





Abstract
The PRoteomics IDEntifications (PRIDE, http://www.ebi.ac.uk/pride) database at the European Bioinformatics Institute is one of the most prominent data repositories of mass spectrometry (MS)-based proteomics data. Here, we summarize recent developments in the PRIDE database and related tools. First, we provide up-to-date statistics in data content, splitting the figures by groups of organisms and species, including peptide and protein identifications, and post-translational modifications. We then describe the tools that are part of the PRIDE submission pipeline, especially the recently developed PRIDE Converter 2 (new submission tool) and PRIDE Inspector (visualization and analysis tool). We also give an update about the integration of PRIDE with other MS proteomics resources in the context of the ProteomeXchange consortium. Finally, we briefly review the quality control efforts that are ongoing at present and outline our future plans.
INTRODUCTION
Mass spectrometry (MS)-based proteomics approaches are widely used in the life sciences. There are three main workflows, with bottom-up proteomics the most extensively used technique (also known as shot-gun proteomics) (1). In this experimental set up, the proteins to be analysed are enzymatically digested by a protease (most often trypsin) into potentially highly complex peptide mixtures, which are then subjected to fractionation by multidimensional liquid chromatography steps before they are measured in the mass spectrometer. Other main approaches are top-down, where intact proteins are measured (2), and targeted proteomics (such as Selected Reaction Monitoring, SRM), where the researcher tries to detect specific proteins in a given sample (3).
The PRoteomics IDEntifications (PRIDE, http://www.ebi.ac.uk/pride) database was originally set up in 2004 (4–8) to enable public data deposition in the MS proteomics field, and to support the experimental data described in publications during the manuscript review process. The main data types stored in PRIDE are protein and peptide identifications (IDs) and quantitative values (including post-translational modifications, PTMs), the analysed mass spectra and the related technical and biological metadata. PRIDE supports bottom-up proteomics approaches, mainly tandem MS (MS/MS) data, but also Peptide Mass Fingerprinting datasets, and presents the data as originally analysed by the researchers, with several popular search engines/analysis workflows fully supported. Unlike other MS proteomics resources, such as PeptideAtlas (9) and the Global Proteome Machine Database (GPMDB) (10), no reprocessing of the data is performed because PRIDE aims to reflect the author’s analysis view on the experimental data. In fact, PRIDE remains as the unique generic resource of this kind since National Center for Biotechnology Information (NCBI) the repository Peptidome (11), its sibling resource in the USA, was discontinued in April 2011. Other MS data repositories, such as MaxQB (12), are more specialized [for an extensive review, see (13)] or are restricted to one particular analysis workflow.
For SRM data, the new PeptideAtlaS SRM Experiment Library (PASSEL) (14) is the main available resource. At present, there is no widely used resource devoted to top-down proteomics approaches. In addition to the ‘pure’ MS proteomics resources, there are other databases that can present an extra layer of information on top of the MS experiments without storing the underlying mass spectra. Some recently developed databases of this kind are the Model Organism Protein Expression Database (MOPED) (15), PaxDB (16) (both of them focused on protein expression information) and neXtProt (17).
Several services have been developed by the PRIDE team, which are heavily used by external users but also by PRIDE itself, especially the ‘Protein Identifier Cross-Reference’ (PICR) service (a protein identifier mapping resource) (18) and the ‘Ontology Lookup Service’ (OLS) (to query, browse and navigate biomedical ontologies) (19). In addition, ‘Database on Demand’ is a service to generate tailored databases for performing proteomics searches (20). As a key point, to improve and make the data submission process easier, several tools have also been made available to the proteomics community, such as the popular PRIDE Converter (21), PRIDE Inspector (22) and the new PRIDE Converter 2 (23). It is important to highlight that all the softwares, including the PRIDE core and web modules (http://code.google.com/p/ebi-pride/), are developed in Java and are open source. PRIDE is a recommended submission site of key journals such as Proteomics, Molecular and Cellular Proteomics and Nature Biotechnology. Currently, scientific journals and funding agencies alike are increasingly mandating public deposition of MS data to support the publication of related proteomics manuscripts.
