





Abstract
Human CST (CTC1–STN1–TEN1) is a ssDNA-binding complex that helps resolve replication problems both at telomeres and genome-wide. CST resembles Replication Protein A (RPA) in that the two complexes harbor comparable arrays of OB-folds and have structurally similar small subunits. However, the overall architecture and functions of CST and RPA are distinct. Currently, the mechanism underlying CST action at diverse replication issues remains unclear. To clarify CST mechanism, we examined the capacity of CST to bind and resolve DNA structures found at sites of CST activity. We show that CST binds preferentially to ss-dsDNA junctions, an activity that can explain the incremental nature of telomeric C-strand synthesis following telomerase action. We also show that CST unfolds G-quadruplex structures, thus providing a mechanism for CST to facilitate replication through telomeres and other GC-rich regions. Finally, smFRET analysis indicates that CST binding to ssDNA is dynamic with CST complexes undergoing concentration-dependent self-displacement. These findings support an RPA-based model where dissociation and re-association of individual OB-folds allow CST to mediate loading and unloading of partner proteins to facilitate various aspects of telomere replication and genome-wide resolution of replication stress.
INTRODUCTION
Mammalian telomeres are bound by a protein complex called shelterin that sequesters the DNA terminus to prevent it from being sensed as DNA damage and activating repair activities (1,2). The shelterin components TRF1 and TRF2 bind the telomeric dsDNA, which is composed of TTAGGG•AATCCC repeats, while POT1 binds to the ssDNA overhang on the G-rich strand. TPP1 links POT1 to TRF1/2 via TIN2. Mammalian cells also contain a second telomere-associated complex called CST (CTC1–STN1–TEN1) that is essential for telomere replication (3,4). Like its yeast counterpart (Cdc13–Stn1–Ten1), mammalian CST helps maintain telomere length by ensuring dsDNA is formed after telomerase elongates the 3′ G-rich strand (5–10). Both yeast and mammalian CST perform this role by enabling DNA polymerase to synthesize the complementary C-rich strand (8,10,11). This process is known as C-strand fill-in. In mammalian cells, CST-mediated C-strand fill-in is absolutely required for telomere length maintenance and cells lacking CST exhibit progressive telomere shortening similar to what is observed in cells that lack telomerase (6).
Mammalian CST has additional roles in replication both at telomeres and elsewhere in the genome. At telomeres, CST also aids in passage of the replication fork through the telomere duplex. Removal of CST slows replication through this region (7,8) and leads to sudden telomere loss and/or a fragile telomere phenotype (7,12,13). The genome-wide roles of CST include facilitating replication through GC-rich regions (14) and promoting firing of late or dormant replication origins after replication fork stalling (13,15). Loss of CST causes an increase in non-telomere associated anaphase bridges, fragile site expression and chromosome breaks, and a decrease in origin firing after HU treatment (12–14). Exactly how CST resolves all the above replication-related issues remains unclear. However multiple studies indicate a link to DNA polymerase α (Pol α) as CST interacts with Pol α and stimulates its activity (7,16–18). During telomeric C-strand fill-in, CST appears unnecessary for telomeric localization of Pol α (7), suggesting that the complex acts to engage Pol α on the ssDNA rather than in Pol α recruitment. As CST preferentially localizes to GC-rich regions of the genome, it has also been suggested that CST may prevent replication fork stalling by melting G-quadruplex (G4) structure (14,19).
Given that CST harbors multiple OB-folds (one each in STN1 and TEN1 and five to six predicted in CTC1) (18,20), we previously suggested that CST might perform its varied roles in replication by utilizing a dynamic binding mechanism similar to that observed for RPA (18,21–23). RPA is a three subunit ssDNA binding complex that is essential for DNA replication, recombination and repair. It functions by directing assembly and disassembly of the multi-protein complexes needed for these reactions. It also removes unwanted DNA secondary structure (22,24). RPA can perform these actions by virtue of having multiple OB-folds, which individually release and rebind ssDNA without causing the entire complex to dissociate (21,22). This microscopic disassociation and re-association of individual OB-folds makes RPA binding very dynamic, enabling it to diffuse along ssDNA and melt unwanted DNA secondary structure. The dynamic binding also exposes regions of ssDNA to nucleate loading of partner proteins (23,25,26). A similar dynamic binding mechanism could enable CST to melt DNA structures at GC-rich regions or load Pol α for C-strand fill-in.
To clarify the mechanism of CST action, we have performed a series of DNA binding studies using substrates that mimic structures commonly found at telomeres or GC-rich regions. Our analysis uncovered ss-dsDNA junction binding and G4 melting activities that are likely to underlie specific aspects of CST function in telomere replication and genome-wide resolution of replication stress. Moreover, smFRET studies provide support for a dynamic binding mechanism that may provide a general foundation for many aspects of CST action.
MATERIALS AND METHODS
CST expression and purification
Sf9 cells were co-infected with baculovirus encoding Flag-CTC1, His-STN1 and TEN1. Two-step protein purification was performed essentially as described (18). Briefly, following cell lysis CST was bound to nickel-Sepharose beads (GE 17526801), eluted with imidazole, bound to FLAG beads (Sigma A2220) and eluted with 3xFLAG-peptide (Sigma F4799). Protein was stored in 1 mg/ml BSA and 15% glycerol at –80°C after flash freezing in liquid nitrogen. The concentration was determined by SDS-PAGE and silver staining using BSA as a control. All experiments were performed with at least 3 independent CST preparations.
DNA constructs
Oligonucleotides used for electrophoretic mobility shift assays (EMSA) were radiolabeled with 32P-ATP (Perkin Elmer BLU002Z250). Single-stranded and fold-back oligonucleotides were boiled for 2 min and fast cooled on ice for 15 min in STE buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA). To prepare partial duplex substrates, the long and short single-stranded oligonucleotides were mixed in a 1:1.1 molar ratio (5 μM long oligonucleotide) in STE buffer, boiled for 2 min and slow cooled to room temperature ∼2 h. Partial duplex substrates for smFRET assays were prepared by mixing Cy3 and Cy5-labelled oligonucleotides (IDT) in a 1:1.5 molar ratio in T50 buffer (10 mM Tris–HCl pH 8.0, 50 mM NaCl), boiled for 2 min and slow cooled to room temperature over ∼2 h. To ensure formation of G4, substrates (ssDNA for EMSA and partial duplex for smFRET) were further diluted to desired concentrations (0.1 nM for EMSA and 5 pM for smFRET) in G4 buffer (20 mM Tris–HCl pH 8.0, 3 mM MgCl2 and 150 mM NaCl (G4 buffer Na) or 100 mM KCl (G4 buffer K)) and incubated for 10 min before addition to the reaction mix for EMSA or immobilization on slides for smFRET.
Binding assays
For Kd analysis, CST (0.01–25 nM) was incubated with 32P-labeled oligonucleotide (0.01 nM) in binding buffer (25 mM Tris pH 8.0, 1 mM DTT, 150 mM NaCl) for 18 h at 4°C to reach reaction equilibrium. For other DNA binding analyses, the indicated amounts of CST were incubated with 0.1 nM 32P-labeled oligonucleotide in 25 mM Tris–HCl pH 8.0, 1 mM DTT, 150 mM NaCl for 30 min at RT. DNA binding was then monitored by EMSA. Samples were separated in 0.7% agarose gels with 1 × TAE for 1 h at 90V and 4°C and then quantified by PhosphorImaging. To determine Kd, app, the amount of bound versus free DNA was quantified using ImageQuantTL software. Data were fit to a one site specific saturation binding equation using Graphpad prism software.
