





Abstract
DrugBank (www.drugbank.ca) is a web-enabled database containing comprehensive molecular information about drugs, their mechanisms, their interactions and their targets. First described in 2006, DrugBank has continued to evolve over the past 12 years in response to marked improvements to web standards and changing needs for drug research and development. This year’s update, DrugBank 5.0, represents the most significant upgrade to the database in more than 10 years. In many cases, existing data content has grown by 100% or more over the last update. For instance, the total number of investigational drugs in the database has grown by almost 300%, the number of drug-drug interactions has grown by nearly 600% and the number of SNP-associated drug effects has grown more than 3000%. Significant improvements have been made to the quantity, quality and consistency of drug indications, drug binding data as well as drug-drug and drug-food interactions. A great deal of brand new data have also been added to DrugBank 5.0. This includes information on the influence of hundreds of drugs on metabolite levels (pharmacometabolomics), gene expression levels (pharmacotranscriptomics) and protein expression levels (pharmacoprotoemics). New data have also been added on the status of hundreds of new drug clinical trials and existing drug repurposing trials. Many other important improvements in the content, interface and performance of the DrugBank website have been made and these should greatly enhance its ease of use, utility and potential applications in many areas of pharmacological research, pharmaceutical science and drug education.
INTRODUCTION
DrugBank is a comprehensive, freely available web resource containing detailed drug, drug-target, drug action and drug interaction information about FDA-approved drugs as well as experimental drugs going through the FDA approval process. The rich, high quality, primary-sourced content found in DrugBank has allowed it become one of the world's most widely used reference drug resources. It is routinely used by the general public, educators, pharmacists, pharmacologists, medicinal chemists, pharmaceutical researchers and the pharmaceutical industry (1). Since its first appearance in 2006, the evolution of DrugBank’s content and interface has largely been directed by the requests of its diverse user community and the efforts of dozens of skilled programmers, domain-specific experts and trained biocurators.
DrugBank 1.0, released in 2006, provided novel (at the time) physico-chemical data on selected FDA-approved drugs and their drug-targets (2). DrugBank 2.0, released in 2008, added pharmacological, pharmacogenomic and molecular biological data (3). DrugBank 3.0, released in 2010, added drug–drug and drug–food interactions, drug transporter data as well as pharmacokinetic information (4). DrugBank 4.0, released in 2014, added significant amounts of drug metabolism data, QSAR (quantitative structure activity relationships) data and ADMET (absorption, distribution, metabolism, excretion and toxicity) data (5). While each of these prior releases provided notable improvements and greatly enriched DrugBank’s data content, this year’s release represents the most significant expansion of DrugBank in more than a decade.
In particular, the quantity of existing data in DrugBank 5.0 has increased enormously. For instance, the number of approved (FDA, Health Canada, EMA, etc.) drugs in the database has grown from 1836 to 2358, the number of reported phase I/II/III investigational drugs has grown from 1219 to 4501, the number of drugs with experimentally acquired mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectra has grown from 690 to 3620, the number of drug-drug interactions has grown from 14 150 to 365 984 and the number of pharmacogenomic and SNP-associated drug effects has grown from 201 to 5993. In addition to this very significant expansion of its existing data, DrugBank 5.0 has added many new datasets or novel kinds of data. This includes information on the influence of hundreds of drugs on metabolite levels (pharmacometabolomics), gene expression levels (pharmacotranscriptomics) and protein expression levels (pharmacoprotoemics). New data have also been added on thousands of investigational drug clinical trials and various drug repurposing trials. Additionally, DrugBank's curation team has greatly improved the quality and consistency of all existing drug indications, enhanced the information on drug-drug and drug-food interactions, filled in data gaps on more than 600 existing drugs and greatly improved the quality and quantity of drug-target binding data. Major improvements to the spectral viewing and spectral search tools, spectral data formats (compatible with SPLASH (6), mzML (7) and nmrML (8)), chemical taxonomies, chemical ontologies (9), as well as text and structure searching/matching have also been made. Further details on the additions and enhancements made to DrugBank 5.0 are described below.
DATABASE ADDITIONS AND IMPROVEMENTS
This section is divided into four subsections: (i) expansion and improvements made to DrugBank's existing data, (ii) the addition of new data content and new data fields to DrugBank, (iii) enhanced DrugBank interface features (iv) a new model for updating and distributing DrugBank.
Enhancement of existing data
Since 2006 DrugBank has seen a progressive expansion in the depth and breadth of its data as well as a significant enhancement to the quality and reliability of its information. The changes over the past 12 years are summarized in Table 1. Compared to the previous release (DrugBank 4.0), DrugBank 5.0 has greatly increased the number of approved drugs, including both small molecule and biotech drugs, from 1836 to 2358. Many drugs exist in a variety of product-specific ingredient forms (salts, esters, etc.) and these forms can significantly affect their efficacy and bioavailability. In response to this, DrugBank 5.0 has worked diligently to capture this information in the DrugCards and to substantially increase the number of product-specific ingredient forms (such as salt forms) contained in the database from 474 (in DrugBank 4.0) to 1551 compounds in DrugBank 5.0 This corresponds to an increase of more than 300%. Given the growing concerns over illicit drugs and designer drugs as well as the continued interest by pharmacologists in understanding the toxicology profiles of withdrawn drugs, the curators for DrugBank 5.0 have also worked hard to expand and enhance this information as well. In particular, we have nearly doubled the number of drug entries in these categories from 268 (in DrugBank 4.0) to 409 compounds in DrugBank 5.0.