In this manuscript, we summarize developments in the PRIDE database and associated tools since the previous Nucleic Acids Research (NAR) database update (8). We will also outline the PRIDE data deposition process, introduce the ProteomeXchange (PX) consortium and quality control (QC) efforts and highlight future developments.
DATA CONTENT IN PRIDE AND HOW TO ACCESS IT
There has been a substantial increase in the amount of stored data in PRIDE during the past years. By September 2012, PRIDE contains 25 853 MS-based proteomics experiments (compared with 9908 when the last NAR manuscript was submitted, in September 2009), around 11.1 million identified proteins (2.5 million in September 2009), 61.9 million identified peptides (11.5 million in September 2009) and 324 million spectra (50.3 million in September 2009). Note that these data holdings are absolute figures, not distinguishing public and pre-publication data. At the moment of writing, 66.7% (17 219) of the experiments were publicly available.
The complete set of data in PRIDE comprises 323 taxonomy identifiers (compared with 60, in September 2009), including human and many model organisms (Table 1). In comparison with figures from 3 years ago, animal species still provide the majority of the data. A total of 89 animal species are represented, contributing 62.2% and 61.9% of all protein and peptide IDs in PRIDE, respectively. Human continues to be the most represented species (28.1% and 37.2% of protein and peptide IDs, respectively). As a matter of fact, human and mouse alone account for almost as many IDs as the other species together: 44.2 % and 49.4 % of the protein and peptide IDs, respectively.
Data content in PRIDE split by taxonomic divisions
| Group of organisms (number of species) | % Protein IDs | % Peptide IDs |
|---|---|---|
| Animals (89) | 62.2 | 61.9 |
| Plants (46) | 19.3 | 14.8 |
| Fungi (22) | 2.7 | 3.0 |
| Bacteria (122) | 12.7 | 17.4 |
| Others (44) | 3.1 | 2.9 |
| Species | ||
| Homo sapiens | 28.1 | 37.2 |
| Mus musculus | 16.1 | 12.2 |
| Zea mays | 9.5 | 2.2 |
| Arabidopsis thaliana | 6.8 | 10.1 |
| B. subtilis | 5.5 | 4.1 |
| Sus scrofa | 5.5 | 1.7 |
| Rattus norvegicus | 3.7 | 2.8 |
| Drosophila melanogaster | 3.3 | 2.3 |
| Danio rerio | 1.7 | 0.9 |
| Puniceispirillum marinum | 1.5 | 1.5 |
| E. coli | 1.2 | 1.7 |
| S. cerevisiae | 1.2 | 1.8 |
| Group of organisms (number of species) | % Protein IDs | % Peptide IDs |
|---|---|---|
| Animals (89) | 62.2 | 61.9 |
| Plants (46) | 19.3 | 14.8 |
| Fungi (22) | 2.7 | 3.0 |
| Bacteria (122) | 12.7 | 17.4 |
| Others (44) | 3.1 | 2.9 |
| Species | ||
| Homo sapiens | 28.1 | 37.2 |
| Mus musculus | 16.1 | 12.2 |
| Zea mays | 9.5 | 2.2 |
| Arabidopsis thaliana | 6.8 | 10.1 |
| B. subtilis | 5.5 | 4.1 |
| Sus scrofa | 5.5 | 1.7 |
| Rattus norvegicus | 3.7 | 2.8 |
| Drosophila melanogaster | 3.3 | 2.3 |
| Danio rerio | 1.7 | 0.9 |
| Puniceispirillum marinum | 1.5 | 1.5 |
| E. coli | 1.2 | 1.7 |
| S. cerevisiae | 1.2 | 1.8 |
Only the top 12 species in terms of protein and peptide identifications are shown.
