 Abdulraouf Ramadan
Abdulraouf Ramadan Sophie Paczesny
Sophie Paczesny- 1Department of Pediatrics, Melvin and Bren Simon Cancer Center, Indiana University, Indianapolis, IN, USA
- 2Department of Microbiology and Immunology, Indiana University, Indianapolis, IN, USA
Hematopoietic stem-cell transplantation (HSCT) is the most potent curative therapy for many malignant and non-malignant disorders. Unfortunately, a major complication of HSCT is graft-versus-host disease (GVHD), which is mediated by tissue damage resulting from the conditioning regimens before the transplantation and the alloreaction of dual immune components (activated donor T-cells and recipient’s antigen-presenting cells). This tissue damage leads to the release of alarmins and the triggering of pathogen-recognition receptors that activate the innate immune system and subsequently the adaptive immune system. Alarmins, which are of endogenous origin, together with the exogenous pathogen-associated molecular patterns (PAMPs) elicit similar responses of danger signals and represent the group of damage-associated molecular patterns (DAMPs). Effector cells of innate and adaptive immunity that are activated by PAMPs or alarmins can secrete other alarmins and amplify the immune responses. These complex interactions and loops between alarmins and PAMPs are particularly potent at inducing and then aggravating the GVHD reaction. In this review, we highlight the role of these tissue damaging molecules and their signaling pathways. Interestingly, some DAMPs and PAMPs are organ specific and GVHD-induced and have been shown to be interesting biomarkers. Some of these molecules may represent potential targets for novel therapeutic approaches.
Introduction
Hematopoietic cells that are capable of self-renewing and reconstituting all types of blood cells along with allogeneic donor T-cells can be used to treat numerous malignant and non-malignant lethal diseases, including leukemias, lymphomas, inherited genetic diseases, and immune deficiencies. However, the success of allogeneic hematopoietic stem-cell transplantation (HSCT) is unfortunately limited by transplant-associated toxicities related to the applied conditioning regimens and the immunologic consequence of donor T-cell recognition of recipient alloantigens, which causes graft-versus-host disease (GVHD). Acute GVHD is characterized by selective tissue damage to the mucosa, particularly of the skin, gastrointestinal (GI) tract, and liver. Other tissues and organs such as the bone marrow, thymus, lungs (1, 2), and brain (3) have also been shown to be potential GVHD targets. Chronic GVHD not only targets organs, including the ones mentioned above, but also can damage the connective tissue and exocrine glands.
The pathogenesis of GVHD can be summarized in three sequential steps: first, the conditioning regimen damages the tissues, causing production of danger signals, which are detailed in this review, and pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, and IL-6. The culmination of these events is what the field refers to as the “cytokine storm,” which activates host antigen-presenting cells (APCs) and the newly infused donor T-cells. The second phase involves proliferation and differentiation of donor T-cells in response to host APCs, which results in rapid intracellular biochemical cascades that induce production of T helper (TH) 1, TH17 (for CD4 T-cells), T cytotoxic (TC) 1, and TC17 cells (for CD8 T-cells) that secrete cytokines such as interferon (IFN)-γ, IL-2, IL-17, and TNF-α. The last step is a complex cascade of cellular mediators and soluble inflammatory molecules that work synergistically to amplify local tissue injury. These mediators further amplify inflammation and target tissue destruction. GVHD is also characterized by an imbalance between the effector T-cells and the regulatory T-cells (Tregs). At all of these steps, the inflammatory cascade and various types of tissue damage lead to the release of biomarkers of GVHD into the blood, the detection of which can be achieved via blood tests. Markers such as elafin (skin-specific), regenerating islet-derived 3-alpha (REG3α, gut-specific), suppressor of tumorigenicity 2 (ST2, a member of the IL-1 receptor family, binding IL-33), and others are detailed in this review. Figure 1 summarizes these events.
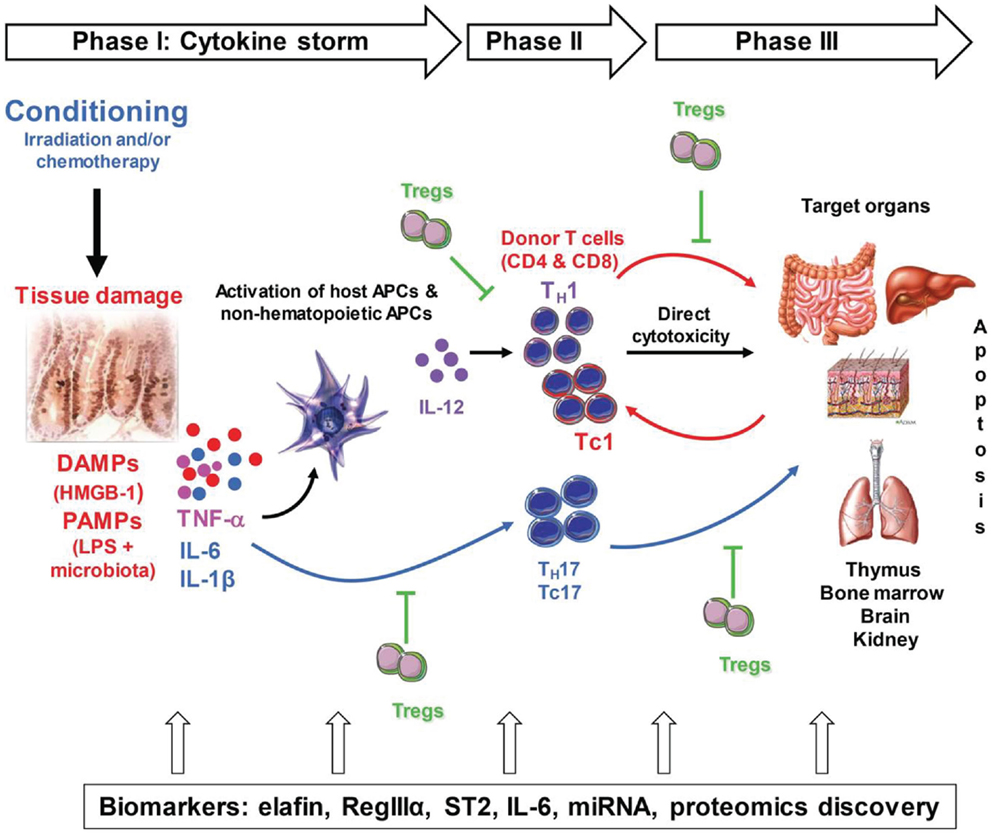
Figure 1. Pathogenesis of acute GVHD. Conditioning by irradiation and/or chemotherapy causes tissue damage. Damaged tissues and cells release DAMPs (HMGB-1), PAMPs (LPS) from gut microbiota as well as inflammatory cytokines such as IL-1β, IL-6, and TNF-α, which contribute to the “cytokine storm.” These are the first danger signals that activate host APCs, which activate and polarized donor T-cells toward pathogenic T-cells (TH1 and TH17 for CD4 and TC1, TC17 for CD8). Activated pathogenic T-cells infiltrate target organs (i.e., GI tract, liver, skin) and amplify local tissue destruction. The presence of regulatory T-cells (Tregs) helps reduce GVHD severity through the inhibition of pathogenic cells activation and/or expansion at early or further phases of GVHD. Some of these DAMPs and PAMPs such as elafin (skin-specific), regenerating islet-derived 3-alpha (REG3α, gut-specific), and suppressor of tumorigenicity 2 (ST2, a member of the IL-1 receptor family, binding IL-33) have been shown to be biomarkers.
Danger Signal Proteins Following HSCT
Following conditioning (radiation and/or chemotherapy), exogenous and endogenous “danger” signals released from damaged tissues orchestrate mesenchymal, epithelial, and immune cellular communications to attempt to restore homeostasis. These danger’ signals induce rapid changes in redox-sensitive proteins, leading to the activation of nuclear transcription factors including nuclear factor (NF)-κb (4), early growth response factor (Egr1), and activator protein (AP)-1 (5), which are heavily involved in inflammatory cytokine production. Indeed, both radiation/chemotherapy effects and pro-inflammatory cytokines generate free reactive oxygen species (ROS) and reactive nitrogen species (RNS) such as superoxide, nitric oxide, hydroxyl radicals, peroxynitrite, and their products (6). Furthermore, inflammatory cytokines, including those of the IL-1 family, and TNF-α (7) require ROS for their activation. In contrast, anti-inflammatory cytokines [transforming growth factor (TGF)-β, IL-10, and IL-4] inhibit ROS/RNS-mediated effects and display anti-oxidative properties (8–10). Therefore, inflammatory and anti-inflammatory cytokines mutually influence each other through the production of ROS/RNS. Recently, it has been shown that mice exhibiting overexpression of ROS (mice deficient for negative regulator of ROS) develop more severe disease in an experimental autoimmune encephalomyelitis model (11), whereas mice deficient in ROS (NOX2 knockout mice) show less infiltration of neutrophils into the ileum and less tissue damage, leading to less severe GVHD (12).
Damage-associated molecular patterns (DAMPs) include exogenous pathogen-associated molecular patterns (PAMPs) as well as endogenous alarmins, each of which play a crucial role in the initiation of GVHD and are described in detail in subsequent paragraphs.
Exogenous PAMPs during Inflammation and GVHD
Early studies in allogeneic murine chimeras induced by radiation showed that the mortality due to “secondary disease,” later called GVHD, was significantly reduced in germ-free mice compared to conventional mice (13). Treating conventional mice with antibiotic prophylaxis also significantly delay mortality in comparison to that in the control group (14). Clinical studies have demonstrated the efficiency of GI decontamination in reducing GVHD (15, 16). PAMPs are conserved microbial molecules released by invading microorganisms (17, 18). They recognize pattern recognition receptors (PRRs), primarily toll-like receptors (TLRs), members of the cytosolic retinoic acid-inducible gene-I-like helicase family (19), and receptors with a nucleotide-binding domain (NOD) and leucine-rich repeats (NOD-like receptors, NLRs). These activate different pathways, resulting in the production of inflammatory cytokines through NF-κb activation. The main link between the PRRs and NF-κb activation/cytokine production during GVHD is the cytoplasmic myeloid differentiation primary response protein 88 (MyD88) in APCs (20). APCs, particularly recipient dendritic cells (DCs), primed by the conditioning are potent sensors of PAMPS, which leads to their activation and augmented major histocompatibility complex (MHC) presentation to T-cells (21). Similar to TLRs, the NLR family has an impact on GVHD. The absence of NOD2 in recipients results in more severe GVHD in both MHC-mismatched and MHC-matched models (22). Single nucleotide polymorphisms of NOD2/CARD15 have also been associated with severe GVHD in patients receiving stem cells from either human leukocyte antigen (HLA)-identical or unrelated donors (23).
Alarmins and Endogenous DAMPs during Inflammation and GVHD
The term “alarmins” is used to describe the endogenous molecules equivalent to PAMPs. They rapidly produce a danger signal after non-programed cell death or a specific modality of programed cell death (24). More recently, they are increasingly referred to as DAMPs, in reference to the term PAMPs, because they share structural and functional similarities with exogenous PAMPS. However, this definition of DAMPs is not used consistently, and sometimes endogenous alarmins and exogenous PAMPs are classified together as DAMPs.
High mobility group box 1
High mobility group box 1 (HMGB-1) is a nuclear protein that binds to nucleosomes and promotes DNA bending (25). It is expressed in most cells but at varying levels, and it is also present in the extracellular milieu after non-programed cell death (26, 27). It can be released in tumors (28). When under oxidative stress induced by irradiation, HMGB-1 acts as a DAMP and can mediate endotoxin lethality in mice (29). Persistence of a high level of HMGB-1 has been reported in chronic inflammatory disorders such as autoimmune disease (30), ischemia, and reperfusion injury (31). Targeted knockout or inhibition of HMGB-1 was shown to be able to increase apoptosis and suppress pancreatic cancer cell growth (32, 33). HMGB-1 has chemotactic activity toward monocytes, macrophage, neutrophils, and DCs (34, 35) in response to inflammatory cytokines such as TNF-α, IL-1, and IFN-γ (29, 36) as well as via reaction with TLR2/4 (37). It has been reported that certain donor HMGB-1 polymorphisms are associated with increased chronic GVHD, whereas recipient with the 2351 insT showed reduced risk of grade II–IV acute GVHD (38). A recent study showed that irradiation downregulates the HMGB-1 receptor (Siglec-G), which makes HSCT recipients more susceptible to GVHD. Moreover, addition of CD24 (Siglec-G ligand) alleviated GVHD in both minor and major-mismatched murine models of allogeneic bone marrow transplantation (39).
S100 proteins
S100 proteins are among more than 20 members of a family of low molecular weight proteins (9–13 kDa). They are produced as monomers and form dimers or multimers spontaneously (40, 41) following calcium binding protein activation (42). The most studied members in this family are S100A7, S100A8, S100A9, S100A12, and S100A15, which are mainly expressed in phagocytes, where they show high antimicrobial activity (43). They are thus released in inflammatory sites (44). Both S100A7 and S100A15 are induced by TH1, TH17, and TH22 cytokines and play an important role in psoriasis pathogenicity and act as alarmins in priming keratinocytes, thereby enhancing IL-6, IL-8, and TNF-α production and amplifying inflammation in the skin (45). On the other hand, S100A8 and S100A9 show a pathogenic role in lung inflammation mediated by neutrophil recruitment (46). Another member, S100A12, has been shown to be positively correlated with increased Escherichia coli colonies in infants via disturbance of the homeostasis between the intestinal microbiome and host immunity (47). No studies have shown the impact of this family in GVHD yet, but all of the above findings suggest that these DAMPs may play a role in different types of tissue damage and the pathology of skin and GI GVHD. Moreover, proteomic analysis of saliva showed that healthy controls have low or non-detectable levels of S100A9 and S100A8 proteins, whereas patients after HSCT without GVHD showed augmented levels of these proteins. Interestingly, patients with GVHD show higher levels of S100A8 and S100A9 than patients without GVHD (48). Moreover, a new study found that released S100 proteins are involved in the pathogenesis of GI GVHD through stimulation of monocytes, which enhance TH17 cells in patients receiving allogeneic HSCT (49).
Elastase inhibitors (endogenous proteases inhibitors)
During infection, the activity of locally produced mucosal alarm antiproteases, such as elafin and secretory leukocyte peptidase inhibitor (SLPI), may add an extra edge to the host defense (50). SLPI and elafin alarm antiproteases have been isolated and characterized under a variety of names in adult and fetal tissues (51). They belong to the family of whey acidic proteins (WAPs). Elafin was isolated from the skin of psoriasis patients (52) and is produced by both epithelial cells and immune cells. Alarm antiproteases are generated locally in areas of infection or neutrophil infiltration and are upregulated by pathogen- and inflammation-associated factors, including cytokines and neutrophil elastases (NEs) (53). Elafin and SLPI have been proposed to possess “defensin/cathelicidin-like” properties (54). It has been shown that in the 117 amino acids encoded by the elafin gene, the first 22 amino acids represent hydrophobic signal peptide. Elafin is produced as a 9.9-kDa full-length non-glycosylated cationic protein (55, 56). Elafin expression in vitro can be enhanced by adding inflammatory cytokines (IL-1 and TNF-α) to cultured bronchial and alveolar epithelial cells (57). These cytokines induce a similar increase in elafin expression by keratinocytes in vitro (58). Interestingly, these cytokines increase expression of elafin more than that of SLPI in vitro (57). Thus, elafin may have greater significance during an inflammatory challenge to the lung, in keeping of the notion that elafin mRNA expression in bronchial epithelial cells is increased by free NE, which is found in abundance during inflammatory events (53, 59). In addition to its NE inhibitory and immunomodulatory activities, elafin possesses broad-spectrum antibacterial, antiviral, and antifungal properties. Elafin expression is increased in the plasma of patients with skin GVHD compared to that of patients without GVHD following allogeneic HSCT without T-cell depletion. Moreover, elafin concentrations have been positively correlated with the grade of skin GVHD. Importantly, elafin is not elevated in rashes caused by conditions other than GVHD, making it a specific biomarker for skin GVHD (60). This is because elafin is induced by inflammatory cytokines, which mediate GVHD by targeting keratinocytes (61).