Comparison between the coverage in DrugBank 1.0, 2.0, 3.0, 4.0 and DrugBank 5.0
| Category | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 |
|---|---|---|---|---|---|
| No. of data fields per DrugCard | 88 | 108 | 148 | 208 | 215 |
| No. of search types | 8 | 12 | 16 | 18 | 20 |
| No. of illustrated drug-action pathways | 0 | 0 | 168 | 232 | 319 |
| No. of illustrated drug metabolism pathways | 0 | 0 | 0 | 53 | 64 |
| No. of drugs with metabolizing enzyme data | 0 | 0 | 762 | 1037 | 3859 |
| No. of drug metabolites with structures | 0 | 0 | 0 | 1239 | 1360 |
| No. of drug-metabolism reactions | 0 | 0 | 0 | 1308 | 1530 |
| No. of drugs with drug transporter data | 0 | 0 | 516 | 623 | 1954 |
| No. of drugs with taxonomic classification information | 0 | 0 | 0 | 6713 | 7387 |
| No. of Inferred SNP-associated drug effects* | 0 | 0 | 0 | 0 | 5993 |
| No. of directly studied SNP-associated drug effects | 0 | 0 | 113 | 201 | 324 |
| No. of drugs with patent/pricing/manufacturer data | 0 | 0 | 1208 | 1450 | 1820 |
| No. of food–drug interactions | 0 | 714 | 1039 | 1180 | 1195 |
| No. of drug–drug interactions | 0 | 13 242 | 13 795 | 14 150 | 365 984 |
| No. of ADMET parameters (Caco-2, LogS) | 0 | 276 | 890 | 6667 | 6700 |
| No. of QSAR parameters per drug | 5 | 6 | 14 | 23 | 23 |
| No. of drugs with drug-target binding constant data | 0 | 0 | 0 | 791 | 1563 |
| No. of drugs with experimental NMR spectra | 0 | 0 | 0 | 306 | 922 |
| No. of drugs with experimental MS spectra | 0 | 0 | 0 | 384 | 2521 |
| No. of drugs with chemical synthesis information | 0 | 38 | 38 | 1285 | 1584 |
| No. of approved small molecule drugs | 841 | 1344 | 1424 | 1552 | 2110 |
| No. of approved drugs with product ingredient structures | 0 | 0 | 0 | 474 | 1551 |
| No. of biotech drugs | 113 | 123 | 132 | 284 | 555 |
| No. of nutraceutical drugs | 61 | 69 | 82 | 87 | 97 |
| No. of withdrawn drugs | 0 | 57 | 68 | 78 | 209 |
| No. of illicit drugs | 0 | 188 | 189 | 190 | 202 |
| No. of experimental drugs | 2894 | 3116 | 5210 | 6009 | 4964 |
| No. of investigational drugs (Phase I, II and III trials) | 0 | 0 | 0 | 1219 | 4501 |
| No. of all drug targets (unique) | 2133 | 3037 | 4326 | 4115 | 4563 |
| No. of approved-drug enzymes/carriers (unique) | 0 | 0 | 164 | 245 | 479 |
| No. of all drug enzymes/carriers (unique) | 0 | 0 | 169 | 253 | 497 |
| No. of external database links | 12 | 18 | 31 | 33 | 35 |
| Total drug product pill images* | 0 | 0 | 0 | 0 | 3600 |
| Number of linked drug indications* | 0 | 0 | 0 | 0 | 3024 |
| Number of clinical trials* | 0 | 0 | 0 | 0 | 245 356 |
| Category | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 |
|---|---|---|---|---|---|
| No. of data fields per DrugCard | 88 | 108 | 148 | 208 | 215 |
| No. of search types | 8 | 12 | 16 | 18 | 20 |
| No. of illustrated drug-action pathways | 0 | 0 | 168 | 232 | 319 |
| No. of illustrated drug metabolism pathways | 0 | 0 | 0 | 53 | 64 |
| No. of drugs with metabolizing enzyme data | 0 | 0 | 762 | 1037 | 3859 |
| No. of drug metabolites with structures | 0 | 0 | 0 | 1239 | 1360 |
| No. of drug-metabolism reactions | 0 | 0 | 0 | 1308 | 1530 |
| No. of drugs with drug transporter data | 0 | 0 | 516 | 623 | 1954 |
| No. of drugs with taxonomic classification information | 0 | 0 | 0 | 6713 | 7387 |
| No. of Inferred SNP-associated drug effects* | 0 | 0 | 0 | 0 | 5993 |
| No. of directly studied SNP-associated drug effects | 0 | 0 | 113 | 201 | 324 |
| No. of drugs with patent/pricing/manufacturer data | 0 | 0 | 1208 | 1450 | 1820 |
| No. of food–drug interactions | 0 | 714 | 1039 | 1180 | 1195 |
| No. of drug–drug interactions | 0 | 13 242 | 13 795 | 14 150 | 365 984 |
| No. of ADMET parameters (Caco-2, LogS) | 0 | 276 | 890 | 6667 | 6700 |
| No. of QSAR parameters per drug | 5 | 6 | 14 | 23 | 23 |
| No. of drugs with drug-target binding constant data | 0 | 0 | 0 | 791 | 1563 |
| No. of drugs with experimental NMR spectra | 0 | 0 | 0 | 306 | 922 |
| No. of drugs with experimental MS spectra | 0 | 0 | 0 | 384 | 2521 |
| No. of drugs with chemical synthesis information | 0 | 38 | 38 | 1285 | 1584 |
| No. of approved small molecule drugs | 841 | 1344 | 1424 | 1552 | 2110 |
| No. of approved drugs with product ingredient structures | 0 | 0 | 0 | 474 | 1551 |
| No. of biotech drugs | 113 | 123 | 132 | 284 | 555 |
| No. of nutraceutical drugs | 61 | 69 | 82 | 87 | 97 |
| No. of withdrawn drugs | 0 | 57 | 68 | 78 | 209 |
| No. of illicit drugs | 0 | 188 | 189 | 190 | 202 |
| No. of experimental drugs | 2894 | 3116 | 5210 | 6009 | 4964 |
| No. of investigational drugs (Phase I, II and III trials) | 0 | 0 | 0 | 1219 | 4501 |
| No. of all drug targets (unique) | 2133 | 3037 | 4326 | 4115 | 4563 |
| No. of approved-drug enzymes/carriers (unique) | 0 | 0 | 164 | 245 | 479 |
| No. of all drug enzymes/carriers (unique) | 0 | 0 | 169 | 253 | 497 |
| No. of external database links | 12 | 18 | 31 | 33 | 35 |
| Total drug product pill images* | 0 | 0 | 0 | 0 | 3600 |