To monitor strand melting, binding reactions were terminated by addition of 6 × helicase buffer (30% glycerol, 50 mM EDTA, 0.9% SDS, 0.25% bromophenol blue and 0.25% xylene cyanol). Reactions were then loaded on 15% native polyacrylamide gels in 1 × TBE and run at 200 V for 1 h. To ensure that melted duplex could not reanneal after reaction termination, a 500 × molar excess of unlabeled short (22 nt) oligonucleotide was added to control reactions in addition to the 6 × helicase buffer. Other samples were boiled in the helicase buffer prior to gel loading and monitored for duplex formation.
Single molecule fluorescence resonance energy transfer (smFRET) assays
Single molecule FRET assays were carried out as previously described (27,28). Briefly, quartz slides and coverslips were sonicated in posassium hydroxide, methanol and acetone, burned on each side using a propane torch (to remove any residual fluorescent molecules), incubated in amino silane solution (150 ml methanol, 7.5 ml acetic acid, 1.5 ml silane), and coated with a mixture of 97% mPEG (to minimize surface interaction with the protein) and 3% biotin PEG. After preparation of the PEG slides, flow chambers were assembled using strips of double-sided tape and epoxy (29). We flowed 30 μl of 0.1 mg/ml NeutrAvidin in T50 buffer into each empty flow chamber and incubated for 5 min. The excess NeutrAvidin was then washed out with T50 buffer. Partial duplex DNA molecules were immobilized on the slides by biotin-NeutrAvidin interaction. Excess oligonucleotide was washed away either with T50 buffer or with G4 buffer. To detect DNA binding and G4 melting, 2 nM CST in binding buffer (25 mM Tris–HCl pH 8.0, 1 mM DTT, 150 mM NaCl) was added and incubated for 10 min at room temperature. Excess CST was washed out with T50 buffer and, to avoid rapid photobleaching of fluorescent dyes, the chambers were supplemented with an image buffer with an oxygen scavenging system (binding buffer + 0.8 mg/ml glucose oxidase, 0.625% glucose, 3 mM Trolox and 0.03 mg/ml catalase) before taking images. For real-time experiments, 2 nM CST in image buffer was added to the oligonucleotide-bound chambers and imaging was initiated immediately. For the real-time experiments with different concentrations of CST, 2 nM CST in image buffer was added to the chambers and incubated for 1 min followed by either washout of excess CST with image buffer, or addition of 2 or 5 nM CST in image buffer, before prompt initiation of imaging. The whole procedure was carried out at room temperature. All smFRET experiments were performed with at least three separate protein preparations.
smFRET data acquisition
A prism-type total internal reflection fluorescence (TIRF) microscopy was used to acquire single-molecule FRET data. The excitation beam was focused into a pellin broca prism (Altos Photonics), which was placed on top of a quartz slide with a thin layer of immersion oil in between to match the index of refraction. Cy3 (donor) and Cy5 (acceptor) dyes were excited through the dual-laser excitation system (532 and 640 nm, Crystal Laser) via TIRF. The fluorescence signals from Cy3 and Cy5 were collected by a water immersion objective lens (60×, 1.2 N.A. Nikon) and then passed through a notch filter to block out excitation beams. The emission signals of Cy3 and Cy5 dyes were separated by a dichroic mirror (FF662-FDi01; Semrock) and detected by the electron-multiplying charge-coupled device camera (iXon 897; Andor Technology). Data were recorded with a time resolution of 100 ms as a stream of imaging frames and analyzed with scripts written in interactive data language to give fluorescence intensity time trajectories of individual molecules. For FRET histograms, the CST-containing binding buffer was washed out of the flow chamber then short movies (2 s) were recorded from 20 to 30 random locations. For real-time measurements, long movies (90 s) were recorded from 5 to 10 random locations immediately after adding imaging buffer (containing CST) to the flow chambers.
smFRET data analysis
Basic data analysis was carried out by the smCamera software written in C++ (Microsoft), with FRET efficiency, E, calculated as the intensity of the acceptor channel divided by the sum of the donor and acceptor intensities. Leakages from the donor channel to the acceptor channel and vice versa were corrected. FRET histograms were generated by using over 4000 molecules and were fitted to Gaussian distributions with an unrestrained peak center position in Prism 7 (GraphPad Software). Traces with or without CST fall off were collected from >2000 traces for each individual experiment. Dwell times were collected by measuring the time the molecule spends in a particular FRET state. The dwell-time histograms were generated from >300 dynamic events.
Circular dichroism to monitor DNA folding
The G4 substrates (10 μM) were heated at 97°C for 2 min and slow cooled to room temperature for ∼2 hrs in 20 mM Tris–HCl pH 8.0, 3 mM MgCl2, 150 mM NaCl. CD spectra were recorded as described (30) at room temperature on a Jasco J-819 CD spectrophotometer over a 340–200 nm wavelength range. CD data were normalized to molar circular dichroism (Δϵ) based on DNA strand concentration using the equation Δϵ = θ/(3298.2cl) where θ is the CD ellipticity in millidegrees, c is DNA strand concentration (mol/l), and l is the path length (cm). Final spectra were plotted as the average of three scans.
RESULTS
CST recognizes ss-dsDNA junctions but does not melt extended stretches of duplex DNA
To better understand the role of CST in telomeric C-strand fill-in, we set out to examine CST binding to substrates mimicking the junction between the telomeric dsDNA and the 3′ overhang. During telomere replication, extension of the G-strand by telomerase occurs processively (31,32). However, the complimentary C-strand is synthesized more gradually (32) by repeated addition of DNA segments to the 5′ terminus in a process similar to Okazaki fragment synthesis. It is not known how DNA polymerase is positioned to generate each new DNA fragment, but one possible mechanism is that CST recognizes the ss-dsDNA junction to direct DNA polymerase to the adjacent region on the overhang.
To explore whether CST recognizes ss-dsDNA junctions, we performed in vitro binding studies using recombinant CST and a mock telomere substrate that harbored both double and single-stranded telomeric DNA. CST was purified from insect cells (Figure 1A) that had been co-infected with baculovirus encoding CTC1, STN1 and TEN1 (18). The mock telomere substrate was generated from a single (fold-back) oligonucleotide that self-hybridized to form 15 nt dsDNA and an 18 nt 3′ telomeric G-strand overhang (Figure 1B, Table 1). We also generated control ss-dsDNA junction substrates that consisted of all non-telomeric sequence or contained telomeric sequence only in the ssDNA. Individual substrates were incubated with various concentrations of CST and binding was analyzed by electrophoretic gel mobility shift assay (EMSA).
