 Emily M. Eshleman
Emily M. Eshleman Laurel L. Lenz
Laurel L. Lenz- 1Department of Immunology and Microbiology, University of Colorado School of Medicine, Aurora, CO, USA
- 2Department of Biomedical Research, National Jewish Health, Denver, CO, USA
Type I interferons (IFNs) were first described for their ability to protect the host from viral infections and may also have beneficial effects under specific conditions within some bacterial infections. Yet, these pleiotropic cytokines are now known to exacerbate infections by numerous life-threatening bacteria, including the intracellular pathogens Listeria monocytogenes and Mycobacterium tuberculosis. The evidence that such detrimental effects occur during bacterial infections in both animals and humans argues for selective pressure. In this review, we summarize the evidence demonstrating a pro-bacterial role for type I IFNs and discuss possible mechanisms that have been proposed to explain such effects. The theme emerges that type I IFNs act to suppress myeloid cell immune responses. The evolutionary conservation of such anti-inflammatory effects, particularly in the context of infections, suggests they may be important for limiting chronic inflammation. Given the effectiveness of type I IFNs in treatment of certain autoimmune diseases, their production may also act to raise the threshold for activation of immune responses to self-antigens.
Introduction
Type I interferons (IFNs) are a class of cytokines that includes numerous IFNα subtypes, IFNβ, IFNδ, IFNε, IFNκ, IFNτ, and IFNω (1, 2). These secreted factors are predominantly produced by innate immune and non-immune cells of humans and other animals in response to recognition of conserved microbial products, rather than specific antigens. The different type I IFNs vary in their sequences but bind and signal using a common, ubiquitously expressed, heteromeric cell surface receptor (IFNAR) comprised of IFNAR1 and IFNAR2 chains. Ligation of IFNAR in diverse cell types activates a canonical JAK/STAT signaling cascade primarily involving JAK1, Tyk2, STAT1, and STAT2 proteins. Activation of these factors leads to induced transcription of numerous type I IFN-stimulated genes (ISGs), the protein products of which largely act to disrupt various stages of viral replication (3, 4). Type I IFNs are thus important for resistance to several viral infections and are used in the clinic for effective antiviral therapy (2). However, type I IFNs also exert a variety of other effects on cellular functions and immune responses. For example, they up or down regulate production of and responsiveness to other cytokines, chemokines, and can stimulate cell growth, cell survival, or apoptosis (2, 5). Consequently, these cytokines are also used for treatment of melanomas, leukemias, and other cancers (6), and as immune modulatory agents to suppress neuroinflammation in patients suffering from relapse-remitting multiple sclerosis (MS) (2). Hence, type I IFNs exert seemingly opposing pro- or anti-inflammatory effects and pro- or anti-apoptotic effects. It is likely that these opposing effects reflect cell type-specific differences in the activation of secondary or “non-canonical” signaling events and/or variations in the dominance of a specific type I IFN species. Indeed, individual type I IFN proteins vary in their ability to elicit specific responses and stimulation of different cell types can cause distinct signaling events (7, 8).
The second class of IFN protein (type II or IFNγ) is more critical for host defense against intracellular bacterial pathogens, such as Mycobacterium tuberculosis and Listeria monocytogenes. IFNγ signals through its own ubiquitously expressed heteromeric receptor (IFNGR), which utilizes IFNGR1 and IFNGR2 chains to activate a canonical JAK/STAT pathway primarily involving JAK1, JAK2, and STAT1. In contrast to type I IFNs, which are broadly expressed, IFNγ is produced primarily by lymphocytes. Antigen-specific IFNγ production occurs when appropriate T lymphocyte populations respond to specific microbial antigens, while antigen non-specific production of IFNγ is stimulated by cytokines such as interleukin (IL)-12 and IL-18. Studies using L. monocytogenes and other bacterial infection models indicate that both T and natural killer (NK) cells are capable of this antigen non-specific IFNγ production (9–11). Myeloid cells such as macrophages and dendritic cells (DCs) are key targets of IFNγ, as shown by the increased susceptibility to L. monocytogenes infection in mice selectively defective for functional IFNGR1 in myeloid cells (12, 13). The expression of numerous IFNγ activated genes (GAGs) is induced by the cytokine. Some of these genes are identical to ISGs and have antiviral effects. However, IFNγ is unique in its ability to elicit a potent anti-microbial state of activation in macrophages. This “M1-type” activation is associated with increased expression of GAGs such as nitric oxide synthase 2 (NOS2) and NADPH oxidase subunits. These enzymes generate nitric oxide (NO) and reactive oxygen species (ROS) that alter cell signaling and under appropriate circumstances can mediate direct killing of bacteria (14, 15). IFNγ-inducible GTPases also promote macrophage resistance to bacterial and parasite infections by increasing the ability of phagosomal compartments to contain and kill engulfed microbes (16–18). IFNγ also upregulates myeloid cell expression of MHC II and other factors important for antigen presentation and T cell activation (19). In addition, IFNγ impacts maintenance and proliferation of hematopoietic stem cells (HSC). Specifically, basal production of IFNγ in the absence of infection drives HSC cycling and the elevated levels occurring during infection can activate HSC proliferation and myelopoiesis to replenish monocytes and other immune cells (20, 21).
Despite the antiviral effects of type I IFNs, and in the context of the antibacterial effects of IFNγ, it is increasingly evident that host responsiveness to type I IFNs correlates with increased host susceptibility to infections by L. monocytogenes, M. tuberculosis, Francisella tularensis, and several other intracellular bacterial pathogens (22–24). Here, relying heavily on the L. monocytogenes model, we review the pathways involved in the induction of type I IFNs by intracellular bacteria and various mechanisms proposed to account for the suppressive effects of type I IFN signaling. A theme that emerges from these studies is that type I IFNs have suppressive effects on anti-microbial and antigen-presenting function of myeloid cells. Such effects may contribute to both the observed ability of these cytokines to increase host susceptibility during bacterial infections and to their effectiveness in therapy of neuroinflammatory disease.
Bacterial Factors Contributing to Type I IFN Production during L. monocytogenes Infection
Listeria monocytogenes is a Gram-positive facultative intracellular bacterium that causes the systemic disease Listeriosis. The mortality rate of Listeriosis is quite high even in hospitalized patients; hence, L. monocytogenes remains a leading cause of death from foodborne illnesses within the United States. L. monocytogenes can infect hematopoietic and non-hematopoietic cell types through phagocytosis or cellular mediated uptake (25, 26). Following systemic infection in the murine model L. monocytogenes localizes to the liver and spleen where resident phagocytes, primarily macrophages and DCs, engulf the bacteria. L. monocytogenes that escape from phagosomal compartments in these cells can replicate within the cell cytosol and further propagate the infection into neighboring cells. To facilitate vacuolar escape, the bacteria secrete a pore-forming hemolysin (hly) known as listeriolysin O (LLO) (27).
Like many other bacteria, L. monocytogenes induces production of pro-inflammatory cytokines such as TNFα and type I IFNs when engulfed by professional phagocytes. Studies of infection in bone marrow derived macrophages (BMM) suggest that there are two waves of the cellular response to L. monocytogenes infection (28, 29). An “early phase” gene expression profile is seen at 1–2 h post infection by both virulent wild-type L. monocytogenes and avirulent Δhly or heat-killed bacteria that cannot escape from vacuolar compartments into the host cytosol (28, 29). Several of these “early phase” genes, including il1b, tnfa, and those encoding several chemokines are induced through the activation of Toll-like receptor (TLRs) and the ensuing activation of NFκB (28, 29). A subsequent “late-phase” response is observed at 4–8 h after infection by wild-type, but not killed or Δhly, L. monocytogenes strains (28, 29). “Late-phase” genes include IFNβ, multiple subtypes of IFNα, and several ISGs (28, 29). The fact that killed and Δhly L. monocytogenes strains fail to induce this late-phase IFN-dominated response supports the interpretation that products from bacteria replicating within the BMM cytosol stimulate cytosolic pathogen recognition receptors (PRR), though TLR stimulation can augment the induction of type I IFNs during L. monocytogenes infection (29, 30). There has been considerable interest in identifying the cytosolic PRRs responsible for type I IFN production during L. monocytogenes infection.
Pathways Leading to the Production of Type I IFNs during Bacterial Infection
Pathways known to be important for induction of type I IFN within L. monocytogenes-infected phagocytes are diagrammed in Figure 1. Amongst the earliest identified cytosolic PRRs were the nucleotide-binding oligomerization domain (NOD)-containing proteins; members of the nucleotide-binding domain, leucine-rich repeat (LRR) protein family referred to as NLRs. NOD1 and NOD2 proteins sense distinct muropeptide fragments from the cell wall from L. monocytogenes and other bacteria (31–33). Recognition of appropriate muropeptides activates a serine/threonine kinase receptor interacting protein (RIP2) to initiate downstream signaling and activation of NFκB (31). With regards to triggering of type I IFN production, NOD1, NOD2, and RIP2 seem to play an ancillary role. They augment the induction of type I IFN and other cytokine expression in response to L. monocytogenes (29, 31–33), but mice and BMM deficient for any one of these proteins retain the ability to mount inflammatory responses and synthesize type I IFNs in response to L. monocytogenes (31, 34, 35). Thus, the recognition of bacterial cell wall components by the cytosolic NOD1 and NOD2 proteins is not crucial for the induction of type I IFNs during L. monocytogenes infection.
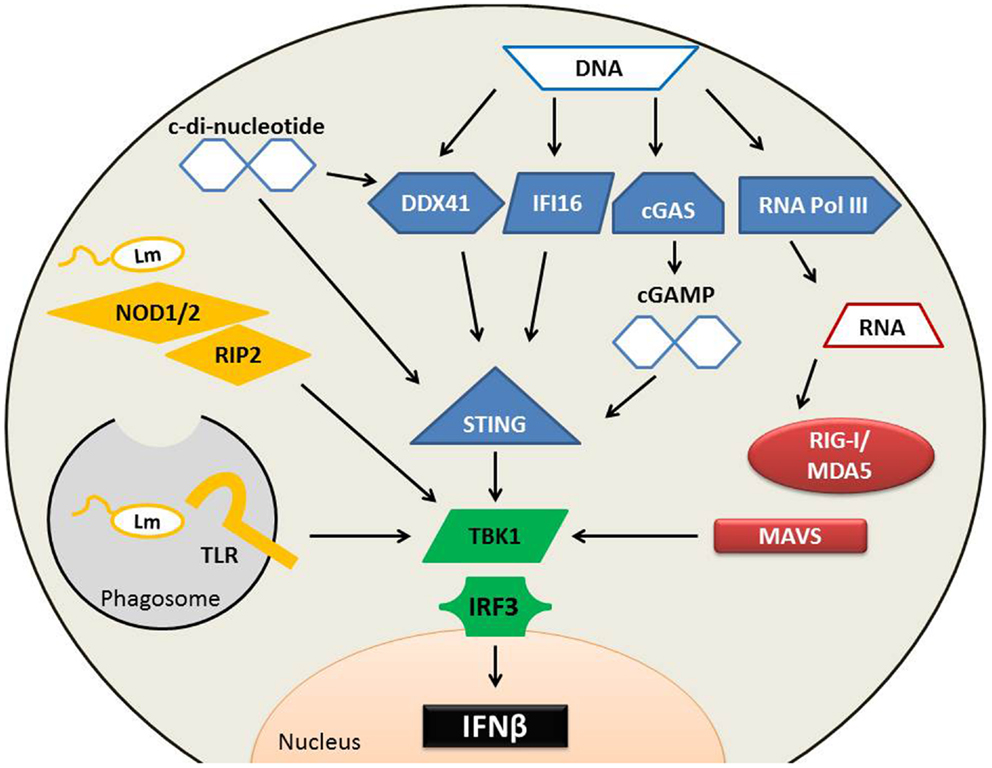
Figure 1. Pathways implicated in type I IFN production following L. monocytogenes infection. Several cytosolic receptors are able to recognize L. monocytogenes (Lm) microbial components to induce type I IFN production. (Yellow) Endosomal TLRs recognize a variety of Lm bacterial patterns including cell wall fragments, which can also stimulate NOD proteins. NOD proteins require association with RIP2 to activate TBK1. Lm secretes RNA, DNA, and cyclic-di-nucleotides. Secreted RNA (red) binds to RIG-I or MDA5, both of which associate with MAVS. Cytosolic DNA (blue) can alternatively be converted into RNA by RNA polymerase III and induce type I IFNs through the RIG-I pathway. DNA is also directly sensed by DDX41 or IFI16 to induce the production of type I IFNs in a STING-dependent mechanism. cGAS can sense DNA and convert it into cGAMP, which binds with high affinity to STING to stimulate IFNβ production. C-di-nucleotides bind to STING directly and to DDX41, either of which may result in IFNβ synthesis. TBK1 and IRF3 are essential for the induction of type I IFN production during Lm infection, and each of these upstream sensing pathways converges on TBK1 activation.
Nucleic acids are potent inducers of type I IFN production and it was shown that extracts from L. monocytogenes induce IFNβ production in a manner sensitive to DNAse treatment of the extracts (36). L. monocytogenes was also reported to actively secrete both RNA and DNA during infection of macrophages (37). Cytosolic RNA is detected by the RNA helicase retinoic acid inducible gene 1 protein (RIG-I), related RIG-I-like (RLR) proteins including melanoma differentiation-associated gene 5 (MDA5), as well as other non-RLR helicases and PRRs (38–40). RNA recognition by RIG-I and MDA5 induces their recruitment to mitochondria, where they encounter an adaptor protein [mitochondrial antiviral signaling (MAVS)] that regulates downstream signaling to induce type I IFNs (41–43). Secreted L. monocytogenes DNA could also be detected using these RNA receptor systems if it is transcribed into RNA by host cell RNA polymerase III (37). However, deficiency in RIG-I, MDA5, or MAVS fails to ablate IFNβ production by L. monocytogenes-infected BMM (37, 44, 45). Thus, it does not appear that RNA sensing is crucial for recognizing cytosolic L. monocytogenes infection in this cell type, although it may play a more important role in sensing L. monocytogenes infection of other cell types and for sensing infection by other bacteria, namely Legionella pneumophila (46, 47).
DNA present in the host cell cytosol can also be detected and trigger the production of type I IFNs (40). Several putative receptors have been identified that might mediate such recognition, including the DNA-dependent activator of IRFs (DAI), IFN-inducible gene (IFI)-16, and LRR flightless-interacting protein (LRRFIP1). Deficiency or knockdown of DAI, IFI16, or LRRFIP1 fails to completely ablate type I IFN production by infected murine BMM (48–50). However, recently IFI16 was shown to have a larger role in the recognition of L. monocytogenes DNA in the human macrophage cell line, THP1. Knockdown of IFI16 in these cells drastically reduced the production of IFNβ in response to L. monocytogenes DNA (51). The DNA-binding DEAD-box helicase DDX41 has also been shown to bind L. monocytogenes DNA. DDX41 elicits type I IFN production through a mechanism requiring the stimulator of interferon genes protein (STING), also called MITA, MPYS, or ERIS (52). STING induces type I IFN production by activating the TNFR-associated NF-κB kinase (TANK)-binding kinase 1 (TBK1), which phosphorylates a C-terminal serine residue on the transcription factor IFN regulatory factor 3 (IRF3) to induce IRF3 dimerization and nuclear translocation (34, 53). IRF3 and other IRF family members bind to the promoters of type I IFN genes and ISGs to regulate and initiate their transcription. TBK1 and IRF3 are thus not surprisingly essential for the production of type I IFNs during L. monocytogenes infection (34, 54). Likewise, deficiency or knockdown of STING significantly decreases IRF3 activation and IFNβ production in BMM, DC, or fibroblasts infected with L. monocytogenes (55–57).
It appears that STING does not respond to intact DNA, but rather to endogenous or exogenous cyclic-di-nucleotides (40). In the presence of cytosolic dsDNA, the enzyme cGAS synthesizes an endogenous cyclic-di-nucleotide, cGAMP (58). Binding of cGAMP to STING occurs with a very high affinity (~4 nM) and induced conformational changes that presumably initiate the downstream events that culminate in type I IFN production (59). Exogenous cyclic-di-nucleotides can also activate STING (56, 57, 60). Both cyclic-di-GMP (cdGMP) and cyclic-di-AMP (cdAMP) are produced by bacteria and function as second messengers. There is evidence that cdAMP is released from replicating L. monocytogenes (61, 62), thus it is conceivable that cdAMP secreted by the bacterium mediates the STING-dependent production of type I IFNs in L. monocytogenes-infected macrophages. However, while the affinity of cdAMP for STING is not known, STING binds cdGMP ~300-fold lower affinity (~1 μM) than cGAMP DNA (59). Thus, it is also conceivable that cGAMP produced by cGAS in response to secreted bacterial DNA contributes to STING-dependent type I IFN production. Regardless, it is important to keep in mind that STING deficient mice showed significantly reduced serum IFNβ only very early (8 h) after L. monocytogenes infection (56, 57). Thus, systemic L. monocytogenes infection can trigger type I IFN through multiple pathways and the impact of STING on overall type I IFN production in this model is limited.
Type I IFN Signaling and Increased Susceptibility to Bacterial Infection
In certain bacterial infection models, protective effects of type I IFNs have been reported. For example, type I IFN can reduce bacterial burdens in cultured cells infected with L. pneumophila or Chlamydia trachomatis and survival of mice is increased in sepsis models with group B Streptococcus and E. coli (63–65). Mice lacking expression of IFNε, which is abundantly expressed within the female reproductive tract, were also reported to be highly susceptible to urogenital infection by C. muridarum (66). The precise mechanisms are not clear in these cases, but the observed protective effects appear to reflect unique aspects of the models and/or pathogens studied since there is considerable evidence to indicate that type I IFNs instead play a deleterious role during infections by numerous other bacterial pathogens (22–24). Specifically, studies with mice lacking IFNAR1 report that bacterial burdens are significantly reduced and survival increased following systemic or mucosal infections with intracellular bacteria that infect the cytosol of host cells, such as L. monocytogenes (54, 67–70) and F. tularensis (71) as well as bacteria like M. tuberculosis (72–74) and C. muridarum (75, 76) that reside within vacuolar compartments. In addition, heightened type I IFN production correlates with increased host susceptibility to several bacterial infections. In mice, examples of this include the correlation of increased type I IFN production in mice with a mutated ubiquitin specific peptidase (USP18) and sensitivity to Salmonella typhimurium (77). Furthermore, isolates of L. monocytogenes and M. tuberculosis that hyper-induce type I IFN production have heightened pathogenicity in animal models (78, 79). The administration of type I IFNs or agents that induce these cytokines also causes increased susceptibility to L. monocytogenes and M. tuberculosis in model infections (54, 78, 80). Type I IFN production is also increased during viral infections. In mice, lymphocytic choriomeningitis virus (LCMV) infection potently induces type I IFNs and leads to ~1000-fold increased susceptibility to a secondary L. monocytogenes infection as measured by bacterial burdens (81). In humans, a similar situation occurs following infection with influenza virus. Influenza infections are often associated with secondary bacterial infections and secondary bacterial pneumonias are estimated to account for up to 25% of the more than 250,000 annual deaths attributed to influenza (82, 83). Such secondary infections are also thought to have caused most of the deaths from the 1918 influenza pandemic (84). Streptococcus pneumoniae is a prevalent bacterial cause of pneumonias and a model of influenza and secondary S. pneumoniae infection showed that increased bacterial burdens and mortality was dependent on IFNAR expression (85). Severe bacterial infections have also been noted in patients receiving prolonged IFNα2 therapy for chronic hepatitis C virus infection (86–88). Moreover, in the absence of obvious viral infections, signatures of type I IFN responses correlate with disease progression in human tuberculosis and leprosy patients (89, 90). Thus, despite numerous differences in the receptors and cytokines themselves, the association of type I IFNs with exacerbated bacterial infections appears to have been conserved in murine and human systems. An improved understanding how these cytokine responses are deleterious to their hosts and what has driven their conservation across this evolutionary span are important questions to address.
Mechanisms Proposed to Account for the Pro-Bacterial Effects of Type I IFNs
A summary of the proposed mechanisms for the deleterious effects of type I IFN signaling during bacterial infections is outlined in Figure 2.
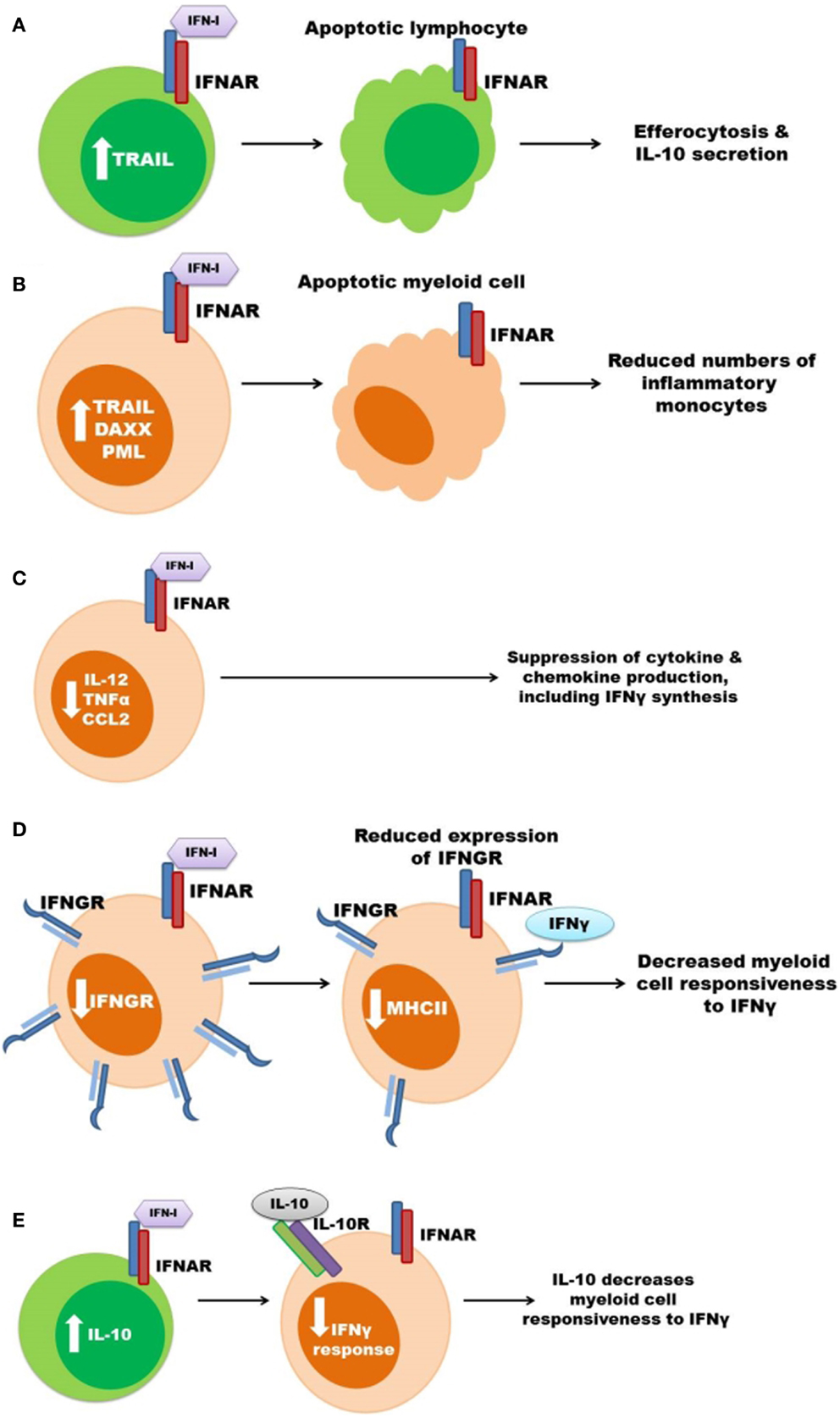
Figure 2. Some mechanisms previously proposed to account for the pro-bacterial effects of type I IFNs. (A) Type I IFNs up regulate pro-apoptotic genes resulting in lymphocyte (green) apoptosis. Apoptotic lymphocytes stimulate myeloid cell IL-10 secretion. (B) Increased apoptosis of myeloid cells (orange) leads to reduced amounts of inflammatory monocytes during infection. (C) Signaling through the IFNAR suppresses myeloid cell secretion of pro-inflammatory cytokines and chemokines, which can result in decreased IFNγ production. (D) Expression of IFNGR is suppressed in response to type I IFN signaling in myeloid cells thus decreasing cellular responsiveness to IFNγ. (E) Type I IFNs induce the production of IL-10 to inhibit IFNγ responsiveness.
Induction of Host Cell Death
It has long been known that bacterial infections can induce death of multiple cell types within tissues of murine hosts. In the systemic L. monocytogenes infection model, this cell death is exacerbated by type I IFNs. O’Connell et al. observed that expression of pro-apoptotic genes such as TNF-related apoptosis-inducing ligand (TRAIL), promyelocytic leukemia (PML), and death-associated protein 6 (Daxx) were increased in the spleens of L. monocytogenes-infected wild-type, but not IFNAR1 deficient, mice (54). Consistent with increased apoptosis in these tissues, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining is also increased in the spleens of infected wild-type mice, when compared to infected IFNAR1−/− or IRF3−/− mice (54, 69). This TUNEL staining was localized to lymphocyte rich follicles within the spleens, suggesting that type I IFN might induce apoptosis of lymphocytes and that such apoptosis could itself be detrimental (54, 69). The possibility that lymphocyte apoptosis is deleterious to the host is also consistent with the observation that mice deficient in lymphocytes or T cells alone are resistant to acute systemic L. monocytogenes infection (91–93). Resistance in T cell-deficient hosts is thought to reflect constitutively heightened macrophage activation (94), and correlates with reduced production of the anti-inflammatory cytokine IL-10 (91). It was thus proposed that in mice responsive to type I IFNs, the uptake of apoptotic cells by macrophages triggers their production of IL-10, which in turn inhibits host resistance (91). Myeloid cells are also sensitive to apoptosis in response to type I IFN and IFNAR expression has also been correlated with increased apoptosis of splenic and pulmonary macrophages during L. monocytogenes and pulmonary C. muridarum infections, respectively (54, 76). As mentioned above, these type I IFNs increase expression of the pro-apoptotic factor TRAIL during L. monocytogenes infection (54). Similar to IFNAR1−/− mice, mice lacking TRAIL demonstrate reduced TUNEL staining and increased resistance during L. monocytogenes infection (95). These effects were further correlated with increased numbers of splenic lymphocytes and monocytes. Type I IFN production induced by LCMV infection also correlates with granulocyte apoptosis and impaired control of L. monocytogenes infection (81). Thus, there is a clear association between type I IFNs, cellular death, and impaired myeloid cell responses during bacterial infections. Nonetheless, it remains unclear whether apoptosis of T or myeloid cells is a primary cause of the increased host susceptibility.
Suppression of Pro-Inflammatory Cytokine and Chemokine Production
Type I IFN production during viral infection is known to suppress production of IL-12 and other pro-inflammatory cytokines (96). Similarly, type I IFN production was associated with reduced secretion of IL-12 and TNFα in both L. monocytogenes and M. tuberculosis infection models (68, 72). Type I IFNs also suppress IL-1β production by inhibiting inflammasome activation (97), and reduced IL-1β secretion correlated with increased host susceptibility in M. tuberculosis infection models (98, 99). Expression of chemokines such as CCL2 is also regulated by type I IFNs (100, 101). CCL2 and its chemokine receptor CCR2 are critical for migration of inflammatory monocytes to sites of infection by L. monocytogenes and other bacteria (102–104). Spleens of IFNAR1−/− mice have increased accumulation of inflammatory monocytes during L. monocytogenes infection (68), however, type I IFNs upregulate CCL2 and recruitment of monocytes into the lung during M. tuberculosis infection (80). In the latter study, accumulation of monocytes correlated with more severe infection. By contrast, type I IFNs were reported to impair production of CXCL1, CXCL2, and neutrophil accumulation in lungs and more severe infection in mice infected with S. pneumoniae (85). Moderately, impaired neutrophil recruitment was also correlated with reduced IL-17 production and increased disease severity in mice in response to type I IFNs during F. tularensis and L. monocytogenes infections (71). However, it is debated whether or not neutrophils are protective during infections by L. monocytogenes and other intracellular bacteria (105, 106). Moreover, the neutropenia seen in patients treated with type I IFNs fails to correlate with their susceptibility to bacterial infections (86, 87). Thus, type I IFNs can alter production of cytokines and chemokines involved in neutrophil or inflammatory macrophage recruitment.
Suppression of Myeloid Cell Responsiveness to IFNγ
IFNγ is critical for the pro-inflammatory anti-microbial (M1) type activation of macrophages and transgenic mice lacking responsiveness to IFNγ selectively in myeloid cells are highly susceptible to L. monocytogenes and other intracellular pathogens (12, 13, 107). The macrophages activated by IFNγ have increased expression of molecules involved in both MHC class I and MHC class II antigen presentation, as well as enzymes producing reactive oxygen and nitrogen species with potential anti-microbial functions and pro-inflammatory cytokines and chemokines, including IL-12 (19). Expression of some, but not all, of these genes can also be induced when macrophages are stimulated with type I IFNs. In contrast, only IFNγ stimulates macrophages to express or upregulate MHC class II molecules (108). Indeed, stimulation of macrophages with type I IFNs suppresses their induction of MHC II expression in response to IFNγ. As mentioned above, type I IFNs also suppress production of IL-12 and CXCL1 and 2 (85, 96). These data suggest that type I IFNs are able to prevent or dampen classical M1-type anti-microbial macrophage activation in response to IFNγ. Consistent with this interpretation, a recent report revealed an inverse correlation between IFNβ and IFNγ gene expression patterns in lesions of human leprosy patients (90). The IFNβ-driven response also correlated with IL-10 production, and IFNβ production contributed to IL-10 secretion, leading the authors to conclude that the impaired IFNγ responses in M. leprae infected macrophages is due to IL-10 production (90). Indeed, IFNβ and IL-10 treatments both impaired the ability of IFNγ to induce expression of the vitamin D receptor, the vitamin D-1a-hydroxylase, and the anti-microbial peptides cathelicidin and DEFB4 in macrophages (90).
Type I IFNs were also associated with the induction of IL-10 secretion and Programed death-ligand 1 (PD-L1) expression by myeloid cells during chronic LCMV infection (109, 110). Experiments using antibody blockade of IFNAR showed reduced expression of these immune suppressive factors and increased clearance of persistent viral infections (109, 110). Interestingly, the blockade of IFNAR also suppressed production of IL-1β and IL-18, arguing against the notion that improved viral clearance was due to increased inflammasome activation. Rather, the improved viral clearance was associated with increased serum IFNγ and blockade of IFNAR failed to improve viral clearance in mice treated with antibody to block IFNγ (109, 110). The therapeutic effects of blocking type I IFN signaling also correlated with an improved ratio of stimulatory versus immune regulatory antigen-presenting cells (APCs) and enhanced antiviral T cell responses. Although the authors of one study further suggested that the suppression of inflammatory and immune responses in these studies reflected chronic type I IFN signaling (109), a second study observed that type I IFNs increased IL-10 secretion and PD-L1 expression by DCs as early as 1 day post infection (110).
Leading up to these recent studies, prior efforts had also demonstrated suppressive effects of type I IFNs on myeloid cell activation during systemic L. monocytogenes infection of mice (70). In this model, the suppressive effects of type I IFNs correlate with reductions in myeloid cell surface IFNGR1. Similarly, surface IFNGR1 staining is significantly reduced on myeloid cells from M. tuberculosis infected patients compared to healthy control and effective treatment of these patients correlates with restored myeloid surface expression of IFNGR1 (111). IFNAR expression is necessary and recombinant type I IFNs are sufficient to trigger IFNGR1 down regulation in mouse and human myeloid, but not T cells (70, 112), suggesting this mechanism might contribute to a selective inhibition of myeloid cell responsiveness to IFNγ. Indeed, the reduced expression of IFNGR1 correlates with decreased responsiveness to IFNγ as indicated by reduced STAT1 phosphorylation and impaired induction of MHC class II expression in the context of L. monocytogenes, M. tuberculosis, and F. novicida infections (70, 111, 113).
Additional mechanistic studies have revealed that type I IFNs suppress myeloid cell surface IFNGR1 within hours of stimulating the IFNAR and that this effect is due to transcriptional silencing of the otherwise constitutively expressed ifngr1 gene (70, 111, 112). The rapid reductions in ifngr1 transcript abundance following IFNβ stimulation are preceded by loss of activated RNA polymerase II at the ifngr1 transcriptional start site and the accumulation of epigenetic marks on nearby histones that are indicative of condensed chromatin (112). The reduction in ifngr1 transcription is also associated with recruitment of the early growth response 3 (Egr3) transcription factor shortly after IFNβ treatment (112). Egr3 is a member of the Egr family of zinc finger transcription factors originally defined for their role in regulation of cell growth and differentiation (114, 115). Egr3 can act as an activator or repressor in response to various stimuli, depending on post-translational modifications and association with various adapter proteins (114–117). One such adaptor protein, the NGFI-A binding protein Nab1 is a known corepressor and is also recruited to the ifngr1 promoter shortly after Egr3 (112). Knockdown of Nab1, but not Nab2, prevented IFNGR1 down regulation in macrophages treated with IFNβ, suggesting that recruitment of a repressive Egr3/Nab1 complex is responsible for rapid silencing of ifngr1 transcription (112). Given that the half-life of IFNGR1 protein is estimated at 3–4 h (118), such transcriptional silencing is sufficient to rapidly reduce myeloid cell responsiveness to IFNγ. Nonetheless, type I IFN stimulation does not appear to cause a complete loss of myeloid cell surface IFNGR1, possibly due to the induction of SOCS proteins and other endogenous negative feedback circuits that attenuate cellular responses to IFNAR signaling. These results suggest that down regulation of IFNGR1 expression might be an early step in the cascade of events leading to the suppression of myeloid cell responses that result in increased bacterial burdens and disease severity during acute and chronic bacterial infections, and the establishment or maintenance of chronic viral infections.
Common Features of Proposed Mechanisms
Given the numerous effects of type I IFNs on various cells of the immune system and on non-immune cells, it is plausible that their pro-pathogen effects vary for different pathogens. This seems particularly likely for pathogens infect different tissue or cell types, where the responses to type I IFNs may differ. For instance, a recent study demonstrated that IFNAR expression on non-hematopoietic cells was required to increase host susceptibility to the intracellular bacterial pathogen Ixodes ovatus Ehrlichia (119). The effects of these cytokines may also differ depending on the route of infection and the presence or nature of competing commensal microbes. For example, Kerbauer et al. suggested that type I IFNs might not be as detrimental to the host following gastric infection of mice with L. monocytogenes (120). Regardless, results from the studies highlighted above clearly implicate myeloid cells/APCs as key targets of type I IFNs in settings where these cytokines are deleterious to the host. Precisely, how these cytokines act to dampen myeloid cell immunity and what selective advantage this confers on the host remains to be discerned.
Apposing Effects of Type I IFNs in Autoimmune Diseases
The role of type I IFN signaling during autoimmune disease remains controversial, possibly indicating that these cytokines have opposing effects in different disease settings. For example, chronic type I IFN production is a hallmark of systemic lupus erythematosus (SLE) and several groups have reported a subset of ISGs upregulated in SLE patients compared to healthy controls (121–124). ISG signatures were also associated with disease severity and progression in SLE patients (121–123). It has thus been suggested that type I IFNs promote SLE pathology through the activation of effector cells. Type I IFNs can paradoxically promote not just death of lymphocytes, but also T cell survival, proliferation, cytotoxicity, and B cell differentiation and antibody production. Any of these effects might conceivably contribute to increased tissue damage and disease progression in SLE patients.
In contrast to the exacerbation of SLE by type I IFNs, these same cytokines confer therapeutic benefits in certain other autoimmune diseases. The most obvious example of this is the neuroinflammatory disease MS. IFNβ is a common therapy and has been shown to reduce the frequency of clinical exacerbations in patients with relapse-remitting MS (125). The mechanisms for these beneficial effects remain uncertain. However, type I IFNs also suppress disease in the murine experimental autoimmune encephalomyelitis (EAE) model of MS. As for MS, IFNβ is therapeutic in the EAE model and deletion of the ifnb gene or IFNAR1 robustly increased EAE pathogenesis in mice (126, 127). Furthermore, using conditional knockouts, it was shown that IFNAR1 expression on myeloid cells was specifically required for the therapeutic effects of IFNβ during EAE (127). Deficiencies in IFNAR1 expression on myeloid cells also severely exacerbated disease and correlated with increased secretion of TNFα and CCL2 as well as increased expression of MHC II (127). These immunosuppressive effects of IFNβ treatment in humans may likewise target myeloid cells.
Other autoimmune diseases where type I IFNs appear to play a protective role include collagen type II induced arthritis in non-human primates (128). Treatment with double-stranded RNA species or recombinant IFNα also lowered the frequency and severity of arthritic symptoms in the murine model of antigen-induced arthritis (129). The pharmacokinetics of IFNβ therapies have shown to be a barrier is translating many of these treatments from animal models to clinical use in humans. However, Mullen et al. engineered a latent form of IFNβ that can only become activated when cleaved by aggrecanase (130). Aggrecanases are highly expressed within the joints and synovial fluid of rheumatoid arthritis and osteoarthritis patients and are responsible for the cleavage of aggrecan, an important component of joint tissue (130). This delivery method allows for temporal and tissue specific release of IFNβ that resulted in a significantly increased half-life of IFNβ as well as reduced pathology and joint swelling from collagen induced arthritis (130).
Humans with autoimmunity often carry a single nucleotide mutation in protein tyrosine phosphatase non-receptor type 22 (PTPN22) (131). PTPN22 is an intracellular protein tyrosine phosphatase that is exclusively found in immune cells (131), and was recently associated with TLR signaling for type I IFN synthesis in myeloid cells (132). Functional PTPN22 was also shown to suppress inflammatory arthritis and promote gut homeostasis (132). Mice deficient in PTPN22 demonstrate increased susceptibility in the dextran sodium sulfate (DSS) mouse model of acute colitis (132). TLR stimulation by microbiota also induced immunosuppressive effects that correlated with type I IFN production and decreased progression of experimental colitis in mice (133). Treatment with recombinant IFNβ phenocopied the decreased colitis achieved through TLR stimulation (133). Moreover, in a randomized placebo controlled study of active ulcerative colitis, a significant clinical response, and in some cases, disease remission, was seen in patients that received IFNβ therapy compared to the patients that received placebo (134, 135). It was noted that the therapeutic effects of IFNβ treatment in this disease correlated with reduced production of IL-13, an effector cytokine driving intestinal inflammation (134). Mice deficient in type I IFN signaling have been shown to have exacerbated DSS-induced acute colitis (136, 137). Furthermore, mice with IFNAR1 deletion specifically in myeloid cells demonstrated significantly increased weight loss and colitis disease activity score when treated with DSS (136). These data suggest that type I IFN signaling specifically in myeloid cell is protective during DSS-induced acute colitis. Interestingly, the authors further showed that IFNAR1−/− mice recovered from DSS treatment more quickly than wild-type mice, suggesting a deleterious role of type I IFNs during the recovery phase of colitis (136).
Conclusion
Interferons are important mediators and regulators of the immune response to viruses, bacteria, and other pathogens. They can suppress inflammatory responses and exacerbate the pathogenesis in certain autoimmune diseases and several intracellular bacterial infections. Indeed, pathogens such as L. monocytogenes may actively promote type I IFN production through secretion of nucleic acids or cyclic-di-nucleotides that are recognized by cytosolic pattern recognition receptors to stimulate a type I IFN response. However, type I IFNs appear to be protective in certain other bacterial infections and in many viral infections, and may exacerbate the autoimmune disease SLE. Thus, blindly blocking their production as a therapy for bacterial infections would likely have severe untoward effects in these other disease settings. It thus remains an important challenge to dissect the mechanisms for the divergent pro- and anti-inflammatory effects of type I IFNs, as well as their paradoxical protective and deleterious effects during infectious and other diseases. As we review here, a number of observations have been correlated with the pro-bacterial effects of type I IFNs. However, while these observations have led to the proposal of several differing mechanisms to explain these detrimental effects of type I IFNs during intracellular bacterial infections, a common theme is the suppression of myeloid cell inflammatory responses. Whether such suppression results from the induction of effector cell death and IL-10 production, suppression of T cell cytokine or chemokine production, suppression of inflammasomes, or down modulation of IFNGR expression remains to be seen. However, even in the absence of experimental proof that points to a specific mechanism, it is attractive to speculate that the deleterious effects of type I IFN signaling during bacterial infections are tolerated because their ability to suppress myeloid cell responses also has a beneficial effect in protecting the host from MS and other autoimmune diseases.
Conflict of Interest Statement
Laurel L. Lenz serves on editorial boards for Frontiers in Immunology and Frontiers in Microbiology. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work on interferon responses and L. monocytogenes is supported by NIH grants AI065638, AI102264, and AI103782. Emily M. Eshleman received support from NIH training grant AI052066.
References
1. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi:10.1038/nri1604
2. Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov (2007) 6:975–90. doi:10.1038/nrd2422
3. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol (2014) 32:513–45. doi:10.1146/annurev-immunol-032713-120231
4. Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem (2007) 282:20047–51. doi:10.1074/jbc.R700004200
5. Kearney S, Delgado C, Lenz LL. Differential effects of type I and II interferons on myeloid cells and resistance to intracellular bacterial infections. Immunol Res (2012) 55:187–200. doi:10.1007/s12026-012-8362-y
6. Bracarda S, Eggermont AMM, Samuelsson J. Redefining the role of interferon in the treatment of malignant diseases. Eur J Cancer (2010) 46:284–97. doi:10.1016/j.ejca.2009.10.013
7. van Boxel-Dezaire AHH, Rani MRS, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity (2006) 25:361–72. doi:10.1016/j.immuni.2006.08.014
8. Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell (2011) 146:621–32. doi:10.1016/j.cell.2011.06.048
9. Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med (2003) 198:1583–93. doi:10.1084/jem.20031051
10. Humann J, Lenz LL. Activation of naive NK cells in response to Listeria monocytogenes requires IL-18 and contact with infected dendritic cells. J Immunol (2010) 184:5172–8. doi:10.4049/jimmunol.0903759
11. Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity (2012) 37:549–62. doi:10.1016/j.immuni.2012.05.029
12. Dighe AS, Campbell D, Hsieh CS, Clarke S, Greaves DR, Gordon S, et al. Tissue-specific targeting of cytokine unresponsiveness in transgenic mice. Immunity (1995) 3:657–66. doi:10.1016/1074-7613(95)90136-1
13. Lee SH, Carrero JA, Uppaluri R, White JM, Archambault JM, Lai KS, et al. Identifying the initiating events of anti-Listeria responses using mice with conditional loss of IFN-γ receptor subunit 1 (IFNGR1). J Immunol (2013) 191:4223–34. doi:10.4049/jimmunol.1300910
14. MacMicking J, Xie Q, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol (1997) 15:323–50. doi:10.1146/annurev.immunol.15.1.323
15. Kumatori A, Yang D, Suzuki S, Nakamura M. Cooperation of STAT-1 and IRF-1 in interferon-gamma-induced transcription of the gp91(phox) gene. J Biol Chem (2002) 277:9103–11. doi:10.1074/jbc.M109803200
16. Hunn JP, Feng CG, Sher A, Howard JC. The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm Genome (2011) 22:43–54. doi:10.1007/s00335-010-9293-3
17. Kim B-H, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. IFN-inducible GTPases in host cell defense. Cell Host Microbe (2012) 12:432–44. doi:10.1016/j.chom.2012.09.007
18. Taylor GA, Feng CG, Sher A. Control of IFN-gamma-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases). Microbes Infect (2007) 9:1644–51. doi:10.1016/j.micinf.2007.09.004
19. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol (2004) 75:164–89. doi:10.1189/jlb.0603252.Journal
20. Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature (2010) 465:793–7. doi:10.1038/nature09135
21. MacNamara KC, Oduro K, Martin O, Jones DD, McLaughlin M, Choi K, et al. Infection-induced myelopoiesis during intracellular bacterial infection is critically dependent upon IFN-γ signaling. J Immunol (2011) 186:1032–43. doi:10.4049/jimmunol.1001893
22. Rayamajhi M, Humann J, Kearney S, Hill KK, Lenz LL. Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence (2010) 1:418–22. doi:10.1084/jem.20091746.ype
23. Decker T, Müller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol (2005) 5:675–87. doi:10.1038/nri1684
24. Malireddi RKS, Kanneganti T-D. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front Cell Infect Microbiol (2013) 3:77. doi:10.3389/fcimb.2013.00077
25. Mostowy S, Cossart P. Cytoskeleton rearrangements during Listeria infection: clathrin and septins as new players in the game. Cell Motil Cytoskeleton (2009) 66:816–23. doi:10.1002/cm.20353
26. Ireton K. Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell Microbiol (2007) 9:1365–75. doi:10.1111/j.1462-5822.2007.00933.x
27. Portnoy D, Jacks P, Hinrichs D. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med (1988) 167:1459–71. doi:10.1084/jem.167.4.1459
28. McCaffrey RL, Fawcett P, O’Riordan M, Lee K-D, Havell EA, Brown PO, et al. A specific gene expression program triggered by Gram-positive bacteria in the cytosol. Proc Natl Acad Sci U S A (2004) 101:11386–91. doi:10.1073/pnas.0403215101
29. Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog (2008) 4:e6. doi:10.1371/journal.ppat.0040006
30. Aubry C, Corr SC, Wienerroither S, Goulard C, Jones R, Jamieson AM, et al. Both TLR2 and TRIF contribute to interferon-β production during Listeria infection. PLoS One (2012) 7:e33299. doi:10.1371/journal.pone.0033299
31. Park J-H, Kim Y-G, McDonald C, Kanneganti T-D, Hasegawa M, Body-Malapel M, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol (2007) 178:2380–6. doi:10.4049/jimmunol.178.4.2380
32. Kobayashi K, Inohara N, Hernandez LD, Galán JE, Núñez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature (2002) 416:194–9. doi:10.1038/416194a
33. Chin AI, Dempsey PW, Bruhn K, Miller JF, Xu Y, Cheng G. Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature (2002) 416:190–4. doi:10.1038/416190a
34. O’Connell R, Vaidya S, Perry AK, Saha SK, Dempsey PW, Cheng G. Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TANK-binding kinase. J Immunol (2005) 174:1602–7. doi:10.4049/jimmunol.174.3.1602
35. Stockinger S, Reutterer B, Schaljo B, Schellack C, Brunner S, Materna T, et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol (2004) 173:7416–25. doi:10.4049/jimmunol.173.12.7416
36. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity (2006) 24:93–103. doi:10.1016/j.immuni.2005.12.003
37. Abdullah Z, Schlee M, Roth S, Mraheil MA, Barchet W, Böttcher J, et al. RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J (2012) 31:4153–64. doi:10.1038/emboj.2012.274
38. Loo Y-M, Gale M. Immune signaling by RIG-I-like receptors. Immunity (2011) 34:680–92. doi:10.1016/j.immuni.2011.05.003
39. Vabret N, Blander JM. Sensing microbial RNA in the cytosol. Front Immunol (2013) 4:468. doi:10.3389/fimmu.2013.00468
40. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol (2014) 32:461–88. doi:10.1146/annurev-immunol-032713-120156
41. Seth RB, Sun L, Ea C-K, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi:10.1016/j.cell.2005.08.012
42. Xu L-G, Wang Y-Y, Han K-J, Li L-Y, Zhai Z, Shu H-B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell (2005) 19:727–40. doi:10.1016/j.molcel.2005.08.014
43. Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature (2005) 437:1167–72. doi:10.1038/nature04193
44. Sun Q, Sun L, Liu H-H, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity (2006) 24:633–42. doi:10.1016/j.immuni.2006.04.004
45. Soulat D, Bauch A, Stockinger S, Superti-Furga G, Decker T. Cytoplasmic Listeria monocytogenes stimulates IFN-beta synthesis without requiring the adapter protein MAVS. FEBS Lett (2006) 580:2341–6. doi:10.1016/j.febslet.2006.03.057
46. Hagmann CA, Herzner AM, Abdullah Z, Zillinger T, Jakobs C, Schuberth C, et al. RIG-I detects triphosphorylated RNA of Listeria monocytogenes during infection in non-immune cells. PLoS One (2013) 8:e62872. doi:10.1371/journal.pone.0062872
47. Monroe KM, McWhirter SM, Vance RE. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog (2009) 5:e1000665. doi:10.1371/journal.ppat.1000665
48. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol (2010) 11:997–1004. doi:10.1038/ni.1932
49. Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, et al. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a beta-catenin-dependent pathway. Nat Immunol (2010) 11:487–94. doi:10.1038/ni.1876
50. Lippmann J, Rothenburg S, Deigendesch N, Eitel J, Meixenberger K, van Laak V, et al. IFNbeta responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell Microbiol (2008) 10:2579–88. doi:10.1111/j.1462-5822.2008.01232.x
51. Hansen K, Prabakaran T, Laustsen A, Jørgensen SE, Rahbæk SH, Jensen SB, et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J (2014) 33:1654–66. doi:10.15252/embj.201488029
52. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu Y-J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol (2011) 12:959–65. doi:10.1038/ni.2091
53. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol (2001) 19:623–55. doi:10.1146/annurev.immunol.19.1.623
54. O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med (2004) 200:437–45. doi:10.1084/jem.20040712
55. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature (2009) 461:788–92. doi:10.1038/nature08476
56. Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, et al. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol (2011) 187:2595–601. doi:10.4049/jimmunol.1100088
57. Sauer J-D, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of STING in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun (2011) 79:688–94. doi:10.1128/IAI.00999-10
58. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (2013) 339:826–30. doi:10.1126/science.1229963
59. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell (2013) 51:226–35. doi:10.1016/j.molcel.2013.05.022
60. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature (2011) 478:515–8. doi:10.1038/nature10429
61. Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science (2010) 328:1703–5. doi:10.1126/science.1189801
62. Witte CE, Whiteley AT, Burke TP, Sauer J-D, Portnoy DA, Woodward JJ. Cyclic di-AMP is critical for Listeria monocytogenes growth, cell wall homeostasis, and establishment of infection. MBio (2013) 4:e282–213. doi:10.1128/mBio.00282-13
63. Plumlee CR, Lee C, Beg AA, Decker T, Shuman HA, Schindler C. Interferons direct an effective innate response to Legionella pneumophila infection. J Biol Chem (2009) 284:30058–66. doi:10.1074/jbc.M109.018283
64. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol (2007) 178:3126–33. doi:10.4049/jimmunol.178.5.3126
65. Ishihara T, Aga M, Hino K, Ushio C, Taniguchi M, Iwaki K, et al. Inhibition of chlamydia trachomatis growth by human interferon-alpha: mechanisms and synergistic effect with interferon-gamma and tumor necrosis factor-alpha. Biomed Res (2005) 26:179–85. doi:10.2220/biomedres.26.179
66. Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA, et al. Interferon-ϵ protects the female reproductive tract from viral and bacterial infection. Science (2013) 339:1088–92. doi:10.1126/science.1233321
67. Fehr BT, Schoedon G, Odermatt B, Holtschke T, Schneemann M, Bachmann MF, et al. Crucial role of interferon consensus sequence binding protein, but neither of interferon regulatory factor 1 nor of nitric oxide synthesis for protection against murine listeriosis. J Exp Med (1997) 185:921–31. doi:10.1084/jem.185.5.921
68. Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med (2004) 200:527–33. doi:10.1084/jem.20040976
69. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med (2004) 200:535–40. doi:10.1084/jem.20040769
70. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med (2010) 207:327–37. doi:10.1084/jem.20091746
71. Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M, et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol (2010) 184:3755–67. doi:10.4049/jimmunol.0902065
72. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interf Cytokine Res (2005) 701:694–701. doi:10.1089/jir.2005.25.694
73. Desvignes L, Wolf AJ, Ernst JD. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J Immunol (2012) 188:6205–15. doi:10.4049/jimmunol.1200255
74. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35:1023–34. doi:10.1016/j.immuni.2011.12.002
75. Nagarajan UM, Prantner D, Sikes JD, Andrews CW, Goodwin AM, Nagarajan S, et al. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun (2008) 76:4642–8. doi:10.1128/IAI.00629-08
76. Qiu H, Fan Y, Joyee AG, Wang S, Han X, Bai H, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol (2008) 181:2092–102. doi:10.4049/jimmunol.181.3.2092
77. Richer E, Prendergast C, Zhang D-E, Qureshi ST, Vidal SM, Malo D. N-ethyl-N-nitrosourea-induced mutation in ubiquitin-specific peptidase 18 causes hyperactivation of IFN-αß signaling and suppresses STAT4-induced IFN-γ production, resulting in increased susceptibility to Salmonella typhimurium. J Immunol (2010) 185:3593–601. doi:10.4049/jimmunol.1000890
78. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A (2001) 98:5752–7. doi:10.1073/pnas.091096998
79. Reutterer B, Stockinger S, Pilz A, Soulat D, Kastner R, Westermayer S, et al. Type I IFN are host modulators of strain-specific Listeria monocytogenes virulence. Cell Microbiol (2008) 10:1116–29. doi:10.1111/j.1462-5822.2007.01114.x
80. Antonelli L, Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, et al. Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest (2010) 120:1674–82. doi:10.1172/JCI40817DS1
81. Navarini AA, Recher M, Lang KS, Georgiev P, Meury S, Bergthaler A, et al. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proc Natl Acad Sci U S A (2006) 103:15535–9. doi:10.1073/pnas.0607325103
82. Bhat N, Wright JG, Broder KR, Murray EL, Greenberg ME, Glover MJ, et al. Influenza-associated deaths among children in the United States, 2003-2004. N Engl J Med (2005) 353:2559–67. doi:10.1056/NEJMoa051721
83. Simonsen L. The global impact of influenza on morbidity and mortality. Vaccine (1999) 17(Suppl 1):S3–10. doi:10.1016/S0264-410X(99)00099-7
84. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis (2008) 198:962–70. doi:10.1086/591708
85. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest (2009) 119:1910–20. doi:10.1172/JCI35412.1910
86. Cooper CL, Al-Bedwawi S, Lee C, Garber G. Rate of infectious complications during interferon-based therapy for hepatitis C is not related to neutropenia. Clin Infect Dis (2006) 42:1674–8. doi:10.1086/504386
87. Yang J-F, Hsieh M-Y, Hou N-J, Dai C-Y, Huang J-F, Lin Z-Y, et al. Bacterial infection and neutropenia during peginterferon plus ribavirin combination therapy in patients with chronic hepatitis C with and without baseline neutropenia in clinical practice. Aliment Pharmacol Ther (2009) 29:1000–10. doi:10.1111/j.1365-2036.2009.03957.x
88. Roomer R, Hansen BE, Janssen HLA, de Knegt RJ. Risk factors for infection during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. Hepatology (2010) 52:1225–31. doi:10.1002/hep.23842
89. Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature (2010) 466:973–7. doi:10.1038/nature09247
90. Teles RMB, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science (2013) 339:1448–53. doi:10.1126/science.1233665
91. Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med (2006) 203:933–40. doi:10.1084/jem.20060045
92. Emmerling P, Finger H, Bockemühl J. Listeria monocytogenes infection in nude mice. Infect Immun (1975) 12:437–9.
93. Sasaki T, Mieno M, Udono H, Yamaguchi K, Usui T, Hara K, et al. Roles of CD4+ and CD8+ cells, and the effect of administration of recombinant murine interferon gamma in listerial infection. J Exp Med (1990) 171:1141–54. doi:10.1084/jem.171.4.1141
94. Nickol AD, Bonventre PF. Anomalous high native resistance to athymic mice to bacterial pathogens. Infect Immun (1977) 18:636–45.
95. Zheng S-J, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol (2004) 173:5652–8. doi:10.4049/jimmunol.173.9.5652
96. Cousens L, Orange J, Su H, Biron C. Interferon-a/b inhibition of interleukin 12 and interferon-g production in vitro and endogenously during viral infection. Proc Natl Acad Sci U S A (1997) 94:634–9. doi:10.1073/pnas.94.2.634
97. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity (2011) 34:213–23. doi:10.1016/j.immuni.2011.02.006
98. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol (2010) 184:3326–30. doi:10.4049/jimmunol.0904189
99. Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1β production in human macrophages. J Immunol (2011) 187:2540–7. doi:10.4049/jimmunol.1100926
100. Brzoza-Lewis KL, Hoth JJ, Hiltbold EM. Type I interferon signaling regulates the composition of inflammatory infiltrates upon infection with Listeria monocytogenes. Cell Immunol (2012) 273:41–51. doi:10.1016/j.cellimm.2011.11.008
101. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest (2011) 121:3657–65. doi:10.1172/JCI57762.of
102. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol (2006) 7:311–7. doi:10.1038/ni1309
103. Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med (1997) 186:1757–62. doi:10.1084/jem.186.10.1757
104. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol (2008) 26:421–52. doi:10.1146/annurev.immunol.26.021607.090326
105. Carr KD, Sieve AN, Indramohan M, Break TJ, Lee S, Berg RE. Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur J Immunol (2011) 41:2666–76. doi:10.1002/eji.201041363
106. Shi C, Hohl TM, Leiner I, Equinda MJ, Fan X, Pamer EG. Ly6G+ neutrophils are dispensable for defense against systemic Listeria monocytogenes infection. J Immunol (2011) 187:5293–8. doi:10.4049/jimmunol.1101721
107. Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, et al. Mice with a selective impairment of IFN-gamma signaling in macrophage lineage cells demonstrate the critical role of IFN-gamma-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol (2010) 184:877–85. doi:10.4049/jimmunol.0902346
108. Reith W, LeibundGut-Landmann S, Waldburger J-M. Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol (2005) 5:793–806. doi:10.1038/nri1708
109. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340:202–7. doi:10.1126/science.1235208
110. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340:207–11. doi:10.1126/science.1235214
111. Singhal A, Jaiswal A, Arora VK, Prasad HK. Modulation of gamma interferon receptor 1 by Mycobacterium tuberculosis: a potential immune response evasive mechanism. Infect Immun (2007) 75:2500–10. doi:10.1128/IAI.01743-06
112. Kearney SJ, Delgado C, Eshleman EM, Hill KK, O’Connor BP, Lenz LL. Type I IFNs downregulate myeloid cell IFN-γ receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences ifngr1 transcription. J Immunol (2013) 191:3384–92. doi:10.4049/jimmunol.1203510
113. Roth KM, Gunn JS, Lafuse W, Satoskar AR. Francisella inhibits STAT1-mediated signaling in macrophages and prevents activation of antigen-specific T cells. Int Immunol (2009) 21:19–28. doi:10.1093/intimm/dxn119
114. O’Donovan KJ, Tourtellotte WG, Millbrandt J, Baraban JM. The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci (1999) 22:167–73. doi:10.1016/S0166-2236(98)01343-5
115. Swirnoff A, Milbrandt J. DNA-binding specificity of NGFI-A and related zinc finger transcription factors. Mol Cell Biol (1995) 15:2275–87.
116. Yu J, de Belle I, Liang H, Adamson ED. Coactivating factors p300 and CBP are transcriptionally crossregulated by Egr1 in prostate cells, leading to divergent responses. Mol Cell (2004) 15:83–94. doi:10.1016/j.molcel.2004.06.030
117. Huang RP, Fan Y, deBelle I, Ni Z, Matheny W, Adamson ED. Egr-1 inhibits apoptosis during the UV response: correlation of cell survival with Egr-1 phosphorylation. Cell Death Differ (1998) 5:96–106. doi:10.1038/sj.cdd.4400322
118. Shah KM, Stewart SE, Wei W, Woodman CBJ, O’Neil JD, Dawson CW, et al. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene (2009) 28:3903–14. doi:10.1038/onc.2009.249
119. Zhang Y, Thai V, McCabe A, Jones M, Macnamara KC. Type I interferons promote severe disease in a mouse model of lethal ehrlichiosis. Infect Immun (2014) 82:1698–709. doi:10.1128/IAI.01564-13
120. Kernbauer E, Maier V, Rauch I, Müller M, Decker T. Route of infection determines the impact of type I interferons on innate immunity to Listeria monocytogenes. PLoS One (2013) 8:e65007. doi:10.1371/journal.pone.0065007
121. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A (2003) 100:2610–5. doi:10.1073/pnas.0337679100
122. Kirou KA, Lee C, George S, Louca K, Peterson MGE, Crow MK. Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum (2005) 52:1491–503. doi:10.1002/art.21031
123. Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med (2003) 197:711–23. doi:10.1084/jem.20021553
124. Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol (2010) 6:40–9. doi:10.1038/nrrheum.2009.237
125. Waubant E, Vukusic S, Gignoux L, Dubief FD, Achiti I, Blanc S, et al. Clinical characteristics of responders to interferon therapy for relapsing MS. Neurology (2003) 61:184–9. doi:10.1212/01.WNL.0000078888.07196.0B
126. Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, et al. IFN-B gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol (2003) 170:4776–84. doi:10.4049/jimmunol.170.9.4776
127. Prinz M, Schmidt H, Mildner A, Knobeloch K-P, Hanisch U-K, Raasch J, et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity (2008) 28:675–86. doi:10.1016/j.immuni.2008.03.011
128. Tak P, Hart B, Kraan M. The effects of interferon beta treatment on arthritis. Rheumatology (1999) 38:362–9. doi:10.1093/rheumatology/38.4.362
129. Ying F, Chalise JP, Narendra SC, Magnusson M. Type I IFN protects against antigen-induced arthritis. Eur J Immunol (2011) 41:1687–95. doi:10.1002/eji.201040956
130. Mullen L, Adams G, Foster J, Vessillier S, Köster M, Hauser H, et al. A comparative study of matrix metalloproteinase and aggrecanase mediated release of latent cytokines at arthritic joints. Ann Rheum Dis (2013) 73(9):1728–36. doi:10.1136/annrheumdis-2013-203513
131. Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol (2012) 13:439–47. doi:10.1038/ni.2246
132. Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity (2013) 39:111–22. doi:10.1016/j.immuni.2013.06.013
133. Lee J, Rachmilewitz D, Raz E. Homeostatic effects of TLR9 signaling in experimental colitis. Ann N Y Acad Sci (2006) 1072:351–5. doi:10.1196/annals.1326.022
134. Mannon PJ, Hornung RL, Yang Z, Yi C, Groden C, Friend J, et al. Suppression of inflammation in ulcerative colitis by interferon-β-1a is accompanied by inhibition of IL-13 production. Gut (2011) 60:449–55. doi:10.1136/gut.2010.226860
135. Nikolaus S, Rutgeerts P, Fedorak R, Steinhart AH, Wild GE, Theuer D, et al. Interferon beta-1a in ulcerative colitis: a placebo controlled, randomised, dose escalating study. Gut (2003) 52:1286–90. doi:10.1136/gut.52.9.1286
136. Rauch I, Hainzl E, Rosebrock F, Heider S, Schwab C, Stoiber D, et al. Type I interferons have opposing effects during the emergence and recovery phases of colitis. Eur J Cancer (2014). doi:10.1002/eji.201344401
Keywords: interferons, interferon receptors, bacterial pathogens, macrophage activation, immune suppression, autoimmunity
Citation: Eshleman EM and Lenz LL (2014) Type I interferons in bacterial infections: taming of myeloid cells and possible implications for autoimmunity. Front. Immunol. 5:431. doi: 10.3389/fimmu.2014.00431
Received: 19 June 2014; Paper pending published: 03 August 2014;
Accepted: 23 August 2014; Published online: 11 September 2014.
Edited by:
Yoichi Furuya, Albany Medical College, USAReviewed by:
Birgit Strobl, University of Veterinary Medicine Vienna, AustriaYubin Zhang, Fudan University, China
Copyright: © 2014 Eshleman and Lenz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurel L. Lenz, Department of Immunology and Microbiology, University of Colorado School of Medicine, Mail Stop 8333, Room P18-9130, 12800 E. 19th Avenue, Aurora, CO 80045-2537, USA e-mail: laurel.lenz@ucdenver.edu