Defensins
The defensins are short peptides with a characteristic β-sheet-rich fold and, like SLPI and elafin, are cysteine-rich, containing multiple disulfide bonds (62, 63). Defensins are classified into three subfamilies (α, β, θ). The α-defensins are neutrophil peptides [human neutrophil peptides (HNPs) 1–4]. In humans, α-defensins [human defensin (HD)-5 and HD-6] are mostly expressed in Paneth cells in the small intestine (64, 65). HNPs 1–3 are expressed in B-cells, γδ T-cells, natural killer (NK) cells, and DCs. α-defensins exhibit wide spectrum antimicrobial coverage against Gram negative and Gram positive organisms and also have some antifungal activity against Candida albicans, as one example. β-defensins (four diverse human β-defensins, HBD 1–4) have been classified based on their function and genomic targeting. They are expressed by epithelial cells, macrophages, macrophage derived DCs, and monocytes (66). In murine models of haploidentical or minor mismatched BMT, it has been shown that the reduction in α-defensins following GVHD damages Paneth cells, resulting in a loss of variation in the gut microbiota composition (67). Consistently, clinical data show significant increases in Lactobacillales and decreases in Clostridiales in patients with GVHD after allogeneic HSCT (68, 69). In this cohort, overall survival was significantly worse in patients with lower intestinal diversity at engraftment as compared to intermediate and high diversity groups, respectively, even after adjustment for other clinical predictors (69).
Cathelicidins
Cathelicidins, which are recognized as a constitutive component of myeloid-derived cells (70), are the second major family of antimicrobial peptides (AMPs). Cathelicidins are highly heterogeneous (71–73), and as mediators of an effective system of host defense, they provide protection to intestinal epithelial cells against invading microorganisms and control the overgrowth of commensal bacteria (54). The human cathelicidin hCAP-18/LL-37 has a C-terminal peptide of α-helical type, which is present in all mammals. The widespread presence of this C-terminus suggests that the α-helical cathelicidin type is the progenitor molecule of this family (74). Even though it is considered a neutrophil-specific constituent distributed in all tissues, which is liberated as LL-37, it has also been shown to be produced by different immune cells, such as NK cells, γδ T-cells, B-cells, monocytes, mast cells, and immature neutrophils (75–77). LL-37 plays an important role in the prevention of oral bacterial infection, and it was found to be down-regulated in gut biopsies of patients infected with Shigella (78, 79). LL-37 expression is also induced after skin injury (80). In addition, its overexpression in human bronchial xenografts preserves the cystic fibrosis-specific bacterial killing defect (81). Recently, LL-37 has been shown to protect against arthritis in murine models through IL-32 suppression, and this observation was confirmed in human peripheral blood mononuclear cells (PBMCs) through decreases in pro-inflammatory cytokines such as TNF-α and IL-1β (82). In contrast, it also mediates immune cell recruitment by promoting chemotaxis, autophagy, and phagocytosis (83–87). It also enhances the adaptive immune responses (88, 89). LL-37 expression is decreased in Crohn’s disease and dermatitis, but elevated in psoriasis and systemic lupus erythematosus (90–92).
The regenerating protein family
The regenerating (Reg) III proteins were discovered in 1984 in pancreatitis experimental models (93). Later studies showed the presence of homologous proteins in human and mice (94–96). There are three different types of Reg III genes in mice (97) and all type III Reg genes appear to have diverged from a common ancestral gene. RegIIIα, RegIIIβ, and RegIIIγ have 60–70% homology and are all expressed in the intestine (97, 98). Reg1α expression is increased in inflamed colonic mucosa and correlates with IL-22 expression (99). In inflammatory bowel disease (100), it has been reported that Reg3α in humans or RegIIIγ, the homologous mouse protein, has an antimicrobial function, controlling bacterial proliferation (101, 102). In addition, following skin injury, RegIIIα expression increases in keratinocytes in response to IL-17 (103). In a haploidentical murine model of GVHD, it has been shown that RegIIIγ is upregulated, and this upregulation is not due to radiation-induced damage but due to the allogeneic response (104). Reg3α concentrations in plasma are increased by threefold in patients with GI GVHD compared to all other patients, including patients with non-GVHD enteritis following allogeneic HSCT. Reg3α expression is also positively correlated with GI GVHD grade and volume of diarrhea, suggesting that Reg3α represents a biomarker for the diagnosis of GI GVHD (105).
Heat shock proteins
The heat shock proteins (HSP) are a family of proteins that have an essential role as molecular chaperones, facilitating protein folding and intracellular transport (106). Expression of these proteins increases under various stress conditions such as infection, hypoxia, trauma, or exposure to toxic drugs, and high levels of HSPs are released by necrotic cells (107, 108). HSP60 (60 kDa) is expressed mainly in mitochondria and on the cell surface of monocytes after IFN-γ stimulation as well as on apoptotic T-cells (109). HSP60 also is overexpressed in intestinal epithelium in Behcet’s disease and in keratinocytes in skin lesions (110, 111). CD14/TLR acts as a coreceptor for HSPs (112). HSP70 expression correlates positively with GVHD grade (113). Another study showed that antibodies to 70 and 90-kDa HSPs are associated with GVHD in patients receiving allogeneic peripheral blood stem-cell (PBSC) transplantation (114).
Heparan sulfate proteoglycans
Heparan sulfate proteoglycans are proteins carrying one or more covalently bound heparan sulfate chain, a large anionic polysaccharide of glycosaminoglycan. These proteins show considerable diversity and interactive properties and are widely found in tissues within the extracellular matrix and are also found intracellularly (115). Functionally, heparin sulfate proteoglycans play critical roles in (i) mediating the formation of chemokine gradients for cell migration (116, 117); (ii) protecting cytokines such as IFN-γ against proteolysis (118); (iii) controlling the diffusion of their ligands (119). Heparan sulfate has also been shown to act as an endogenous TLR4 ligand and is a potent stimulator of T-cell alloreactivity in vitro (120). This action is dependent of the TLR4 pathway in DCs, but not in T-cells. Serum levels of heparan sulfate are elevated at the onset of GVHD and correlate to disease severity in an allogeneic mouse model and in patients received allogeneic HLA-matched HSCT (121).
Adenosine triphosphate
Adenosine triphosphate (ATP) is an essential purine base required for almost all physical responses. Extracellular ATP released from injured but not apoptotic cells is secreted rapidly after irradiation and mediates cellular responses through activation of purinergic receptors, which activate calcium channels (122). ATP activates caspase-1 and produces IL-1β, depending on the NLRP3 inflammasome (123). In GVHD, ATP is an endogenous danger signal released from necrotic cells (124, 125). Accumulation of ATP leads to upregulation of co-stimulatory molecules (e.g., CD80, CD86) in vitro and in vivo, which activates pathogenic donor T-cells and reduces the number of Tregs, resulting in greater production of inflammatory cytokines and aggravation of GVHD severity in an allogeneic murine model (126).
Uric acid
Uric acid is a metabolite of purine nucleotides in humans and has been described as a DAMP released from dying cells (24). Soluble uric acid induces the production of inflammatory cytokines such as monocyte chemotactic protein-1 in rat vascular smooth muscle cells (127) and is recognized by TLR2 and TLR4 and signaled through MyD88 (128). It can also be sensed by NLRP3, a member of the NLRs, and induces IL-1β production via caspase-1 activation (129). Injection of uric acid into mice along with antigen results in activation of CD8 T-cells, whereas abolition of uric acid inhibits the cytotoxicity of T-cells (130). In vitro addition of uric acid upregulates co-stimulatory molecules on bone marrow-derived DCs and leads to T-cell activation (131). In patients with acute GVHD showing high levels of uric acid in serum during the pre-transplantation period, inhibition of uric acid activity may be one aspect of the treatment strategy for reducing the severity of GVHD as is discussed later in this review (132).
Danger Signaling Pathways in GVHD
Individual danger signaling pathways involved in GVHD are detailed below and summarized in Table 1. Figure 2 summarizes TLRs and IL-1 receptor family signaling pathways. Figure 3 summarizes PAMPs and DAMPs common pathogen recognition receptors and their interactions with the signaling pathways.
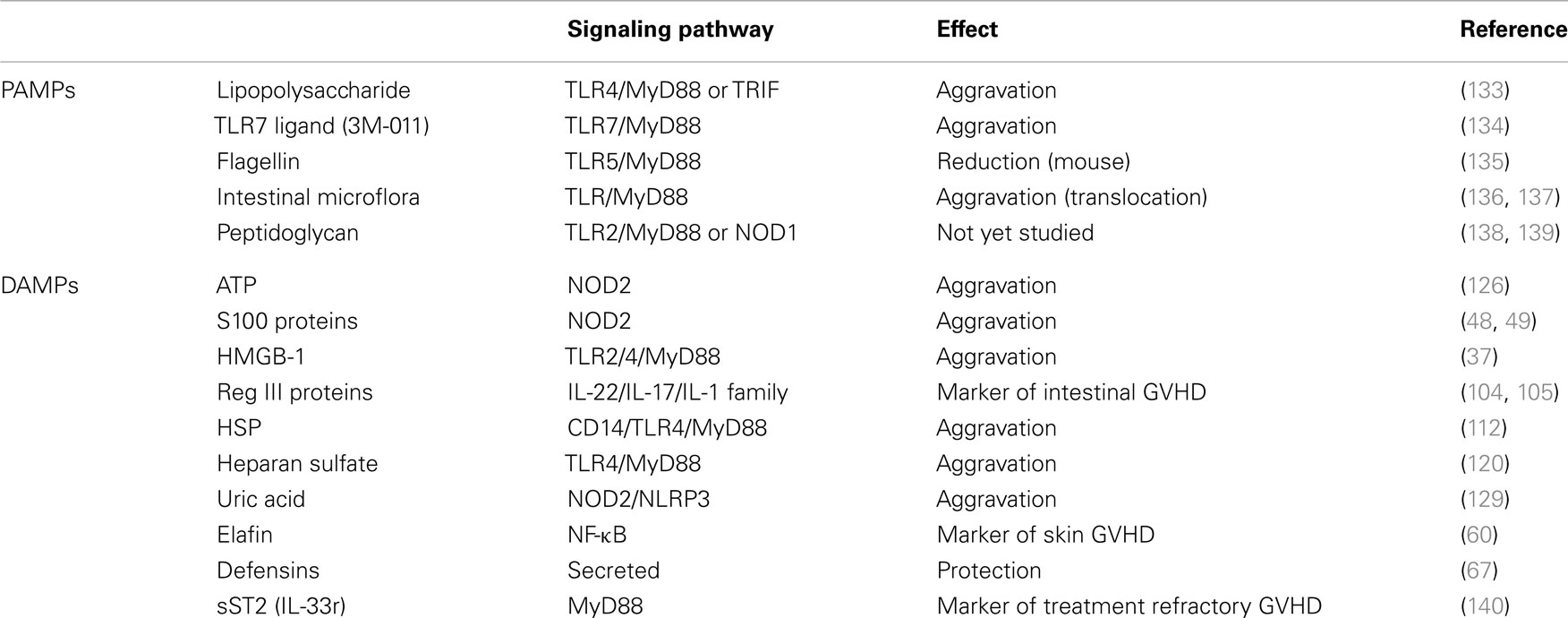
Table 1. GVHD-related PAMPs and DAMPs along with their signaling pathways and effects in GVHD.
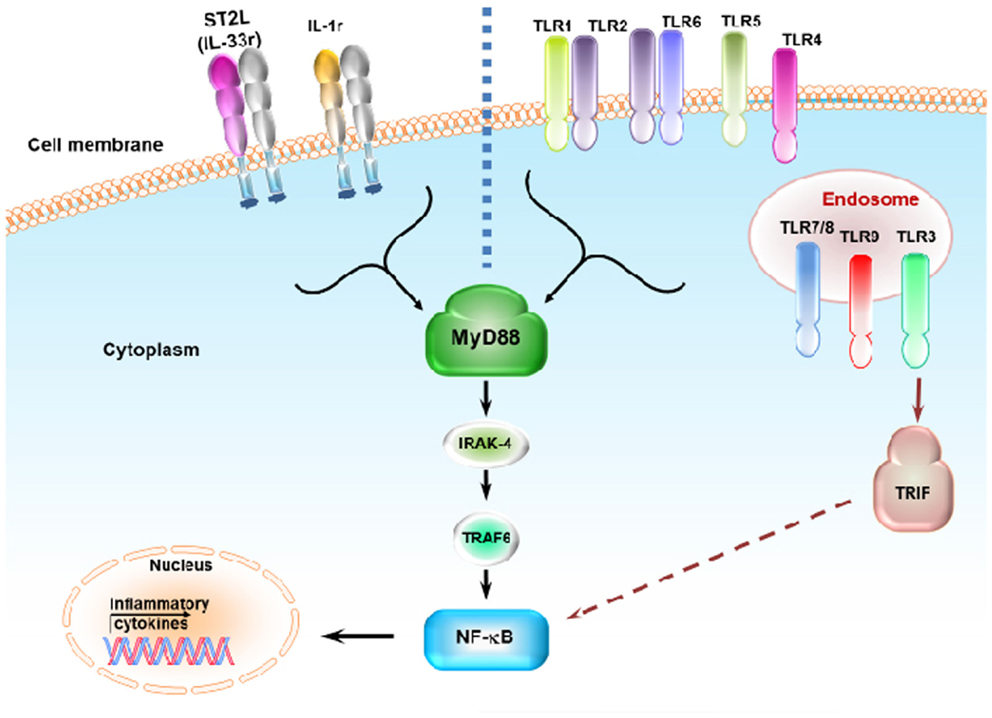
Figure 2. Toll-like receptors and IL-1 receptor family signaling pathways. ST2L (IL-33r) and IL-1r signal through the MyD88, IRAK4, and TRAF6 pathway. ST2L and IL-1r share this pathway with most TLRs. Binding of ST2L, IL-1r, and TLRs activates NF-κb, resulting in the release of inflammatory cytokines. Most TLRs signal through MyD88 expect for TLR3, which signals through the TRIF pathway. TLR4 can signal through both MyD88 and TRIF. TLR3, TLR7/TLR8, and TLR9 are expressed in the endosome while other TLRs are expressed on the cell surface. TLR1 and TLR6 recognize their ligand with TLR2 heterodimers.
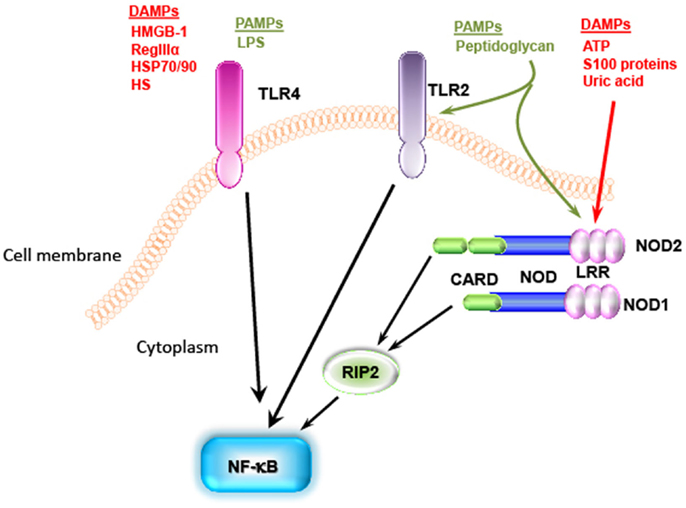
Figure 3. Pathogen-associated molecular patterns and DAMPs share the same pathogen recognition receptors. TLR4 recognizes LPS from Gram negative bacteria as well as DAMPS [i.e., HMGB-1, heat shock proteins (HSP)70/90, and heparan sulfate]. NOD can recognize peptidoglycan, which is known as TLR2 ligand, and some DAMPs such as ATP, S100 proteins, and uric acid. These signaling pathways all end with activation of NF-κb.