Data content in PRIDE split by taxonomic divisions
| Group of organisms (number of species) | % Protein IDs | % Peptide IDs |
|---|---|---|
| Animals (89) | 62.2 | 61.9 |
| Plants (46) | 19.3 | 14.8 |
| Fungi (22) | 2.7 | 3.0 |
| Bacteria (122) | 12.7 | 17.4 |
| Others (44) | 3.1 | 2.9 |
| Species | ||
| Homo sapiens | 28.1 | 37.2 |
| Mus musculus | 16.1 | 12.2 |
| Zea mays | 9.5 | 2.2 |
| Arabidopsis thaliana | 6.8 | 10.1 |
| B. subtilis | 5.5 | 4.1 |
| Sus scrofa | 5.5 | 1.7 |
| Rattus norvegicus | 3.7 | 2.8 |
| Drosophila melanogaster | 3.3 | 2.3 |
| Danio rerio | 1.7 | 0.9 |
| Puniceispirillum marinum | 1.5 | 1.5 |
| E. coli | 1.2 | 1.7 |
| S. cerevisiae | 1.2 | 1.8 |
| Group of organisms (number of species) | % Protein IDs | % Peptide IDs |
|---|---|---|
| Animals (89) | 62.2 | 61.9 |
| Plants (46) | 19.3 | 14.8 |
| Fungi (22) | 2.7 | 3.0 |
| Bacteria (122) | 12.7 | 17.4 |
| Others (44) | 3.1 | 2.9 |
| Species | ||
| Homo sapiens | 28.1 | 37.2 |
| Mus musculus | 16.1 | 12.2 |
| Zea mays | 9.5 | 2.2 |
| Arabidopsis thaliana | 6.8 | 10.1 |
| B. subtilis | 5.5 | 4.1 |
| Sus scrofa | 5.5 | 1.7 |
| Rattus norvegicus | 3.7 | 2.8 |
| Drosophila melanogaster | 3.3 | 2.3 |
| Danio rerio | 1.7 | 0.9 |
| Puniceispirillum marinum | 1.5 | 1.5 |
| E. coli | 1.2 | 1.7 |
| S. cerevisiae | 1.2 | 1.8 |
Only the top 12 species in terms of protein and peptide identifications are shown.
However, the relative proportion of other groups of organisms has increased, especially in the case of plants (46 taxonomy identifiers, 19.3% and 14.8%, respectively) and bacteria (12.7% and 17.4%, respectively). Bacteria are again by far the group of organisms with the highest number of taxonomy identifiers (122). Fungi are also represented (22 taxonomy identifiers, 2.7 and 3.0%, respectively). Apart from human, the most represented organisms in PRIDE are (in this order) mouse, maize, Arabidopsis, Bacillus subtilis, pig, rat, Drosophila, zebrafish, Puniceispirillum marinum, Escherichia coli and Saccharomyces cerevisiae (Table 1).
Table 2 includes the most abundant PTMs present in the database. Not surprisingly, the most often found PTM is oxidation (5.7 million modified sites), mainly due to the high amount of methionine oxidation, a modification that can be biologically relevant (24) but that, in most cases for MS proteomics experiments, is an artifact. Formylation is the second most abundant PTM (around 1.3 million sites), mainly owing to the data present from just one organism (maize). Phosphorylation comes in third place (around 1.1 million sites), with the highest proportion of data coming from human experiments. There is also a considerable amount of other PTMs such as dioxidation, deamidation, acetylation or dehydration, among others (Table 2).