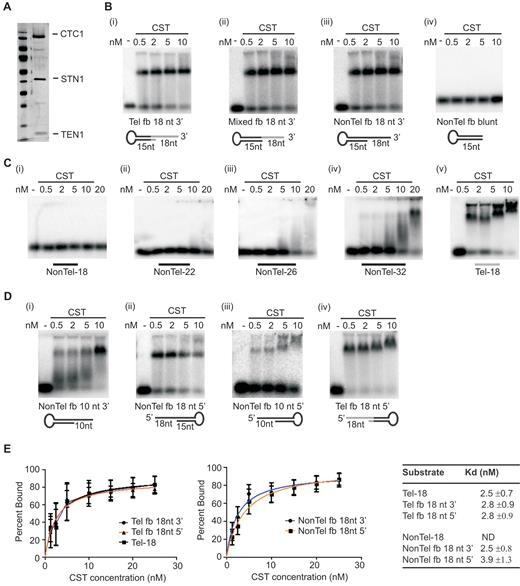
CST binds to ss-dsDNA junctions without melting the DNA duplex. (A) Silver stained polyacrylamide gel (15% on the bottom, 12% on top) showing co-purified CST subunits. (B–D) EMSAs showing CST binding to various substrates. Reactions contained the indicated amounts of CST and 0.1 nM DNA. (B) CST binding to junction substrates with an 18 nt 3′ overhang. Substrates were generated using fold-back oligonucleotides (fb) with: (i) Telomeric sequence at junction, (ii) mixed telomeric and non-telomeric sequence or (iii) non-telomeric sequence. Black lines represent non-telomeric DNA, grey lines indicate telomeric sequence. (iv) Non-telomeric fold-back substrate lacking an overhang. (C) EMSAs showing length of ssDNA substrate needed for CST binding. Non-telomeric substrates were 18, 22, 26 or 32 nt, telomeric substrate was 18 nt. The 18 nt non-telomeric/telomeric substrates had the same sequence as 3′ overhangs of the corresponding fold-back substrates. (D) EMSAs showing CST binding to junction substrates with 10 nt 3′ overhang and 10 or 18 nt 5′ overhangs. (E) Quantification of CST affinity for junction substrates versus ssDNA. Data for binding isotherms to determine apparent dissociation constants (Kd app) were obtained by EMSA and fit to a one site specific binding model. Mean ± SEM, n = 3 independent experiments each with a different protein preparation. ND, Kd not determined as binding undetectable in EMSA.
Oligonucleotides used for EMSA, Strand-melting assay and smFRET. Underline indicates regions of dsDNA
| EMSA | 5′→3′ |
| Non-Tel 18 | AGCGTATCCGTTCAGTTG |
| Non-Tel 26 | GCGTACTCAGTGACTCAGATCGACAG |
| Tel 18 | GGTTAGGGTTAGGGTTAG |
| Telomere fold-back 18 nt 3′ overhang | CTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAGGGTTAGGGTTAGGGTTAG |
| Mixed fold-back 18 nt 3′ overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTGGTTAGGGTTAGGGTTAG |
| Non-Tel fold-back 18nt 3′ nt overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTAGCGTATCCGTTCAGTTG |
| Non-Tel fold-back 10 nt 3′ overhang (15nt ds) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT AGCGTATCCG |
| Tel fold-back 18 nt 5′ overhang | GGTTAGGGTTAGGGTTAGCTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAG |
| Non-Tel fold-back 18 nt 5′ overhang | AGCGTATCCGTTCAGTTGACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| Non-Tel fold-back 10 nt 3′ overhang (22nt ds) | CGATCTGAGTCACTGAGTACGCTTTTTGCGTACTCAGTGACTCAGATCG AGCGTATCCG |
| Non-Tel fold-back (dsDNA) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| EMSA & strand- melting assay | 5′→3′ |
| Non-Tel 22 | CGATCTGAGTCACTGAGTACGC |
| Non-Tel 32 | GCGTACTCAGTGACTCAGATCGAGCGTATCCG |
| EMSA G4 | 5′→3′ |
| 3G4 | TTAGGGTTAGGGTTAGGGTTAGGG |
| 3G5 | TTAGGGTTAGGGTTAGGGTTAGGGTTAGGG |
| 3G46 | TTAGGGTTAGGGTTAGGGTTAGGGAGCGTA |
| G4 | GGGTTAGGGTTAGGGTTAGGG |
| FRET | 5′→3′ |
| Cy5 18 nt anchor | Cy5-GCCTCGCTGCCGTCGCCA-Bio |
| Cy3 18 nt overhang | TGGCGACGGCAGCGAGGCAGCGTATCCGTTCAGTTG-Cy3 |
| Cy3 3G4 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGG-Cy3 |
| Cy3 3G46 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGGAGCGTA-Cy3 |
| EMSA | 5′→3′ |
| Non-Tel 18 | AGCGTATCCGTTCAGTTG |
| Non-Tel 26 | GCGTACTCAGTGACTCAGATCGACAG |
| Tel 18 | GGTTAGGGTTAGGGTTAG |
| Telomere fold-back 18 nt 3′ overhang | CTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAGGGTTAGGGTTAGGGTTAG |
| Mixed fold-back 18 nt 3′ overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTGGTTAGGGTTAGGGTTAG |
| Non-Tel fold-back 18nt 3′ nt overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTAGCGTATCCGTTCAGTTG |
| Non-Tel fold-back 10 nt 3′ overhang (15nt ds) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT AGCGTATCCG |
| Tel fold-back 18 nt 5′ overhang | GGTTAGGGTTAGGGTTAGCTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAG |
| Non-Tel fold-back 18 nt 5′ overhang | AGCGTATCCGTTCAGTTGACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| Non-Tel fold-back 10 nt 3′ overhang (22nt ds) | CGATCTGAGTCACTGAGTACGCTTTTTGCGTACTCAGTGACTCAGATCG AGCGTATCCG |
| Non-Tel fold-back (dsDNA) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| EMSA & strand- melting assay | 5′→3′ |
| Non-Tel 22 | CGATCTGAGTCACTGAGTACGC |
| Non-Tel 32 | GCGTACTCAGTGACTCAGATCGAGCGTATCCG |
| EMSA G4 | 5′→3′ |
| 3G4 | TTAGGGTTAGGGTTAGGGTTAGGG |
| 3G5 | TTAGGGTTAGGGTTAGGGTTAGGGTTAGGG |
| 3G46 | TTAGGGTTAGGGTTAGGGTTAGGGAGCGTA |
| G4 | GGGTTAGGGTTAGGGTTAGGG |
| FRET | 5′→3′ |
| Cy5 18 nt anchor | Cy5-GCCTCGCTGCCGTCGCCA-Bio |
| Cy3 18 nt overhang | TGGCGACGGCAGCGAGGCAGCGTATCCGTTCAGTTG-Cy3 |
| Cy3 3G4 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGG-Cy3 |
| Cy3 3G46 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGGAGCGTA-Cy3 |
| EMSA | 5′→3′ |
| Non-Tel 18 | AGCGTATCCGTTCAGTTG |
| Non-Tel 26 | GCGTACTCAGTGACTCAGATCGACAG |
| Tel 18 | GGTTAGGGTTAGGGTTAG |
| Telomere fold-back 18 nt 3′ overhang | CTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAGGGTTAGGGTTAGGGTTAG |
| Mixed fold-back 18 nt 3′ overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTGGTTAGGGTTAGGGTTAG |
| Non-Tel fold-back 18nt 3′ nt overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTAGCGTATCCGTTCAGTTG |
| Non-Tel fold-back 10 nt 3′ overhang (15nt ds) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT AGCGTATCCG |
| Tel fold-back 18 nt 5′ overhang | GGTTAGGGTTAGGGTTAGCTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAG |
| Non-Tel fold-back 18 nt 5′ overhang | AGCGTATCCGTTCAGTTGACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| Non-Tel fold-back 10 nt 3′ overhang (22nt ds) | CGATCTGAGTCACTGAGTACGCTTTTTGCGTACTCAGTGACTCAGATCG AGCGTATCCG |
| Non-Tel fold-back (dsDNA) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| EMSA & strand- melting assay | 5′→3′ |
| Non-Tel 22 | CGATCTGAGTCACTGAGTACGC |
| Non-Tel 32 | GCGTACTCAGTGACTCAGATCGAGCGTATCCG |
| EMSA G4 | 5′→3′ |
| 3G4 | TTAGGGTTAGGGTTAGGGTTAGGG |
| 3G5 | TTAGGGTTAGGGTTAGGGTTAGGGTTAGGG |