TLR Signaling
The TLR family proteins are transmembrane receptors that were first described in humans in 1994 (141). In 1997, TLR4, which senses lipopolysaccharide (LPS) and activates innate cells, was described (142). TLRs play a key role in innate immunity by recognizing PAMPs. TLRs also recognize endogenous DAMPs (143). This family consists of 10 functional members in humans, where they are expressed on hematopoietic and non-hematopoietic cells (144). Their expression is either on the cell surface (TLR1, TLR2, TLR4, TLR5, and TLR6) or in endosomes (TLR3, TLR7, TLR8, and TLR9). Different TLRs recognize different PAMPs or DAMPS specifically. Among the surface TLRs, TLR2 recognizes Gram positive lipoprotein [such as peptidoglycan (PGN)], and TLR4 with its coreceptor MD-2 recognizes the Gram negative component of the cell wall LPS. TLR5 recognizes bacterial flagellin, and TLR1 and TLR6 form a dimer with TLR2 to recognize PGN. The other members of the family are expressed in endosomes and recognize viral or bacterial nucleic acid components: TLR3 recognizes double-stranded viral RNA; TLR7 and TLR8 recognize single-stranded viral RNA; and TLR9 recognizes bacterial or viral RNA and CpG-containing DNA (145–152). TLR4 also recognizes DAMPs such as the HSP family (HSP70 and HSP90) and heparan sulfate proteoglycans. Upon activation, TLRs transmit the signal through adaptor molecules shared with the IL-1R family myeloid differentiation factor 88 (MyD88) with the exception of TLR3, which signals through toll-IL-1 receptor (TIR) domain-containing adapter inducing IFN-β (TRIF). Only TLR4 can signal through both MyD88 and TRIF (143). Stimulation of TLRs activates NF-κB through a signaling cascade that is mostly MyD88-dependent. TIR adaptor protein (TIRAP) and IL-1 receptor-associated kinase 4 (IRAK4), which interact with TNF receptor-associated protein 6 (TRAF6), result in activation of mitogen-activated protein kinase (MAPK) and NF-κB signaling, leading to inflammatory cytokine production (153). On the other hand, TLR3 signaling, which is independent of MyD88 but dependent on TRIF, requires translocating chain-associating membrane protein (TRAM) for this signaling and activates TRAF3 (154). Stimulation of innate cells (e.g., DCs, macrophage, monocytes, neutrophils, and mast cells) via TLRs results in maturation, upregulation of MHC expression as well as that of co-stimulatory molecules (CD80/CD86, and CD40), and production of pro-inflammatory cytokines such as IL-12, TNF-α, and IL-6 (138, 155–157). This results in T-cell activation and proliferation. The inflammatory environment generated by TLR responses polarizes the response toward TH1, suggesting that the absence of TLR signaling may reduce GVHD severity. This was shown in a murine experimental model; mice deficient for TLR4 showed less severe GVHD than wild-type mice (133). Moreover, clinical studies showed association of TLR4 polymorphisms with GVHD severity (158). Other clinical studies of patients who received Tregs with their allo-HSCT showed significant augmentation of TLR5 expression, which correlated with GVHD severity (159). In contrast, an allogeneic BMT murine model demonstrated that treating mice with flagellin improves overall survival and suppresses GVHD (135). Likewise, TLR7 signaling was shown to increase donor T-cell infiltration when using R-848, a TLR7 ligand. The timing of administration of TLR7 ligands is important for GVHD pathogenesis as when using another TLR7 ligand (3M-011), as repetitive administration aggravates GVHD in an allogeneic mouse model (134). TLR9 signaling was shown to be important for GVHD pathogenicity as TLR9-deficient mice developed less severe GVHD and experienced increased survival. This effect was achieved through the production of less IFN-γ by host APCs (136, 160). Moreover, repeated treatment with CpG (a TLR9 ligand) increased mortality and GVHD severity (134). One clinical study showed no differences in the incidence and severity of GVHD between patients with gene variants associated with TLR9 reduction and controls (161), but another study reported severe acute GVHD when patients received stem cells from an unrelated donor with the A1174 gene. The T1635C variant was found to have a protective effect against GVHD (162). Thus, the roles of TLRs in GVHD remain controversial, according to differences in the timing of administration, experimental settings, microbiota constitution, and other alternative danger signaling pathways.
NLR Signaling
Another family associated with PRRs is the cytoplasmic NLR family, which was first described in 1999 and 2001 studies of NOD1 (CARD 4) (163) and NOD2 (CARD 15) (164), respectively. NOD1 was shown to bind to d-gamma-glutamyl diaminopimelic acid derived from peptidoglycan (139), and NOD2 binds to muramyl dipeptide (MDP) (165). Activation of NOD1 and NOD2 enables the recruitment of kinase receptor-interacting protein 2 (RIP2) (RICK) through caspase recruitment domain (CARD)–CARD homotypic interaction (166). RIP2 engagement by NOD receptors leads to ubiquitination of K63-linked by cellular inhibitors of apoptosis (cIAP)1 and cIAP2 (167), followed by recruitment of the TAK1/TAB2/TAB3 kinase complex to RIP2. X-linked inhibitor of apoptosis protein (XIAP) interacts with RIP2 and results in recruitment of the platform for the linear ubiquitination assembly complex (LUBAC), which mediates NF-κB activation. This signaling pathway converges on the induction of pro-inflammatory cytokines and initiates the innate immune response (168). Mutation in NOD2 leads to an inability for NF-κB activation after MDP stimulation in intestinal epithelial cells (169). However, NOD1 and NOD2 are able to induce direct autophagy through interaction with ATGL16L1, and this mechanism is independent of both RIP2 and NF-κB (170). NOD2 showed the capacity to recognize single-stranded RNA virus and to elicit interferon regulatory factor 3 activation, producing IFN-β through interaction with anti-viral signaling factor mitochondrial adaptor proteins (171). NOD2 also plays an important anti-viral role via CD8 T-cell priming in influenza A virus infection (172). Other studies of the expression of NOD2 on CD4 T-cells showed that the absence of NOD2 signaling impairs TH1 proliferation and response upon Toxoplasma gondii infection (173). Interestingly, the NLR family not only recognizes PAMPs but also recognizes DAMPs, such as ATP and DNA released from dying cells and uric acid (174, 175). Activation of the NLR family occurs via the inflammasome, cleavage of caspase-1, and production of active IL-1β and IL-18 (176). In GVHD models, the absence of NOD2 on donor cells has no impact on GVHD pathogenicity, whereas NOD2 deficiency on only hematopoietic and not on non-hematopoietic recipient cells aggravates GVHD. In vitro experiments showed that the absence of NOD2 on DCs increases the expansion of alloreactive T-cells, indicating that NOD2 negatively regulates DC function and activity (22). Studies in humans proved that the relationship between this signaling pathway and GVHD is more complex. Polymorphisms in NOD2/CARD15 were identified as a risk factor in HSCT involving a HLA-identical sibling donor (177, 178), whereas in other studies of HLA-unrelated transplants, such polymorphisms adversely impacted disease relapse but not GVHD (179–181).
Overall, interactions between TLRs and NLRs have a crucial role in APC stimulation, subsequent alloreactive T-cell recruitment, and activation and aggravation of GVHD.
IL-1 Receptor Signaling
The absence of TLR signaling in TLR-deficient mice does not completely abrogate GVHD (160, 182, 183). The counterpart of TLR in the TIR family is highly involved in different phases of GVHD pathogenesis. IL-1 is one of the most active pro-inflammatory cytokines in inflammatory diseases. The IL-1 receptor family contains 10 members in total. The first two members are IL-1α and IL-1β, both of which are synthesized as 31-kDa precursors (pro-IL-1α and pro-IL-1β). They are enzymatically cleaved into N-terminal prodomains (184) and are agonists for the IL-1 type 1 receptor (IL-1R1) (185). IL-1α and IL-1β seem to activate similar cellular responses upon binding IL-1R1 in vitro, and the main biological difference between these proteins seems to be associated with the source and presentation of the cytokine in vivo.
IL-1α has a central role in mediating sterile inflammation induced by cell necrosis, and it seems to be the dominant agonist in a response dependent on IL-1R1 and MyD88 signaling (186). IL-1α also has been shown to be released from dying cells (187, 188). In HSCT, IL-1α is also a dominant mediator of CD4 T-cell activation mediated by allogeneic endothelial cells expressing HLA-DR (189). IL-1β increases the expression of adhesion molecules on vascular endothelium and enhances the expression of chemokines on T-cells, thus attracting blood-borne inflammatory cells into target tissues. IL-1β also stimulates mucosal myofibroblasts and matrix metalloproteinase (MMP) release, causing tissue mucosal destruction (190–192). The IL-1 receptor antagonist (IL-1Ra) competes with the two agonist molecules for IL-1R1 binding. All three genes are located in a cluster on chromosome 2q. The fourth member of this family is IL-18, the gene for which is located on chromosome 11q (193). IL-18 was identified as a factor promoting IFN-γ production and activates TNF-α receptor-associated factor (194). It has been reported that many hematopoietic and non-hematopoietic cells produce IL-18 in inflammatory conditions (195). In HSCT, it was shown that IL-18 levels in plasma increase with acute GVHD in both human and animal models (196–199).
IL-33 and Its Receptor (ST2) Signaling
IL-33 (also known as IL-1F11) was identified as a new member of the IL-1 cytokine family (200). Similar to IL-1α and HMGB-1, IL-33 has dual functions, acting both as a traditional cytokine and as an intracellular NF with transcriptional regulatory properties (201). IL-33 is widely expressed in tissues, but it appears that its expression in organs is restricted. Human and murine mRNA analysis showed that IL-33 is predominantly expressed in stromal cells including fibroblasts, smooth muscle cells, epithelial cells, and endothelial cells and is largely absent in hematopoietic cells (200). IL-33 may be produced during necrosis. In apoptosis, IL-33 is cleaved by caspases-3/7, leading to inactivation of its pro-inflammatory properties. For this reason, IL-33 is considered an endogenous danger signal or alarmin (202). The only known receptor for IL-33 is ST2 (200). The ST2 gene is now known to encode at least three isoforms of ST2 proteins: the transmembrane form known as ST2L, variant ST2 (ST2V) that is mainly present in human gut (203), and secreted soluble ST2 (sST2), which serves as a decoy receptor for IL-33 to prevent IL-33 binding to and signaling through ST2L (204, 205). The strongest sST2 mRNA expression was detected in heart and lung tissues (206), the cardiovascular system, endothelial cells (207), cardiac myocytes, and fibroblasts (208). The secretory capacity of these cells for sST2 is enhanced by pro-inflammatory cytokines (TNF-α, IL-1β) or LPS (206). sST2 levels in serum were correlated with acute myocardial infarction (209) and pulmonary fibrosis (210). Recently, high levels of plasma sST2 were shown to be a risk factor of GVHD in patients after allogeneic HSCT; patients who were resistant to treatment showed elevated levels of sST2 and had higher mortality regardless of the grade of GVHD (140). This finding may allow physicians to predict disease and apply interventions earlier. It may also represent a novel therapeutic opportunity in GVHD and other related diseases.
Organ-Specific Tissue Damage Following HSCT
Skin
Frequently, the first presentation of GVHD involves the skin, typically manifesting initially as palmar and acral erythema, resembling a sunburn reaction, or an acute symmetric morbilliform eruption (211). The histopathology of GVHD is a lichenoid inflammatory process of the epidermis with variable numbers of lymphocytes arranged in a linear fashion along the basement membrane zone. The hallmark change is dead cells consisting of apoptotic keratinocytes, with tightly adherent lymphocytes observed in the epidermis with associated vacuolar interface changes (212). High-dose radiation activates skin DCs, which upregulates the expression of HLA-DR, adhesion molecules, co-stimulatory molecules (213), and PRRs, producing inflammatory cytokines and danger signals that contribute to skin GVHD by mediating memory T-cell recruitment to the skin (214). In cells undergoing programed cell death, activation of caspase-3 occurs as a downstream event that links both extrinsic (death receptor-mediated) and intrinsic (mitochondrial- or DNA damage-mediated) apoptotic pathways, which means that the presence of caspase-3 in the cell will not only identify it as apoptotic but also will indicate that the apoptotic machinery is involved in the premature demise of target cells. Labeling GVHD lesional skin using antibodies that recognize cleaved caspase-3 (215) identified apoptotic keratinocytes located at the base of rete ridges. Significant elevation of elafin plasma concentrations in patients with skin GVHD is due to keratinocyte damage (60).
GI Tract and Liver
Gut tissue damage may be the first consequence of transplant conditioning and could be of particular significance for GVHD for two main reasons: the transplant conditioning regimen may deplete and/or alter the microbiota and epithelial barrier damage could allow for increased bacterial translocation, specifically in the gut. It is assumed that these processes lead to an increase in inflammation and exacerbate epithelial insult, as shown in an IBD model (216). Certain commensals such as Bifidobacterium strains may protect the host by improving the intestinal barrier. Bifidobacterium have carbohydrate transporters that can generate short-chain fatty acids, particularly acetate, which promotes defense functions in host epithelial cells in the distal colon (217). Recent GVHD studies have begun to analyze the dynamics of the gut flora during HSCT and how the innate immune receptors that recognize microbes may contribute to GVHD pathogenesis. In an experimental irradiation-independent non-myeloablative HSCT model, a gut microbial shift toward pro-inflammatory bacterial species was seen in mice that develop GVHD (136). It is still unclear whether the microbial changes in the gut are the cause or the result of GVHD, and whether these bacterial populations reflect endogenous microflora or overgrowth of pathogenic organisms due to the elimination of benign microbes. Endotoxin is a constituent of normal bowel flora that has the ability to stimulate the release of inflammatory cytokines that are known to be important mediators of clinical GVHD and most likely permeate the systemic circulation through the intestinal barrier, which is disrupted by the conditioning treatment (218). Also, microbial super antigens may activate B-cells by direct stimulation of MHC class II molecules (219). The early phases of changes in the GI tract have been described in animal models that do not use chemotherapy or radiation to condition the host; therefore, direct comparisons to clinical GVHD after bone marrow transplantation are not possible. The initial proliferative phase results in increased crypt cell mitotic activity, crypt lengthening, and the presence of intraepithelial lymphocytes. In experimental systems, this phase seems to be linked to IFN-γ production (220), which increases MHC class II expression and gut permeability by altering tight junction integrity and may modulate crypt stem-cell turnover (221). The histologic features of the GI tract in clinical GVHD and experimental GVHD after myeloablative conditioning are consistent with the destructive and atrophic phases, characterized by villus blunting, lamina propria inflammation, crypt destruction (with crypt stem-cell loss), and mucosal atrophy (222). Cytotoxic T lymphocytes do not appear to play a dominant role in experimental GVHD of the GI tract (215, 223–225), despite the ability of intraepithelial lymphocytes to induce Fas-mediated apoptosis of host-type tumor cells (226). It is clear, when these findings are considered in aggregate, that cytokines and cellular effectors combine to produce the specific damage to target organs as well as the systemic toxicity of acute GVHD. Furthermore, the absence of GVHD toxicity in other visceral organs, such as the kidney (currently debated), argues against circulating cytokines as the sole cause of tissue-specific damage. The infiltrates seen in GVHD target organs are generally thought to consist of T-cells responding to alloantigens presented by host tissues. LPS leakage through the skin or mucosa may act as an adjuvant to the antigens expressed in these tissues, attracting and activating alloreactive donor T-cells. In BMT models, LPS levels increase progressively during the first 4 weeks post-BMT. These levels lead to aggravated disease severity through TLR4 signaling, which induces inflammatory cytokine production. Deficiency of TLR4 on donor bone marrow cells reduces colonic GVHD severity. Interestingly, this reduction in GVHD severity was accompanied with a decrease of IL-23 levels. On the other hand, mice receiving allogeneic bone marrow from IL-23 knockout mice demonstrated less colonic pathology, and low levels of colonic LPS compared to wild-type controls. Interestingly, IL-17 was not detectable in the colon, while IFN-γ was markedly increased, in association with the LPS/IL-23 feedback loop (227). In consequence, IFN-γ activated macrophages after exposure to LPS release a significant amounts of inflammatory cytokines in GI tract but not in other target organs of GVHD (218). IL-23 could also enhance host IL-22-producing type 3 innate lymphoid cells (ILC3s) and promote gut recovery after conditioning. Alloreaction generated by donor T-cells damages the gut stem-cell compartment and eliminates recipient IL-22 producing ILC3, which are the main source of IL-22 in the gut and are known to protect intestinal stem cells (228). A deficiency in ILC3s leads to severe liver and GI GVHD and increases mortality. Moreover, a recent clinical study showed clear correlation between activation and expansion of intestinal ILC3s and the absence of acute GVHD (229). Figure 4 shows a hypothetical model for the roles of IL-23 in GI GVHD. Thus, reductions in the doses of chemoradiotherapy to condition bone marrow transplant recipients have reduced the incidence of GVHD, as demonstrated in experimental models (230, 231). This reduction is the result of reduced priming of mononuclear cells by lower doses of total body irradiation (TBI) and subsequent reductions in TNF-α production (224). Intestinal and liver tissue damage leads to the release of soluble mediators that correlate positively with GI and liver GVHD pathology. Significant augmentation of liver epithelial marker cytokeratin-18 protein (CK18) (232) has been demonstrated in serum from patients with hepatic and GI GVHD. Interestingly, CK18 levels begin to increase before the clinical manifestation of GVHD in some patients. Also, CK18 levels were correlated with bilirubin (liver function marker) levels. This correlation is specific to hepato-intestinal GVHD (233). RegIIIα levels were also significantly increased in patients with GI GVHD as mentioned above (105, 234). It is well known that Reg proteins act downstream of IL-22, which protects the function of intestinal mucosa, intestinal stem cells, and ILC3s (235, 236).