Protein modification content in PRIDE as a whole, and split by species (human and main model organisms represented in PRIDE)
| Modification type | Total | Human | Mouse | Maize | Arabidopsis | Drosophila | E. coli | S. cerevisiae |
|---|---|---|---|---|---|---|---|---|
| Oxidation | 5 707 426 | 2 291 925 | 883 599 | 25 907 | 449 530 | 2400 | 58 921 | 97 803 |
| Deamidation | 663 884 | 333 091 | 48 120 | 30 | 6964 | 83 | 17 335 | 10 859 |
| Phosphorylation | 1 143 766 | 741 619 | 112 910 | 171 539 | 35 521 | 239 | 0 | 33 670 |
| Acetylation | 626 510 | 380 124 | 22 662 | 1419 | 10 614 | 290 | 1356 | 18 762 |
| Dioxidation | 1 123 206 | 31 788 | 1625 | 928 614 | 0 | 0 | 0 | 0 |
| Deamination | 132 145 | 87 016 | 29 374 | 462 | 0 | 262 | 272 | 0 |
| Dehydration | 202 436 | 15 492 | 32 863 | 148 686 | 0 | 30 | 0 | 0 |
| Methylthio | 110 727 | 74 177 | 0 | 0 | 2425 | 0 | 82 | 0 |
| Formylation | 1 297 285 | 5286 | 629 | 1 289 660 | 0 | 0 | 0 | 0 |
| Monomethylation | 299 246 | 261 941 | 48 | 0 | 277 | 0 | 0 | 0 |
| Modification type | Total | Human | Mouse | Maize | Arabidopsis | Drosophila | E. coli | S. cerevisiae |
|---|---|---|---|---|---|---|---|---|
| Oxidation | 5 707 426 | 2 291 925 | 883 599 | 25 907 | 449 530 | 2400 | 58 921 | 97 803 |
| Deamidation | 663 884 | 333 091 | 48 120 | 30 | 6964 | 83 | 17 335 | 10 859 |
| Phosphorylation | 1 143 766 | 741 619 | 112 910 | 171 539 | 35 521 | 239 | 0 | 33 670 |
| Acetylation | 626 510 | 380 124 | 22 662 | 1419 | 10 614 | 290 | 1356 | 18 762 |
| Dioxidation | 1 123 206 | 31 788 | 1625 | 928 614 | 0 | 0 | 0 | 0 |
| Deamination | 132 145 | 87 016 | 29 374 | 462 | 0 | 262 | 272 | 0 |
| Dehydration | 202 436 | 15 492 | 32 863 | 148 686 | 0 | 30 | 0 | 0 |
| Methylthio | 110 727 | 74 177 | 0 | 0 | 2425 | 0 | 82 | 0 |
| Formylation | 1 297 285 | 5286 | 629 | 1 289 660 | 0 | 0 | 0 | 0 |
| Monomethylation | 299 246 | 261 941 | 48 | 0 | 277 | 0 | 0 | 0 |
Protein modification content in PRIDE as a whole, and split by species (human and main model organisms represented in PRIDE)
| Modification type | Total | Human | Mouse | Maize | Arabidopsis | Drosophila | E. coli | S. cerevisiae |
|---|---|---|---|---|---|---|---|---|
| Oxidation | 5 707 426 | 2 291 925 | 883 599 | 25 907 | 449 530 | 2400 | 58 921 | 97 803 |
| Deamidation | 663 884 | 333 091 | 48 120 | 30 | 6964 | 83 | 17 335 | 10 859 |
| Phosphorylation | 1 143 766 | 741 619 | 112 910 | 171 539 | 35 521 | 239 | 0 | 33 670 |
| Acetylation | 626 510 | 380 124 | 22 662 | 1419 | 10 614 | 290 | 1356 | 18 762 |
| Dioxidation | 1 123 206 | 31 788 | 1625 | 928 614 | 0 | 0 | 0 | 0 |
| Deamination | 132 145 | 87 016 | 29 374 | 462 | 0 | 262 | 272 | 0 |
| Dehydration | 202 436 | 15 492 | 32 863 | 148 686 | 0 | 30 | 0 | 0 |
| Methylthio | 110 727 | 74 177 | 0 | 0 | 2425 | 0 | 82 | 0 |
| Formylation | 1 297 285 | 5286 | 629 | 1 289 660 | 0 | 0 | 0 | 0 |
| Monomethylation | 299 246 | 261 941 | 48 | 0 | 277 | 0 | 0 | 0 |
| Modification type | Total | Human | Mouse | Maize | Arabidopsis | Drosophila | E. coli | S. cerevisiae |
|---|---|---|---|---|---|---|---|---|
| Oxidation | 5 707 426 | 2 291 925 | 883 599 | 25 907 | 449 530 | 2400 | 58 921 | 97 803 |
| Deamidation | 663 884 | 333 091 | 48 120 | 30 | 6964 | 83 | 17 335 | 10 859 |
| Phosphorylation | 1 143 766 | 741 619 | 112 910 | 171 539 | 35 521 | 239 | 0 | 33 670 |
| Acetylation | 626 510 | 380 124 | 22 662 | 1419 | 10 614 | 290 | 1356 | 18 762 |
| Dioxidation | 1 123 206 | 31 788 | 1625 | 928 614 | 0 | 0 | 0 | 0 |
| Deamination | 132 145 | 87 016 | 29 374 | 462 | 0 | 262 | 272 | 0 |
| Dehydration | 202 436 | 15 492 | 32 863 | 148 686 | 0 | 30 | 0 | 0 |
| Methylthio | 110 727 | 74 177 | 0 | 0 | 2425 | 0 | 82 | 0 |
| Formylation | 1 297 285 | 5286 | 629 | 1 289 660 | 0 | 0 | 0 | 0 |
| Monomethylation | 299 246 | 261 941 | 48 | 0 | 277 | 0 | 0 | 0 |
This wealth of data can be accessed in different ways (Figure 1):
PRIDE web interface (http://www.ebi.ac.uk/pride). The home page was updated earlier in 2012, but no other major changes have been done to the current web interface. However, it is possible to access all experiments launching the PRIDE Inspector tool using Java Web Start (see below).
PRIDE BioMart (http://www.ebi.ac.uk/pride/prideMart.do). The BioMart interface is useful for batch data retrieval (25). In the current version of the PRIDE BioMart (running on BioMart version 0.7), data integration with Reactome (26) has been extended (27), by enabling the link between phosphorylated proteins present in Reactome pathways and phosphorylated proteins detected by MS approaches stored in PRIDE. The PRIDE BioMart data can also be accessed using a Representational State Transfer web service, which is heavily used. In addition, it is also possible to access the PRIDE BioMart at www.biomart.org, together with many others. In that case, apart from Reactome, it is possible to perform common data searches involving PRIDE and other resources such as UniProt (28), InterPro (29), Ensembl (30) and the Catalogue of Somatic Mutations in Cancer (COSMIC) (31).
PRIDE FTP file server (ftp.ebi.ac.uk/pub/databases/pride/). At present, XML files corresponding to all public experiments in PRIDE can be downloaded in the mzData and PRIDE XML formats.
PRIDE Distributed Annotation System (DAS) server (http://www.ebi.ac.uk/pride-das/das/PrideDataSource/). A PRIDE DAS server (32) was set up following the new specification 1.6. This service is publicly available and can be accessed through DAS clients such as Dasty (33). The PRIDE DAS server has been designed for visualizing protein sequence and annotation, to display the identified peptides for the protein specified in the DAS request, together with the associated PTMs, and total peptide coverage.
A public PRIDE MySQL instance is now available and used, for instance, by the PRIDE Inspector, making this tool be the ideal way to access PRIDE data for most use cases (see next section).
Among the new datasets present in PRIDE, it is important to highlight that by July 2012, all data originally stored in the discontinued NCBI Peptidome (11) had been reannotated, converted into a PRIDE compatible format and made publicly available under experiment accessions 17900-18271.
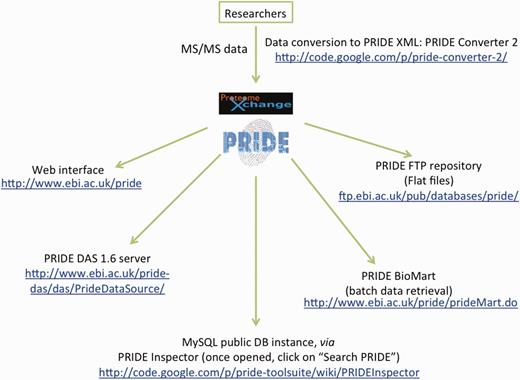
Summary of the ways the user can access and retrieve data from PRIDE. The web links to the existing PRIDE tools are also highlighted, including the PRIDE Converter 2 (needed for data submission).
PRIDE SUBMISSION PROCESS AND RELATED TOOLS
Submission tools
At the moment of writing, submissions to PRIDE are performed using a publicly available XML data format called PRIDE XML, which is derived from mzData. The first step to perform a submission to PRIDE is then to generate PRIDE XML files. Several tools are available to make that process feasible and as straightforward as possible. The new PRIDE Converter 2 (23) is now the recommended submission tool. It can convert a variety of popular proteomics data formats (e.g. Mascot.dat, X!Tandem.xml, OMSSA.csv, Proteome Discoverer.msf, plus all the mass spectral formats, among others) into well-annotated PRIDE XML files.
PRIDE Converter 2 can be used in two modes: (i) a Graphical User Interface mode, suited for most users; and (ii) a Command Line interface mode that makes possible the integration of the conversion process in external pipelines. Batch conversion of files is supported in both modes. Importantly, quantification results for the most popular techniques and 2D gel spot information can now be integrated in PRIDE XML files with PRIDE Converter 2, by providing that information in a new Proteomics Standards Initiative (PSI) tab-delimited standard format called mzTab (http://code.google.com/p/mztab/). Detailed documentation for general users and developers is available at http://code.google.com/p/pride-converter-2/. The PRIDE Converter 2 framework also includes the PRIDE mzTab Generator, PRIDE XML Merger and PRIDE XML Filter (23). A mechanism to make a combined submission to PRIDE and IntAct (34), the molecular interactions resource at the European Bioinformatics Institute, is also present in the PRIDE Converter 2.
The original PRIDE Converter tool (21), one of the main reasons behind the large increase in data contents in PRIDE, is no longer recommended as the main submission tool and will not be maintained any more. Apart from the tools provided by the PRIDE team, there are several existing third-party pipelines/tools that produce PRIDE XML files, such as ProteinLynx Global Server (Waters), hEIDI (http://biodev.extra.cea.fr/docs/heidi), OmicsHub Proteomics (Integromics), PeptideShaker (http://peptide-shaker.googlecode.com) and Proteios (35).
At the moment, we are implementing full support in PRIDE for the new PSI standard formats mzML v1.1 (for MS data) (36) and mzIdentML v1.1 (for protein and peptide IDs) (37), based on the Java libraries jmzML (38), jmzIdentML (39) and jmzReader (40). When this work is complete, data submissions in these formats will be natively supported, rather than having to be run through the PRIDE Converter 2. This will enable us to support in a much better way the reporting of protein inference (41) and ambiguity in modification position.
PRIDE Inspector
PRIDE Inspector is a popular tool introduced in 2011 (>4500 downloads by September 2012), which can be used to visualize and perform an initial quality assessment of the submitted data to PRIDE (22) (http://code.google.com/p/pride-toolsuite/wiki/PRIDEInspector). PRIDE Inspector provides different views on the available data, each focusing on a different aspect: experimental details (biological and technical metadata), protein, peptide, quantification values (if available) and summary charts. This last view is one of the major strengths of the tool because it is possible to perform an initial assessment of data quality, using a variety of simple charts that are generated automatically (22). Using PRIDE Inspector, proteomics researchers can examine their own data sets before the actual submission to PRIDE is performed, and journal editors and reviewers can perform a thorough review of submitted and private data at the pre-publication stage.