| 3G46 | TTAGGGTTAGGGTTAGGGTTAGGGAGCGTA |
| G4 | GGGTTAGGGTTAGGGTTAGGG |
| FRET | 5′→3′ |
| Cy5 18 nt anchor | Cy5-GCCTCGCTGCCGTCGCCA-Bio |
| Cy3 18 nt overhang | TGGCGACGGCAGCGAGGCAGCGTATCCGTTCAGTTG-Cy3 |
| Cy3 3G4 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGG-Cy3 |
| Cy3 3G46 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGGAGCGTA-Cy3 |
| EMSA | 5′→3′ |
| Non-Tel 18 | AGCGTATCCGTTCAGTTG |
| Non-Tel 26 | GCGTACTCAGTGACTCAGATCGACAG |
| Tel 18 | GGTTAGGGTTAGGGTTAG |
| Telomere fold-back 18 nt 3′ overhang | CTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAGGGTTAGGGTTAGGGTTAG |
| Mixed fold-back 18 nt 3′ overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTGGTTAGGGTTAGGGTTAG |
| Non-Tel fold-back 18nt 3′ nt overhang | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGTAGCGTATCCGTTCAGTTG |
| Non-Tel fold-back 10 nt 3′ overhang (15nt ds) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT AGCGTATCCG |
| Tel fold-back 18 nt 5′ overhang | GGTTAGGGTTAGGGTTAGCTAACCGCATCTAGCTTTTTGCTAGATGCGGTTAG |
| Non-Tel fold-back 18 nt 5′ overhang | AGCGTATCCGTTCAGTTGACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| Non-Tel fold-back 10 nt 3′ overhang (22nt ds) | CGATCTGAGTCACTGAGTACGCTTTTTGCGTACTCAGTGACTCAGATCG AGCGTATCCG |
| Non-Tel fold-back (dsDNA) | ACGACTGCATCTAGCTTTTTGCTAGATGCAGTCGT |
| EMSA & strand- melting assay | 5′→3′ |
| Non-Tel 22 | CGATCTGAGTCACTGAGTACGC |
| Non-Tel 32 | GCGTACTCAGTGACTCAGATCGAGCGTATCCG |
| EMSA G4 | 5′→3′ |
| 3G4 | TTAGGGTTAGGGTTAGGGTTAGGG |
| 3G5 | TTAGGGTTAGGGTTAGGGTTAGGGTTAGGG |
| 3G46 | TTAGGGTTAGGGTTAGGGTTAGGGAGCGTA |
| G4 | GGGTTAGGGTTAGGGTTAGGG |
| FRET | 5′→3′ |
| Cy5 18 nt anchor | Cy5-GCCTCGCTGCCGTCGCCA-Bio |
| Cy3 18 nt overhang | TGGCGACGGCAGCGAGGCAGCGTATCCGTTCAGTTG-Cy3 |
| Cy3 3G4 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGG-Cy3 |
| Cy3 3G46 | TGGCGACGGCAGCGAGGCTTAGGGTTAGGGTTAGGGTTAGGGAGCGTA-Cy3 |
The EMSA analysis revealed that CST bound not only the mock telomere substrate but also the non-telomeric and partial telomeric substrates. This result was unexpected because although CST binding to long (35–50 nt) ssDNA substrates is sequence independent, binding to substrates in the 18–30 nt range has only been observed if they resemble telomeric G-strand sequence (3,5,18). In confirmation of previous reports, we were unable to detect CST binding to a fully single-stranded 18 nt oligonucleotide with the same sequence as the overhang on the non-telomeric junction substrate (Figure 1C). We next examined whether CST recognizes a fully non-telomeric junction substrate that has only a 10 nt 3′ overhang. Interestingly, CST also bound this DNA (Figure 1D panel (i)) although with lower efficiency than the substrate with the 18 nt overhang. This result was surprising because CST binds poorly to fully single-stranded DNA ≤12 nt in length even if it consists of TTAGGG repeats (5). Since CST is also unable to bind dsDNA (3,5,18) (Figure 1B panel (iv)), our results suggested that the ss-dsDNA junction stabilized binding.
To quantify this stabilization of CST binding, we performed additional EMSA analysis to measure the apparent dissociation constants (Kd,app) for telomeric and non-telomeric junction substrates with an 18 nt overhang relative to a fully single-stranded 18 nt telomeric oligonucleotide. The 18 nt oligonucleotide had the same sequence as the overhang on the telomeric junction substrate. The Kd analysis revealed that CST bound the two junction substrates and the 18 nt ssDNA with similar affinity (Figure 1E). This result indicates that a region of dsDNA becomes important for stable CST binding when the adjacent ssDNA provides a suboptimal substrate.
Unlike telomeres, which always terminate with a 3′ overhang, DNA replication and repair intermediates can consist of partial duplex DNA with the opposite orientation where the ssDNA extends from the dsDNA junction in a 3′ to 5′ direction (e.g. a stalled leading-strand replication intermediate). We therefore asked whether CST exhibits a preference for junction substrates with 3′ versus 5′ overhangs. We found CST bound substrates with either overhang orientation with similar efficiency (Figure 1B, D and E). This result was again unexpected as CST binds fully ssDNA with 5′ to 3′ directionality with the highest affinity binding sites positioned towards the DNA 3′ end (18). Overall, our results suggested that CST specifically recognizes ss-dsDNA junctions and this junction-binding activity is important for stabilizing CST binding to suboptimal substrates and/or for positioning the complex on the DNA in a non-preferred orientation. However, the above results could also be explained if CST has a helix destabilizing activity similar to that observed for RPA (33,34), instead of a junction binding activity. Since all the junction substrates tested were formed from a single fold-back oligonucleotide, a helix destabilizing activity would allow CST to melt the DNA duplex to give a ssDNA substrate of 53 nt. This would provide sufficient length of non-telomeric ssDNA for CST to bind (18).
To test if CST can destabilize and melt dsDNA, we performed a strand-melting assay using a junction substrate formed from two separate 32P-labeled oligonucleotides (Figure 2A). This two-oligonucleotide substrate consisted of 32 nt and 22 nt non-telomeric oligonucleotides annealed to form a 22 bp duplex with a 10 nt 3′ overhang (Table 1). It was necessary to use 22 bp DNA duplex instead of the 15 bp duplex present in the fold-back substrates to ensure efficient annealing of the two oligonucleotides. The resulting two-oligonucleotide substrate is comparable to those that can be destabilized and melted by RPA (e.g. 23 bp duplex with a 30 nt 5′ overhang or 40 nt duplex (33,34). EMSA analysis indicated that CST bound the two-oligonucleotide junction substrate with similar efficiency to a fold-back substrate with 22 bp duplex and a 10 nt overhang of the same sequence (Figure 2B).
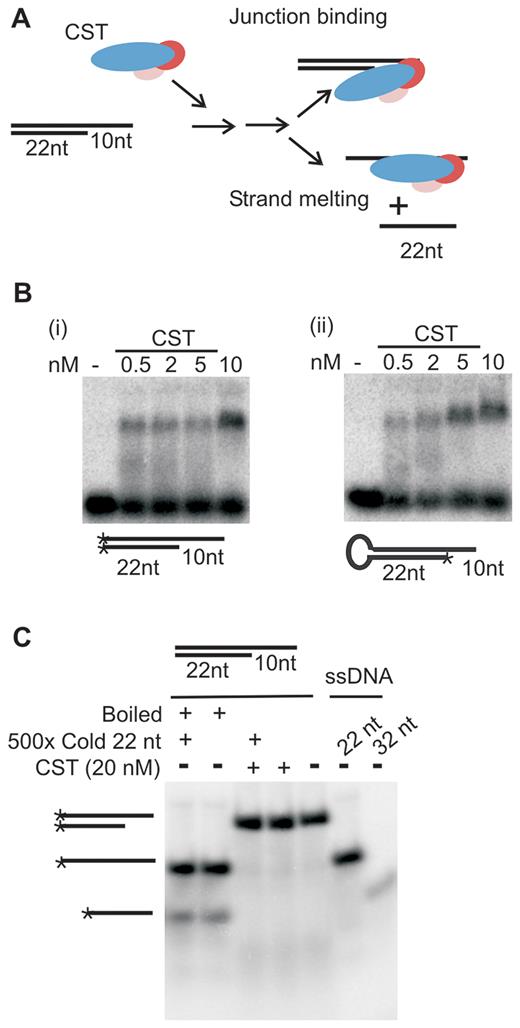
CST binds to junction substrates without melting the DNA duplex. (A) Cartoon of strand-melting assay with junction substrate formed by annealing 32 and 22 nt non-telomeric oligonucleotides. Addition of CST to junction substrate could lead to (i) stable binding to ds/ssDNA junction in which case the strands of the DNA substrate would remain annealed or (ii) transient binding to the 10 nt overhang with subsequent unwinding of the adjacent duplex to give separation of the component 32 and 22 nt oligonucleotides. (B) EMSAs showing CST binding to the junction substrate formed from the 32 and 22 nt oligonucleotides, the 32 nt oligonucleotide alone or a fold-back oligonucleotide with the equivalent 22 nt duplex and 10 nt overhang. (C) Native acrylamide gel showing lack of strand-melting after CST binding to junction substrate. Expected positions of partial duplex and ssDNA are shown to the left. Boiled: samples were boiled to melt DNA just prior to gel loading. * indicates 32P-label.
To test for strand melting, the two-oligonucleotide substrate was incubated with 20 nM CST for 30 min at RT, SDS was then added to denature the protein and samples were analyzed in native acrylamide gels (Figure 2C). The assay provided no evidence for strand melting as the amount of annealed substrate remained unchanged and the amount of unannealed 32 nt or 22 nt DNA did not increase after CST addition. To control for possible re-annealing of separated DNA strands during gel loading, some reactions were terminated by the addition of both SDS and an excess of the unlabeled 22 nt oligonucleotide to serve as a trap for any free 32P-labeled 32 nt oligonucleotide (the 22 nt oligonucleotide is too short to be bound by CST (Figure 1C)). However, there was still no evidence of strand melting as again there was no increase in the amount of un-annealed 32P-labeled 22 nt ssDNA. It therefore appears that CST is unable to melt extended stretches of duplex DNA. In this respect, CST differs from RPA, which exhibits robust helix destabilizing/strand-melting activity (33,34).
To verify that CST recognizes ss-dsDNA junctions but lacks the ability to melt DNA duplex we turned to a single molecule FRET (smFRET) assay. We prepared a non-telomeric partial duplex FRET substrate with an 18 nt 3′ overhang by annealing a 3′ Cy3 (donor dye)-labeled 36 nt oligonucleotide with an 18 nt 5′ Cy5 (acceptor dye)-labeled oligonucleotide (28) (Figure 3A). This non-telomeric ss-dsDNA junction substrate was then anchored to neutravidin-coated slides through a 3′ biotin located on the 18 nt oligonucleotide. The conformational state of the DNA was monitored by smFRET using a prism-type total-internal-reflection configured microscope. The FRET efficiency (E) between Cy3 and Cy5 dyes can report the conformation of ssDNA before and after protein binding. FRET efficiency data were collected from >4000 individual molecules obtained from ≥15 fields of view and plotted as a FRET histogram (Figure 3B). As expected, a high FRET signal was observed in the absence of CST due to the flexibility of the 18 nt ssDNA overhang bringing the Cy3 and Cy5 labels into close proximity. A more minor zero FRET peak reflected the presence of DNA substrate molecules with an inactive or missing Cy3 label. To examine CST binding activity, the protein was added to the immobilized DNA for 10 min and then removed by flushing the slide with imaging buffer prior to data aquisition. Data analysis revealed that CST addition resulted in a FRET efficiency switch from ∼0.72 to ∼0.12. The decrease in FRET efficiency indicated that CST bound the junction substrate and this caused an increase in the time-averaged distance between the Cy3 and Cy5 dyes.
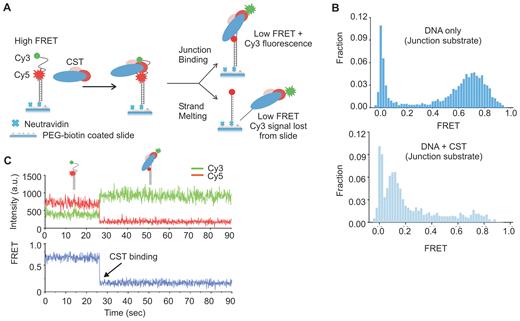
Single molecule FRET showing CST binds to ss-dsDNA junctions. (A) Cartoon showing design of the non-telomeric junction substrate and anticipated FRET signals in the presence or absence of CST. In the absence of CST the flexibility of the 18 nt ssDNA will bring the Cy3 donor and Cy5 acceptor into close proximity. If CST binds without melting the anchoring DNA duplex, the high FRET signal will be lost but emission from the Cy3 donor (green) will be retained. If CST melts the DNA duplex, both the high FRET and the Cy3 donor signal will be lost. (B) FRET histograms generated from FRET measurements of >4000 individual molecules. Top: DNA alone, bottom: DNA + 2 nM CST (C) Representative smFRET real-time trace showing change in individual Cy3 and Cy5 signals (top) and FRET (bottom) with time.
To look more closely at the the effect of CST binding on the junction substrate, we performed a time course analysis to monitor the change in Cy3 and Cy5 signals for individual substrate molecules over time after CST addition (Figure 3C and supplemental Figure S1A). In these experiments, CST was flowed onto the slide in the imaging buffer and imaging was initiated immediately after protein addition so that the initial binding event could be observed. The resulting single-molecule traces revealed a one-step transition to low FRET upon CST binding. The decrease in FRET was not caused by Cy5 photobleaching because Cy5 emission could still be observed if CST-bound substrates were later illuminated with 640 nm light in the absence of Cy3 excitation (supplemental Figure S1B.). Thus, the time course analysis confirmed that CST binding increases the distance between the Cy3 and Cy5 dyes. The analysis also revealed that the Cy3 and Cy5 signals exhibited anti-corrolated fluorescence after CST binding with the Cy3 donor emission increasing as the Cy5 acceptor emission decreased. The increase in Cy3 signal indicated that the Cy3-labeled DNA substrate remained anchored to the slide via base pairing to the biotinylated Cy5-labeled oligonucleotide. This means that CST did not destabilize and melt apart the two strands of the DNA duplex as this would have led to loss of the Cy3-labeled oligonucleotide from the slide (Figure 3A). Thus, the smFRET data confirmed that CST binds to non-telomeric ss-dsDNA junctions without extensive destabilization of the associated DNA dupex.
It remains possible that CST can destabilize a few base pairs of dsDNA at the junction region as such limited strand melting would not have been detected by the above experiments. However, generation of a few extra nucleotides of ssDNA as a result of limited strand melting cannot explain why CST binds efficiently to junction substrates with a 10-18 nt overhang but binds poorly, or not at all, to ssDNA of ≤32 nt (Figure 1B and C). Thus, the above experiments indicate that CST specifically recognizes the ss-dsDNA junction in addition to the adjacent tract of ssDNA. This junction binding activity may be important for CST to engage proteins such as DNA polymerase near primer template junctions during telomere replication and genome-wide resolution of replication stress.
CST can bind and unfold G4 structures
Regions of the genome harboring GC-rich repeats appear to act as barriers to DNA replication due to the single-stranded template DNA forming G-quadruplex (G4) structures which block DNA polymerase (35–37). Since CST binds preferentially to GC-rich DNA (3,5,14,18) and aids in replication through GC-rich regions of the genome (8,14), we next asked whether CST can bind and unfold G4 DNA. To test for G4 binding, we performed EMSAs using a selection of four repeat telomeric oligonucleotides with well characterized G4-forming potential (28,38,39). The oligonucleotides were designed to have 0–6 nt ssDNA located 5′ and/or 3′ of the G4 structure (Figure 4A, Table 1). To ensure folding into G4, each DNA substrate was incubated in 150 mM NaCl, 3 mM MgCl2 prior to use in EMSAs and G4 formation was confirmed by circular dichroism (CD) (Figure 4B). The CD spectra indicated the expected anti-parallel G4 structure as there was a characteristic trough at 260 nm and a positive peak at 290 nm (40).
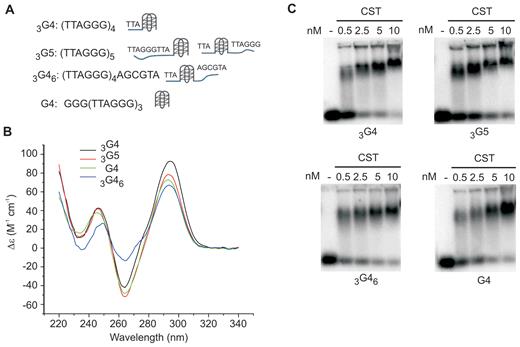
CST binds G4 DNA. (A) Nomenclature, sequence and expected structures of the G4-forming oligonucleotides used in EMSAs. Substrates had 3′ ssDNA (3G4), 3′ or 5′ ssDNA (G5 and 3G46) or no 3′ or 5′ ssDNA (G4). (B) Circular dichroism measurements on G4-forming oligonucleotides to confirm folding into anti-parallel G-quartet. Measurements were in 150 mM NaCl. (C) EMSAs showing CST can bind to telomeric DNA oligonucleotides that form G4 structures. Binding reactions contained the indicated concentrations of CST and 0.1 nM 32P-labeled DNA in 150 mM NaCl, 3 mM MgCl2.
The EMSA analysis revealed that CST bound all of the G4 substrates. Moreover, the efficiency of binding was similar regardless of whether they had ssDNA 3′ or 5′ of the G4 structure (Figure 4C, 3G4, G5, 3G46). These results indicate that CST can bind G4 DNA. However, it was unclear whether CST promotes G4 unfolding thus enabling binding to the resulting ssDNA, or if CST binds directly to the to the G4 structure without unfolding it. The later would be unexpected as OB folds usually bind single-stranded rather than base-paired DNA/RNA (41,42).
To determine if CST can unfold G4 DNA, we again turned to smFRET. The setup was as described above, except the Cy3-labeled oligonucleotide harbored 4 telomeric repeats adjacent to the region annealed to the Cy5-labeled anchor oligonucleotide (Figure 5A). The Cy3 and Cy5 labels were positioned on either side of the telomeric repeats so that they would be in close proximity and give a high FRET signal when the DNA is folded into a G4 structure (28,43–45). Unfolding of the G4 structure would increase the distance between the dyes resulting in a decrease in FRET efficiency. Two different Cy3-labeled substrates were tested: 3G4-Cy3 which terminated with a G4, and 3G46–Cy3 which had an additional 6 nt of non-telomeric ssDNA at the 3′ end (Figure 5C–D, Table 1). The buffers used during slide preparation contained 150 mM NaCl to favor formation of anti-parallel G4 structure.
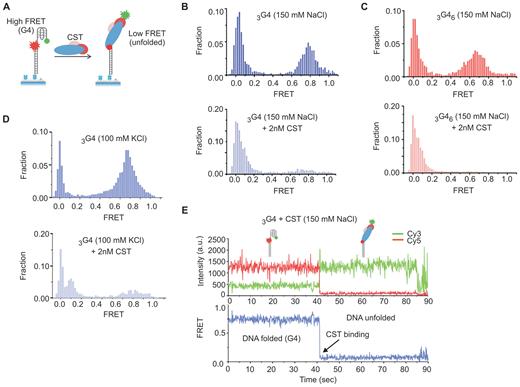
CST unfolds G4 DNA. (A) Schematic of the G4 smFRET substrates showing expected FRET signals with/without G4 unfolding. (B–D) Representative FRET histograms showing CST can bind and unfold G4 DNA. Top, DNA alone; bottom, DNA +2 nM CST. CST was added for 10 min, then excess protein was washed out prior to FRET measurement. (B and C) Unfolding of 3G4 and 3G46 in 150 mM NaCl + 3 mM MgCl2. (D) Unfolding of 3G4 in 100 mM KCl + 3 mM MgCl2. (E) Representative smFRET real-time trace showing a decrease in FRET after CST binding. Top, Cy3 and Cy5 signals showing complementary transition upon CST binding. Bottom, FRET signal. Measurement was performed in the presence of 2 nM CST (no washout).
To assess DNA folding, we analyzed FRET signals from the 3G4-Cy3 substrate in the absence of CST using a range of ionic conditions known to either stabilize (150 mM NaCl) or destabilize G4 structure (0 salt or 50 mM LiCl) (19,28,39,43). In initial experiments, we included 150 mM NaCl in the imaging buffer and FRET signals were quantified from >4000 individual molecules and plotted as a FRET histogram. The histogram revealed a high FRET peak (E = 0.65–0.75) as expected for a folded G4 DNA conformation (Figure 5B). We next introduced imaging buffer containing lower concentrations of NaCl (50 or 0 mM) or LiCl (150 or 50 mM) (Supplementary Figure S2A). The FRET efficiency remained high in 50 mM NaCl and 150 mM LiCl indicating continued DNA folding. However, an intermediate FRET peak appeared in the 50 mM LiCl (E ∼0.5) indicating partial unfolding. In 0 mM salt the high FRET peak disappeared and was replaced by a lower FRET peak (E ∼0.3) characteristic of 24 nt flexible ssDNA. The destabilization of DNA folding in 50 mM LiCl and complete unfolding in 0 mM salt provide strong support for G4 formation (43).
To examine the effect of CST binding, protein was added to the flow chamber and allowed to bind the immobilized DNA for 10 min, then excess CST was flushed out with binding buffer followed by imaging buffer. Imaging of the slides revealed that addition of CST to either the 3G4-Cy3 or the 3G46–Cy3 substrate caused almost complete loss of the high FRET peak with a concomitant increase in the low FRET peak (Figure 5B and C, note difference in scale between top and bottom panels). This decrease in FRET indicates CST can unfold G4 structure and bind the resulting ssDNA. The G4 unfolding and DNA binding appeared to be stable as the loss of the high FRET peak persisted throughout the 10–20 min time period taken to acquire sufficient images to build the FRET histograms. We then repeated the experiment using binding and imaging buffers containing 100 mM KCl + 3 mM MgCl2, conditions that favor formation of a mix of parallel and anti-parallel G4 structure instead of the purely anti-parallel G4 formed in the presence of NaCl (39). We again saw almost complete loss of the high FRET peak indicating G4 unfolding (Figure 5D) (38). We therefore conclude that CST can unfold G4 DNA. Given this capability, we surmise that CST aids in recovery from replication stress by removing G4 structures that form during replication through GC-rich regions of the genome.