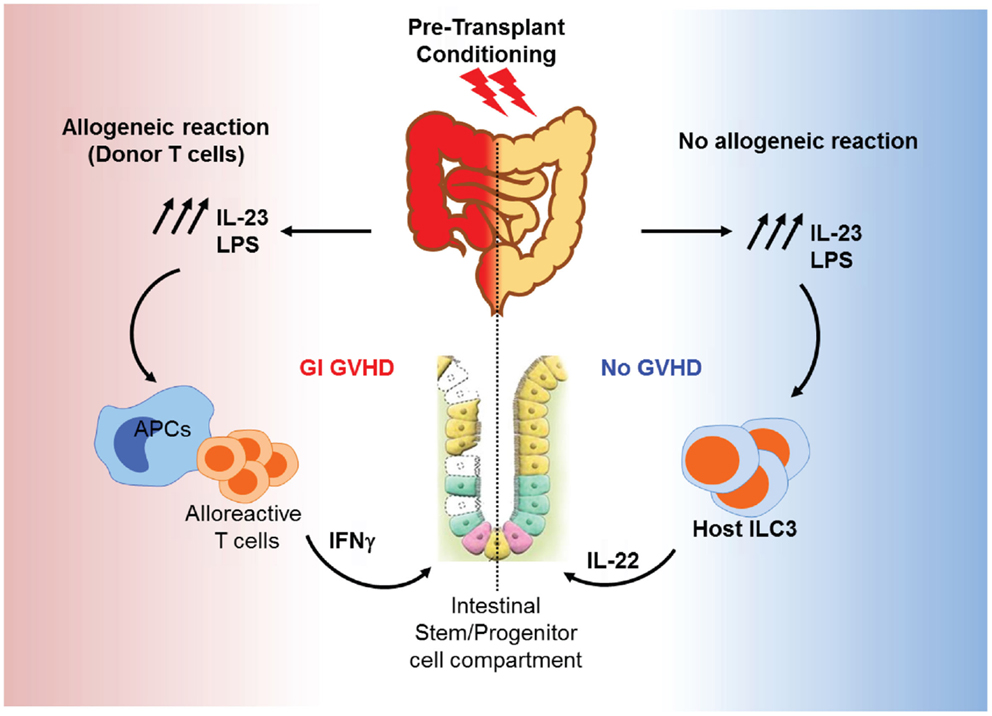
Figure 4. Roles of IL-23 in GI GVHD. Conditioning induces IL-23 production and LPS release from the GI tract. LPS and IL-23 act together to prime APCs to activate alloreactive T-cells. Activated T-cells produce IFN-γ and other inflammatory cytokines, resulting in the elimination of ILC3 and damage of the intestinal stem cells. This leads to more gut injury and GI GVHD (left). In the absence of alloreactive donor T-cells, IL-23 stimulates ILC3 to release IL-22, which protects the intestinal stem-cell compartment and promotes gut recovery from conditioning damage (right).
Thymus
Donor CD8 T-cells can damage medullary epithelial cells in the thymus and cause the generation of donor alloreactive CD4 T-cells (237), which suggests that GVHD autoimmunity can appear during acute GVHD after allogeneic HSCT. The thymic epithelial damage caused by anti-host reactive T-cells impairs negative selection in the thymus, consequently leading to the presence of autoreactive T-cells in the periphery (211, 238, 239). These autoreactive T-cells are specific and able to recognize both self (donor) and host MHC class II antigens. Thus, they may participate in acute allogeneic GVHD and are thought to underlie the pathophysiology of chronic GVHD with its clinical autoimmune manifestations (214, 238). Animal studies revealed that the presence of the thymus is required for the development of autologous GVHD, based on comparisons with thymectomized rats before transplantation (240). The thymus is a primary target organ of GVHD. Thymus toxicity is generated further by regimen preparation (radiation) and age-associated thymus toxicities, resulting in thymus dysfunction and GVHD-induced damage of lymphopoietic microenvironments. However, keratinocyte growth factor (KGF) has promising effects for preventing GVHD-mediated thymic damage (241). Reduced thymic function is detrimental for thymopoiesis recovery, and insufficient recovery of thymopoiesis is directly linked to opportunistic infections and adverse clinical outcomes in recipients (242). Thymic GVHD damages the architecture and composition of the thymic environment (243). In the thymus stromal and epithelial cells, growth factors are produced locally including IL-7 and stem-cell factor (SCF), as are chemokines involved in T-cell precursor migration such as CCL9 and CCL21. Thymic GVHD affects T-cell renewal and differentiation, and in humans, it has been reported that when thymic function is transiently impaired in young patients because of acute GVHD, there are decreases in TCR-β chain rearrangement circles (244, 245).
Oral
The overall incidence of oral complications after bone marrow transplantation is 80%. Most oral complications are resolved in autologous HSCT within 6 months post-transplantation. Oral complications in allogeneic transplantation include mucositis, oral dryness, taste change, and infection, and all symptoms are associated with GVHD (246–248). These oral changes are painful and impair patients’ quality of life (249, 250). The initiation of oral mucositis is induced by DNA and non-DNA damage caused by ROS generated by damaged basal epithelial cells, endothelial cells, and submucosal cells, in particular (251). This leads to NF-κB activation, inducing adhesion molecule expression, and MAPK and COX2 activation, resulting in the generation of IL-1β, IL-6, and TNF-α (252–254). This reinforces NF-κB activation and amplifies the response via the over-production of inflammatory cytokines (255–259). Perturbation in the immune response to the microbiota leads to spontaneous inflammation, and vice versa, changes in microbiota diversity are associated with pro-inflammatory states (260, 261). In HSCT recipients, substitution with coagulase-negative Staphylococci for Streptococci is associated with oral mucositis (137). It was suggested that subcutaneous administration of IL-11 reduces the severity of oral mucositis by maintaining keratin production in epithelial cells and reducing mucosal pro-inflammatory cytokine expression (262). However, it causes severe fluid retention and early mortality in clinical trials (263). Administration of TGF-β3 prior to chemotherapy down-regulates epithelial cell expansion and reduces oral mucositis in hamsters (264). KGF promotes upregulation of Bcl2 and cell survival (265) as well as upregulates IL-13 that attenuates TNF-α (266). KGF has beneficial effects on oral mucositis prevention in high-dose chemotherapy and TBI-treated patients (259, 267).
Therapeutic Approaches to Target Danger Signals
Since the late 1980s, different therapeutic approaches have been established to overcome GVHD (268, 269). These classical therapeutic strategies target only donor T-cells by inhibition or even depletion and may impact the immune reconstitution and graft-versus-leukemia effect. Recent therapeutic strategies have focused on PAMPs and/or DAMPs released by the host after tissue damage, which are the first triggers of activation of host APCs and donor T-cells. Therefore, targeting PAMPs and DAMPs may not impair donor cell function, immune reconstitution, or anti-tumoral activity. Here, we highlight therapeutic strategies targeting danger signal or alarmins.
Siglec-G Ligand
Recently, it has been shown that conditioning inhibits the expression of Siglec-G, which could be activated by HMGB-1. The absence of Siglec-G ligand (CD24) increases susceptibility to GVHD in mice because of the response to DAMPs, but not PAMPs. Administration of CD24 fusion protein led to a reduction in GVHD severity and mortality. This approach reveals the importance of targeting DAMPs separately from exogenous PAMPs in GVHD therapy (39).
Alpha-1 Antitrypsin
Heparan sulfate, as described above, is detected in patient sera following HSCT. Murine studies showed that heparan sulfate levels are reduced significantly upon the use of the elastase inhibitor alpha-1 antitrypsin (270), and this reduction in heparan sulfate was correlated with reduced GVHD severity in murine models. The alteration of heparan sulfate in mice treated with alpha-1 antitrypsin subsequently led to a reduction in inflammatory cytokines such as TNF-α and IL-1β and enhanced IL-10 production and Treg expansion. Another study showed that alpha-1 antitrypsin treatment suppresses IL-32 expression in T-cells (271). Clinical trials using alpha-1 antitrypsin as GVHD prophylaxis are currently underway.
ILC3s
Many recent studies have emphasized the importance of RORγt ILCs in regulating mucosal immune responses via the control of intestinal microflora expansion and composition and regulation of CD4 T-cells (272). Depletion of Nfil3 in mice dramatically impairs the number of IL-22-producing ILC3s, which resulted in compromised innate intestinal immune defense against bacterial infection (273). IL-22-producing ILC3s have a beneficial impact on gut protection in metabolic disorders, improving insulin sensitivity as well as preserving the gut mucosal barrier and endocrine function (274). As mentioned before, IL-22-producing ILC3s have a crucial role in reducing epithelial and intestinal stem-cell damage and reducing GVHD severity and mortality (228). The same research group has demonstrated that daily administration of IL-22 for 3 weeks starting 1 week post-transplantation increases the survival and function of host radio-resistant ILC3s, subsequently reducing apoptosis in host intestinal stem cells and reducing GVHD severity through preservation of host cells from damage without compromising immune function or reconstitution (275). This makes IL-22 administration one of most promising therapies for GI GVHD. The transcription factor aryl hydrocarbon receptor (AhR) is highly expressed in ILC3s and is required for ILC3 development (276), particularly IL-22-producing ILC3s (277). Thus, using small molecules that activate AhR might be a promising future therapeutic strategy in preventing GI GVHD and ameliorating immune functions post-transplantation as well.
IL-1 Receptor Antagonist
Blockade of IL-1 with IL-1 receptor antagonist significantly reduces mortality from experimental GVHD and enhances engraftment of HSCs (278). In a clinical trial, IL-1 receptor antagonist reduced the severity of GVHD (279), but when used as prophylaxis, no significant impact on GVHD was observed (280).
Anti-ST2 or IL-33
A high level of sST2 is a risk factor for GVHD (140), with levels of sST2 positively correlating with high rates of mortality in patients with GVHD. Other studies reported that sST2 is associated with cardiovascular mortality. sST2 concentrations have been linked with inflammatory markers (281), and sST2 has been associated with disease severity in pulmonary arterial hypertension (282). sST2 is rapidly synthesized and released by endothelial cells in inflammatory conditions and in the setting of tissue damage (207). sST2 acts as a decoy receptor for IL-33, inhibiting signaling through membrane ST2 (ST2L) (283). In murine models, exposure of murine T-cells to sST2 inhibits TH2 cytokine production and shifts the cells toward a TH1 response (283). It is possible that high levels of sST2 after tissue damage induced by conditioning leads to activation of donor T-cells toward a type 1 response, thereby increasing GVHD responses. It is also possible that inhibition of sST2 decreases this phenomenon and thus increases type 2 T-cell responses. Another possible therapeutic approach would be to increase the expression of ST2L and make it more available for IL-33 binding and signaling. Both strategies should not impair the immune response of donor cells, but will work by limiting the effects of host tissue damage, opening new robust therapy options for GVHD.
Mesenchymal Stem Cells
Mesenchymal stem cells (MSCs) are multipotent mesenchymal stromal cells with fibroblastic-like morphology that can differentiate into bone, cartilage, or fat cells (284). These cells have the capacity for non-specific immunosuppression and immunomodulation. It has been shown that infusion of MSCs in high-risk major-mismatched transplant recipients reduces the incidence of life-threatening GVHD (285, 286). Recent clinical studies showed that weekly infusion of MSCs with a fixed dose for 3 weeks reduced significantly the severity of the disease; specifically, patients with steroid-refectory acute GVHD experienced a complete response. Clinical response was correlated with a significant decline in RegIIIα and elafin GVHD biomarkers (287).
Anti-TLRs
The expression of PRRs at the epithelial surfaces is equally important as that in immune cells in combating or facilitating entry of organisms into the body, including bacterial translocation from the gut after irradiation (288). Activation of PRRs results in additional vascular damage and infiltration of inflammatory cells that creates a cascade of lesions in a pro-oxidant microenvironment, aggravating tissue damage and causing a “danger” zone (289). An antagonist of LPS, the TLR4 ligand, results in reduced intestinal damage and GVHD severity without altering donor T-cell activity to the host antigen (133). Novel anti-TLR antibodies particularly anti-TLR4 and anti-TLR2 are being developed (290) and will soon represent a novel class of potential therapeutics for GVHD treatment.
Chronic GVHD and Danger Signals
Graft-versus-host disease studies have led to a decrease of early mortality in related-allogeneic HSCTs, but late long-term morbidity and mortality caused by chronic GVHD remains a major challenge (291). The pathogenesis of chronic GVHD is complex and poorly understood, but is likely to involve dysfunction of tolerance determining mechanisms similar to classic autoimmune diseases. Figure 5 summarizes some of the knowledge of the pathophysiology of chronic GVHD. Briefly, negative selection in the thymus is impaired because of thymic epithelial cell (TEC) damage after allogeneic reaction. In addition, the cross talk between alloreactive T-cells and B-cells enhances B-cell activating factor (BAFF) release and production of alloantibodies, which, together with cytokines and chemokines produced by T-cells and B-cells, activates macrophages and induces proliferation and activation of fibroblast and collagen production, resulting in tissue fibrosis. Very few studies showed a direct impact of PAMPs or DAMPs in chronic GVHD. It has been shown that LPS enhances peribronchiolar fibrosis in synergy with TH17 production and leads to chronic pulmonary GVHD (292). However, a clear indirect effect is the altered T- and B-cell homeostasis. Patients with chronic GVHD showed inverted ratio of CD4:CD8 (293). CD4+ regulatory T-cell frequency was dramatically decreased in these patients comparing to patients without active chronic GVHD. This reduction in Tregs:Tcon ratio was explained as following: (i) Tregs acquire a predominately effector memory phenotype (294), (ii) under lymphopenia-induced expansion, CD4+ Tregs proliferate more than conventional T-cells, which increases Treg susceptibility to Fas-mediated apoptosis (295), and (iii) progressive loss of Aire expression by TEC, which is crucial for naive Treg generation (294). In chronic GVHD, critical breakdown in peripheral B-cell tolerance was shown. Among patients with cGVHD, BAFF reaches a persistently high level (296). Chronic exposure to BAFF results in elevated basal expression of the proximal signaling components B-cell linker protein (BLNK) and Syk, which may contribute to increased responsiveness of BCR stimulation (297). Murine models of cGVHD also provided insights showing that unrestrained T follicular helper cells and germinal center B-cells are abnormally increased and strongly correlate with the development of cGVHD (298). Together, chronic GVHD is likely caused by a lack of central tolerance involving thymus dysfunction, disequilibrium of T-reg/Tcon balance, and alloantibodies generated by alloreactive B-cells.
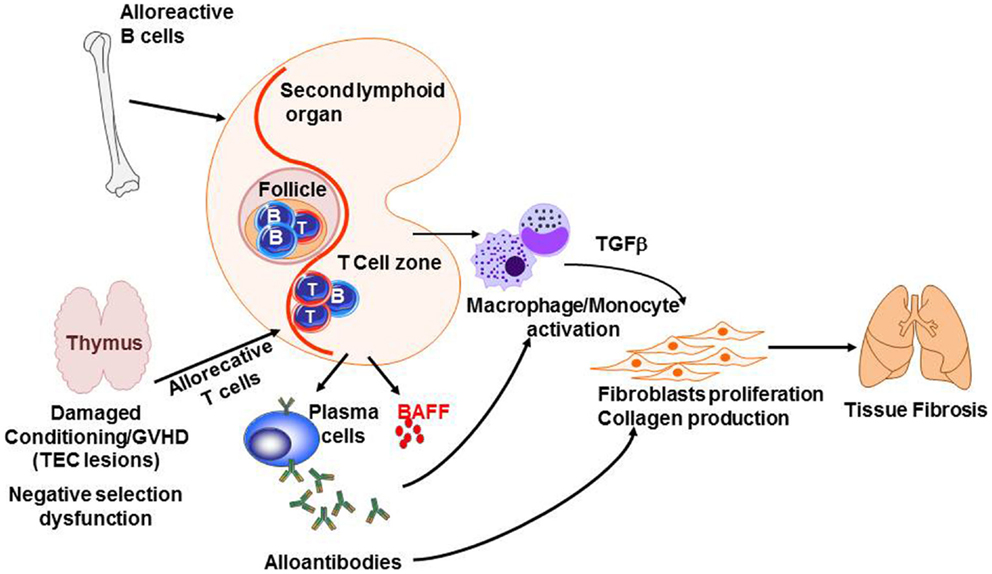
Figure 5. Pathogenesis of chronic GVHD. The thymic epithelial cells (TECs) are damaged by alloreactive T-cells leading to impaired negative selection. In addition, alloreactive B- and T-cells cross talk leading to sBAFF release and production of alloantibodies by plasma cells. At the same time, cytokines and chemokines produced by B- and T-cells activate macrophages and monocytes. Together, antibodies and TGFβ induce fibroblasts proliferation and activation as well as collagen production, which results in fibrosis in target organs such as the lungs.
In a Nutshell: PAMPs and DAMPs in the Pathogenesis of Acute GVHD
Allogeneic HSCT conditioning elicits the first signal of tissue damage by releasing PAMPs, such as intestinal microflora and LPS, and DAMPs, such as S100 proteins, uric acid, HSP, and ATP. These PAMPs and DAMPs are then detected by host innate immune cells, including non-hematopoietic APCs, through PRRs (TLRs and NLRs) leading to downstream signaling through NF-κB, upregulating co-stimulatory molecules, and producing the inflammatory cytokines TNF-α, IL-12, and IL-6. Stimulation of allogeneic donor T-cells by activated host APCs in the proinflammatory environment leads to T-cells expansion and polarization toward TH1, TC1, TH17, and TC17, the key mediators of GVHD pathogenesis. These T-cells produce more inflammatory cytokines, such as IFN-γ, TNF-α, and IL-17, leading to increased tissue damage. As a consequence of damage exacerbation, more DAMPs are released from damaged tissue, such as elafin in skin GVHD, Reg3α in GI GVHD, or soluble ST2, and again newly amplify type 1 response of T-cells creating more severe pathogenicity as shown in Figure 1. Importantly, these molecules can serve as biomarkers for GVHD diagnosis and severity. These molecules may also represent a novel class of therapeutics for GVHD with the possible advantage of not altering the immune reconstitution and T-cell responses against tumors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sophie Paczesny is supported by National Institutes of Health grants R01-CA174667 and R01-CA168814. Sophie Paczesny is an investigator of the Amy Strelzer Manasevit Research Program (grant #200513) and the Lilly Physician Scientist Initiative Program (grant #20091568000).
References
1. Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood (1996) 88(8):3230–9.
2. Yanik GA, Ho VT, Levine JE, White ES, Braun T, Antin JH, et al. The impact of soluble tumor necrosis factor receptor etanercept on the treatment of idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Blood (2008) 112(8):3073–81. doi:10.1182/blood-2008-03-143412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Hartrampf S, Dudakov JA, Johnson LK, Smith OM, Tsai J, Singer NV, et al. The central nervous system is a target of acute graft versus host disease in mice. Blood (2013) 121(10):1906–10. doi:10.1182/blood-2012-09-456590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene (2003) 22(37):5734–54. doi:10.1038/sj.onc.1206663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Granet C, Miossec P. Combination of the pro-inflammatory cytokines IL-1, TNF-alpha and IL-17 leads to enhanced expression and additional recruitment of AP-1 family members, Egr-1 and NF-kappaB in osteoblast-like cells. Cytokine (2004) 26(4):169–77.
6. Bubici C, Papa S, Dean K, Franzoso G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene (2006) 25(51):6731–48. doi:10.1038/sj.onc.1209936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Han D, Ybanez MD, Ahmadi S, Yeh K, Kaplowitz N. Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal (2009) 11(9):2245–63. doi:10.1089/ars.2009.2611
8. Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med (2010) 48(1):1–15. doi:10.1016/j.freeradbiomed.2009.09.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Zhao W, Xie W, Xiao Q, Beers DR, Appel SH. Protective effects of an anti-inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated microglia. J Neurochem (2006) 99(4):1176–87. doi:10.1111/j.1471-4159.2006.04172.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Qian L, Hong JS, Flood PM. Role of microglia in inflammation-mediated degeneration of dopaminergic neurons: neuroprotective effect of interleukin 10. J Neural Transm Suppl (2006) 70:367–71. doi:10.1007/978-3-211-45295-0_56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Noubade R, Wong K, Ota N, Rutz S, Eidenschenk C, Valdez PA, et al. NRROS negatively regulates reactive oxygen species during host defence and autoimmunity. Nature (2014) 509(7499):235–9. doi:10.1038/nature13152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Schwab L, Goroncy L, Palaniyandi S, Gautam S, Triantafyllopoulou A, Mocsai A, et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat Med (2014) 20(6):648–54. doi:10.1038/nm.3517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Jones JM, Wilson R, Bealmear PM. Mortality and gross pathology of secondary disease in germfree mouse radiation chimeras. Radiat Res (1971) 45(3):577–88. doi:10.2307/3573066
14. van Bekkum DW, Knaan S. Role of bacterial microflora in development of intestinal lesions from graft-versus-host reaction. J Natl Cancer Inst (1977) 58(3):787–90.
15. Storb R, Prentice RL, Buckner CD, Clift RA, Appelbaum F, Deeg J, et al. Graft-versus-host disease and survival in patients with aplastic anemia treated by marrow grafts from HLA-identical siblings. Beneficial effect of a protective environment. N Engl J Med (1983) 308(6):302–7. doi:10.1056/NEJM198302103080602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Beelen DW, Elmaagacli A, Muller KD, Hirche H, Schaefer UW. Influence of intestinal bacterial decontamination using metronidazole and ciprofloxacin or ciprofloxacin alone on the development of acute graft-versus-host disease after marrow transplantation in patients with hematologic malignancies: final results and long-term follow-up of an open-label prospective randomized trial. Blood (1999) 93(10):3267–75.
17. Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science (2002) 296(5566):298–300. doi:10.1126/science.1068883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi:10.1146/annurev.immunol.20.083001.084359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Onoguchi K, Yoneyama M, Fujita T. Retinoic acid-inducible gene-I-like receptors. J Interferon Cytokine Res (2011) 31(1):27–31. doi:10.1089/jir.2010.0057
20. Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci (2002) 27(9):474–82. doi:10.1016/S0968-0004(02)02145-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392(6673):245–52. doi:10.1038/32588
22. Penack O, Holler E, van den Brink MR. Graft-versus-host disease: regulation by microbe-associated molecules and innate immune receptors. Blood (2010) 115(10):1865–72. doi:10.1182/blood-2009-09-242784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Holler E, Rogler G, Herfarth H, Brenmoehl J, Wild PJ, Hahn J, et al. Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplantation. Blood (2004) 104(3):889–94. doi:10.1182/blood-2003-10-3543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol (2007) 81(1):1–5. doi:10.1189/jlb.0306164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Agresti A, Bianchi ME. HMGB proteins and gene expression. Curr Opin Genet Dev (2003) 13(2):170–8. doi:10.1016/S0959-437X(03)00023-6
26. Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med (2004) 255(3):332–43. doi:10.1111/j.1365-2796.2003.01296.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature (2002) 418(6894):191–5. doi:10.1038/nature00858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res (2007) 13(10):2836–48. doi:10.1158/1078-0432.CCR-06-1953
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science (1999) 285(5425):248–51. doi:10.1126/science.285.5425.248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Popovic K, Ek M, Espinosa A, Padyukov L, Harris HE, Wahren-Herlenius M, et al. Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum (2005) 52(11):3639–45. doi:10.1002/art.21398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem (2010) 285(51):39888–97. doi:10.1074/jbc.M110.128348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ (2010) 17(4):666–76. doi:10.1038/cdd.2009.149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Brezniceanu ML, Volp K, Bosser S, Solbach C, Lichter P, Joos S, et al. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J (2003) 17(10):1295–7. doi:10.1096/fj.02-0621fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Yang D, Chen Q, Yang H, Tracey KJ, Bustin M, Oppenheim JJ. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J Leukoc Biol (2007) 81(1):59–66. doi:10.1189/jlb.0306180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J Leukoc Biol (2007) 81(1):84–91. doi:10.1189/jlb.0306171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol (2003) 170(7):3890–7. doi:10.4049/jimmunol.170.7.3890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem (2004) 279(9):7370–7. doi:10.1074/jbc.M306793200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kornblit B, Masmas T, Petersen SL, Madsen HO, Heilmann C, Schejbel L, et al. Association of HMGB1 polymorphisms with outcome after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant (2010) 16(2):239–52. doi:10.1016/j.bbmt.2009.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Toubai T, Hou G, Mathewson N, Liu C, Wang Y, Oravecz-Wilson K, et al. Siglec-G-CD24 axis controls the severity of graft-versus-host disease in mice. Blood (2014) 123(22):3512–23. doi:10.1182/blood-2013-12-545335
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Donato R. Intracellular and extracellular roles of S100 proteins. Microsc Res Tech (2003) 60(6):540–51. doi:10.1002/jemt.10296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Heizmann CW, Fritz G, Schafer BW. S100 proteins: structure, functions and pathology. Front Biosci (2002) 7:d1356–68. doi:10.2741/heizmann
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun (2004) 322(4):1111–22. doi:10.1016/j.bbrc.2004.07.096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood (2008) 111(4):2476–84. doi:10.1182/blood-2007-08-109678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol (2007) 81(1):28–37. doi:10.1189/jlb.0306170
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hegyi Z, Zwicker S, Bureik D, Peric M, Koglin S, Batycka-Baran A, et al. Vitamin D analog calcipotriol suppresses the Th17 cytokine-induced proinflammatory S100 “alarmins” psoriasin (S100A7) and koebnerisin (S100A15) in psoriasis. J Invest Dermatol (2012) 132(5):1416–24. doi:10.1038/jid.2011.486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med (2013) 188(9):1137–46. doi:10.1164/rccm.201304-0803OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Jenke AC, Postberg J, Mariel B, Hensel K, Foell D, Dabritz J, et al. S100A12 and hBD2 correlate with the composition of the fecal microflora in ELBW infants and expansion of E. coli is associated with NEC. Biomed Res Int (2013) 2013:150372. doi:10.1155/2013/150372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Chiusolo P, Giammarco S, Fanali C, Bellesi S, Metafuni E, Sica S, et al. Salivary proteomic analysis and acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2013) 19(6):888–92. doi:10.1016/j.bbmt.2013.03.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Reinhardt K, Foell D, Vogl T, Mezger M, Wittkowski H, Fend F, et al. Monocyte-induced development of Th17 cells and the release of S100 proteins are involved in the pathogenesis of graft-versus-host disease. J Immunol (2014) 193(7):3355–65. doi:10.4049/jimmunol.1400983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell (2002) 111(6):867–78. doi:10.1016/S0092-8674(02)01141-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lee SK, Lee SS, Hirose S, Park SC, Chi JG, Chung SI, et al. Elafin expression in human fetal and adult submandibular glands. Histochem Cell Biol (2002) 117(5):423–30. doi:10.1007/s00418-002-0405-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Schalkwijk J, van Vlijmen IM, Alkemade JA, de Jongh GJ. Immunohistochemical localization of SKALP/elafin in psoriatic epidermis. J Invest Dermatol (1993) 100(4):390–3. doi:10.1111/1523-1747.ep12471990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Reid PT, Marsden ME, Cunningham GA, Haslett C, Sallenave JM. Human neutrophil elastase regulates the expression and secretion of elafin (elastase-specific inhibitor) in type II alveolar epithelial cells. FEBS Lett (1999) 457(1):33–7. doi:10.1016/S0014-5793(99)01004-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Kolls JK, McCray PB Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol (2008) 8(11):829–35. doi:10.1038/nri2433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Saheki T, Ito F, Hagiwara H, Saito Y, Kuroki J, Tachibana S, et al. Primary structure of the human elafin precursor preproelafin deduced from the nucleotide sequence of its gene and the presence of unique repetitive sequences in the prosegment. Biochem Biophys Res Commun (1992) 185(1):240–5. doi:10.1016/S0006-291X(05)80981-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Sallenave JM, Silva A. Characterization and gene sequence of the precursor of elafin, an elastase-specific inhibitor in bronchial secretions. Am J Respir Cell Mol Biol (1993) 8(4):439–45. doi:10.1165/ajrcmb/8.4.439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Sallenave JM, Shulmann J, Crossley J, Jordana M, Gauldie J. Regulation of secretory leukocyte proteinase inhibitor (SLPI) and elastase-specific inhibitor (ESI/elafin) in human airway epithelial cells by cytokines and neutrophilic enzymes. Am J Respir Cell Mol Biol (1994) 11(6):733–41. doi:10.1165/ajrcmb.11.6.7946401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Tanaka N, Fujioka A, Tajima S, Ishibashi A, Hirose S. Elafin is induced in epidermis in skin disorders with dermal neutrophilic infiltration: interleukin-1 beta and tumour necrosis factor-alpha stimulate its secretion in vitro. Br J Dermatol (2000) 143(4):728–32. doi:10.1046/j.1365-2133.2000.03766.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. van Wetering S, van der Linden AC, van Sterkenburg MA, Rabe KF, Schalkwijk J, Hiemstra PS. Regulation of secretory leukocyte proteinase inhibitor (SLPI) production by human bronchial epithelial cells: increase of cell-associated SLPI by neutrophil elastase. J Investig Med (2000) 48(5):359–66.
60. Paczesny S, Braun TM, Levine JE, Hogan J, Crawford J, Coffing B, et al. Elafin is a biomarker of graft-versus-host disease of the skin. Sci Transl Med (2010) 2(13):13ra2. doi:10.1126/scitranslmed.3000406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Pfundt R, Wingens M, Bergers M, Zweers M, Frenken M, Schalkwijk J. TNF-alpha and serum induce SKALP/elafin gene expression in human keratinocytes by a p38 MAP kinase-dependent pathway. Arch Dermatol Res (2000) 292(4):180–7. doi:10.1007/s004030050475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Schutte BC, McCray PB Jr. [beta]-defensins in lung host defense. Annu Rev Physiol (2002) 64:709–48. doi:10.1146/annurev.physiol.64.081501.134340
63. Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol (2003) 3(9):710–20. doi:10.1038/nri1180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Ho S, Pothoulakis C, Koon HW. Antimicrobial peptides and colitis. Curr Pharm Des (2013) 19(1):40–7. doi:10.2174/13816128130108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Gwyer Findlay E, Currie SM, Davidson DJ. Cationic host defence peptides: potential as antiviral therapeutics. BioDrugs (2013) 27(5):479–93. doi:10.1007/s40259-013-0039-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Gursoy UK, Pollanen M, Kononen E, Uitto VJ. A novel organotypic dento-epithelial culture model: effect of Fusobacterium nucleatum biofilm on B-defensin-2, -3, and LL-37 expression. J Periodontol (2012) 83(2):242–7. doi:10.1902/jop.2011.110177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Eriguchi Y, Takashima S, Oka H, Shimoji S, Nakamura K, Uryu H, et al. Graft-versus-host disease disrupts intestinal microbial ecology by inhibiting Paneth cell production of alpha-defensins. Blood (2012) 120(1):223–31. doi:10.1182/blood-2011-12-401166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med (2012) 209(5):903–11. doi:10.1084/jem.20112408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Taur Y, Jenq RR, Perales MA, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood (2014) 124(7):1174–82. doi:10.1182/blood-2014-02-554725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett (1995) 374(1):1–5. doi:10.1016/0014-5793(95)01050-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Lehrer RI, Ganz T. Cathelicidins: a family of endogenous antimicrobial peptides. Curr Opin Hematol (2002) 9(1):18–22. doi:10.1097/00062752-200201000-00004
72. Zaiou M, Gallo RL. Cathelicidins, essential gene-encoded mammalian antibiotics. J Mol Med (Berl) (2002) 80(9):549–61. doi:10.1007/s00109-002-0350-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Gennaro R, Zanetti M. Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Biopolymers (2000) 55(1):31–49. doi:10.1002/1097-0282(2000)55:1<31::AID-BIP40>3.3.CO;2-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol (2004) 75(1):39–48. doi:10.1189/jlb.0403147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, et al. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood (2000) 96(9):3086–93.
76. Di Nardo A, Vitiello A, Gallo RL. Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J Immunol (2003) 170(5):2274–8. doi:10.4049/jimmunol.170.5.2274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Sorensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood (1997) 90(7):2796–803.
78. Putsep K, Carlsson G, Boman HG, Andersson M. Deficiency of antibacterial peptides in patients with morbus Kostmann: an observation study. Lancet (2002) 360(9340):1144–9. doi:10.1016/S0140-6736(02)11201-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Islam D, Bandholtz L, Nilsson J, Wigzell H, Christensson B, Agerberth B, et al. Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat Med (2001) 7(2):180–5. doi:10.1038/84627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, et al. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol (2001) 117(1):91–7. doi:10.1046/j.1523-1747.2001.01340.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Bals R, Weiner DJ, Meegalla RL, Wilson JM. Transfer of a cathelicidin peptide antibiotic gene restores bacterial killing in a cystic fibrosis xenograft model. J Clin Invest (1999) 103(8):1113–7. doi:10.1172/JCI6570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Choi KY, Napper S, Mookherjee N. Human cathelicidin LL-37 and its derivative IG-19 regulate interleukin-32-induced inflammation. Immunology (2014) 143(1):68–80. doi:10.1111/imm.12291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Yang D, Chertov O, Oppenheim JJ. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: receptors and activities of human defensins and cathelicidin (LL-37). J Leukoc Biol (2001) 69(5):691–7.
84. Niyonsaba F, Iwabuchi K, Someya A, Hirata M, Matsuda H, Ogawa H, et al. A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology (2002) 106(1):20–6. doi:10.1046/j.1365-2567.2002.01398.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Tjabringa GS, Ninaber DK, Drijfhout JW, Rabe KF, Hiemstra PS. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int Arch Allergy Immunol (2006) 140(2):103–12. doi:10.1159/000092305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Yuk JM, Shin DM, Lee HM, Yang CS, Jin HS, Kim KK, et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe (2009) 6(3):231–43. doi:10.1016/j.chom.2009.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Nagaoka I, Tamura H, Hirata M. An antimicrobial cathelicidin peptide, human CAP18/LL-37, suppresses neutrophil apoptosis via the activation of formyl-peptide receptor-like 1 and P2X7. J Immunol (2006) 176(5):3044–52. doi:10.4049/jimmunol.176.5.3044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Bandholtz L, Ekman GJ, Vilhelmsson M, Buentke E, Agerberth B, Scheynius A, et al. Antimicrobial peptide LL-37 internalized by immature human dendritic cells alters their phenotype. Scand J Immunol (2006) 63(6):410–9. doi:10.1111/j.1365-3083.2006.001752.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Davidson DJ, Currie AJ, Reid GS, Bowdish DM, MacDonald KL, Ma RC, et al. The cationic antimicrobial peptide LL-37 modulates dendritic cell differentiation and dendritic cell-induced T cell polarization. J Immunol (2004) 172(2):1146–56. doi:10.4049/jimmunol.172.2.1146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature (2007) 449(7162):564–9. doi:10.1038/nature06116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Metz-Boutigue MH, Shooshtarizadeh P, Prevost G, Haikel Y, Chich JF. Antimicrobial peptides present in mammalian skin and gut are multifunctional defence molecules. Curr Pharm Des (2010) 16(9):1024–39. doi:10.2174/138161210790963823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kanda N, Hau CS, Tada Y, Sato S, Watanabe S. Decreased serum LL-37 and vitamin D3 levels in atopic dermatitis: relationship between IL-31 and oncostatin M. Allergy (2012) 67(6):804–12. doi:10.1111/j.1398-9995.2012.02824.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Keim V, Rohr G, Stockert HG, Haberich FJ. An additional secretory protein in the rat pancreas. Digestion (1984) 29(4):242–9. doi:10.1159/000199041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Terazono K, Yamamoto H, Takasawa S, Shiga K, Yonemura Y, Tochino Y, et al. A novel gene activated in regenerating islets. J Biol Chem (1988) 263(5):2111–4.
95. Orelle B, Keim V, Masciotra L, Dagorn JC, Iovanna JL. Human pancreatitis-associated protein. Messenger RNA cloning and expression in pancreatic diseases. J Clin Invest (1992) 90(6):2284–91. doi:10.1172/JCI116115
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Itoh T, Teraoka H. Cloning and tissue-specific expression of cDNAs for the human and mouse homologues of rat pancreatitis-associated protein (PAP). Biochim Biophys Acta (1993) 1172(1–2):184–6. doi:10.1016/0167-4781(93)90290-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Narushima Y, Unno M, Nakagawara K, Mori M, Miyashita H, Suzuki Y, et al. Structure, chromosomal localization and expression of mouse genes encoding type III Reg, RegIII alpha, RegIII beta, RegIII gamma. Gene (1997) 185(2):159–68. doi:10.1016/S0378-1119(96)00589-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Abe M, Nata K, Akiyama T, Shervani NJ, Kobayashi S, Tomioka-Kumagai T, et al. Identification of a novel Reg family gene, Reg IIIdelta, and mapping of all three types of Reg family gene in a 75 kilobase mouse genomic region. Gene (2000) 246(1–2):111–22. doi:10.1016/S0378-1119(00)00059-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Sekikawa A, Fukui H, Suzuki K, Karibe T, Fujii S, Ichikawa K, et al. Involvement of the IL-22/REG Ialpha axis in ulcerative colitis. Lab Invest (2010) 90(3):496–505. doi:10.1038/labinvest.2009.147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Granlund A, Beisvag V, Torp SH, Flatberg A, Kleveland PM, Ostvik AE, et al. Activation of REG family proteins in colitis. Scand J Gastroenterol (2011) 46(11):1316–23. doi:10.3109/00365521.2011.605463
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature (2008) 455(7214):804–7. doi:10.1038/nature07250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334(6053):255–8. doi:10.1126/science.1209791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Lai Y, Li D, Li C, Muehleisen B, Radek KA, Park HJ, et al. The antimicrobial protein REG3A regulates keratinocyte proliferation and differentiation after skin injury. Immunity (2012) 37(1):74–84. doi:10.1016/j.immuni.2012.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Eriguchi Y, Uryu H, Nakamura K, Shimoji S, Takashima S, Iwasaki H, et al. Reciprocal expression of enteric antimicrobial proteins in intestinal graft-versus-host disease. Biol Blood Marrow Transplant (2013) 19(10):1525–9. doi:10.1016/j.bbmt.2013.07.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Ferrara JL, Harris AC, Greenson JK, Braun TM, Holler E, Teshima T, et al. Regenerating islet-derived 3-alpha is a biomarker of gastrointestinal graft-versus-host disease. Blood (2011) 118(25):6702–8. doi:10.1182/blood-2011-08-375006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Gething MJ, Sambrook J. Protein folding in the cell. Nature (1992) 355(6355):33–45. doi:10.1038/355033a0
107. Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C. Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol (2007) 81(1):15–27. doi:10.1189/jlb.0306167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Direskeneli H, Saruhan-Direskeneli G. The role of heat shock proteins in Behcet’s disease. Clin Exp Rheumatol (2003) 21(4 Suppl 30):S44–8.
109. Poccia F, Piselli P, Vendetti S, Bach S, Amendola A, Placido R, et al. Heat-shock protein expression on the membrane of T cells undergoing apoptosis. Immunology (1996) 88(1):6–12. doi:10.1046/j.1365-2567.1996.d01-656.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Ergun T, Ince U, Eksioglu-Demiralp E, Direskeneli H, Gurbuz O, Gurses L, et al. HSP 60 expression in mucocutaneous lesions of Behcet’s disease. J Am Acad Dermatol (2001) 45(6):904–9. doi:10.1067/mjd.2001.117728
111. Imamura Y, Kurokawa MS, Yoshikawa H, Nara K, Takada E, Masuda C, et al. Involvement of Th1 cells and heat shock protein 60 in the pathogenesis of intestinal Behcet’s disease. Clin Exp Immunol (2005) 139(2):371–8. doi:10.1111/j.1365-2249.2005.02695.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol (2000) 164(1):13–7. doi:10.4049/jimmunol.164.1.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Jarvis M, Marzolini M, Wang XN, Jackson G, Sviland L, Dickinson AM. Heat shock protein 70: correlation of expression with degree of graft-versus-host response and clinical graft-versus-host disease. Transplantation (2003) 76(5):849–53. doi:10.1097/01.TP.0000083556.73555.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Goral J, Shenoy S, Mohanakumar T, Clancy J Jr. Antibodies to 70 kD and 90 kD heat shock proteins are associated with graft-versus-host disease in peripheral blood stem cell transplant recipients. Clin Exp Immunol (2002) 127(3):553–9. doi:10.1046/j.1365-2249.2002.01770.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harbor Perspect Biol (2011) 3(7):1–33. doi:10.1101/cshperspect.a004952
116. Lortat-Jacob H. The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr Opin Struct Biol (2009) 19(5):543–8. doi:10.1016/j.sbi.2009.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Salanga CL, Handel TM. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res (2011) 317(5):590–601. doi:10.1016/j.yexcr.2011.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Lortat-Jacob H, Baltzer F, Grimaud JA. Heparin decreases the blood clearance of interferon-gamma and increases its activity by limiting the processing of its carboxyl-terminal sequence. J Biol Chem (1996) 271(27):16139–43. doi:10.1074/jbc.271.27.16139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Qu X, Pan Y, Carbe C, Powers A, Grobe K, Zhang X. Glycosaminoglycan-dependent restriction of FGF diffusion is necessary for lacrimal gland development. Development (2012) 139(15):2730–9. doi:10.1242/dev.079236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol (2002) 168(10):5233–9. doi:10.4049/jimmunol.168.10.5233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Brennan TV, Lin L, Huang X, Cardona DM, Li Z, Dredge K, et al. Heparan sulfate, an endogenous TLR4 agonist, promotes acute GVHD after allogeneic stem cell transplantation. Blood (2012) 120(14):2899–908. doi:10.1182/blood-2011-07-368720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, et al. Gamma-irradiation induces P2X(7) receptor-dependent ATP release from B16 melanoma cells. Biochim Biophys Acta (2010) 1800(1):40–6. doi:10.1016/j.bbagen.2009.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430(6996):213–8. doi:10.1038/nature02664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Di Virgilio F. Purinergic mechanism in the immune system: a signal of danger for dendritic cells. Purinergic Signal (2005) 1(3):205–9. doi:10.1007/s11302-005-6312-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Granstein RD, Ding W, Huang J, Holzer A, Gallo RL, Di Nardo A, et al. Augmentation of cutaneous immune responses by ATP gamma S: purinergic agonists define a novel class of immunologic adjuvants. J Immunol (2005) 174(12):7725–31. doi:10.4049/jimmunol.174.12.7725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Wilhelm K, Ganesan J, Muller T, Durr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med (2010) 16(12):1434–8. doi:10.1038/nm.2242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Kanellis J, Watanabe S, Li JH, Kang DH, Li P, Nakagawa T, et al. Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension (2003) 41(6):1287–93. doi:10.1161/01.HYP.0000072820.07472.3B
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum (2005) 52(9):2936–46. doi:10.1002/art.21238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440(7081):237–41. doi:10.1038/nature04516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature (2003) 425(6957):516–21. doi:10.1038/nature01991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Hu DE, Moore AM, Thomsen LL, Brindle KM. Uric acid promotes tumor immune rejection. Cancer Res (2004) 64(15):5059–62. doi:10.1158/0008-5472.CAN-04-1586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Yeh AC, Brunner AM, Spitzer TR, Chen YB, Coughlin E, McAfee S, et al. Phase I study of urate oxidase in the reduction of acute graft-versus-host disease after myeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20(5):730–4. doi:10.1016/j.bbmt.2014.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Cooke KR, Gerbitz A, Crawford JM, Teshima T, Hill GR, Tesolin A, et al. LPS antagonism reduces graft-versus-host disease and preserves graft-versus-leukemia activity after experimental bone marrow transplantation. J Clin Invest (2001) 107(12):1581–9. doi:10.1172/JCI12156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Taylor PA, Ehrhardt MJ, Lees CJ, Panoskaltsis-Mortari A, Krieg AM, Sharpe AH, et al. TLR agonists regulate alloresponses and uncover a critical role for donor APCs in allogeneic bone marrow rejection. Blood (2008) 112(8):3508–16. doi:10.1182/blood-2007-09-113670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Hossain MS, Jaye DL, Pollack BP, Farris AB, Tselanyane ML, David E, et al. Flagellin, a TLR5 agonist, reduces graft-versus-host disease in allogeneic hematopoietic stem cell transplantation recipients while enhancing antiviral immunity. J Immunol (2011) 187(10):5130–40. doi:10.4049/jimmunol.1101334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Heimesaat MM, Nogai A, Bereswill S, Plickert R, Fischer A, Loddenkemper C, et al. MyD88/TLR9 mediated immunopathology and gut microbiota dynamics in a novel murine model of intestinal graft-versus-host disease. Gut (2010) 59(8):1079–87. doi:10.1136/gut.2009.197434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Soga Y, Maeda Y, Ishimaru F, Tanimoto M, Maeda H, Nishimura F, et al. Bacterial substitution of coagulase-negative Staphylococci for Streptococci on the oral mucosa after hematopoietic cell transplantation. Support Care Cancer (2011) 19(7):995–1000. doi:10.1007/s00520-010-0923-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Michelsen KS, Aicher A, Mohaupt M, Hartung T, Dimmeler S, Kirschning CJ, et al. The role of toll-like receptors (TLRs) in bacteria-induced maturation of murine dendritic cells (DCS). Peptidoglycan and lipoteichoic acid are inducers of DC maturation and require TLR2. J Biol Chem (2001) 276(28):25680–6. doi:10.1074/jbc.M011615200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. Nod1 detects a unique muropeptide from Gram-negative bacterial peptidoglycan. Science (2003) 300(5625):1584–7. doi:10.1126/science.1084677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Vander Lugt MT, Braun TM, Hanash S, Ritz J, Ho VT, Antin JH, et al. ST2 as a marker for risk of therapy-resistant graft-versus-host disease and death. N Engl J Med (2013) 369(6):529–39. doi:10.1056/NEJMoa1213299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Nomura N, Nagase T, Miyajima N, Sazuka T, Tanaka A, Sato S, et al. Prediction of the coding sequences of unidentified human genes. II. The coding sequences of 40 new genes (KIAA0041-KIAA0080) deduced by analysis of cDNA clones from human cell line KG-1 (supplement). DNA Res (1994) 1(5):251–62. doi:10.1093/dnares/1.5.223
142. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature (1997) 388(6640):394–7. doi:10.1038/41131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi:10.1146/annurev.immunol.21.120601.141126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. O’Neill LA, Golenbock D, Bowie AG. The history of toll-like receptors – redefining innate immunity. Nat Rev Immunol (2013) 13(6):453–60. doi:10.1038/nri3446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science (1998) 282(5396):2085–8. doi:10.1126/science.282.5396.2085
146. Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science (1999) 285(5428):736–9. doi:10.1126/science.285.5428.736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, et al. A toll-like receptor recognizes bacterial DNA. Nature (2000) 408(6813):740–5. doi:10.1038/35047123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by toll-like receptor 3. Nature (2001) 413(6857):732–8. doi:10.1038/35099560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science (2004) 303(5663):1526–9. doi:10.1126/science.1093620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by toll-like receptor 5. Nature (2001) 410(6832):1099–103. doi:10.1038/35074106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, et al. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol (2002) 168(9):4531–7. doi:10.4049/jimmunol.168.9.4531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, et al. Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol (2005) 174(5):2942–50. doi:10.4049/jimmunol.174.5.2942
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Lee MS, Kim YJ. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annu Rev Biochem (2007) 76:447–80. doi:10.1146/annurev.biochem.76.060605.122847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Khan JA, Brint EK, O’Neill LA, Tong L. Crystal structure of the toll/interleukin-1 receptor domain of human IL-1RAPL. J Biol Chem (2004) 279(30):31664–70. doi:10.1074/jbc.M403434200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Kitamura H, Morikawa H, Kamon H, Iguchi M, Hojyo S, Fukada T, et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat Immunol (2006) 7(9):971–7. doi:10.1038/ni1373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Frleta D, Noelle RJ, Wade WF. CD40-mediated up-regulation of toll-like receptor 4-MD2 complex on the surface of murine dendritic cells. J Leukoc Biol (2003) 74(6):1064–73. doi:10.1189/jlb.0203062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Geisel J, Kahl F, Muller M, Wagner H, Kirschning CJ, Autenrieth IB, et al. IL-6 and maturation govern TLR2 and TLR4 induced TLR agonist tolerance and cross-tolerance in dendritic cells. J Immunol (2007) 179(9):5811–8. doi:10.4049/jimmunol.179.9.5811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Lorenz E, Schwartz DA, Martin PJ, Gooley T, Lin MT, Chien JW, et al. Association of TLR4 mutations and the risk for acute GVHD after HLA-matched-sibling hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2001) 7(7):384–7. doi:10.1053/bbmt.2001.v7.pm11529488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Sawitzki B, Brunstein C, Meisel C, Schumann J, Vogt K, Appelt C, et al. Prevention of graft-versus-host disease by adoptive T regulatory therapy is associated with active repression of peripheral blood toll-like receptor 5 mRNA expression. Biol Blood Marrow Transplant (2014) 20(2):173–82. doi:10.1016/j.bbmt.2013.10.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Calcaterra C, Sfondrini L, Rossini A, Sommariva M, Rumio C, Menard S, et al. Critical role of TLR9 in acute graft-versus-host disease. J Immunol (2008) 181(9):6132–9. doi:10.4049/jimmunol.181.9.6132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Elmaagacli AH, Koldehoff M, Beelen DW. Improved outcome of hematopoietic SCT in patients with homozygous gene variant of toll-like receptor 9. Bone Marrow Transplant (2009) 44(5):295–302. doi:10.1038/bmt.2009.32
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Xiao HW, Luo Y, Lai XY, Shi JM, Tan YM, He JS, et al. Donor TLR9 gene tagSNPs influence susceptibility to aGVHD and CMV reactivation in the allo-HSCT setting without polymorphisms in the TLR4 and NOD2 genes. Bone Marrow Transplant (2014) 49(2):241–7. doi:10.1038/bmt.2013.160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem (1999) 274(21):14560–7. doi:10.1074/jbc.274.21.14560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature (2001) 411(6837):603–6. doi:10.1038/35079114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J, et al. Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J (2004) 23(7):1587–97. doi:10.1038/sj.emboj.7600175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE. The nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol (2007) 29(3):289–301. doi:10.1007/s00281-007-0083-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity (2009) 30(6):789–801. doi:10.1016/j.immuni.2009.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell (2012) 46(6):746–58. doi:10.1016/j.molcel.2012.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Barnich N, Aguirre JE, Reinecker HC, Xavier R, Podolsky DK. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol (2005) 170(1):21–6. doi:10.1083/jcb.200502153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol (2010) 11(1):55–62. doi:10.1038/ni.1823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol (2009) 10(10):1073–80. doi:10.1038/ni.1782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Lupfer C, Thomas PG, Kanneganti TD. Nucleotide oligomerization and binding domain 2-dependent dendritic cell activation is necessary for innate immunity and optimal CD8+ T cell responses to influenza A virus infection. J Virol (2014) 88(16):8946–55. doi:10.1128/JVI.01110-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Shaw MH, Reimer T, Sanchez-Valdepenas C, Warner N, Kim YG, Fresno M, et al. T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol (2009) 10(12):1267–74. doi:10.1038/ni.1816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Franchi L, Warner N, Viani K, Nunez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev (2009) 227(1):106–28. doi:10.1111/j.1600-065X.2008.00734.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi:10.1038/ni.1980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol (2012) 13(4):325–32. doi:10.1038/ni.2231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Granell M, Urbano-Ispizua A, Arostegui JI, Fernandez-Aviles F, Martinez C, Rovira M, et al. Effect of NOD2/CARD15 variants in T-cell depleted allogeneic stem cell transplantation. Haematologica (2006) 91(10):1372–6.
178. van der Velden WJ, Blijlevens NM, Maas FM, Schaap NP, Jansen JH, van der Reijden BA, et al. NOD2 polymorphisms predict severe acute graft-versus-host and treatment-related mortality in T-cell-depleted haematopoietic stem cell transplantation. Bone Marrow Transplant (2009) 44(4):243–8. doi:10.1038/bmt.2009.21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. van der Straaten HM, Paquay MM, Tilanus MG, van Geloven N, Verdonck LF, Huisman C. NOD2/CARD15 variants are not a risk factor for clinical outcome after nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transplant (2011) 17(8):1231–6. doi:10.1016/j.bbmt.2010.12.709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Sairafi D, Uzunel M, Remberger M, Ringden O, Mattsson J. No impact of NOD2/CARD15 on outcome after SCT. Bone Marrow Transplant (2008) 41(11):961–4. doi:10.1038/bmt.2008.9
181. Mayor NP, Shaw BE, Hughes DA, Maldonado-Torres H, Madrigal JA, Keshav S, et al. Single nucleotide polymorphisms in the NOD2/CARD15 gene are associated with an increased risk of relapse and death for patients with acute leukemia after hematopoietic stem-cell transplantation with unrelated donors. J Clin Oncol (2007) 25(27):4262–9. doi:10.1200/JCO.2007.12.1897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol (2007) 7(5):340–52. doi:10.1038/nri2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Garantziotis S, Palmer SM, Snyder LD, Ganous T, Chen BJ, Wang T, et al. Alloimmune lung injury induced by local innate immune activation through inhaled lipopolysaccharide. Transplantation (2007) 84(8):1012–9. doi:10.1097/01.tp.0000286040.85007.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Mizel SB, Kilian PL, Lewis JC, Paganelli KA, Chizzonite RA. The interleukin 1 receptor. Dynamics of interleukin 1 binding and internalization in T cells and fibroblasts. J Immunol (1987) 138(9):2906–12.
185. Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int Rev Immunol (1998) 16(5–6):457–99. doi:10.3109/08830189809043005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med (2007) 13(7):851–6. doi:10.1038/nm1603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Eigenbrod T, Park JH, Harder J, Iwakura Y, Nunez G. Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol (2008) 181(12):8194–8. doi:10.4049/jimmunol.181.12.8194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell (2008) 14(2):156–65. doi:10.1016/j.ccr.2008.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Rao DA, Tracey KJ, Pober JS. IL-1alpha and IL-1beta are endogenous mediators linking cell injury to the adaptive alloimmune response. J Immunol (2007) 179(10):6536–46. doi:10.4049/jimmunol.179.10.6536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Pender SL, MacDonald TT. Matrix metalloproteinases and the gut – new roles for old enzymes. Curr Opin Pharmacol (2004) 4(6):546–50. doi:10.1016/j.coph.2004.06.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Kruidenier L, MacDonald TT, Collins JE, Pender SL, Sanderson IR. Myofibroblast matrix metalloproteinases activate the neutrophil chemoattractant CXCL7 from intestinal epithelial cells. Gastroenterology (2006) 130(1):127–36. doi:10.1053/j.gastro.2005.09.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Heuschkel RB, MacDonald TT, Monteleone G, Bajaj-Elliott M, Smith JA, Pender SL. Imbalance of stromelysin-1 and TIMP-1 in the mucosal lesions of children with inflammatory bowel disease. Gut (2000) 47(1):57–62. doi:10.1136/gut.47.1.57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Busfield SJ, Comrack CA, Yu G, Chickering TW, Smutko JS, Zhou H, et al. Identification and gene organization of three novel members of the IL-1 family on human chromosome 2. Genomics (2000) 66(2):213–6. doi:10.1006/geno.2000.6184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity (1998) 9(1):143–50. doi:10.1016/S1074-7613(00)80596-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol (2001) 19:423–74. doi:10.1146/annurev.immunol.19.1.423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Fujimori Y, Takatsuka H, Takemoto Y, Hara H, Okamura H, Nakanishi K, et al. Elevated interleukin (IL)-18 levels during acute graft-versus-host disease after allogeneic bone marrow transplantation. Br J Haematol (2000) 109(3):652–7. doi:10.1046/j.1365-2141.2000.02095.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Nakamura H, Komatsu K, Ayaki M, Kawamoto S, Murakami M, Uoshima N, et al. Serum levels of soluble IL-2 receptor, IL-12, IL-18, and IFN-gamma in patients with acute graft-versus-host disease after allogeneic bone marrow transplantation. J Allergy Clin Immunol (2000) 106(1 Pt 2):S45–50. doi:10.1067/mai.2000.106774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Zecchina G, Novick D, Rubinstein M, Barak V, Dinarello C, Nagler A. Interleukin-18 binding protein in acute graft versus host disease and engraftment following allogeneic peripheral blood stem cell transplants. J Hematother Stem Cell Res (2001) 10(6):769–76. doi:10.1089/152581601317210863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Itoi H, Fujimori Y, Tsutsui H, Matsui K, Futatsugi S, Okamura H, et al. Fas ligand-induced caspase-1-dependent accumulation of interleukin-18 in mice with acute graft-versus-host disease. Blood (2001) 98(1):235–7. doi:10.1182/blood.V98.1.235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23(5):479–90. doi:10.1016/j.immuni.2005.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A (2007) 104(1):282–7. doi:10.1073/pnas.0606854104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PLoS One (2008) 3(10):e3331. doi:10.1371/journal.pone.0003331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Tago K, Noda T, Hayakawa M, Iwahana H, Yanagisawa K, Yashiro T, et al. Tissue distribution and subcellular localization of a variant form of the human ST2 gene product, ST2V. Biochem Biophys Res Commun (2001) 285(5):1377–83. doi:10.1006/bbrc.2001.5306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest (2007) 117(6):1538–49. doi:10.1172/JCI30634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Palmer G, Lipsky BP, Smithgall MD, Meininger D, Siu S, Talabot-Ayer D, et al. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine (2008) 42(3):358–64. doi:10.1016/j.cyto.2008.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Mildner M, Storka A, Lichtenauer M, Mlitz V, Ghannadan M, Hoetzenecker K, et al. Primary sources and immunological prerequisites for sST2 secretion in humans. Cardiovasc Res (2010) 87(4):769–77. doi:10.1093/cvr/cvq104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. Bartunek J, Delrue L, Van Durme F, Muller O, Casselman F, De Wiest B, et al. Nonmyocardial production of ST2 protein in human hypertrophy and failure is related to diastolic load. J Am Coll Cardiol (2008) 52(25):2166–74. doi:10.1016/j.jacc.2008.09.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Lipsky BP, Toy DY, Swart DA, Smithgall MD, Smith D. Deletion of the ST2 proximal promoter disrupts fibroblast-specific expression but does not reduce the amount of soluble ST2 in circulation. Eur J Immunol (2012) 42(7):1863–9. doi:10.1002/eji.201142274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Weinberg EO, Shimpo M, De Keulenaer GW, MacGillivray C, Tominaga S, Solomon SD, et al. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation (2002) 106(23):2961–6. doi:10.1161/01.CIR.0000038705.69871.D9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Tajima S, Oshikawa K, Tominaga S, Sugiyama Y. The increase in serum soluble ST2 protein upon acute exacerbation of idiopathic pulmonary fibrosis. Chest (2003) 124(4):1206–14. doi:10.1378/chest.124.4.1206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. van den Brink MR, Moore E, Ferrara JL, Burakoff SJ. Graft-versus-host-disease-associated thymic damage results in the appearance of T cell clones with anti-host reactivity. Transplantation (2000) 69(3):446–9. doi:10.1097/00007890-200002150-00026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Meves A, el-Azhary RA, Talwalkar JA, Moore SB, Brewer JD, Motsonelidze C, et al. Acute graft-versus-host disease after liver transplantation diagnosed by fluorescent in situ hybridization testing of skin biopsy specimens. J Am Acad Dermatol (2006) 55(4):642–6. doi:10.1016/j.jaad.2006.04.073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Hofmeister CC, Quinn A, Cooke KR, Stiff P, Nickoloff B, Ferrara JL. Graft-versus-host disease of the skin: life and death on the epidermal edge. Biol Blood Marrow Transplant (2004) 10(6):366–72. doi:10.1016/j.bbmt.2004.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Parkman R. Is chronic graft versus host disease an autoimmune disease? Curr Opin Immunol (1993) 5(5):800–3. doi:10.1016/0952-7915(93)90140-N
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Qin JZ, Chaturvedi V, Denning MF, Bacon P, Panella J, Choubey D, et al. Regulation of apoptosis by p53 in UV-irradiated human epidermis, psoriatic plaques and senescent keratinocytes. Oncogene (2002) 21(19):2991–3002. doi:10.1038/sj.onc.1205404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. McGuckin MA, Eri R, Simms LA, Florin TH, Radford-Smith G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis (2009) 15(1):100–13. doi:10.1002/ibd.20539
217. Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature (2011) 469(7331):543–7. doi:10.1038/nature09646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Nestel FP, Price KS, Seemayer TA, Lapp WS. Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor alpha during graft-versus-host disease. J Exp Med (1992) 175(2):405–13. doi:10.1084/jem.175.2.405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Scholl PR, Geha RS. MHC class II signaling in B-cell activation. Immunol Today (1994) 15(9):418–22. doi:10.1016/0167-5699(94)90271-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest (1989) 83(2):724–7. doi:10.1172/JCI113938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Garside P, Bunce C, Tomlinson RC, Nichols BL, Mowat AM. Analysis of enteropathy induced by tumour necrosis factor alpha. Cytokine (1993) 5(1):24–30. doi:10.1016/1043-4666(93)90020-6
222. Garside P, Reid S, Steel M, Mowat AM. Differential cytokine production associated with distinct phases of murine graft-versus-host reaction. Immunology (1994) 82(2):211–4.
223. Deguchi M, Whitaker-Menezes D, Jones SC, Aiba S, Nakagawa S, Tagami H, et al. 12E2: a cloned murine dermal cell with features of dermal dendrocytes and capacity to produce pathologic changes resembling early Kaposi’s sarcoma. Am J Pathol (2003) 163(5):1817–25. doi:10.1016/S0002-9440(10)63541-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Whitaker-Menezes D, Jones SC, Friedman TM, Korngold R, Murphy GF. An epithelial target site in experimental graft-versus-host disease and cytokine-mediated cytotoxicity is defined by cytokeratin 15 expression. Biol Blood Marrow Transplant (2003) 9(9):559–70. doi:10.1016/S1083-8791(03)00288-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Noga SJ, Vogelsang GB, Seber A, Davis JM, Schepers K, Hess AD, et al. CD34+ stem cell augmentation of allogeneic, elutriated marrow grafts improves engraftment but cyclosporine A is still required to reduce GVHD and morbidity. Transplant Proc (1997) 29(1–2):728–32. doi:10.1016/S0041-1345(96)00442-3
226. Lin T, Brunner T, Tietz B, Madsen J, Bonfoco E, Reaves M, et al. Fas ligand-mediated killing by intestinal intraepithelial lymphocytes. Participation in intestinal graft-versus-host disease. J Clin Invest (1998) 101(3):570–7. doi:10.1172/JCI896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Das R, Chen X, Komorowski R, Hessner MJ, Drobyski WR. Interleukin-23 secretion by donor antigen-presenting cells is critical for organ-specific pathology in graft-versus-host disease. Blood (2009) 113(10):2352–62. doi:10.1182/blood-2008-08-175448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
228. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity (2012) 37(2):339–50. doi:10.1016/j.immuni.2012.05.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
229. Munneke JM, Bjorklund AT, Mjosberg JM, Garming-Legert K, Bernink JH, Blom B, et al. Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease. Blood (2014) 124(5):812–21. doi:10.1182/blood-2013-11-536888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood (1997) 90(8):3204–13.
231. Hill GR, Teshima T, Gerbitz A, Pan L, Cooke KR, Brinson YS, et al. Differential roles of IL-1 and TNF-alpha on graft-versus-host disease and graft versus leukemia. J Clin Invest (1999) 104(4):459–67. doi:10.1172/JCI6896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology (2002) 40(5):403–39. doi:10.1046/j.1365-2559.2002.01387.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Luft T, Conzelmann M, Benner A, Rieger M, Hess M, Strohhaecker U, et al. Serum cytokeratin-18 fragments as quantitative markers of epithelial apoptosis in liver and intestinal graft-versus-host disease. Blood (2007) 110(13):4535–42. doi:10.1182/blood-2006-10-049817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Levine JE, Paczesny S, Sarantopoulos S. Clinical applications for biomarkers of acute and chronic graft-versus-host disease. Biol Blood Marrow Transplant (2012) 18(1 Suppl):S116–24. doi:10.1016/j.bbmt.2011.10.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Sanos SL, Vonarbourg C, Mortha A, Diefenbach A. Control of epithelial cell function by interleukin-22-producing RORgammat+ innate lymphoid cells. Immunology (2011) 132(4):453–65. doi:10.1111/j.1365-2567.2011.03410.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol (2011) 12(1):21–7. doi:10.1038/ni.1962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Wu T, Young JS, Johnston H, Ni X, Deng R, Racine J, et al. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J Immunol (2013) 191(1):488–99. doi:10.4049/jimmunol.1300657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Teshima T, Reddy P, Liu C, Williams D, Cooke KR, Ferrara JL. Impaired thymic negative selection causes autoimmune graft-versus-host disease. Blood (2003) 102(2):429–35. doi:10.1182/blood-2003-01-0266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
239. Tivol E, Komorowski R, Drobyski WR. Emergent autoimmunity in graft-versus-host disease. Blood (2005) 105(12):4885–91. doi:10.1182/blood-2004-12-4980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
240. Sorokin R, Kimura H, Schroder K, Wilson DH, Wilson DB. Cyclosporine-induced autoimmunity. Conditions for expressing disease, requirement for intact thymus, and potency estimates of autoimmune lymphocytes in drug-treated rats. J Exp Med (1986) 164(5):1615–25. doi:10.1084/jem.164.5.1615
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. Reddy P, Arora M, Guimond M, Mackall CL. GVHD: a continuing barrier to the safety of allogeneic transplantation. Biol Blood Marrow Transplant (2009) 15(1 Suppl):162–8. doi:10.1016/j.bbmt.2008.10.014
242. Wils EJ, Rombouts EJ, van Mourik I, Spits H, Legrand N, Braakman E, et al. Stem cell factor consistently improves thymopoiesis after experimental transplantation of murine or human hematopoietic stem cells in immunodeficient mice. J Immunol (2011) 187(6):2974–81. doi:10.4049/jimmunol.1004209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
243. Hauri-Hohl MM, Keller MP, Gill J, Hafen K, Pachlatko E, Boulay T, et al. Donor T-cell alloreactivity against host thymic epithelium limits T-cell development after bone marrow transplantation. Blood (2007) 109(9):4080–8. doi:10.1182/blood-2006-07-034157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
244. Zlotoff DA, Zhang SL, De Obaldia ME, Hess PR, Todd SP, Logan TD, et al. Delivery of progenitors to the thymus limits T-lineage reconstitution after bone marrow transplantation. Blood (2011) 118(7):1962–70. doi:10.1182/blood-2010-12-324954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
245. Clave E, Busson M, Douay C, Peffault de Latour R, Berrou J, Rabian C, et al. Acute graft-versus-host disease transiently impairs thymic output in young patients after allogeneic hematopoietic stem cell transplantation. Blood (2009) 113(25):6477–84. doi:10.1182/blood-2008-09-176594
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
246. Barker GJ, Epstein JB, Williams KB, Gorsky M, Raber-Durlacher JE. Current practice and knowledge of oral care for cancer patients: a survey of supportive health care providers. Support Care Cancer (2005) 13(1):32–41. doi:10.1007/s00520-004-0691-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
247. Brennan MT, Elting LS, Spijkervet FK. Systematic reviews of oral complications from cancer therapies, oral care study group, MASCC/ISOO: methodology and quality of the literature. Support Care Cancer (2010) 18(8):979–84. doi:10.1007/s00520-010-0856-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
248. Barasch A, Epstein JB. Management of cancer therapy-induced oral mucositis. Dermatol Ther (2011) 24(4):424–31. doi:10.1111/j.1529-8019.2011.01434.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
249. Bellm LA, Epstein JB, Rose-Ped A, Martin P, Fuchs HJ. Patient reports of complications of bone marrow transplantation. Support Care Cancer (2000) 8(1):33–9.
250. Schubert MM, Correa ME. Oral graft-versus-host disease. Dent Clin North Am (2008) 52(1):79–109. doi:10.1016/j.cden.2007.10.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
251. Denham JW, Hauer-Jensen M. The radiotherapeutic injury – a complex ‘wound’. Radiother Oncol (2002) 63(2):129–45. doi:10.1016/S0167-8140(02)00060-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
252. Sonis ST. The pathobiology of mucositis. Nat Rev Cancer (2004) 4(4):277–84. doi:10.1038/nrc1318
253. Yeoh AS, Bowen JM, Gibson RJ, Keefe DM. Nuclear factor kappaB (NFkappaB) and cyclooxygenase-2 (Cox-2) expression in the irradiated colorectum is associated with subsequent histopathological changes. Int J Radiat Oncol Biol Phys (2005) 63(5):1295–303. doi:10.1016/j.ijrobp.2005.04.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
254. Sonis ST. The biologic role for nuclear factor-kappaB in disease and its potential involvement in mucosal injury associated with anti-neoplastic therapy. Crit Rev Oral Biol Med (2002) 13(5):380–9. doi:10.1177/154411130201300502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
255. Lima V, Brito GA, Cunha FQ, Reboucas CG, Falcao BA, Augusto RF, et al. Effects of the tumour necrosis factor-alpha inhibitors pentoxifylline and thalidomide in short-term experimental oral mucositis in hamsters. Eur J Oral Sci (2005) 113(3):210–7. doi:10.1111/j.1600-0722.2005.00216.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
256. Freitas AP, Bitencourt FS, Brito GA, de Alencar NM, Ribeiro RA, Lima-Junior RC, et al. Protein fraction of Calotropis procera latex protects against 5-fluorouracil-induced oral mucositis associated with downregulation of pivotal pro-inflammatory mediators. Naunyn Schmiedebergs Arch Pharmacol (2012) 385(10):981–90. doi:10.1007/s00210-012-0778-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
257. Curra M, Martins MA, Lauxen IS, Pellicioli AC, Sant’Ana Filho M, Pavesi VC, et al. Effect of topical chamomile on immunohistochemical levels of IL-1beta and TNF-alpha in 5-fluorouracil-induced oral mucositis in hamsters. Cancer Chemother Pharmacol (2013) 71(2):293–9. doi:10.1007/s00280-012-2013-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
258. Nomura M, Kamata M, Kojima H, Hayashi K, Sawada S. Irsogladine maleate reduces the incidence of fluorouracil-based chemotherapy-induced oral mucositis. Ann Oncol (2013) 24(4):1062–6. doi:10.1093/annonc/mds584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
259. Raber-Durlacher JE, von Bultzingslowen I, Logan RM, Bowen J, Al-Azri AR, Everaus H, et al. Systematic review of cytokines and growth factors for the management of oral mucositis in cancer patients. Support Care Cancer (2013) 21(1):343–55. doi:10.1007/s00520-012-1594-5
260. Maynard CL, Elson CO, Hatton RD, Weaver CT. Reciprocal interactions of the intestinal microbiota and immune system. Nature (2012) 489(7415):231–41. doi:10.1038/nature11551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
261. Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol (2013) 14(7):660–7. doi:10.1038/ni.2611
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
262. Epstein JB, Gorsky M, Guglietta A, Le N, Sonis ST. The correlation between epidermal growth factor levels in saliva and the severity of oral mucositis during oropharyngeal radiation therapy. Cancer (2000) 89(11):2258–65. doi:10.1002/1097-0142(20001201)89:11<2258::AID-CNCR14>3.0.CO;2-Z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
263. Antin JH, Lee SJ, Neuberg D, Alyea E, Soiffer RJ, Sonis S, et al. A phase I/II double-blind, placebo-controlled study of recombinant human interleukin-11 for mucositis and acute GVHD prevention in allogeneic stem cell transplantation. Bone Marrow Transplant (2002) 29(5):373–7. doi:10.1038/sj.bmt.1703394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
264. Sonis ST, Van Vugt AG, Brien JP, Muska AD, Bruskin AM, Rose A, et al. Transforming growth factor-beta 3 mediated modulation of cell cycling and attenuation of 5-fluorouracil induced oral mucositis. Oral Oncol (1997) 33(1):47–54. doi:10.1016/S0964-1955(96)00043-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
265. Farrell CL, Rex KL, Chen JN, Bready JV, DiPalma CR, Kaufman SA, et al. The effects of keratinocyte growth factor in preclinical models of mucositis. Cell Prolif (2002) 35(Suppl 1):78–85. doi:10.1046/j.1365-2184.35.s1.8.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
266. Na DH, Lee JE, Jang SW, Lee KC. Formation of acylated growth hormone-releasing peptide-6 by poly(lactide-co-glycolide) and its biological activity. AAPS PharmSciTech (2007) 8(2):Article43. doi:10.1208/pt0802043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
267. Spielberger R, Stiff P, Bensinger W, Gentile T, Weisdorf D, Kewalramani T, et al. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med (2004) 351(25):2590–8. doi:10.1056/NEJMoa040125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
268. Paczesny S, Duncan C, Jacobsohn D, Krance R, Leung K, Carpenter P, et al. Opportunities and challenges of proteomics in pediatric patients: circulating biomarkers after hematopoietic stem cell transplantation as a successful example. Proteomics Clin Appl (2014) 8:837–50. doi:10.1002/prca.201400033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
269. Choi SW, Reddy P. Current and emerging strategies for the prevention of graft-versus-host disease. Nat Rev Clin Oncol (2014) 11(9):536–47. doi:10.1038/nrclinonc.2014.102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
270. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci U S A (2012) 109(2):564–9. doi:10.1073/pnas.1117665109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
271. Marcondes AM, Li X, Tabellini L, Bartenstein M, Kabacka J, Sale GE, et al. Inhibition of IL-32 activation by alpha-1 antitrypsin suppresses alloreactivity and increases survival in an allogeneic murine marrow transplantation model. Blood (2011) 118(18):5031–9. doi:10.1182/blood-2011-07-365247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
272. Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature (2013) 498(7452):113–7. doi:10.1038/nature12240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
273. Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, et al. Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J Exp Med (2014) 211(9):1723–31. doi:10.1084/jem.20140212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
274. Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature (2014) 514:237–41. doi:10.1038/nature13564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
275. Lindemans C, Mertelsmann A, Dudakov JA, Velardi E, Hua G, O’Connor M, et al. IL-22 administration protects intestinal stem cells from GVHD. Biol Blood Marrow Transplant (2014) 20(2):S53–4. doi:10.1016/j.bbmt.2013.12.056
276. Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science (2011) 334(6062):1561–5. doi:10.1126/science.1214914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
277. Lee JS, Cella M, Colonna M. AHR and the transcriptional regulation of type-17/22 ILC. Front Immunol (2012) 3:10. doi:10.3389/fimmu.2012.00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
278. McCarthy PL Jr, Abhyankar S, Neben S, Newman G, Sieff C, Thompson RC, et al. Inhibition of interleukin-1 by an interleukin-1 receptor antagonist prevents graft-versus-host disease. Blood (1991) 78(8):1915–8.
279. Antin JH, Weinstein HJ, Guinan EC, McCarthy P, Bierer BE, Gilliland DG, et al. Recombinant human interleukin-1 receptor antagonist in the treatment of steroid-resistant graft-versus-host disease. Blood (1994) 84(4):1342–8.
280. Antin JH, Weisdorf D, Neuberg D, Nicklow R, Clouthier S, Lee SJ, et al. Interleukin-1 blockade does not prevent acute graft-versus-host disease: results of a randomized, double-blind, placebo-controlled trial of interleukin-1 receptor antagonist in allogeneic bone marrow transplantation. Blood (2002) 100(10):3479–82. doi:10.1182/blood-2002-03-0985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
281. Chen LQ, de Lemos JA, Das SR, Ayers CR, Rohatgi A. Soluble ST2 is associated with all-cause and cardiovascular mortality in a population-based cohort: the Dallas Heart Study. Clin Chem (2013) 59(3):536–46. doi:10.1373/clinchem.2012.191106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
282. Zheng YG, Yang T, He JG, Chen G, Liu ZH, Xiong CM, et al. Plasma soluble ST2 levels correlate with disease severity and predict clinical worsening in patients with pulmonary arterial hypertension. Clin Cardiol (2014) 37(6):365–70. doi:10.1002/clc.22262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
283. Trajkovic V, Sweet MJ, Xu D. T1/ST2 – an IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev (2004) 15(2–3):87–95. doi:10.1016/j.cytogfr.2004.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
284. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol (2008) 8(9):726–36. doi:10.1038/nri2395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
285. Koc ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, et al. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol (2000) 18(2):307–16.
286. Baron F, Lechanteur C, Willems E, Bruck F, Baudoux E, Seidel L, et al. Cotransplantation of mesenchymal stem cells might prevent death from graft-versus-host disease (GVHD) without abrogating graft-versus-tumor effects after HLA-mismatched allogeneic transplantation following nonmyeloablative conditioning. Biol Blood Marrow Transplant (2010) 16(6):838–47. doi:10.1016/j.bbmt.2010.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
287. Yin F, Battiwalla M, Ito S, Feng X, Chinian F, Melenhorst JJ, et al. Bone marrow mesenchymal stromal cells to treat tissue damage in allogeneic stem cell transplant recipients: correlation of biological markers with clinical responses. Stem Cells (2014) 32(5):1278–88. doi:10.1002/stem.1638
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
288. Santaolalla R, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol (2012) 28(2):124–9. doi:10.1097/MOG.0b013e3283506559
289. Jones GR. Free radicals in immunological killing: the case of tumor necrotising factor (TNF). Med Hypotheses (1986) 21(3):267–71. doi:10.1016/0306-9877(86)90019-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
290. Komai-Koma M, Li D, Wang E, Vaughan D, Xu D. Anti-toll-like receptor 2 and 4 antibodies suppress inflammatory response in mice. Immunology (2014) 143(3):354–62. doi:10.1111/imm.12312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
291. Socie G, Ritz J. Current issues in chronic graft-versus-host disease. Blood (2014) 124(3):374–84. doi:10.1182/blood-2014-01-514752
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
292. Gowdy KM, Nugent JL, Martinu T, Potts E, Snyder LD, Foster WM, et al. Protective role of T-bet and Th1 cytokines in pulmonary graft-versus-host disease and peribronchiolar fibrosis. Am J Respir Cell Mol Biol (2012) 46(2):249–56. doi:10.1165/rcmb.2011-0131OC
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
293. Bruggen MC, Klein I, Greinix H, Bauer W, Kuzmina Z, Rabitsch W, et al. Diverse T-cell responses characterize the different manifestations of cutaneous graft-versus-host disease. Blood (2014) 123(2):290–9. doi:10.1182/blood-2013-07-514372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
294. Krenger W, Blazar BR, Hollander GA. Thymic T-cell development in allogeneic stem cell transplantation. Blood (2011) 117(25):6768–76. doi:10.1182/blood-2011-02-334623
295. Matsuoka K, Kim HT, McDonough S, Bascug G, Warshauer B, Koreth J, et al. Altered regulatory T cell homeostasis in patients with CD4+ lymphopenia following allogeneic hematopoietic stem cell transplantation. J Clin Invest (2010) 120(5):1479–93. doi:10.1172/JCI41072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
296. Allen JL, Fore MS, Wooten J, Roehrs PA, Bhuiya NS, Hoffert T, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood (2012) 120(12):2529–36. doi:10.1182/blood-2012-06-438911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
297. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol (2014) 192(8):3626–36. doi:10.4049/jimmunol.1302062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: graft-versus-host disease, danger signals, tissue damage, alarmins, pathogen-associated molecular patterns, damage-associated molecular patterns, innate immunity, biomarkers
Citation: Ramadan A and Paczesny S (2015) Various forms of tissue damage and danger signals following hematopoietic stem-cell transplantation. Front. Immunol. 6:14. doi: 10.3389/fimmu.2015.00014
Received: 26 September 2014; Paper pending published: 17 November 2014;
Accepted: 08 January 2015; Published online: 28 January 2015.
Edited by:
Attilio Bondanza, S. Raffaele Scientific Institute, ItalyReviewed by:
Rupali Das, Children’s Hospital of Philadelphia, USAJoerg Halter, University Hospital Basel, Switzerland
Copyright: © 2015 Ramadan and Paczesny. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdulraouf Ramadan and Sophie Paczesny, Department of Pediatrics, Melvin and Bren Simon Cancer Center, and Department of Microbiology and Immunology, Indiana University, 1044 West Walnut Street, Room R4-425, Indianapolis, IN 46202, USA e-mail: aramadan@iupui.edu; sophpacz@iu.edu