| Number of linked drug indications* | 0 | 0 | 0 | 0 | 3024 |
| Number of clinical trials* | 0 | 0 | 0 | 0 | 245 356 |
* new data
| Category | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 |
|---|---|---|---|---|---|
| No. of data fields per DrugCard | 88 | 108 | 148 | 208 | 215 |
| No. of search types | 8 | 12 | 16 | 18 | 20 |
| No. of illustrated drug-action pathways | 0 | 0 | 168 | 232 | 319 |
| No. of illustrated drug metabolism pathways | 0 | 0 | 0 | 53 | 64 |
| No. of drugs with metabolizing enzyme data | 0 | 0 | 762 | 1037 | 3859 |
| No. of drug metabolites with structures | 0 | 0 | 0 | 1239 | 1360 |
| No. of drug-metabolism reactions | 0 | 0 | 0 | 1308 | 1530 |
| No. of drugs with drug transporter data | 0 | 0 | 516 | 623 | 1954 |
| No. of drugs with taxonomic classification information | 0 | 0 | 0 | 6713 | 7387 |
| No. of Inferred SNP-associated drug effects* | 0 | 0 | 0 | 0 | 5993 |
| No. of directly studied SNP-associated drug effects | 0 | 0 | 113 | 201 | 324 |
| No. of drugs with patent/pricing/manufacturer data | 0 | 0 | 1208 | 1450 | 1820 |
| No. of food–drug interactions | 0 | 714 | 1039 | 1180 | 1195 |
| No. of drug–drug interactions | 0 | 13 242 | 13 795 | 14 150 | 365 984 |
| No. of ADMET parameters (Caco-2, LogS) | 0 | 276 | 890 | 6667 | 6700 |
| No. of QSAR parameters per drug | 5 | 6 | 14 | 23 | 23 |
| No. of drugs with drug-target binding constant data | 0 | 0 | 0 | 791 | 1563 |
| No. of drugs with experimental NMR spectra | 0 | 0 | 0 | 306 | 922 |
| No. of drugs with experimental MS spectra | 0 | 0 | 0 | 384 | 2521 |
| No. of drugs with chemical synthesis information | 0 | 38 | 38 | 1285 | 1584 |
| No. of approved small molecule drugs | 841 | 1344 | 1424 | 1552 | 2110 |
| No. of approved drugs with product ingredient structures | 0 | 0 | 0 | 474 | 1551 |
| No. of biotech drugs | 113 | 123 | 132 | 284 | 555 |
| No. of nutraceutical drugs | 61 | 69 | 82 | 87 | 97 |
| No. of withdrawn drugs | 0 | 57 | 68 | 78 | 209 |
| No. of illicit drugs | 0 | 188 | 189 | 190 | 202 |
| No. of experimental drugs | 2894 | 3116 | 5210 | 6009 | 4964 |
| No. of investigational drugs (Phase I, II and III trials) | 0 | 0 | 0 | 1219 | 4501 |
| No. of all drug targets (unique) | 2133 | 3037 | 4326 | 4115 | 4563 |
| No. of approved-drug enzymes/carriers (unique) | 0 | 0 | 164 | 245 | 479 |
| No. of all drug enzymes/carriers (unique) | 0 | 0 | 169 | 253 | 497 |
| No. of external database links | 12 | 18 | 31 | 33 | 35 |
| Total drug product pill images* | 0 | 0 | 0 | 0 | 3600 |
| Number of linked drug indications* | 0 | 0 | 0 | 0 | 3024 |
| Number of clinical trials* | 0 | 0 | 0 | 0 | 245 356 |
| Category | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 |
|---|---|---|---|---|---|
| No. of data fields per DrugCard | 88 | 108 | 148 | 208 | 215 |
| No. of search types | 8 | 12 | 16 | 18 | 20 |
| No. of illustrated drug-action pathways | 0 | 0 | 168 | 232 | 319 |
| No. of illustrated drug metabolism pathways | 0 | 0 | 0 | 53 | 64 |
| No. of drugs with metabolizing enzyme data | 0 | 0 | 762 | 1037 | 3859 |
| No. of drug metabolites with structures | 0 | 0 | 0 | 1239 | 1360 |
| No. of drug-metabolism reactions | 0 | 0 | 0 | 1308 | 1530 |
| No. of drugs with drug transporter data | 0 | 0 | 516 | 623 | 1954 |
| No. of drugs with taxonomic classification information | 0 | 0 | 0 | 6713 | 7387 |
| No. of Inferred SNP-associated drug effects* | 0 | 0 | 0 | 0 | 5993 |
| No. of directly studied SNP-associated drug effects | 0 | 0 | 113 | 201 | 324 |
| No. of drugs with patent/pricing/manufacturer data | 0 | 0 | 1208 | 1450 | 1820 |
| No. of food–drug interactions | 0 | 714 | 1039 | 1180 | 1195 |
| No. of drug–drug interactions | 0 | 13 242 | 13 795 | 14 150 | 365 984 |
| No. of ADMET parameters (Caco-2, LogS) | 0 | 276 | 890 | 6667 | 6700 |
| No. of QSAR parameters per drug | 5 | 6 | 14 | 23 | 23 |
| No. of drugs with drug-target binding constant data | 0 | 0 | 0 | 791 | 1563 |
| No. of drugs with experimental NMR spectra | 0 | 0 | 0 | 306 | 922 |
| No. of drugs with experimental MS spectra | 0 | 0 | 0 | 384 | 2521 |
| No. of drugs with chemical synthesis information | 0 | 38 | 38 | 1285 | 1584 |
| No. of approved small molecule drugs | 841 | 1344 | 1424 | 1552 | 2110 |
| No. of approved drugs with product ingredient structures | 0 | 0 | 0 | 474 | 1551 |
| No. of biotech drugs | 113 | 123 | 132 | 284 | 555 |
| No. of nutraceutical drugs | 61 | 69 | 82 | 87 | 97 |
| No. of withdrawn drugs | 0 | 57 | 68 | 78 | 209 |
| No. of illicit drugs | 0 | 188 | 189 | 190 | 202 |
| No. of experimental drugs | 2894 | 3116 | 5210 | 6009 | 4964 |
| No. of investigational drugs (Phase I, II and III trials) | 0 | 0 | 0 | 1219 | 4501 |
| No. of all drug targets (unique) | 2133 | 3037 | 4326 | 4115 | 4563 |
| No. of approved-drug enzymes/carriers (unique) | 0 | 0 | 164 | 245 | 479 |
| No. of all drug enzymes/carriers (unique) | 0 | 0 | 169 | 253 | 497 |
| No. of external database links | 12 | 18 | 31 | 33 | 35 |
| Total drug product pill images* | 0 | 0 | 0 | 0 | 3600 |
| Number of linked drug indications* | 0 | 0 | 0 | 0 | 3024 |
| Number of clinical trials* | 0 | 0 | 0 | 0 | 245 356 |
* new data
Historically, DrugBank has been noted for its extensive and very comprehensive data on drug targets. This continues to be a major focus for the DrugBank team and the number of drug targets (including proteins, RNA, DNA and other macromolecules), has increased from 4115 to 4563 unique molecules. Much of this increase was due to the inclusion of detailed target data for nearly 300 antibiotics. This enriched antibiotic dataset contains information from hundreds of target organisms and their corresponding molecular target data. An even more significant expansion has been seen in the number of known drug metabolizing enzymes and drug transporters, which nearly doubled from 253 (in DrugBank 4.0) to 497 different proteins (in DrugBank 5.0). While drug-target information is important for many pharmaceutical research applications, knowing how strongly certain drugs bind to their targets is often even more important. For this year’s release, the number of compounds with drug-target binding constant data has grown from 791 to 2242. Much of this binding constant data, along with most of the data on drug targets added to this year’s release of DrugBank was acquired from the primary literature. Using primary sources and employing expert biocurators is one of the reasons that DrugBank’s data content has become so unique and so reliable. Over the past 12 years, 27 572 different peer-reviewed papers have been collected and assessed. Those sources meeting the acceptance criteria had their data manually extracted, validated and entered by the DrugBank curation team. DrugBank is also well regarded for its ongoing efforts to compile comprehensive, detailed information on experimental and investigational drugs as well as their protein targets. This kind of information has been used by many researchers to explore new drug leads or to repurpose existing drugs. For DrugBank 5.0 the number of phase I/II/III investigational drugs has grown from 1219 to 4501 compounds. With many compounds moving from the ‘experimental’ category (i.e. drugs that are at the preclinical or animal testing stage) to the ‘investigational’ category (i.e. drugs that are in human clinical trials), the number of experimental drugs in DrugBank 5.0 actually dropped from 6009 to 4964.
With DrugBank 5.0, the quantity and quality of richly illustrated drug action and drug metabolism pathways has continued to grow. In particular, the number of drug action pathways increased from 232 to 312 and the number of drug metabolism pathways expanded from 53 to 64. All of these pathways are illustrated using PathWhiz (10), a JavaScript web-based image rendering and viewing system. PathWhiz allows users to interactively view, zoom and click on colourful pathway images that are richly annotated, heavily hyperlinked and carefully rendered. DrugBank's curation team is also continuing with its increased efforts to capture much more drug action, drug metabolism and drug transport data. This information is key to understanding drug pharmacokinetics, drug bioavailability and drug ADMET characteristics (absorption, distribution, metabolism, excretion and toxicity). For DrugBank 5.0 the number of drugs with metabolizing enzyme data has grown from 1037 (in DrugBank 4.0) to 3859 compounds (nearly a 400% increase) while the number of drugs with drug transporter data has increased from 623 to 1954 molecules (a >300% change).
Perhaps the most significant changes or enhancements of existing data for DrugBank 5.0 have been with DrugBank’s drug–drug interaction data. Drug–drug interaction information is vitally important for patients, physicians and pharmacists, especially given the aging population and the fact that elderly patients in North America consume between 8 and 13 different drugs each year, with each patient reporting 2–3 drug related problems (11). To more fully address this issue, the number of drug-drug interactions in DrugBank 5.0 has grown from 14 150 to 365 984 (a 26-fold increase). This expanded drug-drug interaction dataset has been manually sourced from FDA/Health Canada drug labels as well as primary literature. Drug and target/enzyme categories have also been used to generate drug–drug interactions, especially when a drug label explicitly lists a category (for example, ‘Drug X should not be used with strong CYP3A4 inhibitors’ will create interactions in both directions between drug X and all drugs in DrugBank marked as strong CYP3A4 inhibitors). As a result, the DrugBank curation team was able to capture interactions with drugs that are not yet approved (such as experimental or investigational drugs). Each category contains ‘exclusions’ if they are specifically mentioned in a label. Likewise direct drug-to-drug interactions that are particularly severe will take precedence over any interactions that were generated from a category-derived annotation.