To further characterize the G4 unfolding process, we performed a real-time analysis to examine the change in FRET signals from individual DNA molecules with time after CST addition (Figure 5E, Supplementary Figure S3). As in our earlier real-time experiments, CST was flowed onto the slide in the imaging buffer and imaging was initiated immedetially after protein addition to visualize the initial binding event. The resulting 90 s real-time traces revealed a sharp one-step FRET decrease from the high FRET (E ∼0.75) to the low FRET state (E ∼0.1) upon CST binding. Thus, a single CST binding event causes rapid and complete G4 unfolding. The rapid one step change in FRET efficiency is similar to that observed when RPA or the C. glaubrata Cdc13-Stn1-Ten1 complex unfold G4 structures (19,46) but it differs from the mechanism of G4 unfolding by POT1 which occurs in four steps and requires two POT1 molecules (28). The four FRET transitions seen with POT1 reflect sequential binding of the 4 OB folds (two from each POT1 molecule) to cause gradual G4 unfolding (28). The more rapid G4 unfolding by human CST, CgCST and RPA may reflect the larger number OB folds in these complexes and/or a smoother DNA binding trajectory (18,19,47,48).
CST exhibits facilitated displacement
While analyzing real-time traces showing the change in FRET from individual DNA molecules, we noticed that for both the 3G4 and the non-telomeric junction substrates only ∼75% of the traces showed stable loss of the high FRET signal after the initial CST binding event. The remaining ∼25% of traces showed reformation of the high FRET configuration during the 90 s time course (Figure 6A and B, Supplementary Figure S4)) indicating subsequent CST dissociation. In some traces, multiple binding and dissociation events were apparent during a single time course. The frequent dissociation of CST from the two substrates was striking because filter-binding assays have previously shown that CST binding to telomeric and many non-telomeric substrates is very stable (5,18). For telomeric oligonucleotides, t|$\frac{1}{2}$| for CST dissociation ranges from ∼4.5-8 h for 18–48 nt substrates.
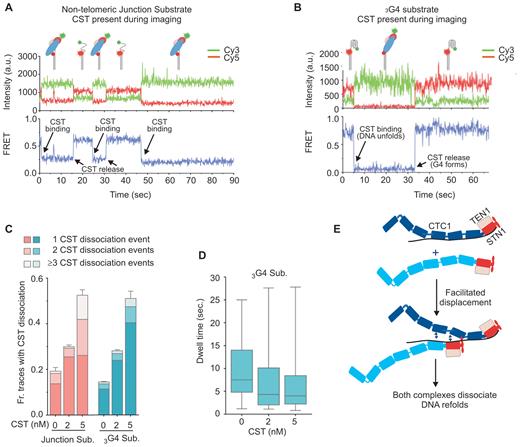
Dissociation of CST from DNA is concentration dependent. (A and B) Representative smFRET real-time traces showing CST dissociation from the non-telomeric junction substrate with 18 nt overhang (A) or the 3G4 G-quadruplex forming substrate (B). 2 nM CST was added to the slide in the imaging buffer and the recording was initiated soon afterwards. T = 0 indicates the start of recording. (C) Fraction of traces showing one or more dissociation events from non-telomeric junction substrate (pink) or 3G4 substrate (blue) after CST removal (0 nM), in the continued presence of 2 nM CST, or after addition of 5 nM CST. N = 3 independent experiments ± S.E.M., 260–560 real-time traces were analyzed for each protein concentration with the junction substrate and >1000 with 3G4. (D) Dwell times for CST binding to 3G4 substrate before protein dissociation. Traces analyzed were as in C, a minimum of 179 dissociation events were analyzed for each CST concentration (B). (E) Model illustrating how unbound CST could cause facilitated displacement of bound CST. The CST complex is shown with a 1:1:1 stoichiometry of CTC1, STN1 and TEN1. Individual OB folds (known for STN1 and TEN1, predicted for CTC1 (18)) are represented as oblongs. Note, the FRET data cannot distinguish between complete CST dissociation from the DNA versus some residual CST binding (e.g. to the ss-dsDNA junction or G4 structure) that exposes sufficient ssDNA to reform the high FRET state.
The discrepancy in the CST binding stability as revealed by the real-time smFRET analysis versus filter-binding assays reminded us of findings with RPA where the stability of RPA binding to ssDNA depends on the concentration of excess unbound RPA (26). Although RPA binds tightly to ssDNA, it dissociates rapidly when free RPA is present thus allowing another RPA molecule to bind. This concentration-dependent protein turnover, known as facilitated exchange, reflects the dynamic nature of RPA binding (see below). The conditions in our real-time FRET analyses of CST binding paralleled those used to show concentration-dependent RPA turnover in that an excess of unbound CST was present during the 90 s taken to acquire the real-time data. In contrast, unbound CST was essentially absent from the filter-binding experiments used to measure CST dissociation rates because an excess of cold competitor DNA was added to samples following the initial binding reaction (5,18). This difference in amount of unbound CST present in the two experiments, suggested to us that the rapid dissociation of CST seen in the smFRET real-time analysis might be directly related to the excess CST present in the flow chamber during data acquisition.
To test whether CST dissociation depends on the concentration of free protein, we obtained real-time traces from slides harboring three separate flow chambers that each contained a different amount of free CST during data acquisition. Either the non-telomeric junction substrate or the 3G4 substrate were immobilized on the slide and 2 nM CST added to each chamber for 1 min. The chambers were then flushed with imaging buffer containing 0, 2 or 5 nM CST and 90 s real-time traces were captured from each portion of the slide. The result was striking as the fraction of traces showing one or more CST dissociation events was directly proportional to the CST concentration present during data acquisition (Figure 6C), with most dissociation events observed at the highest protein concentration. Analysis of the dwell time for CST association with 3G4 indicated that the duration of individual binding events became correspondingly shorter with increased CST concentration (Figure 6D). Interestingly, the frequency of CST dissociation from the non-telomeric junction substrate was similar to that seen for the 3G4 substrate, indicating that the dissociation event does not depend on the ability of the DNA to fold back into an intramolecular G4 structure. Based on the above observations, we conclude that the presence of excess unbound CST increases the likelihood that a bound CST complex will dissociate, i.e. unbound CST facilitates the displacement of bound CST.
The facilitated displacement of CST is comparable to the facilitated exchange observed with RPA (26). In the case of RPA, the concentration-dependent turnover is thought to stem from the dynamic release and re-binding of individual OB folds from the DNA (22,23). This process only leads to macroscopic dissociation of RPA if free RPA is available to occupy the exposed ssDNA. Since CST resembles RPA in harboring multiple ssDNA binding sites/OB-folds, the concentration-dependent self-displacement of CST provides support for a supports a dynamic binding mechanism for CST also. As with RPA, the microscopic dissociation would allow a second CST complex (or another protein) to initiate binding. In some cases, the outcome would be unstable binding and dissociation of both complexes from the DNA (Figure 6E). We term this process ‘facilitated displacement’. In our experiments, this displacement would be observed as reappearance of the high FRET signal.
It may be that CST also undergoes facilitated exchange where a bound CST complex dissociates and is simultaneously replaced by a second complex. We would not expect to detect these events with our experimental set-up because the length of exposed ssDNA would likely be insufficient to flex or fold into the compact state necessary for high FRET. The ability of RPA to undergo facilitated exchange is thought to be extremely important for RPA function because it provides a mechanism to load downstream ssDNA binding proteins during DNA replication and repair (21,22,26). We suggest that facilitated displacement and possibly facilitated exchange also underlie CST function as it would allow CST to load proteins needed to resolve various forms of replication stress (Figure 7).
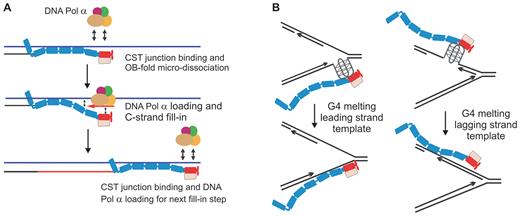
Models for CST mechanism of action. (A) Model illustrating how junction recognition and dynamic binding could lead to Pol α loading on the telomeric G-strand overhang to achieve C-strand fill-in. (B) Model for how CST binding could relieve genome-wide replication stress by promoting G4 unfolding. Oblongs represent individual OB folds in CTC1, STN1 and TEN1
DISCUSSION
Here, we demonstrate that human CST exhibits a series of DNA binding activities that are directly relevant to the various roles of CST genome-wide. These activities include specific recognition of ss-dsDNA junctions, removal of G4 structure and facilitated displacement of protein molecules from a DNA substrate. Together or individually, these activities provide mechanisms for CST to enable DNA replication through GC-rich sequence, regulate C-strand synthesis at telomeres, and support priming of DNA synthesis down-stream of DNA lesions and possibly at late-firing or dormant origins (11,13,49). Our finding that free CST can promote the release of bound CST from ssDNA is particularly interesting because it provides evidence for dynamic DNA binding through dissociation and re-association of individual DNA-binding domains/OB-folds. The dynamic binding is likely to provide a mechanism for regulating the association of partner proteins such as Pol α with ssDNA. While relatively few CST interactions partners have been identified thus far (only Pol α, TPP1 and Rad51) (7,14,50), the large size of CTC1 suggests there are likely to be many others. Hence, the dynamic nature of CST binding to ssDNA may well allow CST to regulate the DNA-association of an array of different proteins depending on the replication issue to be resolved. Currently it is unclear whether CST regulation is restricted to protein displacement or if CST can also promote protein loading by exposing stretches of ssDNA to nucleate protein association. Additional studies are required to clarify this point.
The combined ability of CST to recognize ss-dsDNA junctions and bind DNA dynamically provide a ready explanation for why the process of telomeric C-strand fill-in occurs via incremental extension of the DNA 5′ terminus rather than a single primed DNA synthesis reaction (Figure 7A). While CST appears to coat the newly extended overhang (5,6), it may be that only the complex adjacent to the ss-dsDNA junction is capable of engaging DNA polymerase for C-strand synthesis. This might be because junction recognition somehow alters CST binding dynamics to favor partial CST dissociation from the ssDNA with concomitant DNA polymerase loading. Upon synthesis of one segment of C-strand, a new ss-dsDNA junction would be generated and the adjacent CST complex would become competent to engage DNA polymerase to synthesize the next segment of C-strand DNA. It is possible that the junction recognition on partial duplex DNA also favors loading of CST binding partners at DNA lesions and replication blocks elsewhere in the genome.
The G4-unfolding activity of CST is likely to be important for CST function both during C-strand fill-in and conventional replication of GC-rich dsDNA. At the time of C-strand fill-in, the elongated 3′ overhang can form G4 structures which must be removed to prevent them from impeding DNA synthesis by Pol α (37). During replication of dsDNA, G4 structures also need to be removed to prevent obstruction of DNA polymerase (36,51) (Figure 7B). It is now apparent that in addition to CST, cells harbor a plethora of factors capable of resolving G4 DNA (e.g. RPA, POT1, BLM, WRN, RTEL and other helicases) (35,52,53), raising the question as to why so many apparently redundant activities are needed. Presumably, each activity has specific advantages and disadvantages. We suggest that CST is well suited for efficient G4 removal in situations where RPA binding is undesirable and helicases are unable to act. Such a situation may occur during leading strand replication through GC-rich regions. Formation of G4 structures in these regions (e.g. as a result of transcription (36)) would block replication (51) and if the structure is on the leading strand, there may not be room to load a helicase. In this situation, binding of CST may be an efficient way to remove the G4 (Figure 7B). Moreover, if the G4 causes the replisome to disengage so it becomes necessary to re-prime leading strand synthesis (54,55), CST may enable a new polymerase to load and initiate the re-priming event. At telomeres, G4 structures are likely to form on the lagging strand template. In this situation BLM, WRN and POT1 may all contribute to G4 removal (28,35,56). However, CST unfolds G4 structures more rapidly than POT1 (28), which could explain why the presence of CST makes replication through the telomere duplex more efficient.
It is striking that CST and RPA share many of the same DNA binding activities despite having distinct complex architecture (18) and quite separate roles in DNA replication (8,57). Given that CST is used to resolve replication-related issues that are guaranteed to arise each cell cycle (e.g. telomeric C-strand fill-in and resolution of G4 structure) it may be that cells have evolved this second multi-OB fold ssDNA binding complex to avoid the complication of ATR activation by RPA-coated ssDNA (22,58). For example, CST binding to the G-overhang after telomerase extension prevents RPA association and ATR activation (6) while simultaneously providing a mechanism to engage DNA polymerase for C-strand fill-in. Likewise, CST binding at sites of G4 structure could provide a way to remove the structure and rapidly reinitiate DNA synthesis without affecting the overall replication program through ATR activation. Since the cellular abundance of CST is low relative to RPA, it seems likely that CST would have to be specifically recruited to its sites of action. The recruitment could occur via interaction with other chromosomal proteins to increase local CST concentration. At telomeres this is likely to occur via interaction with TPP1 (18,50).
Mutations in the CTC1 and STN1 subunits of CST cause a severe disease called Coats plus (59–61). Patients suffer from pleiotropic symptoms that include neurological disorders due to brain calcifications, gastrointestinal and retinal bleeding, and bone marrow failure. In some cases, symptoms overlap those seen in patients with the short telomere disorder Dyskeratosis congenita (61). The patient mutations are always biallelic and they result in partial loss of CST function. It is striking that many of the CTC1 mutations lie in the predicted OB fold domains (18,62). This positioning suggests that the mutations are likely to affect DNA binding activities such as junction recognition, G4 unfolding or overall binding dynamics. If this is the case, loss of the CST-based pathway to resolve replication issues may require use of alternative, less benign pathways that result in cellular damage and hence the clinical symptoms of Coats plus. Understanding which activities of CST are altered by specific CTC1 mutations could lead to better disease management in Coats plus patients.
ACKNOWLEDGEMENTS
We are most grateful to Dr Jonathan Chaires and Dr Robert Gray for their help with the CD analysis. We thank Stacey Cranert for preliminary EMSA analysis. Author contributions are as follows: A.B. designed project and performed experiments and wrote sections of the manuscript. Y.W. performed FRET experiments and wrote part of the methods section. J.D. guided smFRET experiments. C.M.P. devised the project, guided experimental design, oversaw the research and wrote sections of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Institute of Health [RO1 GM041803 to C.M.P.]; University of Cincinnati (to J.D.). Funding for open access charge: National Institutes of Health [RO1 GM041803].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments