptide binds to MHC class I protein in the open conformer form. J Immunol (2010) 184(11):6199–208. doi:10.4049/jimmunol.1000078
201. Goodridge JP, Lee N, Burian A, Pyo CW, Tykodi SS, Warren EH, et al. HLA-F and MHC-I open conformers cooperate in a MHC-I antigen cross-presentation pathway. J Immunol (2013) 191(4):1567–77. doi:10.4049/jimmunol.1300080
202. Parham P, Norman PJ, Abi-Rached L, Guethlein LA. Human-specific evolution of killer cell immunoglobulin-like receptor recognition of major histocompatibility complex class I molecules. Philos Trans R Soc Lond B Biol Sci (2012) 367(1590):800–11. doi:10.1098/rstb.2011.0266
203. Jiang YJ, Chen O, Cui C, Zhao B, Han XX, Zhang ZN, et al. KIR3DS1/L1 and HLA-Bw4-80I are associated with HIV disease progression among HIV typical progressors and long-term nonprogressors. BMC Infect Dis (2013) 13:405. doi:10.1186/1471-2334-13-405
204. Pelak K, Need AC, Fellay J, Shianna KV, Feng S, Urban TJ, et al. Copy number variation of KIR genes influences HIV-1 control. PLoS Biol (2011) 9(11):e1001208. doi:10.1371/journal.pbio.1001208
205. Rivero-Juarez A, Neukam K, Labarga P, Camacho A, Macias J, Barreiro P, et al. Sustained virological response in HIV/HCV co-infected patients treated with pegylated interferon/ribavirin can be predicted from the overall rate of viral load decline over the first 4 weeks of therapy. J Infect (2014) 68(4):372–7. doi:10.1016/j.jinf.2013.11.009
206. Larsen MH, Hylenius S, Andersen AMN, Hviid TVF. The 3’-untranslated region of the HLA-G gene in relation to pre-eclampsia: revisited. Tissue Antigens (2010) 75(3):253–61. doi:10.1111/j.1399-0039.2009.01435.x
207. Vianna P, Dalmaz CA, Veit TD, Tedoldi C, Roisenberg I, Chies JAB. Immunogenetics of pregnancy: role of a 14-bp deletion in the maternal HLA-G gene in primiparous pre-eclamptic Brazilian women. Hum Immunol (2007) 68(8):668–74. doi:10.1016/j.humimm.2007.05.006
208. Donadi EA, Castelli EC, Arnaiz-Villena A, Roger M, Rey D, Moreau P. Implications of the polymorphism of HLA-G on its function, regulation, evolution and disease association. Cell Mol Life Sci (2011) 68(3):369–95. doi:10.1007/s00018-010-0580-7
209. Fujii T, Ishitani A, Geraghty DE. A soluble form of the HLA-G antigen is encoded by a messenger-ribonucleic-acid containing intron 4. J Immunol (1994) 153(12):5516–24.
210. Paul P, Cabestre FA, Ibrahim E, Lefebvre S, Khalil-Daher I, Vazeux G, et al. Identification of HLA-G7 as a new splice variant of the HLA-G mRNA and expression of soluble HLA-G5, -G6, and -G7 transcripts in human transfected cells. Hum Immunol (2000) 61(11):1138–49. doi:10.1016/S0198-8859(00)00197-X
211. Rebmann V, Nardi FD, Wagner B, Horn PA. HLA-G as a tolerogenic molecule in transplantation and pregnancy. J Immunol Res (2014) 2014:297073. doi:10.1155/2014/297073
212. Alegre E, Rizzo R, Bortolotti D, Fernandez-Landazuri S, Fainardi E, Gonzalez A. Some basic aspects of HLA-G biology. J Immunol Res (2014) 2014:657625. doi:10.1155/2014/657625
213. Rizzo R, Trentini A, Bortolotti D, Manfrinato MC, Rotola A, Castellazzi M, et al. Matrix metalloproteinase-2 (MMP-2) generates soluble HLA-G1 by cell surface proteolytic shedding. Mol Cell Biochem (2013) 381(1–2):243–55. doi:10.1007/s11010-013-1708-5
214. Zidi I, Guillard C, Marcou C, Krawice-Radanne I, Sangrouber D, Rouas-Freiss N, et al. Increase in HLA-G1 proteolytic shedding by tumor cells: a regulatory pathway controlled by NF-kappa B inducers. Cell Mol Life Sci (2006) 63(22):2669–81. doi:10.1007/s00018-006-6341-y
215. Rouas-Freiss N, Moreau P, Ferrone S, Carosella ED. HLA-G proteins in cancer: do they provide tumor cells with an escape mechanism? Cancer Res (2005) 65(22):10139–44. doi:10.1158/0008-5472.CAN-05-0097
216. Rouas-Freiss N, Naji A, Durrbach A, Carosella ED. Tolerogenic functions of human leukocyte antigen G: from pregnancy to organ and cell transplantation. Transplantation (2007) 84(1):S21–5. doi:10.1097/01.tp.0000269117.32179.1c
217. Chen HX, Chen BG, Shi WW, Zhen R, Xu DP, Lin AF, et al. Induction of cell surface human leukocyte antigen-G expression in pandemic H1N1 2009 and seasonal H1N1 influenza virus-infected patients. Hum Immunol (2011) 72(2):159–65. doi:10.1016/j.humimm.2010.11.009
218. Lajoie J, Loembe MM, Poudrier J, Guedou F, Pepin J, Labbe AC, et al. Blood soluble human leukocyte antigen G levels are associated with human immunodeficiency virus type 1 infection in Beninese commercial sex workers. Hum Immunol (2010) 71(2):182–5. doi:10.1016/j.humimm.2009.11.007
219. Lozano JM, Gonzalez R, Kindelan JM, Rouas-Freiss N, Caballos R, Dausset J, et al. Monocytes and T lymphocytes in HIV-1-positive patients express HLA-G molecule. AIDS (2002) 16(3):347–51. doi:10.1097/00002030-200202150-00005
220. Megret F, Prehaud C, Lafage M, Moreau P, Rouas-Freiss N, Carosella ED, et al. Modulation of HLA-G and HLA-E expression in human neuronal cells after rabies virus or herpes virus simplex type 1 infections. Hum Immunol (2007) 68(4):294–302. doi:10.1016/j.humimm.2006.12.003
221. Onno M, Pangault C, Le Friec G, Guilloux V, Andre P, Fauchet R. Modulation of HLA-G antigens expression by human cytomegalovirus: specific induction in activated macrophages harboring human cytomegalovirus infection. J Immunol (2000) 164(12):6426–34. doi:10.4049/jimmunol.164.12.6426
222. Shi WW, Lin AF, Xu DP, Bao WG, Zhang JG, Chen SY, et al. Plasma soluble human leukocyte antigen-G expression is a potential clinical biomarker in patients with hepatitis B virus infection. Hum Immunol (2011) 72(11):1068–73. doi:10.1016/j.humimm.2011.06.012
223. Tripathi P, Agrawal S. The role of human leukocyte antigen E and G in HIV infection. AIDS (2007) 21(11):1395–404. doi:10.1097/QAD.0b013e32810c8bbc
224. Colonna M, Navarro F, Bellon T, Llano M, Garcia P, Samaridis J, et al. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J Exp Med (1997) 186(11):1809–18. doi:10.1084/jem.186.11.1809
225. Carosella ED, Gregori S, LeMaoult J. The tolerogenic interplay(s) among HLA-G, myeloid APCs, and regulatory cells. Blood (2011) 118(25):6499–505. doi:10.1182/blood-2011-07-370742
226. Gonzalez A, Rebmann V, LeMaoult J, Horn PA, Carosella ED, Alegre E. The immunosuppressive molecule HLA-G and its clinical implications. Crit Rev Clin Lab Sci (2012) 49(3):63–84. doi:10.3109/10408363.2012.677947
227. Le Bouteiller P, Fons P, Herault JP, Bono F, Chabot S, Cartwright JE, et al. Soluble HLA-G and control of angiogenesis. J Reprod Immunol (2007) 76(1–2):17–22. doi:10.1016/j.jri.2007.03.007
228. Shiroishi M, Kuroki K, Ose T, Rasubala L, Shiratori I, Arase H, et al. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J Biol Chem (2006) 281(15):10439–47. doi:10.1074/jbc.M512305200
229. Feger U, Tolosa E, Huang YH, Waschbisch A, Biedermann T, Melms A, et al. HLA-G expression defines a novel regulatory T-cell subset present in human peripheral blood and sites of inflammation. Blood (2007) 110(2):568–77. doi:10.1182/blood-2006-11-057125
230. LeMaoult J, Krawice-Radanne I, Dausset J, Carosella ED. HLA-G1-expressing antigen-presenting cells induce immunosuppressive CD4(+) T cells. Proc Natl Acad Sci U S A (2004) 101(18):7064–9. doi:10.1073/pnas.0401922101
231. Braud VM, Allan DS, O’Callaghan CA, Soderstrom K, D’Andrea A, Ogg GS, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature (1998) 391(6669):795–9.
232. Deschaseaux F, Delgado D, Pistoia V, Giuliani M, Morandi F, Durrbach A. HLA-G in organ transplantation: towards clinical applications. Cell Mol Life Sci (2011) 68(3):397–404. doi:10.1007/s00018-010-0581-6
233. Moreau P, Adrian-Cabestre F, Menier C, Guiard V, Gourand L, Dausset J, et al. IL-10 selectively induces HLA-G expression in human trophoblasts and monocytes. Int Immunol (1999) 11(5):803–11. doi:10.1093/intimm/11.5.803
234. Yang YP, Chu WJ, Geraghty DE, Hunt JS. Expression of HLA-G in human mononuclear phagocytes and selective induction by IFN-gamma. J Immunol (1996) 156(11):4224–31.
235. Gonzalez-Hernandez A, LeMaoult J, Lopez A, Alegre E, Caumartin J, Le Rond S, et al. Linking two immuno-suppressive molecules: indoleamine 2,3 dioxygenase can modify HLA-G cell-surface expression. Biol Reprod (2005) 73(3):571–8. doi:10.1095/biolreprod.105.040089
236. Lila N, Carpentier A, Amrein C, Khalil-Daher I, Dausset J, Carosella ED. Implication of HLA-G molecule in heart-graft acceptance. Lancet (2000) 355(9221):2138. doi:10.1016/S0140-6736(00)02386-2
237. Bahri R, Hirsch F, Josse A, Rouas-Freiss N, Bidere N, Vasquez A, et al. Soluble HLA-G inhibits cell cycle progression in human alloreactive T lymphocytes. J Immunol (2006) 176(3):1331–9. doi:10.4049/jimmunol.176.3.1331
238. Favier B, LeMaoult J, Lesport E, Carosella ED. ILT2/HLA-G interaction impairs NK-cell functions through the inhibition of the late but not the early events of the NK-cell activating synapse. FASEB J (2010) 24(3):689–99. doi:10.1096/fj.09-135194
239. Le Gal FA, Riteau B, Sedlik C, Khalil-Daher I, Menier C, Dausset J, et al. HLA-G-mediated inhibition of antigen-specific cytotoxic T lymphocytes. Int Immunol (1999) 11(8):1351–6. doi:10.1093/intimm/11.8.1351
240. Liang SY, Baibakov B, Horuzsko A. HLA-G inhibits the functions of murine dendritic cells via the PIR-B immune inhibitory receptor. Eur J Immunol (2002) 32(9):2418–26. doi:10.1002/1521-4141(200209)32:9<2418::AID-IMMU2418>3.0.CO;2-L
241. Ristich V, Liang S, Zhang W, Wu J, Horuzsko A. Tolerization of dendritic cells by HLA-G. Eur J Immunol (2005) 35(4):1133–42. doi:10.1002/eji.200425741
242. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
243. Fournel S, Aguerre-Girr M, Huc X, Lenfant F, Alam A, Toubert A, et al. Soluble HLA-G1 triggers CD95/CD95 ligand-mediated apoptosis in activated CD8(+) cells by interacting with CD8. J Immunol (2000) 164(12):6100–4. doi:10.4049/jimmunol.164.12.6100
244. Contini P, Ghio M, Poggi A, Filaci G, Indiveri F, Ferrone S, et al. Soluble HLA-A,-B,-C and -G molecules induce apoptosis in T and NKCD8(+) cells and inhibit cytotoxic T cell activity through CD8 ligation. Eur J Immunol (2003) 33(1):125–34. doi:10.1002/immu.200390015
245. Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4(+)CD25(high)FOXP3(+) regulatory T cells. Stem Cells (2008) 26(1):212–22. doi:10.1634/stemcells.2007-0554
246. Naji A, Menier C, Morandi F, Agaugue S, Maki G, Ferretti E, et al. Binding of HLA-G to ITIM-bearing Ig-like transcript 2 receptor suppresses B cell responses. J Immunol (2014) 192(4):1536–46. doi:10.4049/jimmunol.1300438
247. Qiu J, Terasaki PI, Miller J, Mizutani K, Cai J, Carosella ED. Soluble HLA-G expression and renal graft acceptance. Am J Transplant (2006) 6(9):2152–6. doi:10.1111/j.1600-6143.2006.01417.x
248. Rajagopalan S, Bryceson YT, Kuppusamy SP, Geraghty DE, van der Meer A, Joosten I, et al. Activation of NK cells by an endocytosed receptor for soluble HLA-G. PLoS Biol (2006) 4(1):e9. doi:10.1371/journal.pbio.0040009
249. Baudhuin J, Migraine J, Faivre V, Loumagne L, Lukaszewicz AC, Payen D, et al. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc Natl Acad Sci U S A (2013) 110(44):17957–62. doi:10.1073/pnas.1221535110
250. Chibueze CE, Yoshimitsu M, Arima N. CD160 expression defines a uniquely exhausted subset of T lymphocytes in HTLV-1 infection. Biochem Biophys Res Commun (2014) 453(3):379–84. doi:10.1016/j.bbrc.2014.09.084
251. da Silva GK, Vianna P, Veit TD, Crovella S, Catamo E, Cordero EAA, et al. Influence of HLA-G polymorphisms in human immunodeficiency virus infection and hepatitis C virus co-infection in Brazilian and Italian individuals. Infect Genet Evol (2014) 21:418–23. doi:10.1016/j.meegid.2013.12.013
252. Castelli EC, Moreau P, Chiromatzo AOE, Mendes CT, Veiga-Castelli LC, Yaghi L, et al. In silico analysis of microRNAS targeting the HLA-G 3’ untranslated region alleles and haplotypes. Hum Immunol (2009) 70(12):1020–5. doi:10.1016/j.humimm.2009.07.028
253. Rousseau P, Le Discorde M, Mouillot G, Marcou C, Carosella ED, Moreau P. The 14 bp deletion-insertion polymorphism in the 3’ UT region of the HLA-G gene influences HLA-G mRNA stability. Hum Immunol (2003) 64(11):1005–10. doi:10.1016/j.humimm.2003.08.347
254. Chen XY, Yan WH, Lin A, Xu HH, Zhang JG, Wang XX. The 14 bp deletion polymorphisms in HLA-G gene play an important role in the expression of soluble HLA-G in plasma. Tissue Antigens (2008) 72(4):335–41. doi:10.1111/j.1399-0039.2008.01107.x
255. Veit TD, Chies JAB. Tolerance versus immune response – microRNAs as important elements in the regulation of the HLA-G gene expression. Transpl Immunol (2009) 20(4):229–31. doi:10.1016/j.trim.2008.11.001
256. Tan Z, Randall G, Fan J, Camoretti-Mercado B, Brockman-Schneider R, Pan L, et al. Allele-specific targeting of microRNAs to HLA-G and risk of asthma. Am J Hum Genet (2007) 81(4):829–34. doi:10.1086/521200
257. Castelli EC, Mendes CT, Deghaide NHS, de Albuquerque RS, Muniz YCN, Simoes RT, et al. The genetic structure of 3’ untranslated region of the HLA-G gene: polymorphisms and haplotypes. Genes Immun (2010) 11(2):134–41. doi:10.1038/gene.2009.74
258. Martinetti M, Pacati I, Cuccia M, Badulli C, Pasi A, Salvaneschi L, et al. Hierarchy of baby-linked immunogenetic risk factors in the vertical transmission of hepatitis C virus. Int J Immunopathol Pharmacol (2006) 19(2):369–78. doi:10.1177/039463200601900213
259. Segat L, Zupin L, Kim HY, Catamo E, Thea DM, Kankasa C, et al. HLA-G 14 bp deletion/insertion polymorphism and mother-to-child transmission of HIV. Tissue Antigens (2014) 83(3):161–7. doi:10.1111/tan.12296
260. Segat L, Catamo E, Fabris A, Padovan L, Morgutti M, Crovella S. HLA-G 3’ UTR haplotypes and HIV vertical transmission. AIDS (2009) 23(14):1916–8. doi:10.1097/QAD.0b013e32832f8104
261. Larsen MH, Zinyama R, Kallestrup P, Gerstoft J, Gomo E, Thorner LW, et al. HLA-G 3’ untranslated region 14-base pair deletion: association with poor survival in an HIV-1-infected Zimbabwean population. J Infect Dis (2013) 207(6):903–6. doi:10.1093/infdis/jis924
262. Cordero EAA, Veit TD, da Silva MAL, Jacques SMC, Silla LMDR, Chies JAB. HLA-G polymorphism influences the susceptibility to HCV infection in sickle cell disease patients. Tissue Antigens (2009) 74(4):308–13. doi:10.1111/j.1399-0039.2009.01331.x
263. Celsi F, Catamo E, Kleiner G, Tricarico PM, Vuch J, Crovella S. HLA-G/C, miRNAs, and their role in HIV infection and replication. Biomed Res Int (2013) 2013:693643. doi:10.1155/2013/693643
264. Turk WJR, Kimani J, Bielawny T, Wachihi C, Ball TB, Plummer FA, et al. Associations of human leukocyte antigen-G with resistance and susceptibility to HIV-1 infection in the Pumwani sex worker cohort. AIDS (2013) 27(1):7–15. doi:10.1097/QAD.0b013e32835ab1f2
265. Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Investig (1997) 99(7):1472–7. doi:10.1172/JCI119308
266. Takaki A, Wiese M, Maertens G, Depla E, Seifert U, Liebetrau A, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med (2000) 6(5):578–82. doi:10.1038/75063
267. Weng PJ, Fu YM, Ding SX, Xu DP, Lin AF, Yan WH. Elevation of plasma soluble human leukocyte antigen-G in patients with chronic hepatitis C virus infection. Hum Immunol (2011) 72(5):406–11. doi:10.1016/j.humimm.2011.02.008
268. Khorrami S, Mohammadpour H, Shahzamani K, Zarif MN, Sharifi AH, Merat S, et al. The relationship between HLA-G and viral loads in non-responder HCV-infected patients after combined therapy with IFN-alpha 2 alpha and ribavirin. Hum Immunol (2015) 76(2–3):181–6. doi:10.1016/j.humimm.2014.12.012
269. Bochud PY, Cai T, Overbeck K, Bochud M, Dufours JF, Mullhaupt B, et al. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. J Hepatol (2009) 51(4):655–66. doi:10.1016/j.jhep.2009.05.016
270. Amiot L, Vu N, Rauch M, L’Helgoualc’h A, Chalmel F, Gascan H, et al. Expression of HLA-G by mast cells is associated with hepatitis C virus-induced liver fibrosis. J Hepatol (2014) 60(2):245–52. doi:10.1016/j.jhep.2013.09.006
271. Carosella ED. The tolerogenic molecule HLA-G. Immunol Lett (2011) 138(1):22–4. doi:10.1016/j.imlet.2011.02.011
272. Kanai T, Fujii T, Unno N, Yamashita T, Hyodo H, Miki A, et al. Human leukocyte antigen-G-expressing cells differently modulate the release of cytokines from mononuclear cells present in the decidua versus peripheral blood. Am J Reprod Immunol (2001) 45(2):94–9. doi:10.1111/j.8755-8920.2001.450205.x
273. Kapasi K, Albert SE, Yie SM, Zavazava N, Librach CL. HLA-G has a concentration-dependent effect on the generation of an allo-CTL response. Immunology (2000) 101(2):191–200. doi:10.1046/j.1365-2567.2000.00109.x
274. Morandi F, Ferretti E, Castriconi R, Dondero A, Petretto A, Bottino C, et al. Soluble HLA-G dampens CD94/NKG2A expression and function and differentially modulates chemotaxis and cytokine and chemokine secretion in CD56bright and CD56dim NK cells. Blood (2011) 118(22):5840–50. doi:10.1182/blood-2011-05-352393
275. Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Risch NJ, et al. A novel MHC class I-like gene is mutated in patients with hemochromatosis. Blood (1996) 88(10):2579.
276. Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell (1998) 93(1):111–23. doi:10.1016/S0092-8674(00)81151-4
277. Costa M, Cruz E, Oliveira S, Benes V, Ivacevic T, Silva MJ, et al. Lymphocyte gene expression signatures from patients and mouse models of hereditary hemochromatosis reveal a function of HFE as a negative regulator of CD8(+) T-lymphocyte activation and differentiation in vivo. PLoS One (2015) 10(4):e0124246. doi:10.1371/journal.pone.0124246
278. Reuben A, Phenix M, Santos MM, Lapointe R. The WT hemochromatosis protein HFE inhibits CD8+T-lymphocyte activation. Eur J Immunol (2014) 44(6):1604–14. doi:10.1002/eji.201343955
279. Rohrlich PS, Fazilleau N, Ginhoux F, Firat H, Michel F, Cochet M, et al. Direct recognition by alphabeta cytolytic T cells of Hfe, a MHC class Ib molecule without antigen-presenting function. Proc Natl Acad Sci U S A (2005) 102(36):12855–60. doi:10.1073/pnas.0502309102
280. Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem (2006) 281(39):28494–8. doi:10.1074/jbc.C600197200
281. Pascolo S, Ginhoux F, Laham N, Walter S, Schoor O, Probst J, et al. The non-classical HLA class I molecule HFE does not influence the NK-like activity contained in fresh human PBMCs and does not interact with NK cells. Int Immunol (2005) 17(2):117–22. doi:10.1093/intimm/dxh191
282. Wang CY, Babitt JL. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol (2016) 23(3):189–97. doi:10.1097/MOH.0000000000000236
283. Enns CA. Possible roles of the hereditary hemochromatosis protein, HFE, in regulating cellular iron homeostasis. Biol Res (2006) 39(1):105–11. doi:10.4067/S0716-97602006000100013
284. Muckenthaler MU. How mutant HFE causes hereditary hemochromatosis. Blood (2014) 124(8):1212–3. doi:10.1182/blood-2014-07-581744
285. Kenneth NS, Mudie S, Naron S, Rocha S. TfR1 interacts with the IKK complex and is involved in IKK-NF-kappaB signalling. Biochem J (2013) 449(1):275–84. doi:10.1042/BJ20120625
286. Ghatreh-Samani M, Esmaeili N, Soleimani M, Asadi-Samani M, Ghatreh-Samani K, Shirzad H. Oxidative stress and age-related changes in T cells: is thalassemia a model of accelerated immune system aging? Cent Eur J Immunol (2016) 41(1):116–24. doi:10.5114/ceji.2015.56973
287. Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol (2001) 33(10):940–59. doi:10.1016/S1357-2725(01)00063-2
288. Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell (2004) 117(3):285–97. doi:10.1016/S0092-8674(04)00343-5
289. Kehl-Fie TE, Skaar EP. Nutritional immunity beyond iron: a role for manganese and zinc. Curr Opin Chem Biol (2010) 14(2):218–24. doi:10.1016/j.cbpa.2009.11.008
290. Nekhai S, Kumari N, Dhawan S. Role of cellular iron and oxygen in the regulation of HIV-1 infection. Future Virol (2013) 8(3):301–11. doi:10.2217/fvl.13.6
291. Greaves DE, Griffiths WJ, Lever AM. Does venesection reduce HIV viral load in patients with hereditary haemochromatosis? Antivir Ther (2013) 18(1):135–8. doi:10.3851/IMP2316
292. Sikorska K, Romanowski T, Stalke P, Izycka Swieszewska E, Bielawski KP. Association of hepcidin mRNA expression with hepatocyte iron accumulation and effects of antiviral therapy in chronic hepatitis C infection. Hepat Mon (2014) 14(11):e21184. doi:10.5812/hepatmon.21184
293. Palmer CS, Cherry CL, Sada-Ovalle I, Singh A, Crowe SM. Glucose metabolism in T cells and monocytes: new perspectives in HIV pathogenesis. EBioMedicine (2016) 6:31–41. doi:10.1016/j.ebiom.2016.02.012
294. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest (2015) 125(1):194–207. doi:10.1172/JCI76012
295. Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab (2011) 14(4):443–51. doi:10.1016/j.cmet.2011.07.014
296. Agarwal N, Balasubramanyam A. Viral mechanisms of adipose dysfunction: lessons from HIV-1 Vpr. Adipocyte (2015) 4(1):55–9. doi:10.4161/adip.29852
297. Ma R, Yang L, Niu F, Buch S. HIV tat-mediated induction of human brain microvascular endothelial cell apoptosis involves endoplasmic reticulum stress and mitochondrial dysfunction. Mol Neurobiol (2016) 53(1):132–42. doi:10.1007/s12035-014-8991-3
298. Cardoso EM, Hagen K, de Sousa M, Hultcrantz R. Hepatic damage in C282Y homozygotes relates to low numbers of CD8+ cells in the liver lobuli. Eur J Clin Invest (2001) 31(1):45–53. doi:10.1046/j.1365-2362.2001.00744.x
299. Macedo MF, Porto G, Costa M, Vieira CP, Rocha B, Cruz E. Low numbers of CD8+ T lymphocytes in hereditary haemochromatosis are explained by a decrease of the most mature CD8+ effector memory T cells. Clin Exp Immunol (2010) 159(3):363–71. doi:10.1111/j.1365-2249.2009.04066.x
300. Arosa FA, Oliveira L, Porto G, da Silva BM, Kruijer W, Veltman J, et al. Anomalies of the CD8+ T cell pool in haemochromatosis: HLA-A3-linked expansions of CD8+CD28− T cells. Clin Exp Immunol (1997) 107(3):548–54. doi:10.1046/j.1365-2249.1997.d01-967.x
301. Rubio JP, Bahlo M, Tubridy N, Stankovich J, Burfoot R, Butzkueven H, et al. Extended haplotype analysis in the HLA complex reveals an increased frequency of the HFE-C282Y mutation in individuals with multiple sclerosis. Hum Genet (2004) 114(6):573–80. doi:10.1007/s00439-004-1095-9
302. Drakesmith H, Chen N, Ledermann H, Screaton G, Townsend A, Xu XN. HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proc Natl Acad Sci U S A (2005) 102(31):11017–22. doi:10.1073/pnas.0504823102
303. de Monye C, Karcher DS, Boelaert JR, Gordeuk VR. Bone marrow macrophage iron grade and survival of HIV-seropositive patients. AIDS (1999) 13(3):375–80. doi:10.1097/00002030-199902250-00010
304. Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature (1987) 326(6114):711–3. doi:10.1038/326711a0
305. She H, Xiong S, Lin M, Zandi E, Giulivi C, Tsukamoto H. Iron activates NF-kappaB in Kupffer cells. Am J Physiol Gastrointest Liver Physiol (2002) 283(3):G719–26. doi:10.1152/ajpgi.00108.2002
306. Lagathu C, Eustace B, Prot M, Frantz D, Gu Y, Bastard JP, et al. Some HIV antiretrovirals increase oxidative stress and alter chemokine, cytokine or adiponectin production in human adipocytes and macrophages. Antivir Ther (2007) 12(4):489–500.
307. Gao Y, Li Z, Gabrielsen JS, Simcox JA, Lee SH, Jones D, et al. Adipocyte iron regulates leptin and food intake. J Clin Invest (2015) 125(9):3681–91. doi:10.1172/JCI81860
308. Rumberger JM, Peters T Jr, Burrington C, Green A. Transferrin and iron contribute to the lipolytic effect of serum in isolated adipocytes. Diabetes (2004) 53(10):2535–41. doi:10.2337/diabetes.53.10.2535
309. Grinspoon S, Thomas L, Miller K, Pitts S, Herzog D, Klibanski A. Changes in regional fat redistribution and the effects of estrogen during spontaneous weight gain in women with anorexia nervosa. Am J Clin Nutr (2001) 73(5):865–9.
310. Hegele RA. Familial partial lipodystrophy: a monogenic form of the insulin resistance syndrome. Mol Genet Metab (2000) 71(4):539–44. doi:10.1006/mgme.2000.3092
311. Reitman ML, Arioglu E, Gavrilova O, Taylor SI. Lipoatrophy revisited. Trends Endocrinol Metab (2000) 11(10):410–6. doi:10.1016/S1043-2760(00)00309-X
312. Choi S, Choi Y, Choi Y, Kim S, Jang J, Park T. Piperine reverses high fat diet-induced hepatic steatosis and insulin resistance in mice. Food Chem (2013) 141(4):3627–35. doi:10.1016/j.foodchem.2013.06.028
313. Hulgan T, Tebas P, Canter JA, Mulligan K, Haas DW, Dube M, et al. Hemochromatosis gene polymorphisms, mitochondrial haplogroups, and peripheral lipoatrophy during antiretroviral therapy. J Infect Dis (2008) 197(6):858–66. doi:10.1086/528697
314. Armitage AE, Stacey AR, Giannoulatou E, Marshall E, Sturges P, Chatha K, et al. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. Proc Natl Acad Sci U S A (2014) 111(33):12187–92. doi:10.1073/pnas.1402351111
315. Minchella PA, Armitage AE, Darboe B, Jallow MW, Drakesmith H, Jaye A, et al. Elevated hepcidin is part of a complex relation that links mortality with iron homeostasis and anemia in men and women with HIV infection. J Nutr (2015) 145(6):1194–201. doi:10.3945/jn.114.203158
316. Kerkhoff AD, Meintjes G, Opie J, Vogt M, Jhilmeet N, Wood R, et al. Anaemia in patients with HIV-associated TB: relative contributions of anaemia of chronic disease and iron deficiency. Int J Tuberc Lung Dis (2016) 20(2):193–201. doi:10.5588/ijtld.15.0558
317. Makui H, Soares RJ, Jiang W, Constante M, Santos MM. Contribution of Hfe expression in macrophages to the regulation of hepatic hepcidin levels and iron loading. Blood (2005) 106(6):2189–95. doi:10.1182/blood-2005-02-0629
318. Sini M, Sorbello O, Civolani A, Demelia L. Hemochromatosis gene mutations: prevalence and effects on pegylated-interferon and ribavirin therapy response in chronic hepatitis C in sardinia. J Clin Exp Hepatol (2012) 2(3):211–7. doi:10.1016/j.jceh.2012.06.004
320. Thursz M. Iron, haemochromatosis and thalassaemia as risk factors for fibrosis in hepatitis C virus infection. Gut (2007) 56(5):613–4. doi:10.1136/gut.2006.113076
321. Matas M, Picornell A, Cifuentes C, Payeras A, Homar F, Gonzalez-Candelas F, et al. Relating the outcome of HCV infection and different host SNP polymorphisms in a Majorcan population coinfected with HCV-HIV and treated with pegIFN-RBV. Int Microbiol (2014) 17(1):11–20. doi:10.2436/20.1501.01.203
322. Coelho-Borges S, Cheinquer H, Wolff FH, Cheinquer N, Krug L, Ashton-Prolla P. Effect of HFE gene polymorphism on sustained virological response in patients with chronic hepatitis C and elevated serum ferritin. Arq Gastroenterol (2012) 49(1):9–13. doi:10.1590/S0004-28032012000100003
323. Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol (2013) 4:321. doi:10.3389/fimmu.2013.00321
324. Fevrier M, Dorgham K, Rebollo A. CD4+ T cell depletion in human immunodeficiency virus (HIV) infection: role of apoptosis. Viruses (2011) 3(5):586–612. doi:10.3390/v3050586
325. Lewis DE, Ulrich RG, Atassi H, Atassi MZ. HLA-DR peptide inhibits HIV-induced syncytia. Immunol Lett (1990) 24(2):127–31. doi:10.1016/0165-2478(90)90023-J
326. Iouzalen N, Andreae S, Hannier S, Triebel F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur J Immunol (2001) 31(10):2885–91. doi:10.1002/1521-4141(2001010)31:10<2885::AID-IMMU2885>3.0.CO;2-2
327. Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur J Immunol (2003) 33(4):970–9. doi:10.1002/eji.200323382
328. Keet IP, Tang J, Klein MR, LeBlanc S, Enger C, Rivers C, et al. Consistent associations of HLA class I and II and transporter gene products with progression of human immunodeficiency virus type 1 infection in homosexual men. J Infect Dis (1999) 180(2):299–309. doi:10.1086/314862
329. Malhotra U, Holte S, Dutta S, Berrey MM, Delpit E, Koelle DM, et al. Role for HLA class II molecules in HIV-1 suppression and cellular immunity following antiretroviral treatment. J Clin Investig (2001) 107(4):505–17. doi:10.1172/JCI11275
330. Ranasinghe S, Cutler S, Davis I, Lu R, Soghoian DZ, Qi Y, et al. Association of HLA-DRB1-restricted CD4(+) T cell responses with HIV immune control. Nat Med (2013) 19(7):930–3. doi:10.1038/nm.3229
331. Julg B, Moodley ES, Qi Y, Ramduth D, Reddy S, Mncube Z, et al. Possession of HLA class II DRB1*1303 associates with reduced viral loads in chronic HIV-1 clade C and B infection. J Infect Dis (2011) 203(6):803–9. doi:10.1093/infdis/jiq122
332. Hohler T, Gerken G, Notghi A, Lubjuhn R, Taheri H, Protzer U, et al. HLA-DRB1*1301 and *1302 protect against chronic hepatitis B. J Hepatol (1997) 26(3):503–7. doi:10.1016/S0168-8278(97)80414-X
333. Kummee P, Tangkijvanich P, Poovorawan Y, Hirankarn N. Association of HLA-DRB1*13 and TNF-alpha gene polymorphisms with clearance of chronic hepatitis B infection and risk of hepatocellular carcinoma in Thai population. J Viral Hepat (2007) 14(12):841–8. doi:10.1111/j.1365-2893.2007.00880.x
334. Apple RJ, Erlich HA, Klitz W, Manos MM, Becker TM, Wheeler CM. HLA Dr-Dq associations with cervical-carcinoma show papillomavirus-type specificity. Nat Genet (1994) 6(2):157–62. doi:10.1038/ng0294-157
335. Fainboim L, Marcos Y, Pando M, Capucchio M, Reyes GB, Galoppo C, et al. Chronic active autoimmune hepatitis in children. Strong association with a particular HLA-DR6 (DRB1*1301) haplotype. Hum Immunol (1994) 41(2):146–50. doi:10.1016/0198-8859(94)90008-6
336. Lundstrom E, Kallberg H, Smolnikova M, Ding B, Ronnelid J, Alfredsson L, et al. Opposing effects of HLA-DRB1*13 alleles on the risk of developing anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum (2009) 60(4):924–30. doi:10.1002/art.24410
337. Brown MA, Wordsworth BP, Reveille JD. Genetics of ankylosing spondylitis. Clin Exp Rheumatol (2002) 20(6 Suppl 28):S43–9.
338. Fainboim L, Velasco MCC, Marcos CY, Ciocca M, Roy A, Theiler G, et al. Protracted, but not acute, hepatitis a virus infection is strongly associated with HLA-DRB1*1301, a marker for pediatric autoimmune hepatitis. Hepatology (2001) 33(6):1512–7. doi:10.1053/jhep.2001.24562
339. Bevilacqua E, Fabris A, Floreano P, Pembrey L, Newell ML, Tovo PA, et al. Genetic factors in mother-to-child transmission of HCV infection. Virology (2009) 390(1):64–70. doi:10.1016/j.virol.2009.05.007
340. Cangussu LO, Teixeira R, Campos EF, Rampim GF, Mingoti SA, Martins-Filho OA, et al. HLA class II alleles and chronic hepatitis C virus infection. Scand J Immunol (2011) 74(3):282–7. doi:10.1111/j.1365-3083.2011.02568.x
341. Mangia A, Gentile R, Cascavilla I, Margaglione M, Villani MR, Stella F, et al. HLA class II favors clearance of HCV infection and progression of the chronic liver damage. J Hepatol (1999) 30(6):984–9. doi:10.1016/S0168-8278(99)80250-5
342. Vejbaesya S, Songsivilai S, Tanwandee T, Rachaibun S, Chantangpol R, Dharakul T. HLA association with hepatitis C virus infection. Hum Immunol (2000) 61(3):348–53. doi:10.1016/S0198-8859(99)00131-7
343. Ali L, Mansoor A, Ahmad N, Siddiqi S, Mazhar K, Muazzam AG, et al. Patient HLA-DRB1* and -DQB1* allele and haplotype association with hepatitis C virus persistence and clearance. J Gen Virol (2010) 91(Pt 8):1931–8. doi:10.1099/vir.0.018119-0
344. Corghi DB, Goncales NS, Marques SB, Goncales FL Jr. Distribution of the human leukocyte antigen class II alleles in Brazilian patients with chronic hepatitis C virus infection. Braz J Med Biol Res (2008) 41(10):884–9. doi:10.1590/S0100-879X2008005000041
345. Biberfeld G, Biberfeld P, Buonaguro F, Charpak N, de The G, Rea MF, et al. Mother-to-child transmission of HIV-1 – meeting of World Federation of Scientists in Erice, Italy, August 2001. Joint Working Group – report of AIDS and infectious diseases PMP, and mother and child health PMP – plea for action with special emphasis on antiretroviral therapy: a scientific and community challenge. Acta Paediatr (2001) 90(11):1337–9.
346. Thobakgale CF, Prendergast A, Crawford H, Mkhwanazi N, Ramduth D, Reddy S, et al. Impact of HLA in mother and child on disease progression of pediatric human immunodeficiency virus type 1 infection. J Virol (2009) 83(19):10234–44. doi:10.1128/JVI.00921-09
347. Mphatswe W, Blanckenberg N, Tudor-Williams G, Prendergast A, Thobakgale C, Mkhwanazi N, et al. High frequency of rapid immunological progression in African infants infected in the era of perinatal HIV prophylaxis. AIDS (2007) 21(10):1253–61. doi:10.1097/QAD.0b013e3281a3bec2
348. Muenchhoff M, Prendergast AJ, Goulder PJ. Immunity to HIV in early life. Front Immunol (2014) 5:391. doi:10.3389/fimmu.2014.00391
349. Prendergast A, Tudor-Williams G, Jeena P, Burchett S, Goulder P. International perspectives, progress, and future challenges of paediatric HIV infection. Lancet (2007) 370(9581):68–80. doi:10.1016/S0140-6736(07)61051-4
350. Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature (2004) 432(7018):769–75. doi:10.1038/nature03113
351. Goulder PJR, Pasquier C, Holmes EC, Liang B, Tang YH, Izopet J, et al. Mother-to-child transmission of HIV infection and CTL escape through HLA-A2-SLYNTVATL epitope sequence variation. Immunol Lett (2001) 79(1–2):109–16. doi:10.1016/S0165-2478(01)00272-3
Keywords: major histocompatibility complex, human leukocyte antigen, classical human leukocyte antigens, non-classical human leukocyte antigens, human immunodeficiency virus, hepatitis C virus and immune regulation
Citation: Crux NB and Elahi S (2017) Human Leukocyte Antigen (HLA) and Immune Regulation: How Do Classical and Non-Classical HLA Alleles Modulate Immune Response to Human Immunodeficiency Virus and Hepatitis C Virus Infections? Front. Immunol. 8:832. doi: 10.3389/fimmu.2017.00832
Received: 18 May 2017; Accepted: 30 June 2017;
Published: 18 July 2017
Edited by:
Guido Poli, Vita-Salute San Raffaele University, ItalyReviewed by:
Roberto Biassoni, Istituto Giannina Gaslini (IRCCS), ItalyMario (Mago) Clerici, Università degli Studi di Milano, Italy
Copyright: © 2017 Crux and Elahi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shokrollah Elahi, elahi@ualberta.ca