Other datasets that have seen significant expansion in DrugBank 5.0 include the number of compounds with chemical synthesis information (which grew by 25%), the number of compounds with experimentally collected NMR spectra (which increased by 90%), the number of compounds with experimentally collected MS spectra (which expanded by 700%), and the number of drugs with patent, pricing and manufacturer data (which increased by nearly 30%). Likewise, with the finalization of the ClassyFire chemical classification system (9) and its chemical ontology (ChemOnt) in 2016, all 8099 drug structures within DrugBank 5.0 have been reclassified and updated to comply with the latest ClassyFire/ChemOnt release.
Another very significant effort was undertaken to expand and enrich DrugBank’s pharmacogenomics data. With the rapid growth in pharmaceutically related gene testing and the increased use of pharmacogenomic labeling, the DrugBank team decided that much more pharmacogenomic information needed to be captured and displayed in DrugBank 5.0. This effort involved adding many more SNP-associated drug effects, capturing more detailed genotypic information, building out a number of inferred SNP-drug effects and expanding the content and capability of the GenoBrowse tool for extracting/viewing pharmacogenomics data. Much of the new data were assembled from the primary literature (peer-reviewed papers, data sources such as CPIC.org and FDA labels), as well as material collated from sources such as The Human Cytochrome P450 (CYP) Allele Nomenclature Database (12) and the NHGRI-EBI GWAS Catalog (13). Altogether this work led to the number of SNP-associated drug effects in DrugBank 5.0, growing from just 201 (in DrugBank 4.0) to 5993 – a 30-fold increase.
Data quality and data currency continue to be two of the top priorities for the DrugBank curation team. Because new drugs are constantly being approved, and old drugs are perpetually being re-characterized, re-purposed or removed, the DrugBank curation team continuously combs the literature to update existing DrugCards or to add new DrugCards. As a result, hundreds of drug descriptions, drug targets, drug categories, indications, pharmacodynamics data, mechanisms of action and metabolism data fields have been added, re-written and/or expanded over the past three years.
New data fields and data content
Both the positive and negative effects of drugs are a result of altering the function of not only the intended drug target(s) but also the unintended targets. These on- and off-target actions also lead to a cascade of events that can substantially change the expression of downstream genes, proteins and metabolites, leading to potentially even more significant consequences. Traditionally, DrugBank has focused only on capturing information about the direct (intended and unintended) targets of drug action. Capturing the downstream effects of drug action was initially viewed as being too sparse to provide meaningful information. However, this has now changed with the emergence of several new fields in pharmaceutical science: pharmacometabolomics, pharmacoproteomics and pharmacotranscriptomics. Pharmacometabolomics involves the analysis of how metabolite levels are changed in tissues, cells or biofluids as a consequence of drug dosing (14). Similarly, the analysis of how protein and gene expression is changed upon drug dosing is called pharmacoproteomics and pharmacotranscriptomics (15,16). The data being captured through these pharmaco-omic studies represents new and important information that could go a long way toward the understanding of both drug action and adverse drug reactions.
For DrugBank 5.0, the curation team has compiled a significant quantity of ‘qualitative’ pharmacometabolomic, pharmacoproteomic and pharmacotranscriptomic information from the primary literature. Manual literature searches for pharmacometabolomic data were guided by PolySearch2 (17), a text mining tool developed for DrugBank and other large database annotation projects. Additional pharmacoproteomic and pharmacotranscriptomic data were obtained from the CTD (18). Pharmacoproteomic binding data were subsequently manually verified and expanded by the DrugBank curation team. The inclusion of the pharmacometabolomic, pharmacoproteomic and pharmacotranscriptomic data led to the addition of three new data fields for each drug in DrugBank 5.0 and up to 140 drug-metabolite interactions, 70 drug–protein interactions and 10 000 drug–transcript interactions for any given drug. Each interaction identifies whether the drug increases or decreases the expression of the metabolite, protein or gene and each entry is linked to one or more literature references (via a PMID hyperlink). Each pharmaco-omic entry also captures information on the tissue or biofluid where the measurements were performed (if available). In total, 729 drugs in DrugBank 5.0 have pharmacometabolomic data, 319 drugs have pharmacoproteomic data and 825 drugs have pharmacotranscriptomic data. The total number of drug–metabolite interactions captured in DrugBank 5.0 is 3093, while the total number of drug–protein and drug–transcript interactions is 1302 and 127 383, respectively. As these datasets are quite new and as these fields continue to evolve, it is expected that the amount and type of information captured in these pharmaco-omic data fields will change. Hopefully, as most pharmaco-omic studies steadily become more quantitative, much more quantitative pharmaco-omic data will become available.
In addition to this brand new pharmaco-omic data, DrugBank 5.0 also features a great deal of new data on clinical and pre-clinical drug trials. Many existing drugs are undergoing drug repurposing trials and in many cases the original purpose of a drug can evolve substantially over time. For instance, drugs such as Rogaine (Minoxidil) and Viagra (Sildenafil) were originally developed as (unsuccessful) anti-hypertensive medications but they have been repurposed to become very successful treatments for hair loss (Minoxidil) and erectile dysfunction (Sildenafil). A significant number of existing drugs are assigned new indications by the FDA through drug repurposing trials each year. In addition to these drug re-purposing trials, hundreds of phase I (safety), phase II (pilot efficacy), and phase III (efficacy) trials are conducted each year on new and emerging drugs. Likewise, more and more phase IV (long term risk) trials are now being done on many previously-approved drugs. The drug trial information captured in DrugBank 5.0 now includes the drug, the trial phase, the status, the type of trial, the trial title and the indication/disease for which the trial is being conducted. In total, 3080 drugs in DrugBank 5.0 have phase I trial data, 3450 drugs have phase II trial data, 2523 drugs have phase III data, and 1741 drugs have phase IV data. This information was captured through primary reference searches, text mining and through a variety of online databases including ClinicalTrials.gov.
