
Abstract
Background
Lysophosphatidic acid (LPA) is a signalling phospholipid with multiple biological functions, mainly mediated through specific G protein-coupled receptors. Aberrant LPA signalling is being increasingly implicated in the pathology of common human diseases, such as arteriosclerosis and cancer. The lifetime of the signalling pool of LPA is controlled by the equilibrium between synthesizing and degradative enzymatic activity. In the current study, we have characterized these enzymatic pathways in rat brain by pharmacologically manipulating the enzymatic machinery required for LPA degradation.
Results
In rat brain cryosections, the lifetime of bioactive LPA was found to be controlled by Mg2+-independent, N-ethylmaleimide-insensitive phosphatase activity, attributed to lipid phosphate phosphatases (LPPs). Pharmacological inhibition of this LPP activity amplified LPA1 receptor signalling, as revealed using functional autoradiography. Although two LPP inhibitors, sodium orthovanadate and propranolol, locally amplified receptor responses, they did not affect global brain LPA phosphatase activity (also attributed to Mg2+-independent, N-ethylmaleimide-insensitive phosphatases), as confirmed by Pi determination and by LC/MS/MS. Interestingly, the phosphate analog, aluminium fluoride (AlFx-) not only irreversibly inhibited LPP activity thereby potentiating LPA1 receptor responses, but also totally prevented LPA degradation, however this latter effect was not essential in order to observe AlFx--dependent potentiation of receptor signalling.
Conclusions
We conclude that vanadate- and propranolol-sensitive LPP activity locally guards the signalling pool of LPA whereas the majority of brain LPA phosphatase activity is attributed to LPP-like enzymatic activity which, like LPP activity, is sensitive to AlFx- but resistant to the LPP inhibitors, vanadate and propranolol.
Similar content being viewed by others


Background
Lysophosphatidic acid (LPA, 1- or 2-acyl-sn-glycero-3-phosphate) is a signalling phospholipid mediating multiple biological responses, such as cellular proliferation, prevention of apoptosis, and platelet aggregation, and is involved in the development and function of the nervous, cardiovascular, immune, and reproductive systems [1, 2]. Aberrant LPA signalling has been claimed to be associated with the pathology of common human diseases, such as arteriosclerosis [3] and cancer [4]. Signalling by LPA is mainly mediated through specific G protein-coupled receptors (GPCRs) [5–7]. The receptors for LPA are widely expressed, being found in the brain, circulation and digestive tract. Currently, there are five GPCRs that have been identified as bona fide receptors for LPA (LPA1-5) along with a putative sixth receptor (LPA6) [8].
Physiologically relevant levels of LPA can be found in serum and other body fluids. In addition, several cell types, including platelets, adipocytes, and ovarian cancer cells, can produce and release LPA. Significant amounts of LPA have been detected from brain tissue [9–11]. It is postulated that the majority of bioactive LPA is generated extracellularly from lysophospholipids, such as lysophosphatidylcholine (LPC), by the plasma ecto-enzyme, lysophospholipase D, identical to autotaxin, an autocrine motility factor originally isolated from the conditioned medium of cancer cells [12]. Intracellularly, LPA can be generated by phospholipase A1/A2 (PLA1/PLA2, respectively) -catalyzed deacylation of phosphatidic acid (PA) [13, 14]. Other proposed pathways for LPA generation include de novo biosynthesis either from glycerol-3-phosphate (GP) by glycerol-3-phosphate acyltransferase or from monoacylglycerol (MAG) by monoacylglycerol kinase [15].
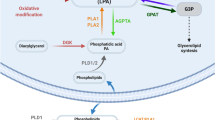
After being produced, the lifetime of the signalling pool of LPA is thought to be controlled by enzymatic degradation. LPA is rapidly dephosphorylated by a family of integral membrane proteins known as lipid phosphate phosphatases (LPPs) [16–18]. The LPP family (also known as PAP2) comprises four members (LPP1, LPP2, LPP3 and a splice variant LPP1a) which dephosphorylate their lipid substrates, namely LPA, PA, sphingosine 1-phosphate (S1P), and ceramide 1-phosphate [19–21]. All the LPP subtypes are expressed in the brain [18, 19, 22] but very little is known about their functional roles. A hallmark of LPP activity is that it does not require Mg2+ and is resistant to the alkylating agent, N-ethylmaleimide (NEM) [23]. The other known pathways for LPA metabolism include de novo formation of PA by lysophosphatidic acid acyltransferase and lysophospholipase -catalyzed hydrolysis of the acyl group to form glycerophosphate [15]. The two degradative pathways generating inorganic phosphate (Pi) as a result of LPA degradation are depicted in Figure 1.

The two enzymatic pathways generating inorganic phosphate (P i ) as a result of LPA degradation. In the LPA → MAG → G pathway, lipid phosphate phosphatases (LPPs) dephosphorylate LPA resulting in the generation of equimolar amounts of monoacylglycerol (MAG) and Pi. MAG is further hydrolyzed by monoglyceride lipase (MGL), resulting in cleavage of the acyl moiety and the formation of glycerol (G). In the LPA → GP → G pathway, lysophospholipases (LPLs) catalyze the deacylation of LPA with the concomitant formation of glycerophosphate (GP). GP can be further metabolized by GP phosphatase (GPase) activity thereby generating equimolar amounts of Pi and G.
35 S]GTPγS autoradiography represents a powerful functional approach to anatomically localize receptor-dependent Gi/o protein activity directly in brain cryosections (for reviews, see [24, 25]). In addition to a receptor’s anatomical distribution, 35 S]GTPγS autoradiography can monitor the receptor-G protein axis in its native cellular microenvironment and is therefore often referred to as functional autoradiography. Our previous studies have demonstrated that brain sections retain the capacity to generate endogenous GPCR agonists, such as adenosine and LPA, during incubation. This results in tonic adenosine A1 and LPA receptor activity in anatomically defined brain regions and therefore serves as a convenient functional readout to monitor agonist activity at the two receptors [26–28]. The LPA-evoked 35 S]GTPγS binding response in rat brain sections reflects LPA1 receptor activity, as it is sensitive to the LPA1/3-selective antagonist Ki16425 and is restricted to the developing white matter tracts [24, 26, 29]. This labelling pattern faithfully mirrors the known expression pattern of LPA1 receptors in the developing rat brain [30–33].
Our previous studies indicated that the enzymatic machinery generating and metabolizing membrane-derived lipid mediators was well preserved in brain cryosections. We recently demonstrated that a comprehensive elimination of the enzymatic hydrolysis of the endocannabinoid 2-arachidonoylglycerol (2-AG) in brain sections leads to 2-AG accumulation and subsequent cannabinoid CB1 receptor activation, as successfully revealed using functional autoradiography [34]. Using this approach, we show here that pharmacological inhibition of LPP activity in brain sections with vanadate or propranolol results in amplification of LPA1 receptor signalling with no net effect on global LPA phosphatase activity at the bulk brain level, also attributable to Mg2+ -independent, NEM-resistant LPP-like phosphatases. We show further that the phosphate analog AlFx- not only potentiates LPA1 receptor signalling, but also totally prevents LPA degradation, resulting in the accumulation of several LPA species in brain sections, as demonstrated by LC/MS/MS measurements. The presently described approach offers a versatile tool to monitor the strength of lipid-GPCR signalling axis in anatomically defined brain structures and may prove useful also for further studies exploring enzymatic pathways estimating the lifetime of still uncharacterized endogenous signalling lipids.
Results
The LPP inhibitors Na3VO4 and propranolol locally amplify LPA1 receptor signalling without affecting global LPA degradation
The LPP-mediated degradation of LPA is susceptible to the phosphatase inhibitor sodium orthovanadate (Na3VO4) [35] and to propranolol [20, 36], better known as a classical β-adrenoceptor blocking agent e.g. used in the treatment of hypertension. Another β-blocker, nadolol, has no demonstrable LPP inhibiting capacity [36] and therefore can serve as a useful control compound. We tested the effects of these compounds on basal LPA1 receptor signalling. Treatment of brain sections with propranolol (1 mM) or Na3VO4 (100 μM) resulted in stimulated 35 S]GTPγS binding responses that were restricted to the white matter areas of 4 week-old rat brain (Figure 2a). The observed labelling pattern was fully reproduced by the addition of exogenous LPA (50 μM) and all the evoked responses, including the tonic LPA1 receptor activity observed under basal conditions, were abolished by treatment with the LPA1/3 receptor selective antagonist Ki16425 (5 μM) (Figure 2a and b). Previously, we demonstrated that Ki16425 dose-dependently diminished the basal and LPA-evoked 35 S]GTPγS binding responses in the white matter tracts (IC50 values 35 ± 9 nM and 87 ± 19 nM, respectively) [28]. Dose–response studies revealed that the maximal effective concentrations for Na3VO4 and propranolol were 100 μM and 1 mM, respectively (See Additional file 1: The maximal effective concentrations for Na3VO4 and propranolol). As expected, treatment with nadolol (1 mM) had no effect on basal LPA1 receptor signalling (Figure 2c, Additional file 2: Propranolol, but not nadolol, induces LPA mimicking binding response). Both Na3VO4 and propranolol, but not nadolol, amplified the LPA-evoked (0.5 μM to 50 μM) binding responses (Figure 2c). To rule out the direct agonism of propranolol and vanadate at the LPA1 receptor, we performed classical filtration-based 35 S]GTPγS binding assay and found that neither compound was able to stimulate 35 S]GTPγS binding to the rat cerebellar membranes, whereas exogenous LPA evoked a dose-dependent response (See Additional file 3: Propranolol and vanadate do not activate LPA1 receptors). We additionally observed that the LPA3 receptor-preferring agonist (2 S)-OMPT (0.5 μM to 50 μM) induced only a weak response in the brain of a 4-week old rat when compared to signal achieved with exogenous LPA (10 μM) (See Additional file 4: Comparison of the 35 S]GTPγS binding responses between LPA and (2 S)-OMPT). This lends further support to the argument that observed LPA-evoked signalling is reflecting the activity of the myelin enriched LPA1 receptors instead of LPA3 receptors that are expressed to a lesser degree in the brain [5]. Finally, we found that exogenously added autotaxin substrate LPC did not boost tonic LPA1 receptor activity suggesting that tonic LPA1 activity in brain sections is not due to LPA formed as a result of autotaxin activity (See Additional file 5: Autotaxin is not responsible for tonic LPA1 activity).

Vanadate and propranolol evoke LPA-mimicking [35 S]GTPγS binding response that is sensitive to LPA 1/3 receptor antagonist. (a) Coronal brain sections were incubated using a three-step autoradiography protocol as detailed in Methods. Test chemicals were included during the [35 S]GTPγS labelling step (step 3) in the presence of 0.1% BSA. Treatment with propranolol (1 mM) or Na3VO4 (100 μM), results in G protein activity in the LPA1 receptor enriched white matter tracts, a response that is mimicked by exogenous LPA (50 μM). The LPA1/3 receptor antagonist Ki16425 (5 μM) abolishes all the evoked responses in the white matter regions, including the tonic LPA1 signal observed under basal conditions (cc, corpus callosum). Scale bar = 2 mm. The eight-point [14 C] standard used in the quantification is shown at the bottom of image. (b) and (c) Quantitative data of the binding responses including the dose response for exogenous LPA (0.5 μM to 50 μM) with or without the simultaneous treatment with the inhibitors. Autoradiography films were digitized and [35 S]GTPγS binding was quantified from the corpus callosum of coronal sections of 4 week-old rat brain, as described in Methods. Quantitative data are calculated as nCi/g equivalents with non-specific binding subtracted from total binding. The data are expressed as a percentage of basal binding (mean + SEM) from six individual animals (n = 6). Significance level: (b) ***p < 0.001 compared to the treatment without Ki16425 (c) ***p < 0.001 compared to control in each LPA concentration.
The LPPs catalyze the hydrolysis of the phosphate group of their lipid substrates resulting in the generation of inorganic phosphate (Pi). The measurement of the released Pi offers a straightforward way to monitor LPP activity [37]. In preliminary experiments, incubation of brain sections with exogenously added LPA, PA and S1P resulted in Pi formation, indicating that lysophospholipid-degrading phosphatases were active under the assay conditions employed (See Additional file 6: Phosphate generation from exogenous LPA, PA, and S1P). To confirm that Pi generation was dependent on brain tissue, empty slides were incubated under identical conditions but in this case, there was no generation of Pi from LPA (data not shown). Further studies with LPA (50 μM) indicated, that LPP-like phosphatase activity accounted for the majority of LPA degradation, as ~ 93% of LPA-derived Pi was formed as a result of Mg2+-independent, NEM-resistant phosphatase activity (Figure 3). In the routine assay buffer containing Mg2+, 46 ± 1% (mean ± SEM, n = 3) of exogenous LPA (50 μM, corresponding to 47.5 nmol potentially available Pi per slide) was degraded during the 90 min incubation whereas in the Mg2+-free assay buffer supplemented with NEM (5 mM), the respective figure was 44 ± 3%. However, neither the LPP inhibitors Na3VO4 (100 μM), propranolol (1 mM), nor nadolol (1 mM) affected total LPA phosphatase activity in a statistically significant manner, assessed at the bulk level of brain sections (Figure 3). In line with this, when the LPA content of brain sections treated with Na3VO4 or propranolol was analyzed using LC/MS/MS, there was no significant accumulation of LPA when compared to control sections that were incubated in the absence of these inhibitors (data not shown). We undertook a search for additional inhibitors in an attempt to identify compounds that could comprehensively target the global pool of LPA phosphatases in brain sections.

Brain tissue dephosphorylates LPA through LPP-like phosphatases that are resistant to propranolol and Na 3 VO 4 . Slides with three horizontal rat brain sections underwent the autoradiography mimicking incubation protocol, as detailed in Methods (a), or were pretreated with NEM (5 mM) followed by the autoradiography mimicking protocol with the exception that Mg2+ was omitted from the incubation buffer (b). Following 90 min incubation in the presence of 0.1% BSA together with the indicated combinations and concentrations of the compounds, the assay buffer was quantitatively collected and the Pi content was determined as described in Methods. Note that the bulk of LPA degradation is due to NEM resistant and Mg2+ independent phosphatase activity (compare a and b) and that Na3VO4 (100 μM), propranolol (1 mM) or nadolol (1 mM) do not affect basal or LPA-derived Pi generation in a statistically significant manner. The data are expressed as nmol Pi generated per slide (mean + SEM) and were obtained from three independent experiments performed in triplicate (n = 3). Significance level: Bars indicated with ¤ represent significant difference (p < 0.001) to bars indicated with # but there is no significant difference between the bars marked with the same symbol.
The phosphate analog aluminium fluoride amplifies LPA1 receptor signalling and totally prevents LPA degradation resulting in bulk accumulation of endogenous LPA species
Sodium fluoride (NaF) is commonly used as a phosphatase inhibitor. Aluminium fluoride (AlFx-) acts as a transition stage phosphate analog also capable of inhibiting several phosphatases [38]. AlFx- forms spontaneously in aqueous solutions in the presence of aluminium (Al3+) and fluoride ions. We pretreated brain sections with NaF or AlFx- in order to test whether these compounds could affect LPA receptor activity and/or LPA degradation. Interestingly, when sections were pretreated with AlFx- (AlCl3 50 μM + NaF 10 mM), a stimulated 35 S]GTPγS binding response throughout the LPA1 receptor-enriched white matter regions was evident (Figure 4a and b). This labelling pattern was fully mimicked by the addition of exogenous LPA (0.5 μM to 50 μM) (Figure 4a and b), and was blocked by Ki16425 (5 μM) (Figure 4a and c). Similarly, pretreatment of brain sections with NaF (10 mM) resulted in stimulated 35 S]GTPγS binding responses throughout the white matter regions (Figure 4a). Pretreatment with these compounds was sufficient to evoke LPA1 receptor signalling, suggesting that in contrast to the reversibly acting inhibitors Na3VO4 and propranolol (that needed to be present during the 35 S]GTPγS labelling step), AlFx- and NaF had inhibited LPP activity in an irreversible manner.

AlF x -and NaF evoke LPA-mimicking [35 S]GTPγS binding response that is sensitive to LPA 1/3 receptor antagonist. (a) Horizontal brain sections were incubated using a three-step autoradiography protocol as described in Methods. AlFx- (NaF 10 mM + AlCl3 50 μM) was included during the 40 min preincubation step (step 1), NaF (10 mM) was included in step 2, and LPA (50 μM) in step 3, whereas Ki16425 (5 μM) was present throughout all steps. The buffer in all the steps additionally contained 0.1% BSA. Treatment with AlFx- and NaF results in region-specific G protein activity that is restricted to the LPA1 receptor enriched white matter tracts, a response that is mimicked by exogenous LPA and is sensitive to Ki16425 (cc, corpus callosum; fi, fimbria of the hippocampus). Scale bar = 5 mm. (b) and (c) Quantitative data of the binding responses. Autoradiography films were digitized and [35 S]GTPγS binding was quantified from corpus callosum of the coronal sections of 4 week-old rat brain, as detailed in Methods. Data are calculated as nCi/g equivalents with non-specific binding subtracted from the total and are expressed as a percentage of basal binding (mean + SEM) from six individual animals (n = 6). Significance level: (b) ***p < 0.001 compared to control in each LPA concentration (c) ***p < 0.001 compared to the treatment without Ki16425.
We wished to explore in more detail the mode of inhibition of these compounds, as well as the behaviour of NaF in our experimental setting. This was justified by the fact that aluminium is a common constituent of glassware and F- can etch it from the glass. Deforoxamine mesylate (DFOM) is an aluminium and iron(III) chelator that can be used in experimental settings to reveal if aluminium is present in the system. When brain sections were treated with DFOM (50 μM), the responses to AlFx- and NaF (10 mM) in functional autoradiography were totally abolished (Figure 5a). It is noteworthy that DFOM needed to be added together with AlFx- or NaF in order to achieve this reversal; if added after pretreatment with AlFx- or NaF, DFOM was ineffective (data not shown). These studies indicate that AlFx- acted as an irreversible inhibitor of brain LPP activity thereby amplifying tonic LPA1 receptor activity. The general phosphatase inhibitor, NaF per se, did not inhibit LPPs, but mimicked the action of AlFx- based on the ability of F- to etch Al3+ from the glass slides.

AlF x -(not NaF) generates the LPA 1 receptor-mediated response; AlF x -also blocks the LPA phosphatase activity. (a) Functional autoradiography using horizontal brain sections was performed using a three-step protocol as detailed in Methods. AlFx- (NaF 10 mM + AlCl3 50 μM) was included during the 40 min pre-incubation step (step 1), NaF (10 mM) was present in step 2 whereas deferoxamine mesylate (DFOM, 50 μM) was present throughout steps 1–3 and in addition during a 10 min pre-incubation prior to AlFx- treatment. The buffer in step 3 additionally contained 0.1% BSA. Both NaF and AlFx- induce [35 S]GTPγS binding in the LPA1 receptor enriched white matter areas. The responses to AlFx- and NaF are totally abolished with the aluminium and iron(III) chelator DFOM (cc, corpus callosum; fi, fimbria of the hippocampus). Scale bar = 5 mm. (b) Slides with two horizontal brain sections underwent the three-step autoradiography mimicking protocol as detailed in Methods. AlFx- (NaF 10 mM + AlCl3 50 μM) and NaF (10 mM) were present during the 90 min incubation step (step 3). DFOM (50 μM) was present in steps 2 and 3. The buffer in step 3 additionally contained 0.1% BSA. After the final incubation step, the assay buffer was quantitatively collected and the Pi content was determined as described in Methods. Note that NaF partially and AlFx- totally inhibit LPA-derived Pi formation and that DFOM reverses these actions. The data are expressed as nmol Pi generated per slide (mean + SEM) and are derived from three independent experiments performed in triplicate (n = 3). Significance level: ***p < 0.001 and **p < 0.01 compared to the treatment with LPA alone.
We were curious to examine whether AlFx- could also inhibit LPA degradation at the bulk brain level. When sections were pretreated with AlFx- but then omitted from all subsequent steps, AlFx- readily facilitated LPA1 receptor signalling (Figure 4, Figure 5a), but such a pretreatment did not inhibit LPA degradation in a statistically significant manner. Degradation of exogenous LPA (50 μM) alone yielded 22.6 ± 1.2 nmol Pi per slide whereas pretreatment with AlFx- followed by incubation with exogenous LPA yielded 21.4 ± 0.4 nmol Pi per slide (mean ± SEM, n = 3). However, when added together with LPA, AlFx- totally (and NaF partially) blocked the formation of LPA-derived Pi, thus providing evidence of the ability of these compounds to inhibit the vanadate- and propranolol-insensitive pool of LPA phosphatases in a reversible manner (Figure 5b). Treatment with DFOM (50 μM) totally prevented the ability of AlFx- and NaF to inhibit the degradation of LPA (Figure 5b), indicating that AlFx-, rather than NaF, was the active compound.
To further explore the consequences of total inhibition of LPA phosphatase activity, AlFx- -treated brain sections were incubated for 40 min in autoradiography buffer, the buffer was removed and tissue LPA content extracted using chloroform-methanol, followed by LC/MS/MS analysis. Four LPA species with different acyl substitutions (16:0 LPA, 18:1 LPA, 18:0 LPA and 20:4 LPA) were examined in the present study. The relative abundances of the four LPA species in brain sections incubated under control conditions were 20:4 LPA ≈ 16:0 LPA < 18:1 LPA < 18:0 LPA. The amounts of three of these species (16:0 LPA, 18:1 LPA and 20:4 LPA) were significantly increased after AlFx- treatment when compared to control sections (Figure 6). These experiments indicate that total blockade of LPA phosphatase activity with AlFx- treatment resulted in accumulation of several endogenous LPA species at the bulk brain level. However, no such bulk LPA accumulation was required to observe the AlFx- -evoked potentiation of LPA1 receptor signalling.

Treatment with AlF x -results in accumulation of several endogenous LPA species in rat brain sections. The slides with two horizontal brain sections were treated with AlFx- (NaF 10 mM + AlCl3 50 μM) for 40 min followed by extraction of LPA species, as described in Methods. The samples were analyzed with LC/MS/MS for four endogenous LPA species, 16:0 LPA, 18:0 LPA, 18:1 LPA and 20:4 LPA. The amounts of three of these LPA species are increased in a statistically significant manner in AlFx- treated sections as compared to control sections. The results are expressed as nmol/g of tissue protein (mean + SEM) and are derived from three individual experiments performed in triplicate (n = 3). Significance level: ***p < 0.001 and **p < 0.01 compared to control.
The LPA → MAG → G pathway efficiently degrades exogenous LPA whereas the LPA → GP → G pathway is inactive
In addition to the LPA → MAG → G pathway (Figure 1), another Pi generating pathway for LPA degradation involves its deacylation to form glycerophosphate which is further dephosphorylated by glycerophosphatases to glycerol (the LPA → GP → G pathway) (Figure 1). Since there are two degradative pathways for LPA that potentially release Pi, we wished to clarify which pathway accounts for LPA degradation in our model. Rat cerebellar membranes have been extensively used in our laboratory to study degradation of labile lipid messengers such as endocannabinoids [34, 39]. Since LPA1 receptors are known to be present in the cerebellum [30] we performed kinetic monitoring of Pi production from exogenous LPA and glycerol 3-phosphate (GP) after incubation with rat cerebellar membranes in 96-well-format. In this setting, cerebellar membranes generated Pi from LPA (10 μM) but there was no Pi generation from GP (10 μM) (Figure 7a), indicating that the Pi formed from LPA in our experimental setting is principally due to LPP/LPP-like activity. As a further proof, we used the dephosphorylation-resistant thio-analog of LPA, (2 S)-OMPT, that is also the LPA3 receptor.-preferring agonist. As expected, there was no Pi generation from (2 S)-OMPT (10 μM) (Figure 7a).

Rat cerebellar membranes principally metabolize LPA via the LPA → MAG → G pathway. (a) Rat cerebellar membranes (1 μg/well) were incubated in the absence (basal) or presence of LPA, glycerol 3-phosphate (GP), or the phosphatase-resistant LPA analog (2 S)-OMPT (10 μM final concentration each). Pi generation was kinetically monitored for 90 min using a fluorescent assay as described in Methods. Rat cerebellar membranes generate Pi from exogenous LPA but not from exogenous GP or (2 S)-OMPT. The data are expressed as nmol of Pi generated per mg of protein during the 90 min incubation (mean + SEM, n = 4 for basal and LPA, n = 3 for GP and (2 S)-OMPT). Significance level: ***p < 0.001 compared to basal. (b) Rat cerebellar membranes were pretreated for 30 min with DMSO or the broadly-acting serine hydrolase inhibitor MAFP (1 μM) or the MGL-specific inhibitor JZL184 (100 μM). This was followed by 90 min incubation in the absence (basal) or presence of LPA (10 μM final concentration). Glycerol generation was kinetically monitored for 90 min using a fluorescent assay as described in Methods. Rat cerebellar membranes readily generate glycerol from exogenous LPA; this response is largely blocked by MAFP and partially so by JZL184. The data are expressed as nmol of glycerol generated per mg of protein during the 90 min incubation (mean + SEM, n = 4). Significance level: ***p < 0.001 compared to either basal or particular treatment. (c) Simultaneous kinetic monitoring of Pi and glycerol generation from exogenous LPA by rat cerebellar membranes during 90 min incubation. Note that Pi generation precedes and exceeds that of glycerol during the early time-points (10–30 min) suggesting temporal first phosphatase (LPA → MAG) then lipase (MAG → G) action. The data are expressed as nmol of Pi or glycerol generated per mg of protein and are means of duplicate wells from one representative experiment.
Since both of the two Pi releasing pathways for LPA degradation finally produce glycerol, we assessed cerebellar membrane-dependent glycerol generation from exogenous LPA in brain tissue. Monoglyceride lipase (MGL) is believed to be mainly responsible for the MAG → G conversion. In addition, two novel α/β-hydrolase domain containing proteins, ABHD6 and ABHD12, have been identified to hydrolyze brain endocannabinoid 2-arachidonoylglycerol (2-AG) [40] and together the three serine hydrolases account for ~99% of brain 2-AG hydrolase activity [41]. It is therefore likely that in addition to MGL, ABHD6 and ABHD12 are involved in the degradation of both 1- and 2-monoacylglycerols. To delineate the relative contributions of the three hydrolases, we pretreated cerebellar membranes with two serine hydrolase inhibitors, methylarachidonoylfluorophosphonate (MAFP) and compound JZL184. The former is a potent, non-selective inhibitor of MGL [39, 42] that also inhibits ABHD6/ABHD12, whereas the latter is a MGL-selective inhibitor [43]. As expected, in rat cerebellar membranes incubated together with LPA (10 μM), glycerol production closely matches with Pi generation (Figure 7a and b), indicating that MAG → G conversion takes place under the assay conditions employed. With MAFP pretreatment (1 μM), LPA-derived glycerol production was decreased by 91% (Figure 7b). With JZL184 pretreatment (100 μM), the corresponding reduction was 71% (Figure 7b). The selectivity of the inhibitors towards MGL likely explains the difference in the inhibition of glycerol production from LPA between the two inhibitors. The time-dependent generation of Pi and glycerol from exogenous LPA is presented in Figure 7c. It appears that Pi generation precedes that of G, a finding supporting sequential actions of phosphatases and lipases on the LPA → MAG → G pathway.
Discussion
Functional autoradiography provides a straightforward approach to study the proximal step of signalling of various Gi/o-coupled receptors in brain cryosections. We recently demonstrated that brain sections retain the capacity to generate endocannabinoids during incubations i.e. evidence of the ability of the brain sections to preserve sufficient functional enzymatic machinery to generate endogenous GPCR activating ligands [34]. The lifetime of the signalling pool of LPA is thought to be controlled by the equilibrium between synthesizing and degradative enzymatic activity. In the current study, we have characterized these enzymatic pathways and their role in tonic LPA1 receptor activity by pharmacologically manipulating the enzymatic machinery required for LPA degradation. We observed that in brain sections, the lifetime of bioactive LPA is controlled by Mg2+-independent, NEM-insensitive phosphatase activity attributable to LPPs. Pharmacological inhibition of this LPP activity by AlFx-, propranolol or sodium orthovanadate amplified LPA1 receptor signalling, as revealed using functional autoradiography. We provided further evidence to show that the majority of brain LPA phosphatase activity seems to be carried out by LPP-like enzymatic activity which like LPP activity, is sensitive to AlFx- but appears to be resistant to the two other LPP inhibitors, vanadate and propranolol. Finally, we demonstrated that degradation of exogenous LPA is almost entirely channelled via the LPA → MAG → G pathway and that MGL accounts for the majority of oleylglycerol-hydrolyzing activity in brain tissue.
All the three subtypes of Mg2+-independent/NEM resistant LPPs are expressed in the brain, yet very little is known about the role of the LPPs as regulators of LPA receptor signalling in the nervous system. Knockout studies of all the LPP subtypes have been reported [44–46]. Study with LPP1 knockout mice indicated that LPP1 plays a role in regulating the degradation of circulating LPA in vivo but that study failed to disrupt the LPP1 encoding gene in the brain, obscuring the function of LPP1 in the nervous system [46]. Knockout of LPP3 turned out to be embryonically lethal [45] whereas in vitro studies using cell lines lacking LPP3 address involvement of LPP3 in early neural development [47]. The LPPs are likely to be involved in LPA dephosphorylation in brain cryosections, as brain sections efficiently generate Pi from exogenous LPA largely in a NEM resistant and Mg2+-independent way. Propranolol and vanadate have been demonstrated to inhibit LPPs in various cell types [20, 35, 36, 48], vanadate also in the rat brain [49]. Propranolol has been shown to act as a moderately effective inhibitor of LPPs [20] supporting our finding where the vanadate-induced response is relatively stronger when compared to the response observed with propranolol. Since propranolol and vanadate amplified LPA1 receptor signalling only when present in the 35 S]GTPγS labelling step, these drugs presumably inhibit LPPs in a reversible manner. In brain sections, LPP activity appears to locally control the lifetime of the signalling pool of LPA and LPPs must therefore reside in close proximity to the LPA1 receptors, as propranolol and vanadate had no effect on LPA degradation when assessed at the bulk brain level.
In functional autoradiography, AlFx- more efficiently induced the LPA1 receptor-mediated signal as compared to the signals observed with vanadate or propranolol. Since AlFx- is able to induce the LPA1 receptor-mediated signal when present only in the pre-incubation step, it appears to inhibit LPPs in an irreversible manner. This proposal is supported by the finding that the Al3+ chelator DFOM failed to reverse AlFx- -evoked response, if added only after pretreatment of brain sections with AlFx- (and NaF). AlFx- is known to mimic the chemical structure of phosphate and therefore affects the activity of several phosphoryl transfer enzymes [38]. As a phosphate analog, AlFx- might bind to the Pi recognizing binding pocket of the LPPs and by this mechanism lead to irreversible inhibition. All the studied inhibitors evoked 35 S]GTPγS binding responses that were largely restricted to the white matter areas of the brain when compared to grey matter (See Additional file 7: Inhibitor-evoked 35 S]GTPγS binding responses are restricted to the white matter areas of the brain) reflecting to selectivity towards the myelin-enriched LPA1 receptors. This also provides evidence to show, that though AlFx- is known to act as a general activator of heterotrimeric G proteins, it seems not to induce global binding response in the grey matter areas and therefore seems not to act as a general G protein activator in functional autoradiography. It is notable that in contrast to propranolol and vanadate, when present in the latter step together with exogenous LPA, AlFx- totally prevented the degradation of LPA at the bulk brain level, suggesting that in addition to irreversibly inhibiting LPPs, AlFx- can inhibit other LPP-like phosphatases in a reversible manner. Based on the present findings, the major portion of brain LPA phosphatase activity appears to be attributable to the LPP-like phosphatases which in a similar manner as LPPs, are sensitive to AlFx- but resistant to the LPP inhibitors, vanadate and propranolol.
Since there was no Pi generation from exogenous glycerol 3-phosphate, it seems that LPA is predominantly degraded by the LPA → MAG → G pathway in our experimental setting whereas the LPA → GP → G pathway plays a minor role. According to our findings, both phosphohydrolases (LPPs/LPP-like) and MGL and related hydrolases (ABHD6/ABHD12) seem to be active. The Pi- and glycerol -generating enzymatic routes involved in LPA degradation are summarized in Additional file 8: Summary of enzymatic routes generating Pi and glycerol. Previously, NEM-insensitive LPA phosphohydrolase activity was studied in the nuclear fraction isolated from rabbit cerebral cortex [50]. This activity was found to be present also in the microsomal fraction. In the nuclear fraction, phosphohydrolase activity was found to be sensitive to NaF (50 mM) but virtually insensitive to propranolol (0.5 mM). Dephosphorylation by phosphohydrolases was found to be more active route for LPA degradation when compared to deacylation by lysophospholipases. It was also indicated that followed by dephosphorylation of LPA, monoacyl product is rapidly converted to glycerol by monoglyceride lipase. These findings support our present findings concerning active pathways involved in LPA degradation in brain as well as about the existence of LPP-like, propranolol and vanadate -insensitive, phosphohydrolase activity.
The LPP-like phosphatases remain to be characterized in future experiments. One interesting group of brain-specific membrane proteins are plasticity related genes (PRGs) that have recently been identified and were originally proposed to act as LPA phosphatases [51]. Among the family of PRGs, PRG-1 shares close homology to the LPPs, having three conserved integral domains facing the extracellular side of the plasma membrane, the feature that enables LPPs to dephosphorylate their lipid substrates. However, the catalytic residues responsible for LPP activity are not fully conserved in PRGs [51] suggesting that PRGs might not act as LPA phosphatases. Instead, PGR-1 was recently demonstrated to act at the postsynaptic side of the excitatory glutamatergic synapse where it could mediate the uptake of bioactive lipids [52]. PRG-1 was found to effectively control the levels of LPA in the synapse though its mechanism of action seems to be more transporter-like than dephosphorylating. The transporter mechanism is not expected to be active in our experimental setting and therefore we hypothesize that PRGs are not controlling LPA levels in our model.
Conclusions
We demonstrate that the lifetime of bioactive LPA is controlled by LPPs in rat brain cryosections and that pharmacological inhibition of this LPP activity results in amplification of basal and LPA-stimulated LPA1 receptor activity. We conclude that LPP acts locally to control the lifetime of the signalling pool of LPA in the vicinity of LPA1 receptors whereas the majority of brain LPA phosphatase activity is attributable to additional LPP-like enzymatic activity. Functional autoradiography represents a valuable tool for studies into LPA degradation by LPPs and LPP-like enzymatic activity. Compounds affecting LPA degradation could prove to be attractive targets for drug development, since altered LPA levels are associated with common human diseases, e.g. several forms of cancer. The approach described in this paper may also prove useful for further studies elucidating enzymatic pathways regulating the lifetime of still uncharacterized endogenous signalling lipids.
Methods
Chemicals
Propranolol was purchased from Biomol (Plymouth Meeting, PA, USA). AlCl3 and NaF were from Merck (Darmstat, Germany). 1-oleoyl-2-methyl-sn-glycero-3-phosphothionate ((2S)-OMPT), 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphate (16:0 LPA), 1-heptadecanoyl-2-hydroxy-sn-glycero-3-phosphate (17:0 LPA), 1-stearoyl-2-hydroxy-sn-glycero-3-phosphate (18:0 LPA), 1-arachidonoyl-2-hydroxy-sn-glycero-3-phosphate (20:4 LPA) were from Avanti Polar Lipids (Alabaster, AL, USA). 1-oleyl-2-hydroxy-sn-glycero-3-phosphate (18:1 LPA), sodium orthovanadate (Na3VO4), nadolol, bovine serum albumin (BSA, fatty acid free), dithiothreitol (DTT), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), guanosine diphosphate (GDP), 3-(4-[4-([1-(2 chlorophenyl)ethoxy] carbonyl amino)-3-methyl-5-isoxazolyl]benzylsulfanyl) propanoic acid (Ki16425), GTPγS, N-ethylmaleimide (NEM), glycerol 3-phosphate, and deferoxamine mesylate (DFOM) were from Sigma (St Louis, MO, USA). Methylarachidonoylfluorophosphonate (MAFP) and compound JZL184 were from Cayman Chemical (Ann Arbor, MI, USA). [35 S]GTPγS (initial specific activity 1250 Ci/mmol) was purchased from NEN Life Science Products Inc. (Boston, MA, USA). All other chemicals were of the finest purity available.
Animals
Experiments were performed using 4-week old male Wistar rats obtained from the National Laboratory Animal Centre, University of Eastern Finland, Kuopio, Finland. Approval for the harvesting of animal tissue was applied, registered and obtained from the local welfare officer of the University of Eastern Finland. The experiments did not involve any in vivo treatment. The animals were housed in groups of five to ten individuals per cage under standard laboratory conditions (12–12 h light–dark cycle, food and water ad libitum, 60% relative humidity). The rats were decapitated 8–9 h after lights on, and within the next 5 min, the whole brain was dissected out, dipped briefly in isopentane (chilled on dry ice) and stored at −80 °C. Horizontal, coronal or sagittal brain sections (20 μm thick) were cut at −19 °C to −21 °C using a Leica cryostat, thaw-mounted onto Superfrost®Plus slides (Menzel-Gläser, Germany), dried for 1–4 h at room temperature under a constant stream of air and stored thereafter at −80 °C.
[35 S]GTPγS autoradiography
35 S]GTPγS autoradiography was performed as previously described [26, 28, 34]. Briefly, the brain sections were processed in three sequential steps consisting of pre-incubation for 20/40 min (step 1), GDP-loading for 50/60 min (step 2), and 35 S]GTPγS labelling for 90 min (step 3). For some treatments, a brief additional incubation (10 min) was performed prior to step 1, as detailed in Results. All the steps were performed at 20 °C using Tris-based buffer (50 mM Tris–HCl, pH 7.4, 1 mM EDTA, 100 mM NaCl, 5 mM MgCl2). Steps 2 and 3 included 1 μM DPCPX (to block the tonic adenosine A1 receptor signal) and 2 mM GDP and step 3 additionally included 1 mM DTT and 40–250 pM 35 S]GTPγS. Chemicals being investigated (LPA species used was 18:1 LPA) and 0.1% BSA (fatty acid free) were included in the assay during steps 1,2 or 3 as described in Results. In addition, some slides in each experiment were incubated in the presence of 10 μM GTPγS to determine non-specific binding. After the 90 min autoradiography step, the slides were rinsed twice (5 min each time) in ice-cold washing buffer (50 mM Tris–HCl, pH 7.4 and 5 mM MgCl2), dipped for 30 s in ice-cold deionized water, and air-dried. The slides were arranged into a cassette together with 14 C] standard (Amersham, Little Chalfont, Bucks, UK) and exposed against a radiosensitive film (BioMax MR™, Kodak Scientific Imaging Film) for 2–5 days. After exposure, the films were developed for 3–4 min at 4 °C with Kodak D-19 developer.
[35 S]GTPγS membrane binding assay
A previously described method was utilized for preparation of membranes [39]. Briefly, eight cerebella were homogenized in nine volumes of ice-cold 0.32 M sucrose. The homogenate was centrifuged at 1000 × g for 10 min at 4 °C and the resulting supernatant was centrifuged at 100 000 × g for 30 min at 4 °C. The high speed centrifugation was repeated twice, resuspending the pellet in ice-cold deionised water. After the final centrifugation, the membranes were suspended in Tris–HCl (50 mM, pH 7.4) supplemented with EDTA (1 mM) and stored thereafter at −80 °C.
The 35 S]GTPγS membrane binding assay was performed as previously described [28, 34], with minor modifications. Briefly, the final incubation volume (400 μl) contained 5 μg membrane protein in the incubation buffer (50 mM Tris–HCl, pH 7.4, 1 mM EDTA, 100 mM NaCl, 5 mM MgCl2, 10 μM GDP, 1 mM DTT, 0.5% BSA, and ~ 150 pM 35 S]GTPγS) plus the chemicals of interest. The incubation buffer was supplemented with 1 μM DPCPX to suppress basal 35 S]GTPγS binding due to endogenous adenosine. The membranes were pre-incubated for 30 min prior to conducting the 35 S]GTPγS binding step. The incubations were stopped after 90 min by the addition of 4 ml ice-cold washing buffer (50 mM Tris–HCl, pH 7.4 and 5 mM MgCl2), followed by filtration through glass fibre filters (Whatman GF/B) and by two additional washes with the buffer. The filters were transferred into scintillation vials along with HiSafe3 scintillation liquid (Wallac, Turku, Finland). After vertical shaking for 15 min to extract radioactivity trapped in filters, the tubes were counted the next day with Wallac LKB 1213 Rackbeta, Wallac, Turku, Finland.
Determination of Pi and glycerol
To estimate LPA-degrading phosphatase activity under the assay conditions mimicking functional autoradiography, triplicate slides with two or three horizontal brain sections underwent the autoradiography mimicking protocol (except that DPCPX, GDP and radioligand were omitted). The experiments with Na3VO4, propranolol, and nadolol (as a negative control for propranolol) were performed with and without NEM pretreatment. The protocol consisted of 10 min pre-incubation in the assay buffer, 30 min incubation in the assay buffer with or without 5 mM NEM, then a washing step with assay buffer, and finally 90 min incubation in the presence of chemicals in interest as well as 0.1% BSA and 1 mM DTT. In NEM-treated sections, Mg2+ was omitted from the assay buffer. In experiments with NaF and AlFx-, the protocol consisted of two sequential 40 min incubations (steps 1 and 2) and finally 90 min incubation (step 3) in the presence of the chemicals of interest as well as 0.1% BSA and 1 mM DTT. LPA species used was 18:1 LPA. In all experiments, after the final 90 min incubation step, the postincubation buffer was collected quantitatively and the Pi content was determined in duplicate using the Fiske-Subbarow method, as described in Esmann (1988) [53] after TCA-precipitation of BSA which interfered with the Pi determinations. Absorbances (λ 700 nm) were read with a Tecan Spectrafluor microplate reader.
To clarify the enzymatic routes responsible for LPA degradation in our experimental setting, Pi formation was determined using enzyme-coupling fluorescent method [54]. The glycerol content was determined using a coupled enzyme reaction (Free Glycerol Reagent, Sigma, Cat.# F6428) with the exception that H2O2 production was coupled to peroxidase-dependent formation of the fluorescent dye resorufin. Briefly, rat cerebellar membranes (1 μg/well of 96-well plate), prepared as described in [39], were pretreated with the serine hydrolase inhibitors MAFP (1 μM) or JZL184 (100 μM) for 30 min and then incubated with or without 18:1 LPA, GP, or (2 S)-OMPT (10 μM final concentration). The fluorescence (λex 530 nm, λem 590 nm) was monitored kinetically for 90 min at 10 min intervals using Tecan Infinite M200 fluorometer.
Extraction of LPA for mass spectrometric determination
Slides with two horizontal brain sections were incubated for 40 min in the presence of AlFx- (NaF 10 mM + AlCl3 50 μM) mimicking the 35 S]GTPγS autoradiography. The control slides were incubated similarly in the assay buffer. After 40 min incubation, slides were rinsed twice (2 min each time) in ice-cold washing buffer, dipped for 30s in ice-cold deionized water and air-dried. The modified extraction method of Bligh and Dyer [55] was applied for the isolation of LPA from the tissue matrix. One sample consisted of pooled tissue obtained from four slides. The brain tissue was scraped manually from the slides with a spatula using the mixture of 50 mM Tris–HCl, pH 7.40 and methanol with a ratio of 1:4 (v/v); this mixture also included an internal standard (17:0 LPA) used in the quantification. The tissue was transferred to a screw-capped Pyrex® borosilicate glass test tube. The mixture of 50 mM Tris–HCl, pH 7.40 and methanol (1:4, v/v) was added to the test tube to bring the volume up to 200 μl. Chloroform was added to yield a water/methanol/chloroform ratio of 1:4:2 (v/v/v) and the samples were shaken for 1 h with a vertical shaker (Heidolph Multi Reax, Heidolph Instruments GmbH & Co, Schwabach, Germany). To achieve the phase separation, 80 μl of chloroform and 80 μl of water were added. After vortexing for 1 min, the samples were centrifuged at 1800 x g for 15 min at room temperature. The upper aqueous layer was transferred to an HPLC sample vial.
Liquid chromatography/tandem mass spectrometry (LC/MS/MS)
The method for LC/MS/MS determination of LPAs with varying acyl chains has been previously described [9]. The HPLC system comprised of an Agilent 1200 Series Rapid Resolution LC System (Agilent Technologies, Waldbronn, Germany) consisting of a solvent micro vacuum degasser, a binary pump, a thermostatted column compartment SL, and an autosampler SL. Ten microliter of sample solution were injected onto a reversed phase HPLC column (XBridge™ C8 2.1x50 mm, 2.5 μm) (Waters, Ireland) using gradient elution with 50 μM ammonium acetate + 1% triethylamine (TEA) (A) and 1% TEA in 90% methanol (B) as follows: 0–6.0 min 20% B → 90% B, 6.0-10.0 min 90% B, 10.0-10.1 min 90% B → 20% B, 10.0-15.0 min 20% B. The flow rate was 0.3 ml/min, column temperature was maintained at 40 °C and the autosampler tray temperature was set to 10 °C.
The mass spectrometric analysis was carried out with an Agilent 6410 Triple Quadrupole LC/MS equipped with an electrospray ionization source (Agilent Technologies, Palo Alto, CA, USA). The following ionization conditions were used: ESI negative ion mode, drying gas (nitrogen) temperature 300 °C, drying gas flow rate 8 l/min, nebulizer pressure 40 psi and capillary voltage 4000 V. Analyte detection was performed using multiple reaction monitoring (MRM) with the following transitions: m/z 409 → 153 for 16:0 LPA, m/z 437 → 153 for 18:0 LPA, m/z 435 → 153 for 18:1 LPA, m/z 457 → 153 for 20:4 LPA, and m/z 423 → 153 for 17:0 LPA. The fragmentor voltage was 160 V and collision energy 20 V except 23 V for 17:0 LPA. Data were acquired by Agilent MassHunter Workstation Acquisition software (Agilent Technologies, Data Acquisition for Triple Quad., version B.01.03).
Data analysis
Autoradiography films were digitized using a HP scanjet 7400c scanner. For the quantitative data, optical densities on the autoradiograms were measured using ImageJ, a freely available java-based image analysis software system developed in the National Institutes of Health, USA (http://rsb.info.nih.gov/ij/). Optical densities were converted to nCi/g using nonlinear transformation built by the greyscale values of [14 C] standards. In LC/MS/MS experiments, an internal standard (17:0 LPA) was used for quantification, and peak area ratios of the analyte to the IS were calculated as a function of the concentration ratios of the analyte to the internal standard using Agilent MassHunter software (Quantitative Analysis Version B.01.03). The protein content of brain sections was determined by the Pierce BCA Protein Assay Kit with BSA as the standard. The statistical differences were determined either using one-way ANOVA with Tukey’s multiple comparison post hoc test or t test (LC/MS/MS experiments) with ***p < 0.001, **p < 0.01, or *p < 0.05 considered as statistically significant. All statistical data analyses were conducted using GraphPad Prism for Windows.
Abbreviations
- 2-AG:
-
2-arachidonoylglycerol
- AlFx-:
-
Aluminium fluoride
- BSA:
-
Bovine serum albumin
- DFOM:
-
Deferoxamine mesylate
- GP:
-
Glycerol phosphate
- GPCR:
-
G protein-coupled receptor
- LC/MS/MS:
-
Liquid chromatography/tandem mass spectrometry
- LPA:
-
Lysophosphatidic acid
- LPP:
-
Lipid phosphate phosphatase
- MAG:
-
Monoacylglycerol
- MGL:
-
Monoglyceride lipase
- NaF:
-
Sodium fluoride
- Na3VO4:
-
Sodium orthovanadate
- NEM:
-
N-ethylmaleimide
- PA:
-
Phosphatidic acid
- Pi:
-
Inorganic phosphate
- S1P:
-
Sphingosine 1-phosphate.
References
Birgbauer E, Chun J: New developments in the biological functions of lysophospholipids. Cell Mol Life Sci. 2006, 63: 2695-2701.
Moolenaar WH, van Meeteren LA, Giepmans BN: The ins and outs of lysophosphatidic acid signaling. BioEssays. 2004, 26: 870-881.
Siess W: Athero- and thrombogenic actions of lysophosphatidic acid and sphingosine-1-phosphate. Biochim Biophys Acta. 2002, 1582: 204-215.
Mills GB, Moolenaar WH: The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003, 3: 582-591.
Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, Chun J: LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010, 50: 157-186.
Ishii S, Noguchi K, Yanagida K: Non-Edg family lysophosphatidic acid (LPA) receptors. Prostaglandins Other Lipid Mediat. 2009, 89: 57-65.
zu Heringdorf D: Lysophospholipid receptors: Signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta. 2007, 1768: 923-940.
Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH: International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid Receptor Nomenclature. Pharmacol Rev. 2010, 62: 579-587.
Aaltonen N, Laitinen JT, Lehtonen M: Quantification of lysophosphatidic acids in rat brain tissue by liquid chromatography-electrospray tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010, 878: 1145-1152.
Nakane S, Oka S, Arai S, Waku K, Ishima Y, Tokumura A, Sugiura T: 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch Biochem Biophys. 2002, 402: 51-58.
Sugiura T, Nakane S, Kishimoto S, Waku K, Yoshioka Y, Tokumura A, Hanahan DJ: Occurrence of lysophosphatidic acid and its alkyl ether-linked analog in rat brain and comparison of their biological activities toward cultured neural cells. Biochim Biophys Acta. 1999, 1440: 194-204.
van Meeteren LA, Moolenaar WH: Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007, 46: 145-160.
Aoki J: Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol. 2004, 15: 477-489.
Aoki J, Inoue A, Okudaira S: Two pathways for lysophosphatidic acid production. Biochim Biophys Acta. 2008, 1781: 513-518.
Tigyi G, Parrill AL: Molecular mechanisms of lysophosphatidic acid action. Prog Lipid Res. 2003, 42: 498-526.
Brindley DN, Pilquil C: Lipid phosphate phosphatases and signaling. J Lipid Res. 2009, 50 (Suppl): 225-230.
Pyne S, Kong KC, Darroch PI: Lysophosphatidic acid and sphingosine 1-phosphate biology: the role of lipid phosphate phosphatases. Semin Cell Dev Biol. 2004, 15: 491-501.
Sigal YJ, McDermott MI, Morris AJ: Integral membrane lipid phosphatases/phosphotransferases: common structure and diverse functions. Biochem J. 2005, 387: 281-293.
Kai M, Wada I, Imai S, Sakane F, Kanoh H: Cloning and characterization of two human isozymes of Mg2 + −independent phosphatidic acid phosphatase. J Biol Chem. 1997, 272: 24572-24578.
Roberts R, Sciorra VA, Morris AJ: Human type 2 phosphatidic acid phosphohydrolases. Substrate specificity of the type 2a, 2b, and 2c enzymes and cell surface activity of the 2a isoform. J Biol Chem. 1998, 273: 22059-22067.
Waggoner DW, Gomez-Munoz A, Dewald J, Brindley DN: Phosphatidate phosphohydrolase catalyzes the hydrolysis of ceramide 1-phosphate, lysophosphatidate, and sphingosine 1-phosphate. J Biol Chem. 1996, 271: 16506-16509.
Hooks SB, Ragan SP, Lynch KR: Identification of a novel human phosphatidic acid phosphatase type 2 isoform. FEBS Lett. 1998, 427: 188-192.
Jamal Z, Martin A, Gomez-Munoz A, Brindley DN: Plasma membrane fractions from rat liver contain a phosphatidate phosphohydrolase distinct from that in the endoplasmic reticulum and cytosol. J Biol Chem. 1991, 266: 2988-2996.
Laitinen JT: [35 S]GTPgammaS autoradiography: A powerful functional approach with expanding potential for neuropharmacological studies on receptors coupled to Gi family of G proteins. Curr Neuropharmacol. 2004, 2: 191-206.
Sovago J, Dupuis DS, Gulyas B, Hall H: An overview on functional receptor autoradiography using [35 S]GTPgammaS. Brain Res Rev. 2001, 38: 149-164.
Aaltonen N, Palomaki VA, Lecklin A, Laitinen JT: Neuroanatomical mapping of juvenile rat brain regions with prominent basal signal in [35 S]GTPγS autoradiography. J Chem Neuroanat. 2008, 35: 233-241.
Laitinen JT: Selective detection of adenosine A1 receptor-dependent G-protein activity in basal and stimulated conditions of rat brain [35 S]guanosine 5'-(gamma-thio)triphosphate autoradiography. Neuroscience. 1999, 90: 1265-1279.
Palomaki VA, Laitinen JT: The basic secretagogue compound 48/80 activates G proteins indirectly via stimulation of phospholipase D-lysophosphatidic acid receptor axis and 5-HT1A receptors in rat brain sections. Br J Pharmacol. 2006, 147: 596-606.
Waeber C, Chiu ML: In vitro autoradiographic visualization of guanosine-5'-O-(3-[35 S]thio)triphosphate binding stimulated by sphingosine 1-phosphate and lysophosphatidic acid. J Neurochem. 1999, 73: 1212-1221.
Allard J, Barron S, Diaz J, Lubetzki C, Zalc B, Schwartz JC, Sokoloff P: A rat G protein-coupled receptor selectively expressed in myelin-forming cells. Eur J Neurosci. 1998, 10: 1045-1053.
Cervera P, Tirard M, Barron S, Allard J, Trottier S, Lacombe J, Daumas-Duport C, Sokoloff P: Immunohistological localization of the myelinating cell-specific receptor LP(A1). Glia. 2002, 38: 126-136.
Handford EJ, Smith D, Hewson L, McAllister G, Beer MS: Edg2 receptor distribution in adult rat brain. NeuroReport. 2001, 12: 757-760.
Weiner JA, Hecht JH, Chun J: Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. J Comp Neurol. 1998, 398: 587-598.
Palomaki VA, Lehtonen M, Savinainen JR, Laitinen JT: Visualization of 2-arachidonoylglycerol accumulation and cannabinoid CB1 receptor activity in rat brain cryosections by functional autoradiography. J Neurochem. 2007, 101: 972-981.
Simon MF, Rey A, Castan-Laurel I, Gres S, Sibrac D, Valet P, Saulnier-Blache JS: Expression of ectolipid phosphate phosphohydrolases in 3T3F442A preadipocytes and adipocytes. Involvement in the control of lysophosphatidic acid production. J Biol Chem. 2002, 277: 23131-23136.
Holinstat M, Voss B, Bilodeau ML, Hamm HE: Protease-activated receptors differentially regulate human platelet activation through a phosphatidic acid-dependent pathway. Mol Pharmacol. 2007, 71: 686-694.
McDermott MI, Sigal YJ, Crump JS, Morris AJ: Enzymatic analysis of lipid phosphate phosphatases. Methods. 2006, 39: 169-179.
Li L: The biochemistry and physiology of metallic fluoride: action, mechanism, and implications. Crit Rev Oral Biol Med. 2003, 14: 100-114.
Saario SM, Savinainen JR, Laitinen JT, Järvinen T, Niemi R: Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem Pharmacol. 2004, 67: 1381-1387.
Blankman JL, Simon GM, Cravatt BF: A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007, 14: 1347-1356.
Savinainen JR, Saario SM, Laitinen JT: The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf). 2012, 204: 267-276.
Savinainen JR, Yoshino M, Minkkilä A, Nevalainen T, Laitinen JT: Characterization of binding properties of monoglyceride lipase inhibitors by a versatile fluorescence-based technique. Anal Biochem. 2010, 399: 132-134.
Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, FJ Pavo´ n, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF: Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009, 5: 37-44.
Zhang N, Sundberg JP, Gridley T: Mice mutant for Ppap2c, a homolog of the germ cell migration regulator wunen, are viable and fertile. Genesis. 2000, 27: 137-140.
Escalante-Alcalde D, Hernandez L, Le Stunff H, Maeda R, Lee HS, Cheng Gang, Sciorra VA, Daar I, Spiegel S, Morris AJ, Stewart CL: The lipid phosphatase LPP3 regulates extra-embryonic vasculogenesis and axis patterning. Development. 2003, 130: 4623-4637.
Tomsig JL, Snyder AH, Berdyshev EV, Skobeleva A, Mataya C, Natarajan V, Brindley DN, Lynch KR: Lipid phosphate phosphohydrolase type 1 (LPP1) degrades extracellular lysophosphatidic acid in vivo. Biochem J. 2009, 419: 611-618.
Sánchez-Sánchez R, Morales-Lázaro SL, Baizabal JM, Sunkara M, Morris AJ, Escalante-Alcalde D: Lack of lipid phosphate phosphatase-3 in embryonic stem cells compromises neuronal differentiation and neurite outgrowth. Dev Dyn. 2012, 241: 953-964.
English D, Cui Y, Siddiqui R, Patterson C, Natarajan V, Brindley DN, Garcia JG: Induction of endothelial monolayer permeability by phosphatidate. J Cell Biochem. 1999, 75: 105-117.
Gaveglio VL, Pasquare SJ, Giusto NM: Metabolic pathways for the degradation of phosphatidic acid in isolated nuclei from cerebellar cells. Arch Biochem Biophys. 2011, 507: 271-280.
Baker RR, Chang HA: Metabolic path for the degradation of lysophosphatidic acid, an inhibitor of lysophosphatidylcholine lysophospholipase, in neuronal nuclei of cerebral cortex. Biochim Biophys Acta. 2000, 1483: 58-68.
Brauer AU, Nitsch R: Plasticity-related genes (PRGs/LRPs): a brain-specific class of lysophospholipid-modifying proteins. Biochim Biophys Acta. 2008, 1781: 595-600.
Trimbuch T, Beed P, Vogt J, Schuchmann S, Maier N, Kintscher M, Breustedt J, Schuelke M, Streu N, Kieselmann O, Brunk I, Laube G, Strauss U, Battefeld A, Wende H, Birchmeier C, Wiese S, Sendtner M, Kawabe H, Kishimoto-Suga M, Brose N, Baumgart J, Geist B, Aoki J, Savaskan NE, Bräuer AU, Chun J, Ninnemann O, Schmitz D, Nitsch R: Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell. 2009, 138: 1222-1235.
Esmann M: ATPase and phosphatase activity of Na+, K + −ATPase: molar and specific activity, protein determination. Methods Enzymol. 1988, 156: 105-115.
Vazquez MJ, Rodriguez B, Zapatero C, Tewb DG: Determination of phosphate in nanomolar range by an enzyme-coupling fluorescent method. Anal Biochem. 2003, 320: 292-298.
Bligh EG, Dyer WJ: A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959, 37: 911-917.
Acknowledgements
The authors wish to thank M.Sc. Ville Palomäki for his participation in the early stages of this study and for some autoradiography images used in this manuscript. We wish to acknowledge Ms. Pirjo Hänninen, Ms. Satu Marttila, Ms. Taija Hukkanen, and Ms. Minna Glad for their highly competent technical assistance in the laboratory, and Dr. Ewen MacDonald for revising the language of this manuscript. We also thank Ms. Elena Fonalleras Lozano and Ms. Casandra Riera Ribas for performing additional experiments relevant to this paper as Erasmus exchange students. The MAG lipase part of this work (depicted in Figure 7) was supported by the Academy of Finland (grant 139620 to JTL).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NA carried out the LC/MS/MS determinations, supervised and partly conducted the autoradiography experiments and quantification of the results, and drafted the manuscript. ML provided the facilities and carried out the design of the LC/MS/MS determinations. M.Sc. student KV for the most part carried out the autoradiography experiments and quantification of the results, and participated in the Pi determinations. Erasmus exchange student GAG for the most part carried out the Pi determinations. JTL initiated the study, carried out its design and coordination and helped to draft the manuscript as a senior author. All authors read and approved the final manuscript.
Electronic supplementary material
12869_2011_971_MOESM1_ESM.pdf
Additional file 1 :The maximal effective concentrations for Na 3 VO 4 and propranolol. (Autoradiography image) (PDF 290 kb) (PDF 290 KB)
12869_2011_971_MOESM2_ESM.pdf
Additional file 2 :Propranolol, but not nadolol, induces LPA mimicking binding response. (Autoradiography image) (PDF 164 kb) (PDF 165 KB)
12869_2011_971_MOESM4_ESM.pdf
Additional file 4 :Comparison of the [35 S]GTPγS binding responses between LPA and (2 S)-OMPT. (Autoradiography image) (PDF 248 kb) (PDF 248 KB)
12869_2011_971_MOESM5_ESM.pdf
Additional file 5 :Autotaxin is not responsible for tonic LPA 1 activity. (Autoradiography image) (PDF 102 kb) (PDF 103 KB)
12869_2011_971_MOESM7_ESM.pdf
Additional file 7 :Inhibitor-evoked [35 S]GTPγS binding responses are restricted to the white matter areas of the brain. (Graph) (PDF 60 kb) (PDF 61 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Aaltonen, N., Lehtonen, M., Varonen, K. et al. Lipid phosphate phosphatase inhibitors locally amplify lysophosphatidic acid LPA1 receptor signalling in rat brain cryosections without affecting global LPA degradation. BMC Pharmacol 12, 7 (2012). https://doi.org/10.1186/1471-2210-12-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2210-12-7