In addition, through the ‘Search PRIDE Database’ option, PRIDE Inspector can access data already in PRIDE for data mining purposes using the PRIDE public MySQL instance, which is updated regularly. Apart from being a stand-alone tool, as mentioned before, PRIDE Inspector can also be accessed using Java Web Start at the PRIDE web page.
PRIDE and the ProteomeXchange consortium
PRIDE is a founding member of the PX consortium (http://www.proteomexchange.org) (42). The members of the consortium, led by PRIDE and PeptideAtlas (9), are implementing a system to enable the automated and standardized sharing of MS-based proteomics data between the main existing MS proteomics repositories. In the first implementation of the data workflow, PRIDE acts as the initial submission point for MS/MS data. At the moment of writing, around 50 PX datasets have been submitted (see updated list of publicly available datasets at http://proteomecentral.proteomexchange.org). As a result of the PX efforts, PRIDE has started to accept raw data (in either binary or an open XML format) because it is a mandatory component of a PX submission. The files are accessible through FTP and are stored at the EBI raw file repository (43).
QUALITY CONTROL IN PRIDE
A major focus of PRIDE development in the past 2 years was to ensure at the very least minimal annotation of experiments and to perform basic quality checks of the submitted data to PRIDE. The development of PRIDE Inspector was the key step forward in that direction, and many of its components have been used in the internal PRIDE submission pipeline. The automated pipeline allows the detection of clear errors in the submitted data that are notified to the submitter and can then be corrected (44). Finally, the development of the PRIDE Converter 2 as the new submission tool has improved consistency at the level of experimental annotation.
In 2013, we will finalize and release a new resource called PRIDE-Q, as a quality-controlled subset of PRIDE, which can fulfil both a minimum level of annotation and Peptide Spectrum Match quality standards.
DISCUSSION
In the past 3 years, PRIDE has worked on two main tasks: (i) development of robust data submission pipelines (such as the development of the PRIDE Converter 2), including the initial implementation of the PX consortium data workflow, and the possibility to capture quantification information in a standardized way; and (ii) establishment of QC checks, including the development of PRIDE Inspector and an internal data submission pipeline, able to flag obvious errors that can be then communicated to the submitters.
However, many more future efforts will be needed in both directions. We are working on the development of the new PRIDE system (including a new database schema and web interface) that will fully support the PSI standards mzML and mzIdentML. This will be a gradual process, and support for these formats will be added sequentially to the PRIDE system (also as submission formats) and tools, while we will also keep supporting PRIDE XML.
On the other hand, the release of PRIDE-Q, envisioned to help non-expert proteomics biologists to ‘digest’ the potentially complex information coming from MS data, will happen in 2013. However, the quality requirements will need to be refined and will evolve dynamically over time.
It is worth highlighting that PRIDE has been used for research purposes in several recent studies involving the meta-analysis of combined data coming from very different proteomics experimental setups (45–47), the improvement and assessment of existing protein sequence databases (48,49) or to support genomics-related findings (50,51). Some research has also been performed to demonstrate the usefulness of data in PRIDE to perform a posteriori QC of the stored data (52). We expect that this trend will continue to grow in the near future, and that PRIDE continues on a trajectory from a publication-centric repository to an integrative resource for MS-based protein expression data. PRIDE will keep playing an important role for the community, also in the context of the nascent Human Proteome Project (53). We invite interested parties in PRIDE developments (including the associated software and tools) to follow the PRIDE Twitter account (@pride_ebi).
FUNDING
The PRIDE team is funded by the Wellcome Trust [WT085949MA]; EU FP7 grants ‘Sling’ [226073]; ‘ProteomeXchange’ [260558]; ‘PRIME-XS’ [262067]; ‘LipidomicNet’ [202272]; BBSRC grant ‘PRIDE Converter’ [reference BB/I024204/1] and EMBL core funding. Funding for open access charge: Wellcome Trust [WT085949MA].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The PRIDE team would like to thank all data submitters for their contributions.

{kind=link}
Comments