In addition to the usual quantitative, textual data found throughout DrugBank, this year’s release also includes pictures for the first time. In particular coloured, zoomable/clickable ‘pill pictures’ have been integrated into DrugBank 5.0. Requests for pill images have been increasing over the years. This appears to be due to the growing role that DrugBank is playing in pharmacist/physician/patient education as well as its frequent use as reference resource for pharmacists. The pill pictures include close-up shots of the medication (to show branding labels) as well as information on the size, dose, shape and color of the pills. DrugBank 5.0 contains pill or medication images for a total of 533 drugs (38% of all FDA-approved active ingredients). Each image may be clicked to zoom in and view a larger image of the pill as well as specific product details. All of the pill images were obtained from the RxImage API provided by the US NLM. A screenshot montage of a number of pill images from DrugBank 5.0 is shown in Figure 1.
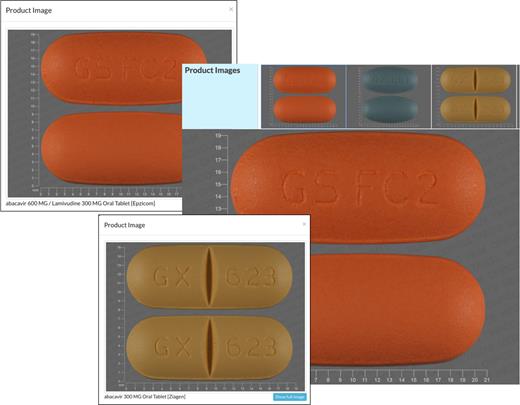
A screenshot montage of DrugBank's new pill images.
Over the past 5 years DrugBank has been steadily adding to its collection of experimentally measured NMR, ESI-MS/MS and GC-MS spectra—for both approved and experimental drugs. DrugBank 5.0 now contains 1861 experimentally measured NMR spectra (for 922 drugs), 16 449 ESI-MS/MS spectra (for 1662 drugs) and 1444 GC-MS spectra (for 859 drugs and their TMS-derivatives). These reference spectra have proven to be very popular within the medicinal chemistry, analytical chemistry and pharmacokinetics communities. However, in many cases it is difficult or impossible to acquire NMR, MS/MS or GC-MS spectra of many compounds due to the cost, stability, solubility, illicit nature or status of the drug. Over the past three years our group has been actively developing software to accurately predict the MS/MS and GC-MS spectra of chemicals based on their known structure. In particular, CFM-ID (19) and CFM-ID 2.0 (20) are two tools we developed that are able to accurately predict ESI-MS/MS spectra (under multiple collision energies) and GC-MS spectra. In particular, CFM-ID is able to predict ESI-MS/MS spectra with a weighted precision of 0.49 and a weighted recall of 0.61 when compared to experimental ESI-MS/MS spectra (19). CFM-ID 2.0 is able to predict GC-MS spectra with a weighted precision of 0.88, a weighted recall of 0.76 and a Stein dot-product score of 0.53 compared to experimental GC-MS spectra (20). For this year’s release, all drug compounds in DrugBank 5.0 have had their ESI-MS/MS spectra predicted (at collision energies of 10, 20 and 40 eV) with CFM-ID and their GC-MS spectra predicted (of appropriate TMS-derivatized compounds) with CFM-ID 2.0. All of the predicted spectra are labeled as ‘predicted’ and they are intended to serve as guides to help with the identification or characterization of known drug compounds. In total there are 60 155 predicted ESI-MS/MS spectra (for 10 008 drugs) and 5241 predicted GC/MS spectra (for 5241 drugs).
Similar challenges with collecting experimental spectral data on drugs also persist with collecting data on drug metabolites. While the DrugBank curation team has spent a great deal of time and effort over the past five years acquiring drug metabolite and drug metabolism data, the quantity of experimental information has proven to be quite limited. This is because drug metabolite information is often treated as proprietary information by many drug companies. Given the limited pool of experimentally validated data, in silico prediction of drug metabolism has become increasingly popular. It has also become much more robust and sophisticated (21). There are now several well-regarded commercial programs that are able to accurately predict drug metabolites from query drug or xenobiotic structures. Well known examples include Meteor Nexus, MetaboExpert, JChem Metabolizer and MetaDrug. However, a number of these tools place restrictions on the use or distribution of the predicted compounds, most do not predict microbial metabolism and many substantially over-predict the number of metabolites. Recently, our team developed a free, open-access, in silico metabolism prediction tool called BioTransformer (22) to specifically address the limitations of these commercial programs. In particular, BioTransformer predicts phase I, phase II, microbial and promiscuous enzyme reactions as well as appropriate combinations of each. Extensive testing indicates that BioTransformer outperforms existing commercial in silico metabolism programs in terms of precision, recall and overall accuracy. In particular, when tested on >100 test molecules, BioTransformer achieved an average precision of 0.46, and an average recall of 0.66, compared to an average precision of 0.45 and an average recall of 0.55 for one of the best commercial programs (Meteor Nexus). Consequently, the DrugBank curation team has applied BioTransformer to predict potential drug metabolites for all drugs in DrugBank without any listed or experimentally observed drug metabolites. As a result, DrugBank 5.0 now contains 470 drugs with predicted drug metabolism products covering a total of 1500 compounds. All of the predicted drug metabolites are labeled as ‘predicted by BioTransformer’ and they are intended to serve as vehicles for hypothesis testing or the identification of previously unidentified drug metabolites.
New and enhanced interface features
These days the quality of an online database is determined not only by the quality of its content but also by quality of its interface. As always, the DrugBank team continues to strive to improve the database’s user interface, to enhance its search utilities and to respond to user suggestions. For DrugBank 5.0 we have introduced a number of interface improvements including: (i) mobile device compatibility; (ii) improved spectral viewing/browsing tools; (iii) improved spectral searching; (iv) enhanced advanced search; (v) new search tools for pharmaco-omic queries; (vi) better drug indications and drug categories and (vii) better resources for data exchange and interoperability.
With regard to mobile device compatibility, the DrugBank interface has been re-designed to support facile viewing of its pages and content on the smaller screens of smartphones and tablets. Given that mobile devices now account for a rapidly growing segment of the computer market and the fact that web traffic from mobile devices exceeds that from laptops and desktops, it is clear that DrugBank (and other scientific websites) must adapt to this trend. Previously much of the content in DrugBank (especially with the DrugCards) could not be easily viewed on devices with smaller screens. The switch to mobile device compatibility is based on modern HTML and CSS technologies and frameworks, and has been tested on a number of mobile device platforms including iOS and Android. Now essentially all pages in DrugBank are formatted for viewing and searching with mobile devices.
The significant additions of both predicted and experimentally collected spectral data to DrugBank have also precipitated a need to improve DrugBank’s spectral viewing tools. Previously, static PDF images or relatively crude ‘stick’ spectrum renderings were the norm for most of DrugBank’s spectral data. With DrugBank 5.0 all MS and NMR spectra are now viewable with JSpectraViewer, a locally developed JavaScript spectral viewing tool that is able to read and render nmrML and mzML formatted spectra. For NMR spectra, JSpectraViewer allows users to interactively hover over spectral peaks and to see 1H spectral assignments and the corresponding protons on a rendered image of the molecule. For MS spectra, JSpectraViewer allows users to interactively hover over spectral peaks and to see the structures of the fragment ions that are predicted (via CFM-ID) to correspond to the observed m/z value. Screenshots of DrugBank’s new spectral viewing tools are shown in Figure 2.
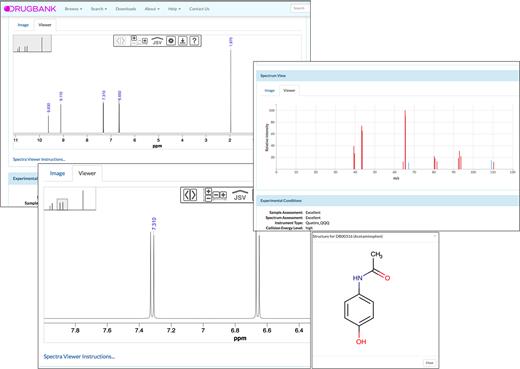
A screenshot montage of DrugBank's new spectral viewing features showing experimentally acquired NMR and ESI-MS/MS spectra for acetaminophen.
The improvements to DrugBank’s spectral viewing tools have also led to a number of improvements to DrugBank's spectral searching tools. In particular, DrugBank’s MS spectral search and MS spectral results page has been redesigned to better reflect the needs and expectations of mass spectroscopists. In particular, the MS Search query page has been modified in terms of ‘Adduct Type’ field and the results page has been modified to display ‘Formula’, ‘Monoisotopic Mass’, ‘Delta(ppm)’ and ‘m/z calculator’. Furthermore, all MS spectra in DrugBank are now mapped to a SPLASH key (6). This spectral hash identifier allows MS spectra within DrugBank to be easily searched or identified via the web through a standard Google (or other search engine) query.
With the continued additions of new data fields and growing requests for new kinds of queries from its users, the DrugBank team has also been actively improving and correcting DrugBank's Advanced Search. Users can now extract data from Drugs (active ingredients) and Drug Targets (associated proteins including known targets, metabolizing enzymes, and transporters/carriers) using all available fields and place them into an HTML table or into CSV format. The Advanced Search now allows users to build structured queries containing predicates such as ‘matches/does not match’, ‘starts/ends with’, ‘greater/less than’, and ‘is/is not present’. Queries can be set to match only if all fields match, or if any fields match, the search criteria.
The addition of large amounts of brand new pharmaco-omic data is required for the development of a new query tool for performing pharmaco-omic searches. DrugBank’s new Pharmco-omic Search allows users to easily construct queries such as ‘Find all metabolites that are increased with aspirin intake’ or ‘Find which drugs that decrease the levels of insulin’ or ‘Find all genes that are decreased with antihypertensive medications’ or ‘Find all metabolite and proteins that are increased with antifungal medications’ or ‘Find all drugs that lower plasma levels of histidine’. By allowing users to search for classes of drugs and to query multiple types of omics data at the same time, it is hoped that DrugBank 5.0’s pharmaco-omic data may be more fully exploited and mined by the community.
DrugBank's original data models along with its original classification scheme for drug indications and drug categories have been starting to show their age. In particular, these schemas made certain types of searches difficult or confusing for many users. Consequently, with the new release, and thanks to user feedback, our team has actively worked to update these components. The new drug categorization data model integrates various classifications and ontologies to produce a much more useful set of categories for each drug. The WHO/ATC classification system and the MeSH classifications for drugs can now be browsed and searched, while the FDA Established Pharmacological Class system has also be integrated into DrugBank. Drug indications have also been updated, providing a list of linked indications on each drug card (above the original text indication). These drug indications have been manually curated from drug labels and have undergone a thorough expert review process. Although the new indications are only available for currently marketed drug products, they will be extended to all approved drug products in the future.
Because much of the data in DrugBank are downloadable and much of its data are being used to create or supplement other drug resources, the need for improved data exchange standards and improved interoperability within DrugBank has become much greater. In response to these needs, all of DrugBank’s chemical structures are now accessible in canonical SMILES, SDF, MOL, PDB, InChI and InChIKey formats. Furthermore, all experimentally observed and theoretically predicted MS/MS, GC-MS and NMR spectra are stored and downloadable in mzML (7) and nmrML (8) formats and all MS/MS and GC-MS spectra are assigned SPLASH keys (6) for rapid spectral querying and matching. Likewise, all sequence (DNA and protein) data are stored in FASTA format and all remaining textual data are stored in XML and JSON format. All of these DrugBank ‘component’ files can be freely downloaded (for academics) from the DrugBank 5.0 website.
A new model of data updating and distribution
The size, scope and level of use of DrugBank has grown enormously since it was first released in 2006. In many respects, it has grown far beyond the capabilities of a single, well-meaning academic lab. With more than 12 million hits/year and an average of 30 user queries a week, the maintenance and upkeep of DrugBank has become very taxing. Numerous requests from large corporations for free datasets or free database customizations were drawing valuable resources from the Wishart lab’s normal academic research activities. Furthermore, the free-to-use, open access model for all of DrugBank's data was leading to a number of situations where these data were being used for substantial financial gain by other parties from other countries. Based on recommendations from our funders and our university, we have created a new model that should allow DrugBank to be much more sustainable, more responsive to user needs and much more up-to-date. In particular, DrugBank will be moving forward through a public-private partnership with a university spin-off company (OMx Personal Health Analytics Inc.) and the University of Alberta.
As part of this arrangement the DrugBank website will continue to be freely accessible to all. Furthermore, all of the DrugBank data will be freely downloadable for academics, pharmacists, physicians, educators or other individuals who have no intent to use the data internally for commercial product development or to re-sell or incorporate the data into a commercial product. Additionally, two datasets containing the DrugBank identifiers have been released under an unrestricted license (Creative Commons CC0) to permit linking to DrugBank in an unrestricted manner. New CC0 datasets will continue to be released in the future.
Furthermore, updates, improvements and enhancements to DrugBank, its content and its interface will continue to be done at increasingly regular intervals and with shorter response times. Indeed, most of the DrugBank curation team (which consists of trained pharmacists and individuals with graduate degreees in pharmacology or biochemistry) is now managed and employed by OMx Inc. OMx has implemented a rigorous curator training and monitoring program that involves a week of supervised curation, followed by a familiarization period with the DrugBank system, followed by additional training regarding review protocols, and continuous training on quality standards and quality controls. All information entered by curators undergoes a secondary review by the curation team lead or other senior curators. Likewise, all changes are tracked in OMx's database annotation system, and tagged to specific curators so that feedback can be provided in the case of error reports. Regular audits are also applied to various aspects of the database, and an automated system to flag various issues is under development.
Beginning in 2016, all commercial queries (from companies or corporations) along with specific database and data customization requests requiring substantial programming efforts have been handled through OMx Inc. In addition, an enhanced version of DrugBank (DrugBank-Plus) is now being developed and maintained by OMx Inc. to handle the many commercial requests and customization needs of large pharma and big data companies. A portion of the revenues from the sales of DrugBank-Plus are being used to support and sustain the DrugBank research activities at the University of Alberta. This arrangement allows the staff and students in the Wishart lab to focus on grant-driven research and for OMx Inc. to address the specific needs of the pharma and big data industries. Overall, we believe this partnership will allow the vast majority (>99.9%) of users of DrugBank to continue to access and use DrugBank as they always have. It will also ensure the sustainability of this resource and to help keep it useful, up-to-date, informative and cutting-edge for everyone for many years to come.
CONCLUSION
Over the past 12 years DrugBank has grown from a small, somewhat specialized drug database to a large, comprehensive drug data resource covering almost all aspects of drug function, formulation, mechanism and metabolism. Throughout this evolution, the DrugBank team has strived to maintain the highest quality and currency of data, to continuously improve the quality of its user interface and to attentively listen to all members of its user community. With this latest release of DrugBank we have undertaken the most significant and large-scale update in DrugBank’s history. Many existing datasets have grown in size by factors of 3- to 30-fold. In addition to this significant enhancement to DrugBank’s existing data collection, we have also added substantial quantities of new and, what we believe will be, highly relevant data. This includes new pharmaco-omic data covering the influence of drugs on metabolite levels, gene expression levels and protein expression levels. New data have also been added on hundreds of investigational drug clinical trials and various drug repurposing trials along with thousands of up-to-date drug images of approved drugs. New data have also been added on (predicted) drug metabolites as well as (predicted) drug MS and NMR spectra. These additions and enhancements are intended to facilitate research in pharmacogenomics, pharmacoproteomics, pharmacotranscriptomics, pharmacometabolomics, pharmacokinetics, pharmacodynamics, pharmaceutics and drug design/discovery. They are also intended to make the data within DrugBank relevant to a far wider audience including chemists, pharmacists, physicians, educators and the general public. With our new public–private partnership involving OMx, Inc. we are confident that DrugBank will continue to be relevant, referential and responsive for the foreseeable future.
ACKNOWLEDGEMENTS
The authors would like to thank ChemAxon, Inc. for their continued support as well as the many users of DrugBank for their valuable feedback and suggestions.
FUNDING
Genome Alberta (a division of Genome Canada); Canadian Institutes of Health Research; Western Economic Diversification; Alberta Innovates Health Solutions. Funding for open access charge: Genome Canada.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments