Beyond Paralogs: The Multiple Layers of Redundancy in Bacterial Pathogenesis
 Soma Ghosh
Soma Ghosh Tamara J. O'Connor
Tamara J. O'Connor- Department of Biological Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Redundancy has been referred to as a state of no longer being needed or useful. Microbiologists often theorize that the only case of true redundancy in a haploid organism would be a recent gene duplication event, prior to divergence through selective pressure. However, a growing number of examples exist where an organism encodes two genes that appear to perform the same function. For example, many pathogens translocate multiple effector proteins into hosts. While disruption of individual effector genes does not result in a discernable phenotype, deleting genes in combination impairs pathogenesis: this has been described as redundancy. In many cases, this apparent redundancy could be due to limitations of laboratory models of pathogenesis that do not fully recapitulate the disease process. Alternatively, it is possible that the selective advantage achieved by this perceived redundancy is too subtle to be measured in the laboratory. Moreover, there are numerous possibilities for different types of redundancy. The most common and recognized form of redundancy is functional redundancy whereby two proteins have similar biochemical activities and substrate specificities allowing each one to compensate in the absence of the other. However, redundancy can also exist between seemingly unrelated proteins that manipulate the same or complementary host cell pathways. In this article, we outline 5 types of redundancy in pathogenesis: molecular, target, pathway, cellular process, and system redundancy that incorporate the biochemical activities, the host target specificities and the impact of effector function on the pathways and cellular process they modulate. For each type of redundancy, we provide examples from Legionella pathogenesis as this organism employs over 300 secreted virulence proteins and loss of individual proteins rarely impacts intracellular growth. We also discuss selective pressures that drive the maintenance of redundant mechanisms, the current methods used to resolve redundancy and features that distinguish between redundant and non-redundant virulence mechanisms.
Redundancy—Biology's Contingency Plan
Bacteria are one of nature's ultimate survivalists, able to adapt to extreme and dynamic environmental conditions. One of the reasons for their robustness is redundancy, contingency plans for a given process that enhances their fitness. Genetic redundancy describes two copies of the same gene whereby the protein encoded by one can function in place of the other. A classic example of genetic redundancy occurs in metabolism, where two genes encode proteins that catalyze the same reaction (Toda et al., 1987). However, redundancy extends beyond gene duplication. Two proteins or sets of proteins with different catalytic activities can generate the same product (Wagner, 2000). The ability to synthesize a molecule de novo and the ability to acquire that molecule from the environment is also a form redundancy. In this case, the proteins and their functions are completely unrelated but they serve a common goal. Thus, redundancy can occur at multiple levels within a system and is largely defined by what a bacterium is trying to accomplish.
Redundancy in Microbial Pathogenesis
Koch's postulates outline a set of criteria to define causal relationships between pathogens and disease (Koch, 1891; Evans, 1976). With advances in molecular biology techniques and bacterial genetics, Stanley Falkow proposed a molecular version of Koch's postulates to define virulence factors responsible for the pathogenesis of an individual microorganism (Falkow, 1988). The postulate sets an exclusive condition where disruption of a gene should result in a virulence defect and that phenotype should be reversed upon allelic replacement of the gene. For decades, the postulate has been used to identify many virulence factors in numerous pathogens (Isberg et al., 1987; Hersh et al., 1999). At the same time however, a growing number of genes that failed Falkow's criteria but played important roles in disease began to emerge (Falkow, 2004; Choy et al., 2012; Gaspar and Machner, 2014). The lack of phenotypes associated with genetic mutations was attributed to redundancy amongst virulence factors. While redundancy is not the only explanation for this phenomenon (discussed below), it is becoming a common feature in microbial pathogenesis with examples from Legionella (Luo and Isberg, 2004; Belyi et al., 2006), Pseudomonas (Kvitko et al., 2009; Cunnac et al., 2011), Yersinia (Ratner et al., 2016), Chlamydia (Cocchiaro and Valdivia, 2009), Salmonella (Zhou et al., 2001), and Mycobacterium (Downing et al., 2005; Ganapathy et al., 2015). While an exciting challenge for microbiologists, redundancy is a major obstacle in identifying virulence factors, deciphering their roles in disease and developing new therapeutic agents to combat infection.
Redundancy in Legionella Pathogenesis
Legionella pneumophila is an intracellular bacterial pathogen with a broad host range spanning over 15 species of amoebae and ciliated protozoa (Rowbotham, 1980) to mammalian macrophages (Horwitz and Silverstein, 1980). Intracellular growth of L. pneumophila requires a number of key events be accomplished. L. pneumophila must disrupt endocytic and autophagic targeting of its membrane-bound compartment, termed the Legionella-containing vacuole (LCV) to avoid digestion in the lysosome (Horwitz, 1983; Berger et al., 1994; Swanson and Isberg, 1995; Wiater et al., 1998; Choy et al., 2012); transform the phagosome into a replication-permissive compartment (Kagan and Roy, 2002; Derre and Isberg, 2004; Kagan et al., 2004); acquire nutrients to grow (Sauer et al., 2005; Allard et al., 2009; Isaac et al., 2015); expand and maintain the integrity of the replication vacuole to accommodate increasing bacterial numbers (Laguna et al., 2006; Creasey and Isberg, 2012); avoid detection by host innate immune recognition (Laguna et al., 2006; Zamboni et al., 2006; Coers et al., 2007; Fontana et al., 2011; Pereira et al., 2011; Creasey and Isberg, 2012; Barry et al., 2013); inhibit host cell death to maintain an intracellular environment that supports replication (Losick and Isberg, 2006; Abu-Zant et al., 2007); and eventually, exit from the host cell (Horwitz and Silverstein, 1980). As it turns out, L. pneumophila employs multiple strategies to accomplish each of these tasks.
With Falkow's molecular Koch's postulates in mind, several genetic screens to correlate gene disruptions with virulence defects have been employed to identify L. pneumophila virulence genes (Berger and Isberg, 1993; Sadosky et al., 1993; VanRheenen et al., 2004; Laguna et al., 2006). Parallel genetic screens independently identified a collection of 26 genes encoding components of a Type IVb secretion system, subsequently named Icm/Dot (Marra et al., 1992; Berger and Isberg, 1993; Brand et al., 1994). Mutations in icm/dot genes abolish L. pneumophila intracellular growth in macrophages (Berger and Isberg, 1993; Brand et al., 1994) and amoebal hosts (Segal and Shuman, 1999) demonstrating a critical role for the Icm/Dot complex in L. pneumophila pathogenesis. The identification of Icm/Dot was not surprising as numerous pathogens employ secretion systems to deploy proteins, termed effectors to the host cell to establish growth. Yet the search for Icm/Dot translocated substrates (IDTS) using similar genetic screening strategies was relatively unsuccessful, identifying only a small handful of IDTS-encoding genes that were important for L. pneumophila pathogenesis (VanRheenen et al., 2004; Laguna et al., 2006; Isaac et al., 2015). As a consequence, more creative genetic screening strategies were implemented (Luo and Isberg, 2004; Campodonico et al., 2005): not only did this lead to the identification of the first set of IDTS but also the presence of multiple paralogs of many IDTS in the L. pneumophila genome (Luo and Isberg, 2004). As a result, the lack of phenotypes associated with genetic mutations in a single IDTS was attributed to redundancy.
The presence of multiple IDTS paralogs was the first evidence of redundancy in L. pneumophila pathogenesis. However, the simultaneous deletion of all paralogs from a single family of IDTS did not impair L. pneumophila intracellular growth (Bardill et al., 2005). The simplest explanation was that these genes were dispensable under the experimental conditions tested. However, the subsequent use of biochemical and bioinformatics-based approaches had begun to define a collection of 270 translocated proteins (de Felipe et al., 2005; Huang et al., 2011; Zhu et al., 2011). In the process, pairs of IDTS that modulate the same host protein via different mechanisms or different components of the same pathway were identified (Nagai et al., 2002; Machner and Isberg, 2006; Murata et al., 2006; Belyi et al., 2008). In parallel, genetic screens in host cells to identify host factors important for L. pneumophila pathogenesis demonstrated that while depletion of a single host factor rarely impaired L. pneumophila replication, the combined deletion of pairs of host proteins that function in common processes significantly disrupted intracellular growth of the bacterium (Dorer et al., 2006). Collectively, these results suggested that redundancy extends beyond paralogs to more complex mechanisms that function at pathway and system levels, providing an explanation for the lack of phenotypes for mutants lacking all members of a paralogous family of IDTS. In support of this, it was subsequently shown that deleting specific combinations of unrelated IDTS impairs L. pneumophila intracellular growth while deletion of each gene individually failed to elicit a phenotype (O'Connor et al., 2011, 2012). Thus, redundancy appeared to be a multi-tiered phenomenon integrating many different forms.
Types of Redundancy
Redundancy in microbial pathogenesis manifests in many forms that encompass a broad spectrum of functional relationships and multiple levels of biological systems: this complexity necessitates a structured nomenclature to define the different types of redundancy. Genetic and functional redundancy are often used interchangeably, defining compensatory roles for two proteins with the same biochemical activities that allow one to substitute in place of the other. However, the use of function can be somewhat subjective, as it can refer to a precise biochemical activity or more generally, to the impact of that activity on a particular pathway or cellular process. As a consequence, the term functional redundancy has been omitted here as it could be used to describe more than one type of redundancy outlined below. Instead, we propose 5 types of redundancy (Figure 1A): molecular, target, pathway, cellular process, and system redundancy that incorporate the biochemical activities of effectors, their host target specificities, their impact on host cell biology and their contributions to pathogenesis. In many cases, virulence strategies are multi-tiered encompassing several types of redundancy.
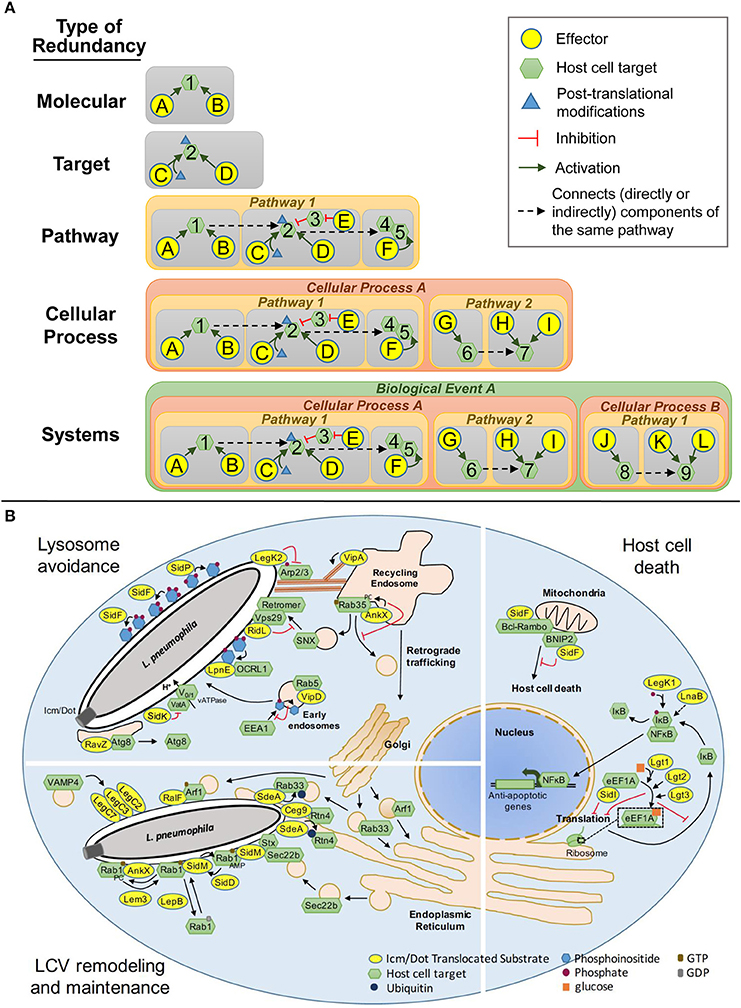
Figure 1. Types of Redundancy. (A) Schematic representations of the 5 classes of redundancy: Molecular, two or more effectors that modify the same host target using the same molecular mechanism; Target, effectors that modulate the same host protein using different molecular mechanisms; Pathway, effectors that modulate a single host pathway but target different components of that pathway; Cellular Process, effectors that target redundant or complementary host pathways that collectively govern a single cellular process; System, effectors that modulate more than one host cellular process to accomplish a common goal. (B) Redundant Icm/Dot translocated substrates that modulate lysosomal trafficking, vacuole remodeling and maintenance and host cell death in Legionella pathogenesis.
Molecular Redundancy
Molecular redundancy defines two or more effectors that modify the same host target using the same molecular mechanism (Figure 1A). In this case, one effector can function in place of the other because it has the same activity and target specificity as its counterpart(s). In some cases, molecular redundancy is likely to be a byproduct of gene duplication however, this is not the sole source with examples of horizontal gene transfer and convergent evolution leading to the presence of molecularly redundant proteins.
Molecular redundancy is exemplified by the L. pneumophila SidE family of IDTS, SidE, SdeA, SdeB, and SdeC (Luo and Isberg, 2004). Individual paralogs consist of a mono-ADP-ribosyltransferase domain, a deubiquitylation domain, and a phosphohydrolase domain that collectively catalyze the ubiquitination of the host proteins Reticulon 4 (Rtn4) (Kotewicz et al., 2016) and Rab33b (Qiu et al., 2016) (Figure 1B). While each member of this family is individually dispensable for intracellular replication, the simultaneous deletion of all four members impairs growth of L. pneumophila in the amoebal hosts Acanthamoebae castellanii (Bardill et al., 2005) and Dictyostelium discoideum (Qiu et al., 2016). The virulence defects can be rescued by SdeA alone (Bardill et al., 2005; Qiu et al., 2016) demonstrating that, at least in these two hosts, a single paralog is sufficient for L. pneumophila intracellular replication.
A second example of molecular redundancy in L. pneumophila is the Lgt family of proteins consisting of Lgt1, Lgt2/LegC8, and Lgt3/LegC5 (de Felipe et al., 2005; Belyi et al., 2006) (Figure 1B). Each paralog is a functional glucosyltransferase that covalently modifies the host protein elongation factor 1A (eEF1A) at serine 53 via mono-O-glycosylation (Belyi et al., 2006, 2008). Modification of eEF1A by any of the three paralogs impairs host protein synthesis (Belyi et al., 2006, 2008). The simultaneous deletion of all three 3 paralogs does not impair L. pneumophila intracellular growth nor does it completely abolish host protein translation in infected cells (Belyi et al., 2006, 2008) suggesting that L. pneumophila encodes additional IDTS that modulate this process.
Effectors with Similar Activities Can Be Misinterpreted as Redundant
Effector paralogs are most readily identified by sequence and/or structural similarities. However, homology does not necessarily indicate that two proteins perform the same function and therefore have molecular redundancy. For example, the IDTS VipD is targeted to early endosomes through its interaction with the host protein Rab5 (Gaspar and Machner, 2014). Binding to Rab5 activates VipD phospholipase activity resulting in dephosphorylation of phosphoinositol 3-phosphate on endosomes (Gaspar and Machner, 2014). Sequence homology comparisons identified three paralogs of VipD encoded in the L. pneumophila genome: VdpA, VpdB, and VpdC, each consisting of a functional phospholipase domain based on the conservation of all active site and catalytic residues (VanRheenen et al., 2006; Gaspar and Machner, 2014). However, in vitro binding assays demonstrated that unlike VipD, neither VpdA nor VpdB bind Rab5 (Gaspar and Machner, 2014). Thus, while it appears that the catalytic activities of these four proteins are conserved and they are all likely to alter host phosphoinositide pools, their respective binding partners and the host pathways they modulate may vary significantly.
The unlikelihood of redundancy amongst larger families of effectors with conserved activities or domains is more apparent, as demonstrated by the five F-box domain-containing proteins of the E3 ubiquitin ligase family in L. pneumophila (Ensminger and Isberg, 2010). The F-box protein provides substrate specificity to the E3 ubiquitin ligase complex, typically Skp-Cullin-F-box (SCF) (Zheng et al., 2002). Despite their common F-box domain, only LegU1, LegAU13/AnkB, and LicA interact with Skp1 while only LegU1 and LegAU13/AnkB interact with CUL1 (Ensminger and Isberg, 2010). Moreover, the pattern of host protein ubiquitination varies significantly between the five IDTS (Ensminger and Isberg, 2010). Thus, despite similar biochemical activities, the host proteins they target for ubiquitination and the corresponding host processes they impact are likely to differ. Indeed, many families of IDTS with common functional domains such as kinases, phosphatases, ankyrin-repeat, or coil-coil domains (de Felipe et al., 2005) typically have additional, unrelated functional domains that set them apart.
Effectors that exhibit similar activities or target specificities in vitro or in vivo outside the context of an infection can also be misinterpreted as redundant, as these similarities may not translate to redundancy in the context of a host under native conditions. Additionally, enzymatic functions and/or target specificities defined in vitro could be biased based on the substrates and/or assays used to investigate them and thus, misleading as to their true functions within a host. As a consequence, in vitro studies may suggest redundancy between two proteins that are in fact quite distinct in the context of an infection.
Target Redundancy
Target redundancy defines effectors that modulate the same host protein using different molecular mechanisms (Figure 1A). In this case, the activity of one effector cannot replace the other but can have a similar impact on the function of the targeted host protein, and its component pathway. Thus, contrary to molecular redundancy, target redundancy defines redundant strategies rather than redundant activities. Effectors that are redundant at the target level are more difficult to identify because they typically lack sequence, structural and functional similarity.
Target redundancy is exemplified by the IDTS SidM/DrrA (Machner and Isberg, 2006; Murata et al., 2006) and AnkX/LegA8/AnkN (Pan et al., 2008; Mukherjee et al., 2011; Allgood et al., 2017), herein after referred to as AnkX (Figure 1B). Both effectors modulate the activity of the host small GTPase Rab1 to remodel the LCV but do so by different molecular mechanisms. SidM/DrrA AMPylates the GTP-bound form of Rab1 preventing GTP to GDP exchange by its cognate GAP protein (Muller et al., 2010), whereas AnkX phosphocholinates GTP-bound Rab1, locking it in the active state (Mukherjee et al., 2011). Two additional effectors SidD and Lem3 reverse Rab1 constitutive activation by de-AMPylation and de-phosphocholination, respectively (Neunuebel et al., 2011; Tan and Luo, 2011). Thus, L. pneumophila encodes two sets of IDTS, SidM-SidD and AnkX-Lem3 that are both able to regulate Rab1 activity but do so through different mechanisms.
A second example of target redundancy is observed between the glucosyltransferases Lgt1, Lgt2, Lgt3 (Belyi et al., 2008), and SidI (Shen et al., 2009) (Figure 1B). While the Lgt proteins and SidI both impair host protein synthesis by targeting eEFA1, SidI appears to do so by an alternative mechanism. Similar to Lgt1, Lgt2, and Lgt3, SidI directly interacts with eEFA1 to impair its function however, direct binding is not solely responsible for this effect (Shen et al., 2009). If SidI inactivates eEF1A through modification, the lack of a glycosyltransferase domain suggests it is likely to differ from glycosylation. In addition to Lgt1, Lgt2, Lgt3, and SidI, a fifth effector, SidL has been implicated in impairing host protein synthesis (Fontana et al., 2011), although the mechanism has yet to be elucidated including whether this occurs through eEF1A or another component of the translation machinery. Moreover, the deletion of all five of these IDTS only partially restores host protein synthesis (Fontana et al., 2011), suggesting that additional IDTS regulate this process. Thus, this example encompasses multiple types of redundancy from molecular and target redundancy to pathway and possibly, cellular process redundancy (see below). We predict that many virulence strategies will similarly consist of more than one type of redundancy.
Effectors with Similar Targets but Different Activities Can Be Misinterpreted as Redundant
It is important to distinguish between effectors that modulate the activity of common host proteins but do not achieve the same effect on the component host pathway: these types of effectors are not redundant. An example of non-redundant IDTS with a common target is SidM/DrrA and LepB. SidM/DrrA functions as a GDI displacement factor to recruit Rab1 to the Legionella vacuole, then constitutively activates Rab1 by locking it in the GTP bound form via covalent modification (Machner and Isberg, 2007; Muller et al., 2010). Upon de-AMPylation of Rab1 by SidD (Neunuebel et al., 2011), LepB acts as a Rab1 GTPase activating protein (GAP) promoting GTP hydrolysis and release of Rab1 from the LCV (Ingmundson et al., 2007). Although, SidM and LepB both target Rab1, they have opposite effects on its activity and distribution which differentially impacts Rab1-mediated vesicle trafficking events. Thus, SidM/DrrA and LepB are not target redundant.
Pathway Redundancy
Pathway redundancy defines effectors that modulate a single host pathway but target different components of that pathway (Figure 1A). Sets of effectors that belong to this category can manipulate different proteins in a single complex, different components at various steps along the pathway or regulators of the pathway. However, while the mechanisms and the host proteins used to modulate the pathway differ, the outcome of that modulation is the same and these effectors collectively serve to achieve a common goal.
Pathway redundancy is illustrated by the IDTS VipD and SidK that both modulate the endocytic pathway but do so by targeting different components at different stages of LCV maturation on the way to the lysosome (Figure 1B). On early endosomes, VipD dephosphorylates PI3P, which functions as an anchor for the tethering protein EEA1 (Gaspar and Machner, 2014). The lack of EEA1 at endosomal surfaces prevents endosome fusion with the LCV (Gaspar and Machner, 2014). Vacuole acidification occurs downstream of early endosome fusion events and is mediated by vATPases, multi-component proton pumps (Forgac, 2007). SidK directly binds VatA, a component of the vATPase to inhibit its function (Xu et al., 2010). While VipD can impair early endosome fusion with the LCV, it is not sufficient to avoid endosomal fusion completely as 40% of LCVs containing wild type bacteria stain positive for the early endosomal marker Rab5 (Gaspar and Machner, 2014). SidK acts as part of a contingency plan when endocytic maturation of the LCV is not completely thwarted. Moreover, while vacuoles containing the ΔvipD mutant are more likely to accumulate Rab5 than those containing wild type bacteria, the frequency is significantly lower than that observed for a dot- mutant (Gaspar and Machner, 2014): this suggests that other effectors function to modulate the endocytic pathway. Several effectors including VipA, VipF, SetA, and Ceg19 are likely candidates based on their ability to disrupt trafficking along the vacuole sorting pathway in yeast (Shohdy et al., 2005; Franco et al., 2012).
A second example of pathway redundancy is observed between the effectors RidL, LpnE, and AnkX which target separate mediators of retrograde trafficking between endosomes and the trans-Golgi network to alter the fate of the LCV (Figure 1B). RidL directly interacts with the retromer complex subunit Vps29 to compete with endosome sorting nexins for retromer and PI3P binding (Finsel et al., 2013). LpnE directly interacts with OCRL1 (Weber et al., 2009), a phosphoinositol 5-phosphatase that regulates retrograde trafficking by altering phosphoinositide phosphate pools. AnkX phosphocholinates Rab35 (Mukherjee et al., 2011), a regulator of cargo sorting and recycling from recycling endosomes. Modification of Rab35 prevents microtubule-dependent endosomal vesicle transport to the LCV (Pan et al., 2008). Loss of RidL, LpnE, or AnkX moderately increases the frequency of LAMP1 staining of LCVs demonstrating that the all three effectors independently contribute to disrupting maturation of the LCV along the endocytic pathway (Newton et al., 2007; Pan et al., 2008; Finsel et al., 2013).
Cellular Process Redundancy
Cellular process redundancy occurs when sets of effectors compensate for one another by targeting redundant or complementary host pathways that collectively govern a single cellular process (Figure 1A). An example of a cellular process that is mediated by multiple pathways is the unfolded protein response (UPR). The UPR is activated through three separate sensory pathways: inositol requiring enzyme-1 (IRE1), protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Walter and Ron, 2011). Distinct pathways allow the cell to respond to multiple signs of ER stress enhancing the sensitivity and breadth of the sensory system but all pathways lead to a common response that includes global translation inhibition, upregulation of ER stress proteins, ER membrane expansion and under extreme conditions, activation of pro-apoptotic pathways (Walter and Ron, 2011). While multiple pathways provide robustness to the host, it affords pathogens multiple ways to hijack a cellular process and when necessary, the ability to do so without completely abolishing the cellular process, which can have negative, even detrimental effects on the pathogen itself.
A critical event in Legionella pathogenesis is remodeling and maintenance of the LCV to support bacterial replication: this is accomplished through the recruitment of ER-derived membrane material. Three parallel mechanisms by which Legionella achieves this have been described (Figure 1B). The SdeA, SdeB, and SdeC family of IDTS drives rearrangement of tubular ER and its association with the LCV through ubiquitination of Rtn4 (Kotewicz et al., 2016), a regulator of tubular ER dynamics (Zurek et al., 2011) (Figure 1B). SidM/DrrA and RalF target components of the early secretory pathway to redirect vesicles trafficking between the ER and the Golgi to the LCV. SidM/DrrA does so by recruiting and activating Rab1 at the LCV and promoting non-canonical functional pairing between the plasma membrane tSNARE syntaxins at the LCV and the ER-derived vesicle vSNARE Sec22b (Arasaki et al., 2012). RalF does so by recruiting and activating the host protein ARF1 at the LCV (Nagai et al., 2002) (Figure 1B). The loss of either SidM/DrrA or RalF alters the timing and efficiency of ER protein accumulation at the LCV (Nagai et al., 2002; Ingmundson et al., 2007) demonstrating redundant roles for these proteins in LCV remodeling.
A second example of redundancy at the level of cellular processes is SidF and SidP (Figure 1B). Each effector contributes to modulation of host lipid metabolism to modulate the relative abundance of phosphoinositides (PIs) at the LCV, specifically conversion from a PI(3)P rich environment to a PI(4)P rich environment. SidF is a phosphoinositide 3-dephosphatase with specificity for PI(3,4)P2 and PI(3,4,5)P3 preventing PI(3)P accumulation at the LCV (Banga et al., 2007) while SidP is a phosphoinositide 3-phosphatase that hydrolyzes PI(3)P and PI(3,5)P2 removing PI(3)P from the LCV (Toulabi et al., 2013). The host protein ORCL1, a PI(4,5)P2 5-phosphatase also localizes to the LCV and thus may also promote PI(4)P accumulation at the surface (Weber et al., 2009) (Figure 1B). OCRL1 targeting to the LCV is Icm/Dot-dependent but the specific IDTS required for this has yet to be determined (Weber et al., 2009). Phosphoinositides distinguish individual organelle membranes in the host cell and serve as anchors for organelle-specific host proteins. Several IDTS exploit PIs decorating the LCV to anchor themselves to the surface. Many of these IDTS have been implicated in LCV remodeling including SidM/DrrA, SidC, LidA, and RidL (Machner and Isberg, 2006; Murata et al., 2006; Finsel et al., 2013; Hsu et al., 2014). Loss of SidF impairs Rab1 recruitment to the LCV (Toulabi et al., 2013), likely as a consequence of the inability of SidM to attach itself to the LCV surface. Thus, PI dynamics play a central role determining the repertoires of IDTS at the vacuole surface and thus the fate of the Legionella vacuole.
System Redundancy
System redundancy defines effectors that modulate more than one host cellular process to accomplish a single task (Figure 1A). An example in biology of a single event that is governed by multiple host cellular processes is cell death. Cell death can be achieved through apoptosis, necrosis, pyroptosis, or autophagy. Each process may be triggered by different cues and the mechanisms by which the cell is terminated may vary but the result is the same—death. In some cases, components mediating these pathways are completely distinct; in other cases they may overlap. For a pathogen, the more options at its disposal for manipulating the host cell to accomplish a specific goal, the greater the likelihood of its success. System redundancy provides yet another layer of insurance by allowing a pathogen to tap into multiple cellular processes to ensure completion of a critical event.
Intracellular growth of L. pneumophila requires the viability of the host cell but cell death is induced by host cells when bacteria cannot be eradicated through lysosomal targeting. L. pneumophila regulates host cell death by targeting host signal transduction, translation, and apoptosis (Figure 1B). The IDTS LnaB and LegK1 activate the host transcription factor, nuclear factor κB (NFκB) causing upregulation of anti-apoptotic pathway-associated genes (Losick and Isberg, 2006; Ge et al., 2009). While the mechanism of action of LnaB is unknown, in vitro studies suggest that LegK1 promotes the degradation of the NFκB inhibitor IκB through direct phosphorylation (Ge et al., 2009). Lgt1, Lgt2, Lgt3, SidI, and SidL promote prolonged NFκB signaling by blocking host protein synthesis and thus cellular levels of IκB from being replenished (Fontana et al., 2011). SidF promotes host cell survival by inhibiting the activity of the pro-apoptotic proteins BNIP3 and Bcl-Rambo through direct binding (Banga et al., 2007). Thus, L. pneumophila orchestrates the induction of host cell survival mechanisms while simultaneously obstructing host cell death pathways by targeting distinct cellular processes.
Lysosomal avoidance by L. pneumophila is orchestrated through four separate cellular processes: the endocytic pathway using VipD (Gaspar and Machner, 2014) and SidK (Xu et al., 2010); retrograde transport via RidL (Finsel et al., 2013), LpnE (Weber et al., 2009), and AnkX (Mukherjee et al., 2011); actin cytoskeleton dynamics through LegK2 (Michard et al., 2015) and VipA (Franco et al., 2012); and autophagy by RavZ (Choy et al., 2012) (Figure 1B). The mechanisms of action of VipD, SidK, RidL, LpnE, and AnkX have been discussed previously (see section Pathway Redundancy). Altering endosome transport to the LCV is also achieved by manipulating the actin cytoskeleton. LegK2 phosphorylates the Arp2/3 complex subunits ARPC1B and ARP3 (Michard et al., 2015): this prevents actin nucleation at the site of the LCV thus perturbing endosome trafficking to the LCV (Michard et al., 2015). In contrast, VipA localizes to endosomes and promotes actin polymerization by directly binding to actin. In yeast, VipA impairs vacuole sorting and thus is predicted to similarly alter organelle trafficking during infection (Shohdy et al., 2005; Franco et al., 2012). Host cells are not without their own forms of redundancy. When bacteria fail to be delivered to the lysosome, host cells can also target pathogens to the lysosome via autophagy (Xie and Klionsky, 2007). The IDTS RavZ localizes to the LCV where it irreversibly deconjugates the autophagic protein Atg8 thereby preventing autophagasome membrane nucleation at the site of the LCV (Choy et al., 2012).
System redundancy is also exemplified by SidM/DrrA, SdeA, SdeB, SdeC, RalF, and a functional complex formed by LegC2/YlfB, LegC3, and LegC7 (Figure 1B). SidM/DrrA, SdeABC, and RalF modulate LCV remodeling by hijacking tubular ER dynamics and vesicle trafficking along the early secretory pathway (see section Cellular Process Redundancy). LegC2/YlfB, LegC3, and LegC7 collectively mimic Q-SNARE proteins and directly bind the R-SNARE protein VAMP4 (Shi et al., 2016). LegC2/YlfB-LegC3-LegC7/YlfA complex pairing with VAMP4 diverts VAMP4-containing vesicle trafficking along the retrograde transport pathway between endosomes and the trans-Golgi network to the LCV (Shi et al., 2016). L. pneumophila mutants lacking LegC2/YlfB and LegC7/YlfA show reduced accumulation of the ER marker calnexin at the LCV but do not exhibit an increase in LAMP1 staining (Campodonico et al., 2016). Thus, recruitment of VAMP4-containing vesicles serves to remodel and maintain the LCV but does not impact LCV trafficking to the lysosome (Campodonico et al., 2016; Shi et al., 2016). Differential targeting of endosomes to the LCV suggests the existence of distinct populations of endosomal vesicles, some of which are actively recruited to the LCV to enable L. pneumophila replication while others are actively excluded because they promote L. pneumophila trafficking to the lysosome.
Effectors and Host Target Specificity
Several studies have identified a number of effectors capable of interacting with more than one host target that often function in more than one host pathway or host cellular process. The most common example in L. pneumophila pathogenesis is IDTS that target host Rab proteins, the gatekeepers of membrane transport and trafficking. In addition to Rab1, SidM/DrrA also binds Rab8B, Rab10, and Rab27A (Machner and Isberg, 2006; Yu et al., 2015). Similarly, the IDTS LidA binds activated Rab1 and Rab6A (Machner and Isberg, 2006; Murata et al., 2006; Chen and Machner, 2013) but has also been shown to interact with Rab8B, Rab10, and Rab27A (Yu et al., 2015). Lpg0393 is a guanine nucleotide exchange factor for Rab5, Rab21, and Rab22, all of which are associated with endosomal trafficking (Sohn et al., 2015) while PieE can interact with Rab1, Rab2, Rab5c, Rab6a, and Rab7 (Mousnier et al., 2014) which encompass various stages of secretory, endocytic, and endosome recycling pathways as well as late endosome- and autophagosome-lysosome fusion events (Stenmark, 2009). While overlapping functions between IDTS may provide a source of redundancy and thus, insurance against failure to complete critical events in the infection cycle, it can also be a potential source of decreased specificity. In cases where L. pneumophila has to exploit subtle differences in host cellular pathways, for instance to discriminating between subpopulations of endosomal vesicles, redundancy may be less beneficial. Importantly, many of the Rab protein targets were identified using in vitro systems or in vivo systems outside the context of infection. In the case of SidM/DrrA, initial screening experiments identified seven putative Rab protein targets but subsequent validation experiments narrowed the list down to only two (Yu et al., 2015). Thus, extreme caution has to be exercised in assigning redundant functions before the biological relevance of effector-host target interactions is determined.
Selective Pressures Driving the Maintenance of Redundant Virulence Proteins
Genetic redundancy is unstable over time. Genes performing similar functions tend to experience genetic drift, unless each gene undergoes independent selective pressure (Clark, 1994; Force et al., 1999; Bergthorsson et al., 2007). So how are redundant proteins maintained? The simplest explanation is that so-called redundant effectors have both overlapping and distinct functions and that selection for their independent activities drives the maintenance of their redundant functions. For example, within the Lgt1/Lgt2/Lgt3/SidI/SidL family of IDTS that inhibit protein synthesis by targeting eEF1A, SidI also interacts with eEF1Bγ (Shen et al., 2009) another component of the translation machinery (Browne and Proud, 2002). Similarly, Lgt1 has a second putative binding partner, Hsb1 that plays a role in mRNA surveillance during translation (Belyi et al., 2009). In addition, members of this family vary in their ability to block the unfolded protein response during L. pneumophila infection (Hempstead and Isberg, 2015; Treacy-Abarca and Mukherjee, 2015). While the significance of these differences has not been elucidated, the independent activities of individual members of this group may be responsible for their maintenance in the genome despite their apparent redundant functions.
Redundancy amongst effectors may compensate for temporal or regulatory differences in gene expression. For instance, Lgt1 is expressed early in the infection cycle while Lgt3 is expressed at later stages prior to bacterial egress (Belyi et al., 2008). The overlapping functions of effectors may allow a specific host processes to be modulated throughout the infection cycle despite differences in their individual expression patterns. Differences in gene expression between redundant effectors may correlate with requirements for their non-overlapping functions at different stages of the infection cycle or differences in the regulatory mechanisms controlling their expression. While redundancy resulting from gene duplication is likely to establish common regulatory networks for individual paralogs, this is unlikely for independently acquired redundant effector genes that are dispersed throughout the genome. Indeed, the mechanisms by which newly acquired effector genes are integrated into existing regulatory networks are not well established. Conservation of redundant effectors may ensure their functions are fulfilled despite variations in their respective gene expression patterns.
Redundancy between effectors may drive the maintenance of redundant virulence strategies when a host protein, pathway or process is impaired by a single effector but not completely abolished. For instance, while VipD can impair fusion of early endosomes with the LCV, it is not sufficient to avoid it completely (Gaspar and Machner, 2014). Variations in the numbers of endosomes in a host cell, the timing and amount of VipD translocated into the host cell, the efficiency of VipD targeting to endosomes, variations in substrate abundance and/or rates of catalysis or the efficiency of endosome fusion with the LCV may render VipD insufficient to avoid downstream events of the endocytic pathway. SidK (Xu et al., 2010) is part of a back-up plan when inhibition of endosome fusion with the LCV is incomplete. As many IDTS, including VipD are toxic when expressed at high levels, the need to limit effector abundance may restrict the ability of any one effector to completely control a particular event. Additional IDTS like LegK2 (Michard et al., 2015), AnkX (Mukherjee et al., 2011), RidL (Finsel et al., 2013), and VipA (Franco et al., 2012) allow L. pneumophila to impair endocytic maturation of the LCV at different points without obliterating major cellular processes.
Redundancy can provide an advantage when enhanced fidelity is required for critical functions (Thomas, 1993). Variations in the host cell type or fluctuations in their external environment may necessitate redundant virulence strategies. In its natural habitat, L. pneumophila is destined to encounter many amoebal species thus, the greater the number of amoebae L. pneumophila can survive and replicate within, the greater its fitness. The importance of individual IDTS could be impacted by multiple factors: differences in amoebal cell biology, nutrient availability, variations in host targets that impact their recognition or manipulation by IDTS or differences in the components or pathways governing cellular processes targeted by L. pneumophila. Maintaining a large cohort of IDTS arms the bacterium with the specific combinations of IDTS necessary for optimal growth in multiple hosts but as a consequence may indirectly result in the accumulation of IDTS that perform overlapping or redundant functions under certain circumstances. Redundancy may also provide a means for pathogens to evolve virulence strategies without compromising fitness. For pathogens that are subject to dynamic and unpredictable environments, have broad host ranges, or find themselves in a perpetual co-evolutionary arms race with their host, this is particularly important.
When Redundancy is not Redundancy at All
Redundancy is not the only explanation for the absence of phenotypes associated with genetic mutations. Whether a gene is required for pathogenesis can vary depending on the host examined, the conditions under which gene requirements are assessed or the type and sensitivity of the assay used. As a consequence, what may be perceived as redundancy is instead an inability to detect phenotypes using a particular experimental system. For example, the IDTS SdhA is essential for L. pneumophila replication in bone marrow-derived primary macrophages but not in cultured U937 cells, a monocyte-derived macrophage cell line (Laguna et al., 2006). Loss of SdhA causes the induction of host cell death in response to L. pneumophila challenge (Laguna et al., 2006; Creasey and Isberg, 2012), which is likely to differ between primary and immortalized cells. Similarly, the SidE family of IDTS is important for L. pneumophila growth in amoebal hosts but is dispensable in primary macrophages (Bardill et al., 2005; Qiu et al., 2016). In these two cases, the host cell type greatly impacts whether a gene is designated as important for L. pneumophila pathogenesis. While redundancy is becoming the default justification for a lack of phenotypes, it is not always the culprit. As more effectors and the host processes they modulate are characterized, key differences between effectors that appear to be redundant will most certainly be revealed.
Methods to Resolve Redundancy
A significant body of work has focused on defining the role of individual virulence factors in isolation, yet understanding how these components coordinately contribute to pathogenesis is necessary to define key determinants of disease. This is particularly important for pathogens that employ compensatory virulence strategies, as redundancy can greatly impact the ability to define what a pathogen requires to survive and grow within a host. A number of strategies have emerged to address redundancy in bacterial pathogenesis that encompass genetic, biochemical and bioinformatics-based techniques. While many of the approaches do not specifically determine redundant mechanisms at a molecular level, they do define functional relationships between individual proteins, and in some cases the host pathways they target, enabling targeted analyses to decipher the basis of redundancy at multiple levels.
Brute-Force Characterization One Effector at a Time
Combined biochemical, molecular, and cell biological characterization of effectors is the most comprehensive way to identify redundant proteins. It provides detailed information about their mechanism of action, their host cell targets and their direct impact on host cellular processes. Moreover, deciphering the intricate details of an effector's function can define subtle distinctions between effectors with overlapping functions and thus circumvent their improper classification as redundant. However, this method is not without its drawbacks. The amount of time required to exhaustively characterize protein function can be lengthy, especially if the techniques to do so are not available or the function of the host target protein or its component pathway have yet to be characterized. For a pathogen like L. pneumophila that employs at least 270 IDTS, such an endeavor would be an arduous one. In addition, for many effectors, sequence homology and structure prediction tools are not always informative. For L. pneumophila, as many as one third of all IDTS lack domain homology to any other protein characterized to date and often very little is learned from structural predictions. Finally, while there are several methods to define host targets, including more recently adapted high throughput methods (Mousnier et al., 2014; Yu et al., 2015), this can be challenging as host targets can range from proteins to lipids to small molecules (Machner and Isberg, 2006; Toulabi et al., 2013; Isaac et al., 2015). Thus, while characterizing individual effector functions can be highly informative, the road to defining redundant virulence mechanisms can be bumpy and painstakingly slow.
Insertional Mutagenesis and Depletion (iMAD)
iMAD is a genetic screening strategy developed to resolve redundancy amongst effectors by defining sets of bacterial proteins that target common host pathways and parallel pathways exploited by a pathogen to accomplish a single task (O'Connor et al., 2012; O'Connor and Isberg, 2014). To do so, iMAD integrates bacterial mutagenesis and host RNA interference to systematically identify genetic interactions between a pathogen gene and a host gene based on impaired replication of the pathogen (O'Connor et al., 2012; O'Connor and Isberg, 2014). In the case of L. pneumophila, a library of transposon mutants were assessed for their ability to replicate within host cells depleted of one of five early secretory proteins that promote L. pneumophila intracellular growth (O'Connor et al., 2012). Hierarchical clustering of bacterial gene mutations in IDTS with similar behavioral patterns across all host conditions examined revealed several important functional relationships: (1) Common phenotypic signatures identified sets of bacterial proteins that target common host pathways: these sets of proteins defined distinct functional groups; (2) Deleting pairs of bacterial genes from separate functional groups impaired intracellular growth of L. pneumophila: these functional groups defined separate but redundant host pathways targeted by L. pneumophila to generate a replication vacuole; (3) Specific defects in host cell biology resulting from loss of bacterial proteins could be predicted for genes based on the characterization of other members of its group: this identified three sets of proteins that independently contribute to the maintenance of replication vacuole membrane integrity; (4) Different combinations of bacterial genes were required for optimal growth in different hosts defining sources of adaptation to host variation. By grouping individual effectors that commonly manipulate a single host pathway and redundant pathways that contribute to a single process, iMAD defines functional relationships between effectors at the target, pathway, cellular process, and system levels. With more efficient methods for generating arrayed bacterial mutant libraries, commercially available RNAi libraries and the development of CRISPR technology to facilitate host cell protein depletion, and the replacement of DNA microarrays with massively parallel sequencing techniques to monitor bacterial mutant populations, more comprehensive, high-throughput iMAD screens are now possible.
Genome Reduction and Minimal Effector Repertoires
Genome reduction followed by effector repertoire reconstitution is another strategy used to identify redundant effectors and the host pathways they target (Cunnac et al., 2011). Progressive removal of all 28 effector genes from the plant pathogen Pseudomonas syringae Pto DC3000 determined 15 of the effector genes are collectively dispensable for growth in the plant host Nicotiana benthamiana (Kvitko et al., 2009). The subsequent reintroduction of different combinations of effectors defined several redundant-effector groups (REGs) that promote P. syringae growth in N. benthamiana, two of which were determined to mediate resistance to independent arms of plant innate immunity (Block and Alfano, 2011; Cunnac et al., 2011). By analyzing correlates between effector combinations and rescued P. syringae growth during infection, a minimal set of 8 effectors was defined that was sufficient to promote growth of P. syringae to near wild type levels. The effector repertoire reconstitution linked individual REGs with the host pathways they target, identified specific combinations of effectors that are sufficient to cause disease and demonstrated the ability to swap different members of individual REGs and still achieve robust P. syringae growth. For genetically amenable pathogens with manageable sizes of effector repertoires, preferably clustered in a minimal set of genetic loci to facilitate combinatorial effector reintroduction into the genome, effector repertoire reduction, and reconstitution strategies provides a comprehensive method to define redundant effectors, the cellular processes they modulate and the minimum set of effector functions required for pathogenesis.
Effector Interactome Mapping
Proteome-based analyses that use high throughput mass spectrometry allow the interactomes of entire effector subfamilies to be mapped. Affinity purified-mass spectrometry has been used to define the host interacting partners of 58 secreted virulence factors in Chlamydia trachomatis called inclusion membrane proteins (Incs) (Mirrashidi et al., 2015). The results not only allowed groups of effectors to be assigned to specific cellular processes but identified sets of Inc proteins that target the same host proteins or different members of the same multiprotein complex. Mapping the interactome network of all 58 Inc proteins revealed sets of Inc proteins that converge on common targets, pathways and cellular processes: this defined focal points of host modification by C. trachomatis and thus, potential sources of redundancy. Moreover, the C. trachomatis Inc-human interactome had significant overlap with that of other pathogens. Comparisons with three viral-human interactomes (Jager et al., 2011; Davis et al., 2015; Ramage et al., 2015) identified 98 shared host targets between C. trachomatis and at least one of the three viruses. Similarly, a number of the Inc host protein targets are also common targets of L. pneumophila IDTS including Rtn4 (Kotewicz et al., 2016), vATPases (Xu et al., 2010), and the retromer complex (Finsel et al., 2013). The lack of similarly between the respective C. trachomatis Inc proteins and L. pneumophila IDTS and differences in the host protein complex subunits targeted demonstrates that each bacterial pathogen has acquired or evolved different mechanisms to modulate the same host proteins, pathways and/or cellular processes. Comparing effector functions between pathogens not only allows additional redundant virulence mechanisms to be defined but establishes a critical set of events central to microbial pathogenesis.
Comparative and Functional Genomics Can Predict Redundant Virulence Mechanisms
Comparative genomics combined with phenotypic analyses provide a means to define correlates between effector conservation and redundant virulence mechanisms (Baltrus et al., 2011). The P. syringae pan-genome effector repertoire consists of 57 effectors but different subsets of effectors are sufficient for growth in the same plant host. Computational analyses that correlate specific combinations of effectors and host tropism (Baltrus et al., 2011) allow redundant virulence mechanisms to be elucidated on a global scale. Alternatively, comparative genomes can be used as a more targeted approach. For example, in L. pneumophila SdhA is a critical virulence determinant in macrophages (Laguna et al., 2006). While the precise function of SdhA is still unclear, the severe growth defect of the ΔsdhA mutant is due to loss of vacuole integrity that leads to a robust host innate immune response and consequently either bacterial or host cell death (Laguna et al., 2006; Creasey and Isberg, 2012). L. pneumophila encodes two paralogs of SdhA, SidH, and SdhB, but their deletion only moderately enhances the already severe intracellular growth defect of the ΔsdhA mutant (Laguna et al., 2006). Legionella feeleii lacks a sdhA paralog but grows almost as well as the wild type strain of L. pneumophila in macrophages (Figure 2). While the presence of sidH in L. feeleii may compensate for the absence of sdhA, deletion of sidH does not impair L. feeleii growth in macrophages (Figure 2). The dispensability of SdhA (and SidH) in L. feeleii is not due to the lack of plaA and/or traI, which suppresses the ΔsdhA mutant phenotype in L. pneumophila (Creasey and Isberg, 2012). Thus, while SdhA plays a critical role in L. pneumophila pathogenesis, the entire family of SidH paralogs is dispensable in L. feeleii. While there are a number of explanations for this discrepancy, L. feeleii encodes 27 additional putative IDTS that are not conserved in L. pneumophila (Burstein et al., 2016), one or more of which may compensate for the absence of SdhA despite their lack of homology. As more genomes of pathogen isolates are sequenced, correlates between effector conservation and phenotypes will allow alternate virulence mechanisms employed by pathogens to be defined.
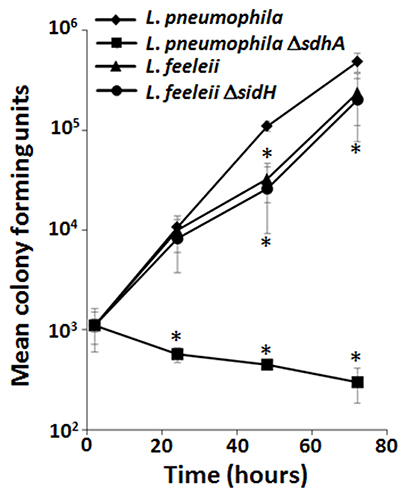
Figure 2. Lack of the SidH family of Dot/Icm translocated substrates does not impair growth of L. feeleii in macrophages despite being indispensable in L. pneumophila. Growth of wild type L. pneumophila, L. pneumophila ΔsdhA, wild type L. feeleii and L. feeleii ΔsidH in A/J mouse bone marrow-derived macrophages, based on recovered colony forming units (CFU) on solid media from lysed host cells, was monitored over 72 h encompassing 3 consecutive rounds of infection (Supplemental Material). Plotted is the total bacterial yield at the indicated time points normalized to the L. pneumophila wild-type strain by the number of intracellular bacteria 2 h post infection. Data are representative of at least 2 independent experiments ± standard deviation of 3 replicates. An asterisk indicates a P < 0.05 based on a Student's t-test relative to the L. pneumophila wild type strain.
Future Directions
Much of the research in microbial pathogenesis employs a reductionist's approach, where the individual components are investigated in isolation. While this strategy has proven extremely useful in identifying key players and their functions, it does not offer tremendous insight into the complex interactions that exist at the systems level. Pathogens invest an incredible amount of resources to build a robust virulence strategy. Redundancy allows pathogens to rapidly adapt to frequently changing environments and the elaborate, multi-tiered antimicrobial strategies employed by their hosts. As more and more effectors are characterized, a striking pattern of redundancy is beginning to emerge. In this review, we establish a structured nomenclature for the different forms of redundancy observed across multiple levels of biological organization. The types of redundancy defined here are not mutually exclusive nor are they expected to be exhaustive as more virulence factors are characterized. Instead, we offer a framework to generate a broader, more dynamic view of the mechanisms governing microbial pathogenesis.
Author Contributions
SG generated data presented in the manuscript. SG and TO wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Joseph Vogel, Kim Davis, Mohammad Hossain, Sara Rego, and Jason Park for thoughtful review of the manuscript. This work was supported by the National Institutes of Health grant 1R01AI125402 (TO).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2017.00467/full#supplementary-material
References
Abu-Zant, A., Jones, S., Asare, R., Suttles, J., Price, C., Graham, J., et al. (2007). Anti-apoptotic signalling by the Dot/Icm secretion system of L. pneumophila. Cell Microbiol. 9, 246–264. doi: 10.1111/j.1462-5822.2006.00785.x
Allard, K. A., Dao, J., Sanjeevaiah, P., McCoy-Simandle, K., Chatfield, C. H., Crumrine, D. S., et al. (2009). Purification of Legiobactin and importance of this siderophore in lung infection by Legionella pneumophila. Infect. Immun. 77, 2887–2895. doi: 10.1128/IAI.00087-09
Allgood, S. C., Romero Dueñas, B. P., Noll, R. R., Pike, C., Lein, S., and Neunuebel, M. R. (2017). Legionella effector AnkX disrupts host cell endocytic recycling in a phosphocholination-dependent manner. Front. Cell. Infect. Microbiol. 7:397. doi: 10.3389/fcimb.2017.00397
Arasaki, K., Toomre, D. K., and Roy, C. R. (2012). The Legionella pneumophila effector DrrA is sufficient to stimulate SNARE-dependent membrane fusion. Cell Host Microbe 11, 46–57. doi: 10.1016/j.chom.2011.11.009
Baltrus, D. A., Nishimura, M. T., Romanchuk, A., Chang, J. H., Mukhtar, M. S., Cherkis, K., et al. (2011). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLoS Pathog. 7:e1002132. doi: 10.1371/journal.ppat.1002132
Banga, S., Gao, P., Shen, X., Fiscus, V., Zong, W. X., Chen, L., et al. (2007). Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc. Natl. Acad. Sci. U.S.A. 104, 5121–5126. doi: 10.1073/pnas.0611030104
Bardill, J. P., Miller, J. L., and Vogel, J. P. (2005). IcmS-dependent translocation of SdeA into macrophages by the Legionella pneumophila type IV secretion system. Mol. Microbiol. 56, 90–103. doi: 10.1111/j.1365-2958.2005.04539.x
Barry, K. C., Fontana, M. F., Portman, J. L., Dugan, A. S., and Vance, R. E. (2013). IL-1alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J. Immunol. 190, 6329–6339. doi: 10.4049/jimmunol.1300100
Belyi, Y., Niggeweg, R., Opitz, B., Vogelsgesang, M., Hippenstiel, S., Wilm, M., et al. (2006). Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. U.S.A. 103, 16953–16958. doi: 10.1073/pnas.0601562103
Belyi, Y., Stahl, M., Sovkova, I., Kaden, P., Luy, B., and Aktories, K. (2009). Region of elongation factor 1A1 involved in substrate recognition by Legionella pneumophila glucosyltransferase Lgt1: identification of Lgt1 as a retaining glucosyltransferase. J. Biol. Chem. 284, 20167–20174. doi: 10.1074/jbc.M109.008441
Belyi, Y., Tabakova, I., Stahl, M., and Aktories, K. (2008). Lgt: a family of cytotoxic glucosyltransferases produced by Legionella pneumophila. J. Bacteriol. 190, 3026–3035. doi: 10.1128/JB.01798-07
Berger, K. H., and Isberg, R. R. (1993). Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7, 7–19. doi: 10.1111/j.1365-2958.1993.tb01092.x
Berger, K. H., Merriam, J. J., and Isberg, R. R. (1994). Altered intracellular targeting properties associated with mutations in the Legionella pneumophila dotA gene. Mol. Microbiol. 14, 809–822. doi: 10.1111/j.1365-2958.1994.tb01317.x
Bergthorsson, U., Andersson, D. I., and Roth, J. R. (2007). Ohno's dilemma: evolution of new genes under continuous selection. Proc. Natl. Acad. Sci. U.S.A. 104, 17004–17009. doi: 10.1073/pnas.0707158104
Block, A., and Alfano, J. R. (2011). Plant targets for Pseudomonas syringae type III effectors: virulence targets or guarded decoys? Curr. Opin. Microbiol. 14, 39–46. doi: 10.1016/j.mib.2010.12.011
Brand, B. C., Sadosky, A. B., and Shuman, H. A. (1994). The Legionella pneumophila icm locus: a set of genes required for intracellular multiplication in human macrophages. Mol. Microbiol. 14, 797–808. doi: 10.1111/j.1365-2958.1994.tb01316.x
Browne, G. J., and Proud, C. G. (2002). Regulation of peptide-chain elongation in mammalian cells. Eur. J. Biochem. 269, 5360–5368. doi: 10.1046/j.1432-1033.2002.03290.x
Burstein, D., Amaro, F., Zusman, T., Lifshitz, Z., Cohen, O., Gilbert, J. A., et al. (2016). Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat. Genet. 48, 167–175. doi: 10.1038/ng.3481
Campodonico, E. M., Chesnel, L., and Roy, C. R. (2005). A yeast genetic system for the identification and characterization of substrate proteins transferred into host cells by the Legionella pneumophila Dot/Icm system. Mol. Microbiol. 56, 918–933. doi: 10.1111/j.1365-2958.2005.04595.x
Campodonico, E. M., Roy, C. R., and Ninio, S. (2016). Legionella pneumophila type IV effectors YlfA and YlfB are SNARE-like proteins that form homo- and heteromeric complexes and enhance the efficiency of vacuole remodeling. PLoS ONE 11:e0159698. doi: 10.1371/journal.pone.0159698
Chen, Y., and Machner, M. P. (2013). Targeting of the small GTPase Rab6A' by the Legionella pneumophila effector LidA. Infect. Immun. 81, 2226–2235. doi: 10.1128/IAI.00157-13
Choy, A., Dancourt, J., Mugo, B., O'Connor, T. J., Isberg, R. R., Melia, T. J., et al. (2012). The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076. doi: 10.1126/science.1227026
Clark, A. G. (1994). Invasion and maintenance of a gene duplication. Proc. Natl. Acad. Sci. U.S.A. 91, 2950–2954. doi: 10.1073/pnas.91.8.2950
Cocchiaro, J. L., and Valdivia, R. H. (2009). New insights into Chlamydia intracellular survival mechanisms. Cell. Microbiol. 11, 1571–1578. doi: 10.1111/j.1462-5822.2009.01364.x
Coers, J., Vance, R. E., Fontana, M. F., and Dietrich, W. F. (2007). Restriction of Legionella pneumophila growth in macrophages requires the concerted action of cytokine and Naip5/Ipaf signalling pathways. Cell. Microbiol. 9, 2344–2357. doi: 10.1111/j.1462-5822.2007.00963.x
Creasey, E. A., and Isberg, R. R. (2012). The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl. Acad. Sci. U.S.A. 109, 3481–3486. doi: 10.1073/pnas.1121286109
Cunnac, S., Chakravarthy, S., Kvitko, B. H., Russell, A. B., Martin, G. B., and Collmer, A. (2011). Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae. Proc. Natl. Acad. Sci. U.S.A. 108, 2975–2980. doi: 10.1073/pnas.1013031108
Davis, Z. H., Verschueren, E., Jang, G. M., Kleffman, K., Johnson, J. R., Park, J., et al. (2015). Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 57, 349–360. doi: 10.1016/j.molcel.2014.11.026
de Felipe, K. S., Pampou, S., Jovanovic, O. S., Pericone, C. D., Ye, S. F., Kalachikov, S., et al. (2005). Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J. Bacteriol. 187, 7716–7726. doi: 10.1128/JB.187.22.7716-7726.2005
Derré, I., and Isberg, R. R. (2004). Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect. Immun. 72, 3048–3053. doi: 10.1128/IAI.72.5.3048-3053.2004
Dorer, M. S., Kirton, D., Bader, J. S., and Isberg, R. R. (2006). RNA interference analysis of legionella in drosophila cells: exploitation of early secretory apparatus dynamics. PLoS Pathogens 2:e34. doi: 10.1371/journal.ppat.0020034
Downing, K. J., Mischenko, V. V., Shleeva, M. O., Young, D. I., Young, M., Kaprelyants, A. S., et al. (2005). Mutants of Mycobacterium tuberculosis lacking three of the five rpf-like genes are defective for growth in vivo and for resuscitation in vitro. Infect. Immun. 73, 3038–3043. doi: 10.1128/IAI.73.5.3038-3043.2005
Ensminger, A. W., and Isberg, R. R. (2010). E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infect. Immun. 78, 3905–3919. doi: 10.1128/IAI.00344-10
Evans, A. S. (1976). Causation and disease: the Henle-Koch postulates revisited. Yale J. Biol. Med. 49, 175–195.
Falkow, S. (1988). Molecular Koch's postulates applied to microbial pathogenicity. Rev. Infect. Dis. 10 (Suppl. 2), S274–S276. doi: 10.1093/cid/10.Supplement_2.S274
Falkow, S. (2004). Molecular Koch's postulates applied to bacterial pathogenicity–a personal recollection 15 years later. Nat. Rev. Microbiol. 2, 67–72. doi: 10.1038/nrmicro799
Finsel, I., Ragaz, C., Hoffmann, C., Harrison, C. F., Weber, S., van Rahden, V. A., et al. (2013). The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe 14, 38–50. doi: 10.1016/j.chom.2013.06.001
Fontana, M. F., Banga, S., Barry, K. C., Shen, X., Tan, Y., Luo, Z. Q., et al. (2011). Secreted bacterial effectors that inhibit host protein synthesis are critical for induction of the innate immune response to virulent Legionella pneumophila. PLoS Pathog. 7:e1001289. doi: 10.1371/journal.ppat.1001289
Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y. L., and Postlethwait, J. (1999). Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545.
Forgac, M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929. doi: 10.1038/nrm2272
Franco, I. S., Shohdy, N., and Shuman, H. A. (2012). The Legionella pneumophila effector VipA is an actin nucleator that alters host cell organelle trafficking. PLoS Pathog. 8:e1002546. doi: 10.1371/journal.ppat.1002546
Ganapathy, U., Marrero, J., Calhoun, S., Eoh, H., de Carvalho, L. P., Rhee, K., et al. (2015). Two enzymes with redundant fructose bisphosphatase activity sustain gluconeogenesis and virulence in Mycobacterium tuberculosis. Nat. Commun. 6, 7912. doi: 10.1038/ncomms8912
Gaspar, A. H., and Machner, M. P. (2014). VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc. Natl. Acad. Sci. U.S.A. 111, 4560–4565. doi: 10.1073/pnas.1316376111
Ge, J., Xu, H., Li, T., Zhou, Y., Zhang, Z., Li, S., et al. (2009). A Legionella type IV effector activates the NF-kappaB pathway by phosphorylating the IkappaB family of inhibitors. Proc. Natl. Acad. Sci. U.S.A. 106, 13725–13730. doi: 10.1073/pnas.0907200106
Hempstead, A. D., and Isberg, R. R. (2015). Inhibition of host cell translation elongation by Legionella pneumophila blocks the host cell unfolded protein response. Proc. Natl. Acad. Sci. U.S.A. 112, E6790–E6797. doi: 10.1073/pnas.1508716112
Hersh, D., Monack, D. M., Smith, M. R., Ghori, N., Falkow, S., and Zychlinsky, A. (1999). The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. U.S.A. 96, 2396–2401. doi: 10.1073/pnas.96.5.2396
Horwitz, M. A. (1983). The Legionnaires' disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J. Exp. Med. 158, 2108–2126. doi: 10.1084/jem.158.6.2108
Horwitz, M. A., and Silverstein, S. C. (1980). Legionnaires' disease bacterium (Legionella pneumophila) multiplies intracellularly in human monocytes. J. Clin. Invest. 66, 441. doi: 10.1172/JCI109874
Hsu, F., Luo, X., Qiu, J., Teng, Y. B., Jin, J., Smolka, M. B., et al. (2014). The Legionella effector SidC defines a unique family of ubiquitin ligases important for bacterial phagosomal remodeling. Proc. Natl. Acad. Sci. U.S.A. 111, 10538–10543. doi: 10.1073/pnas.1402605111
Huang, L., Boyd, D., Amyot, W. M., Hempstead, A. D., Luo, Z. Q., O'Connor, T. J., et al. (2011). The E Block motif is associated with Legionella pneumophila translocated substrates. Cell. Microbiol. 13, 227–245. doi: 10.1111/j.1462-5822.2010.01531.x
Ingmundson, A., Delprato, A., Lambright, D. G., and Roy, C. R. (2007). Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450, 365–369. doi: 10.1038/nature06336
Isaac, D. T., Laguna, R. K., Valtz, N., and Isberg, R. R. (2015). MavN is a Legionella pneumophila vacuole-associated protein required for efficient iron acquisition during intracellular growth. Proc. Natl. Acad. Sci. U.S.A. 112, E5208–E5217. doi: 10.1073/pnas.1511389112
Isberg, R. R., Voorhis, D. L., and Falkow, S. (1987). Identification of invasin: a protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell 50, 769–778. doi: 10.1016/0092-8674(87)90335-7
Jäger, S., Cimermancic, P., Gulbahce, N., Johnson, J. R., McGovern, K. E., Clarke, S. C., et al. (2011). Global landscape of HIV-human protein complexes. Nature 481, 365–370. doi: 10.1038/nature10719
Kagan, J. C., and Roy, C. R. (2002). Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4, 945–954. doi: 10.1038/ncb883
Kagan, J. C., Stein, M. P., Pypaert, M., and Roy, C. R. (2004). Legionella subvert the functions of Rab1 and Sec22b to create a replicative organelle. J. Exp. Med. 199, 1201–1211. doi: 10.1084/jem.20031706
Koch, R. (1891). Über bakteriologische Forschung Verhandlung des X Internationalen Medichinischen Congresses. Berlin.
Kotewicz, K. M., Ramabhadran, V., Sjoblom, N., Vogel, J. P., Haenssler, E., Zhang, M., et al. (2016). A single legionella effector catalyzes a multistep ubiquitination pathway to rearrange tubular endoplasmic reticulum for replication. Cell Host Microbe. 21, 169–181. doi: 10.1016/j.chom.2016.12.007
Kvitko, B. H., Park, D. H., Velásquez, A. C., Wei, C. F., Russell, A. B., Martin, G. B., et al. (2009). Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 5:e1000388. doi: 10.1371/journal.ppat.1000388
Laguna, R. K., Creasey, E. A., Li, Z., Valtz, N., and Isberg, R. R. (2006). A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18745–18750. doi: 10.1073/pnas.0609012103
Losick, V. P., and Isberg, R. R. (2006). NF-kappaB translocation prevents host cell death after low-dose challenge by Legionella pneumophila. J. Exp. Med. 203, 2177–2189. doi: 10.1084/jem.20060766
Luo, Z.-Q., and Isberg, R. R. (2004). Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc. Natl. Acad. Sci. U.S.A. 101, 841–846. doi: 10.1073/pnas.0304916101
Machner, M. P., and Isberg, R. R. (2006). Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev. Cell 11, 47–56. doi: 10.1016/j.devcel.2006.05.013
Machner, M. P., and Isberg, R. R. (2007). A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science 318, 974–977. doi: 10.1126/science.1149121
Marra, A., Blander, S. J., Horwitz, M. A., and Shuman, H. A. (1992). Identification of a Legionella pneumophila locus required for intracellular multiplication in human macrophages. Proc. Natl. Acad. Sci. U.S.A. 89, 9607–9611. doi: 10.1073/pnas.89.20.9607
Michard, C., Sperandio, D., Baïlo, N., Pizarro-Cerdá, J., LeClaire, L., Chadeau-Argaud, E., et al. (2015). The legionella kinase LegK2 targets the ARP2/3 complex to inhibit actin nucleation on phagosomes and allow bacterial evasion of the late endocytic pathway. MBio 6, e00354–e00315. doi: 10.1128/mBio.00354-15
Mirrashidi, K. M., Elwell, C. A., Verschueren, E., Johnson, J. R., Frando, A., Von Dollen, J., et al. (2015). Global mapping of the inc-human interactome reveals that retromer restricts chlamydia infection. Cell Host Microbe 18, 109–121. doi: 10.1016/j.chom.2015.06.004
Mousnier, A., Schroeder, G. N., Stoneham, C. A., So, E. C., Garnett, J. A., Yu, L., et al. (2014). A new method to determine in vivo interactomes reveals binding of the Legionella pneumophila effector PieE to multiple Rab GTPases. MBio 5:e01148-14. doi: 10.1128/mBio.01148-14
Mukherjee, S., Liu, X., Arasaki, K., McDonough, J., Galán, J. E., and Roy, C. R. (2011). Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477, 103–106. doi: 10.1038/nature10335
Müller, M. P., Peters, H., Blümer, J., Blankenfeldt, W., Goody, R. S., and Itzen, A. (2010). The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science 329, 946–949. doi: 10.1126/science.1192276
Murata, T., Delprato, A., Ingmundson, A., Toomre, D. K., Lambright, D. G., and Roy, C. R. (2006). The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8, 971–977. doi: 10.1038/ncb1463
Nagai, H., Kagan, J. C., Zhu, X., Kahn, R. A., and Roy, C. R. (2002). A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295, 679–682. doi: 10.1126/science.1067025
Neunuebel, M. R., Chen, Y., Gaspar, A. H., Backlund, P. S. Jr., Yergey, A., and Machner, M. P. (2011). De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science 333, 453–456. doi: 10.1126/science.1207193
Newton, H. J., Sansom, F. M., Dao, J., McAlister, A. D., Sloan, J., Cianciotto, N. P., et al. (2007). Sel1 repeat protein LpnE is a Legionella pneumophila virulence determinant that influences vacuolar trafficking. Infect. Immun. 75, 5575–5585. doi: 10.1128/IAI.00443-07
O'Connor, T. J., Adepoju, Y., Boyd, D., and Isberg, R. R. (2011). Minimization of the Legionella pneumophila genome reveals chromosomal regions involved in host range expansion. Proc. Natl. Acad. Sci. U.S.A. 108, 14733–14740. doi: 10.1073/pnas.1111678108
O'Connor, T. J., Boyd, D., Dorer, M. S., and Isberg, R. R. (2012). Aggravating genetic interactions allow a solution to redundancy in a bacterial pathogen. Science 338, 1440–1444. doi: 10.1126/science.1229556
O'Connor, T. J., and Isberg, R. R. (2014). iMAD, a genetic screening strategy for dissecting complex interactions between a pathogen and its host. Nat. Protoc. 9, 1916–1930. doi: 10.1038/nprot.2014.133
Pan, X., Lührmann, A., Satoh, A., Laskowski-Arce, M. A., and Roy, C. R. (2008). Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320, 1651–1654. doi: 10.1126/science.1158160
Pereira, M. S., Morgantetti, G. F., Massis, L. M., Horta, C. V., Hori, J. I., and Zamboni, D. S. (2011). Activation of NLRC4 by flagellated bacteria triggers caspase-1-dependent and -independent responses to restrict Legionella pneumophila replication in macrophages and in vivo. J. Immunol. 187, 6447–6455. doi: 10.4049/jimmunol.1003784
Qiu, J., Sheedlo, M. J., Yu, K., Tan, Y., Nakayasu, E. S., Das, C., et al. (2016). Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533, 120–124. doi: 10.1038/nature17657
Ramage, H. R., Kumar, G. R., Verschueren, E., Johnson, J. R., Von Dollen, J., Johnson, T., et al. (2015). A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 57, 329–340. doi: 10.1016/j.molcel.2014.12.028
Ratner, D., Orning, M. P., Starheim, K. K., Marty-Roix, R., Proulx, M. K., Goguen, J. D., et al. (2016). Manipulation of interleukin-1beta and interleukin-18 production by Yersinia pestis effectors YopJ and YopM and redundant impact on virulence. J. Biol. Chem. 291, 9894–9905. doi: 10.1074/jbc.M115.697698
Rowbotham, T. J. (1980). Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33, 1179–1183. doi: 10.1136/jcp.33.12.1179
Sadosky, A. B., Wiater, L. A., and Shuman, H. A. (1993). Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect. Immun. 61, 5361–5373.
Sauer, J. D., Bachman, M. A., and Swanson, M. S. (2005). The phagosomal transporter A couples threonine acquisition to differentiation and replication of Legionella pneumophila in macrophages. Proc. Natl. Acad. Sci. U.S.A. 102, 9924–9929. doi: 10.1073/pnas.0502767102
Segal, G., and Shuman, H. A. (1999). Legionella pneumophila utilizes the same genes to multiply within Acanthamoeba castellanii and human macrophages. Infect. Immun. 67, 2117–2124.
Shen, X., Banga, S., Liu, Y., Xu, L., Gao, P., Shamovsky, I., et al. (2009). Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell. Microbiol. 11, 911–926. doi: 10.1111/j.1462-5822.2009.01301.x
Shi, X., Halder, P., Yavuz, H., Jahn, R., and Shuman, H. A. (2016). Direct targeting of membrane fusion by SNARE mimicry: convergent evolution of Legionella effectors. Proc. Natl. Acad. Sci. U.S.A. 113, 8807–8812. doi: 10.1073/pnas.1608755113
Shohdy, N., Efe, J. A., Emr, S. D., and Shuman, H. A. (2005). Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc. Natl. Acad. Sci. U.S.A. 102, 4866–4871. doi: 10.1073/pnas.0501315102
Sohn, Y. S., Shin, H. C., Park, W. S., Ge, J., Kim, C. H., Lee, B. L., et al. (2015). Lpg0393 of Legionella pneumophila is a guanine-nucleotide exchange factor for Rab5, Rab21 and Rab22. PLoS ONE 10:e0118683. doi: 10.1371/journal.pone.0118683
Stenmark, H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525. doi: 10.1038/nrm2728
Swanson, M. S., and Isberg, R. R. (1995). Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect. Immun. 63, 3609–3620.
Tan, Y., and Luo, Z. Q. (2011). Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature 475, 506–509. doi: 10.1038/nature10307
Thomas, J. H. (1993). Thinking about genetic redundancy. Trends Genet. 9, 395–399. doi: 10.1016/0168-9525(93)90140-D
Toda, T., Cameron, S., Sass, P., Zoller, M., and Wigler, M. (1987). Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 50, 277–287. doi: 10.1016/0092-8674(87)90223-6
Toulabi, L., Wu, X., Cheng, Y., and Mao, Y. (2013). Identification and structural characterization of a Legionella phosphoinositide phosphatase. J. Biol. Chem. 288, 24518–24527. doi: 10.1074/jbc.M113.474239
Treacy-Abarca, S., and Mukherjee, S. (2015). Legionella suppresses the host unfolded protein response via multiple mechanisms. Nat. Commun. 6, 7887. doi: 10.1038/ncomms8887
VanRheenen, S. M., Dumenil, G., and Isberg, R. R. (2004). IcmF and DotU are required for optimal effector translocation and trafficking of the Legionella pneumophila vacuole. Infect. Immun. 72, 5972–5982. doi: 10.1128/IAI.72.10.5972-5982.2004
VanRheenen, S. M., Luo, Z. Q., O'Connor, T., and Isberg, R. R. (2006). Members of a Legionella pneumophila family of proteins with ExoU (phospholipase A) active sites are translocated to target cells. Infect. Immun. 74, 3597–3606. doi: 10.1128/IAI.02060-05
Wagner, A. (2000). Robustness against mutations in genetic networks of yeast. Nat. Genet. 24, 355–361. doi: 10.1038/74174
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Weber, S. S., Ragaz, C., and Hilbi, H. (2009). The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell. Microbiol. 11, 442–460. doi: 10.1111/j.1462-5822.2008.01266.x
Wiater, L. A., Dunn, K., Maxfield, F. R., and Shuman, H. A. (1998). Early events in phagosome establishment are required for intracellular survival of Legionella pneumophila. Infect. Immun. 66, 4450–4460.
Xie, Z., and Klionsky, D. J. (2007). Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102–1109. doi: 10.1038/ncb1007-1102
Xu, L., Shen, X., Bryan, A., Banga, S., Swanson, M. S., and Luo, Z. Q. (2010). Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6:e1000822. doi: 10.1371/journal.ppat.1000822
Yu, X., Decker, K. B., Barker, K., Neunuebel, M. R., Saul, J., Graves, M., et al. (2015). Host-pathogen interaction profiling using self-assembling human protein arrays. J. Proteome Res. 14, 1920–1936. doi: 10.1021/pr5013015
Zamboni, D. S., Kobayashi, K. S., Kohlsdorf, T., Ogura, Y., Long, E. M., Vance, R. E., et al. (2006). The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 7, 318–325. doi: 10.1038/ni1305
Zheng, N., Schulman, B. A., Song, L., Miller, J. J., Jeffrey, P. D., Wang, P., et al. (2002). Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416, 703–709. doi: 10.1038/416703a
Zhou, D., Chen, L. M., Hernandez, L., Shears, S. B., and Galán, J. E. (2001). A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 39, 248–259. doi: 10.1046/j.1365-2958.2001.02230.x
Zhu, W., Banga, S., Tan, Y., Zheng, C., Stephenson, R., Gately, J., et al. (2011). Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS ONE 6:e17638. doi: 10.1371/journal.pone.0017638
Keywords: redundancy, pathogenesis, effector, functional redundancy, genetic redundancy, Legionella
Citation: Ghosh S and O'Connor TJ (2017) Beyond Paralogs: The Multiple Layers of Redundancy in Bacterial Pathogenesis. Front. Cell. Infect. Microbiol. 7:467. doi: 10.3389/fcimb.2017.00467
Received: 01 September 2017; Accepted: 20 October 2017;
Published: 15 November 2017.
Edited by:
Matthias P. Machner, National Institutes of Health (NIH), United StatesReviewed by:
Anne-Marie Krachler, University of Texas Health Science Center at Houston, United StatesMichael L. Vasil, University of Colorado Denver School of Medicine, United States
Copyright © 2017 Ghosh and O'Connor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tamara J. O'Connor, toconno7@jhmi.edu