





Abstract
We present the Database of Disordered Protein Prediction (D2P2), available at http://d2p2.pro (including website source code). A battery of disorder predictors and their variants, VL-XT, VSL2b, PrDOS, PV2, Espritz and IUPred, were run on all protein sequences from 1765 complete proteomes (to be updated as more genomes are completed). Integrated with these results are all of the predicted (mostly structured) SCOP domains using the SUPERFAMILY predictor. These disorder/structure annotations together enable comparison of the disorder predictors with each other and examination of the overlap between disordered predictions and SCOP domains on a large scale. D2P2 will increase our understanding of the interplay between disorder and structure, the genomic distribution of disorder, and its evolutionary history. The parsed data are made available in a unified format for download as flat files or SQL tables either by genome, by predictor, or for the complete set. An interactive website provides a graphical view of each protein annotated with the SCOP domains and disordered regions from all predictors overlaid (or shown as a consensus). There are statistics and tools for browsing and comparing genomes and their disorder within the context of their position on the tree of life.
INTRODUCTION
Intrinsically disordered or unstructured proteins (IDPs) exist as highly flexible polypeptide chains in vivo behaving as an ensemble of conformational states with no stable tertiary structure (1). Regions of IDP can exist as unfolded chains or molten globules with well-developed secondary structure and often function through transition between differently folded states (2). Mechanisms for functional conformational transition include binding with other proteins, nucleic acids, various small molecules and numerous posttranslational modifications, such as phosphorylation, which has been shown to be especially important (3,4). Biological functions of known IDPs are varied and their roles include the following: instigation of protein complex formation, molecular recognition as seen in nucleoporins of the nuclear pore complex (5), signal transduction, transcriptional regulation and many other functions (6,7) too numerous to list here.
Much work has been done on producing classification and annotation of known unstructured regions from 3D experimental data found in the PDB (8), DisProt (9) and IDEAL (10) resources. However, the past focus on structured protein domains have limited the total number of described IDP regions. For example: the current release of DisProt (v6.00; 1 July 2012) describes 667 proteins containing 1467 verified disordered regions; and the IDEAL database (as of 9 May 2012) describes 209 disordered proteins in detail, 97 of which have been experimentally verified to be structured and disordered over the same region under different conditions; and also MobiDB (11) has applied a method for identifying mobile regions from nuclear magnetic resonance (NMR) structures (12) to 26 933 proteins (v1.2.1 as of 1 November 2012). Because of the biases of structural resolution and the relative ease of task for disorder prediction methods compared with de novo fold prediction, many algorithms have been developed to discover novel regions of disorder from amino acid sequence alone (13,14).
Disorder prediction has been proven effective even in novel protein space, starting from the first predictor of protein disorder (15) in 1997 followed with over a decade of comparative study since the CASP5 assessment in 2002. The Database of Disordered Protein Prediction (D2P2) database aims to provide unified and exhaustive disorder predictions for all currently sequenced genomes with protein annotations. These data will both further development of current and future predictors; in addition to providing the biological community with ready access to results from predictors not chosen just for availability and ease of use, but for their predictive efficacy and coverage of protein disorder properties.
Two major classes of protein disorder can be described; short regions, typically <∼15–20 residues often serving as flexible linkers between or within domains, and long regions of >30–50 residues. These two classes have different amino acid propensities and frequently two prediction methods or variants are required to get full coverage (16). This sort of behavior can be a barrier to the biological investigator who wishes to have quick access to results. Resources such as MetaDisorder (17) go some way to resolve this issue providing the naïve user with reliable results from a similar spectrum of predictors as D2P2. However, such meta-submission tools are of limited use if your study involves protein sequences at the scale of a whole genome or clade. The MobiDB resource has some similar goals to D2P2 including predictions from Espritz and IUPred pre-computed for 4 662 776 sequences (MobiDB v1.2.1 as of 1 November 2012) from UniProt. D2P2 provides a library of predictions (Figure 1 for example) for a set of 10 429 761 protein sequences from 1765 complete genomes (Table 1) and a growing collection of predictors. A focus of the D2P2 data is providing access to SCOP domain prediction alongside disorder to show the interplay of known structure and disorder. The Dichot system (18) provides complimentary disordered/structured data for UniProt Human sequences and the DISOPRED2 (19) disorder predictor: deemed too computationally expensive to be included with the full D2P2 dataset. Conclusions on the relation of structure and disorder made with Dichot on human sequence and those made with D2P2 are discussed later.
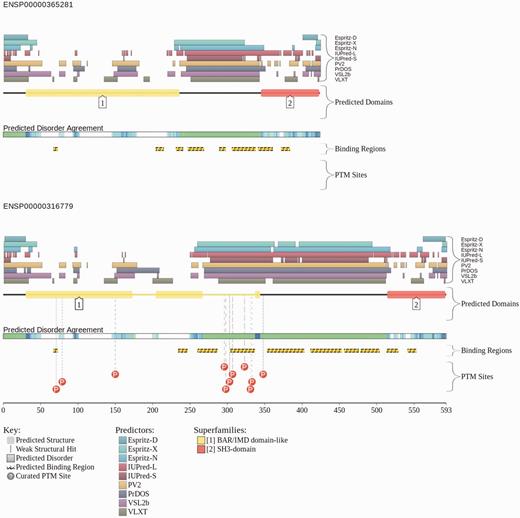
An example graphical report from the D2P2 website for two transcripts of the human gene BIN1. All disorder predictions (pastel-colored blocks) are stacked and aligned against the polypeptide chain in black. Their interplay with the predicted SCOP domains (bright-colored rounded blocks) is shown. The level of agreement between all of the disorder predictors is shown as color intensity in an aligned gradient bar below the stack of predictions. The green segments represent disorder that is not found within a predicted SCOP domain. The blue segments are where the disorder predictions intersect the SCOP domain prediction. Below the disorder agreement line, ANCHOR binding region predictions are displayed (yellow blocks with zigzag infill), along with PTM sites from PhosphoSitePlus when known (shown as lettered spheres hanging below other predictions).
The number of genomes and sequences included in the database at the time of writing
| Domain | Number of genomes | Reference species | Strains | Total sequences |
|---|---|---|---|---|
| Eukarya | 352 | 298 | 54 | 5 746 620 |
| Bacteria | 1305 | 862 | 443 | 4 216 314 |
| Archaea | 108 | 96 | 12 | 238 232 |
| Total | 1765 | 1256 | 509 | 10 429 761 |
| Domain | Number of genomes | Reference species | Strains | Total sequences |
|---|---|---|---|---|
| Eukarya | 352 | 298 | 54 | 5 746 620 |
| Bacteria | 1305 | 862 | 443 | 4 216 314 |
| Archaea | 108 | 96 | 12 | 238 232 |
| Total | 1765 | 1256 | 509 | 10 429 761 |
The intention is to expand this over time as new genomes are described.
The number of genomes and sequences included in the database at the time of writing
| Domain | Number of genomes | Reference species | Strains | Total sequences |
|---|---|---|---|---|
| Eukarya | 352 | 298 | 54 | 5 746 620 |
| Bacteria | 1305 | 862 | 443 | 4 216 314 |
| Archaea | 108 | 96 | 12 | 238 232 |
| Total | 1765 | 1256 | 509 | 10 429 761 |
| Domain | Number of genomes | Reference species | Strains | Total sequences |
|---|---|---|---|---|
| Eukarya | 352 | 298 | 54 | 5 746 620 |
| Bacteria | 1305 | 862 | 443 | 4 216 314 |
| Archaea | 108 | 96 | 12 | 238 232 |
| Total | 1765 | 1256 | 509 | 10 429 761 |
The intention is to expand this over time as new genomes are described.
Users of D2P2 will be those asking basic science questions at the scale of whole genomes or the whole tree of life, or those seeking to develop methods for prediction and wishing to know the specific behaviors of each predictor over a large library of sequence. Additionally, D2P2 data highlight the inverse of well-folded structure and could be informative for developing better approaches to fold prediction, as well as screening novel domain families in conserved protein sequence awaiting crystallographic study.
MATERIALS AND METHODS
Sequences
The sequence library of complete genomes from SUPERFAMILY 1.75 as of 8 November 2011 was used as the basis for all prediction results. This includes 1765 complete genomes from 1256 distinct species from across the whole tree of cellular life (Table 1). Currently, viral genomes are not included in D2P2 but they will be included in future updates.
Predictions
D2P2 currently includes the following: PONDR VL-XT, PONDR VSL2b, PrDOS, PV2, Espritz (all variants) and IUPred (all variants) along with ANCHOR to predict disordered regions that undergo binding transitions during protein–protein interaction.
PONDR® VL-XT: PONDR (Predictor Of Natural Disordered Regions) is a set of neural network predictors of disordered regions on the basis of local amino acid composition, flexibility, hydropathy, coordination number and other factors. These predictors classify each residue within a sequence as either ordered or disordered. PONDR VL-XT integrates three feed forward neural networks: the Variously characterized Long, version 1 (VL1) predictor from Romero et al. (20), which predicts non-terminal residues, and the X-ray characterized N- and C- terminal predictors (XT) from Li et al. (21), which predicts terminal residues. Output for the VL1 predictor starts and ends 11 amino acids from the termini. The XT predictors output provides predictions up to 14 amino acids from their respective ends. A simple average is taken for the overlapping predictions; and a sliding window of 9 amino acids is used to smooth the prediction values along the length of the sequence. Unsmoothed prediction values from the XT predictors are used for the first and last four sequence positions.
PONDR® VSL2 is a combination of neural network predictors for both short and long disordered regions (16). A length limit of 30 residues divides short- and long-disordered regions. Each individual predictor is trained by the dataset containing sequences of that specific length. The final prediction is a weighted average determined by a second layer predictor (16). PONDR® VSL2 applies not only the sequence profile but also the result of sequence alignments from PSI-BLAST (22) and secondary structure prediction from PHD (23) and PSIPRED (24). This predictor is so far the most accurate predictor in the PONDR family (25).
PrDOS is composed of two predictors. The first predictor is implemented using a support vector machine with a position-specific profile of local amino acid sequence. A similar concept to how PSIPRED (24) predicts local secondary structure features. The second predictor assumes the conservation of intrinsic disorder in homologous protein domain families (19,26) and is implemented using PSI-BLAST (22) and a novel measure of disorder (27). The final prediction is taken as the combination of the results of the two predictors described.
PV2 is a meta-predictor that was built upon five prediction methodologies trained on different disordered protein datasets: logistic regression, a neural network, a support vector machine, a conditional random field and finally VSL2B to capture the correlation between the neighboring residues. The PV2 meta-prediction reports a residue as disordered if any two of the underlying methods agree on a disordered state (28). The meta-predictor PV2 achieved either higher or comparable accuracy with other methods in both CASP8 and CASP9 sequences and it had a good balance between sensitivity and specificity. The PV2 meta-predictor was also reliable on the structured domains predictions and it was in the top eight disorder predictors in CASP9 (29) for balanced accuracy.
Espritz predicts three variants of disorder using bidirectional recursive neural networks trained on the following datasets: PDB X-ray crystallography of short disorder (Espritz-X), NMR mobility (Espritz-N) and DisProt data for long disorder (Espritz-D). Either method can be run with a fast or slower variant of the algorithm (30). Because of the wide genomic scale of this database the fast variant was used. Additionally, the following cut-offs were used for the scores (probabilities) generated by each Espritz flavor to yield 5% false positive rate: Esprit-X 0.1434, Espritz-N 0.3089 and Espritz-D 0.5072.
IUPred assumes that the core of a well-structured globular protein has amino acids that can make enough favorable contacts to form a stable 3D structure. A matrix of amino acid pairs holds estimates of their interaction energies which is then used with a position-specific scoring method to predict when stretches of amino acids are not contributing to a stable structure (31). Additionally, IUPred includes both a short (IUPred-S) and long (IUPred-L) variant of its scoring method.
ANCHOR is a predictor of binding sites within disordered regions. It uses the same energy estimation method that underlies IUPred to predict disordered regions in general. ANCHOR finds regions that cannot form enough favorable interactions on their own to form a stable structure, but could gain energy by interacting with a globular structure (32). These sites are often the basis of short linear motifs important in binding to the surface of partner proteins or structured domains in the same polypeptide chain. This property can be functional for both inhibition of an active site or for mediating dynamic protein–protein interactions and complex formation.
SUPERFAMILY is a library of hidden Markov models (HMMs) for SCOP structural domain classifications at the superfamily and family level (33). Assignments from the 1.75 version of the HMM library (34) were used to provide predictions of SCOP structural domains (35,36). E value cut offs used were identical to those in the SUPERFAMILY online resource with the assignments coming directly from a mirror of the source database. When new HMM models and SCOP classifications are added to SUPERFAMILY new annotations will automatically be shown in D2P2.
D2P2 Consensus was calculated at 25, 50, 75 and 100% agreement between all of the prediction methods and stored in the database. This allows a user to filter results based on conservation between prediction methodologies and for outputting likely regions of interest in query sequences online (taken at 75%). For a description of the consensus calculation see Figure 2.
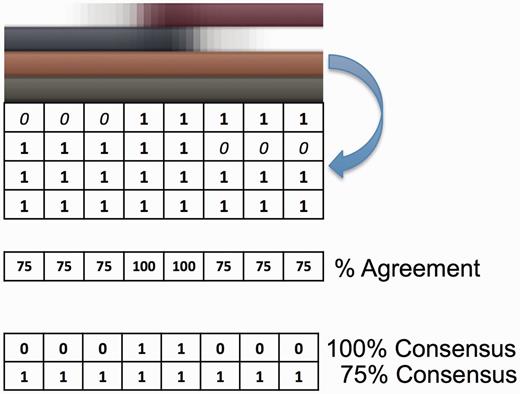
Toy example of the D2P2 predictor consensus calculation (see Figure 1 for a real example). The colored bars (top) represent real valued and binary disorder prediction output for four imagined predictors. Any real valued output is converted to a binary form by thresholding at a cut-off of 0.5 (as per CASP requirements) or at each prediction methods’ advised cut-off minimizing false-positive rate. Next, a binary N × M matrix of per residue (N) and per predictor (M) results is created (blue arrow). The percentage from full agreement of a disordered state is calculated for each column of the binary matrix. This is then re-encoded as a binary matrix (bottom) for each threshold of agreement (or consensus) and further run-length encoded for storage in the database as a set of agreed upon regions of disorder. Taking a higher percentage cut-off of consensus yields a more conservative result with 100% likely under predicting. When searching online with D2P2 75% consensus is used to highlight regions of sequence that are likely disordered.
DATABASE
Data from the database are made available as tab-delimited files for maximum accessibility along with a MySQL schema file for anyone wishing to reconstruct the relational database tables.
Sequence
All protein sequences included in the database are provided along with their mapping to each genome with any comments from the source genome project made available.
Predictions
Disorder predictions for each predictor are available as well as per genome. SUPERFAMILY assignments are available direct from the SUPERFAMILY resource, but derived statistics from these assignments are included in the available data. All predictor outputs were consolidated into a single format in the database by thresholding any real valued result to a binary prediction, all predicted regions were then run-length encoded. Original real valued results are also included in the database for interested parties in the form of JSON arrays. A simple web service is available to obtain all binary predictions for a sequence as JSON by sequence ID query.
Search
Search for disorder in sequences of interest is provided through queries using lists of sequence ID either from the originating genome project or UniProt ID where applicable, free text search of the protein’s comments and sequence IDs from the genome project, as well as exactly matching whole sequences to all genomes. Included with finding exactly matched sequences CS-BLAST (37) is available to find the nearest matching protein in the database to identify likely disordered regions of novel sequence, though for this task some prediction methods included in D2P2 provide their own online prediction portals that are linked from the database online. For investigating disorder on a larger scale where a user does not have a protein of interest, a browse page is provided. The whole database can be inspected for proteins that come from a specific genome or taxon, as well as those that match specific content such as SCOP superfamily assignment, domain-centric Gene Ontology assignment, DisProt and IDEAL curated validation, the percentage of disorder content in the protein and the percentage agreement of all predictors agreeing on a given disordered region.
Statistics
Several pre-computed statistics are included in the database per predictor for each sequence, these include the following: the number of residues predicted disordered; the percentage of protein predicted disordered; the number of residues predicted disordered in a predicted SCOP domain; the percentage of the predicted disorder that lies in a SCOP domain and the percentage of the whole protein predicted disordered and inside a SCOP domain. Additionally, per sequence and per predictor pair-wise comparison statistics are included for the purpose of future predictor development: the number of residues both methods agree are disordered; the percentage of each methods total disorder that agrees with another method; the percentage of the whole protein the methods agree are disordered; the number of residues predicted with one method but not another; the percentage of all residues predicted in one method not found in another and the percentage of the whole protein one method predicts to be disordered that another method does not.
Reports
Graphical reports are available via the web of all disorder, SCOP structure, ANCHOR binding region and PhosphoSitePlus (38) post-translational modification (PTM) site assignments for a given set of sequences of interest. Additionally, where relevant experimental annotations and cross references are provided by the DisProt and IDEAL curated databases along with predicted disorder. Dependent on browser functionality a scalable vector graphics figure is made available with all prediction data embedded, mouse popups provide direct access to each region of interest. Additionally, publication ready figures are also one-click downloadable for any search result. In Figure 1, we see an example of such a report.
Source code
Perl source code for the website is available through Git at: https://github.com/MattOates/d2p2.pro.
RESULTS
The real product of the work is the database itself, but we describe briefly below a first global look at the data.
Global comparison of disorder predictors
One aim of the D2P2 database is to provide statistics for improving disorder prediction. In Figure 3, we show each prediction method’s coverage compared over all sequences in the database. Certain features are immediately apparent. At 0–1%, all predictors avoid stable globular structures, with a rapid change to a regime of unstructured regions covering 10–50% of a given protein being common. All prediction methods change trend toward higher frequencies at >98% coverage mark, representing families of profoundly unstructured proteins. IUPred-S (short variant) as expected has higher frequency of short sub-regions and lower frequency in longer regions, so too does VL-XT. PrDOS and VSL2b are relatively balanced toward long and short regions of disorder predicted, with PV2 predicting greater numbers of long disordered regions over short. An avenue of improvement might be to investigate the production of a meta-predictor that better handles short and long regions of disorder, perhaps including IUPred-S and PV2 with VL-XT to avoid over prediction; feasibility of such approaches was discussed recently by Peng and Kurgan (39). The aim of this work is not to develop a meta-predictor but to empower the prediction community to use D2P2 as a key information resource driving methods development.
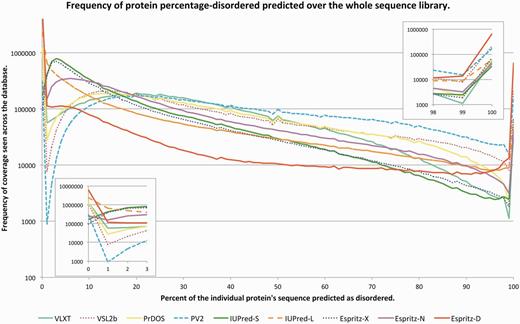
A graph showing the distribution of total disorder coverage per-protein over the whole database of protein sequences for each predictor. The X axis shows the percentage of a protein sequence that was covered with disorder prediction from a given predictor, binned at 1% intervals. The Y axis shows the frequency of observed sequences with a given percentage coverage of disorder, log10 scaled for ease of comparison. The inset (left) shows the first 3% zoomed for clarity of how each predictor treats more structured proteins, the inset (right) shows the final 3% where proteins are predicted to be profoundly disordered with little to no stable tertiary structure.
Prediction by domain of cellular life
Figure 4 shows global statistics per predictor for each domain of cellular life. The general trend for all disorder predicted is that Eukarya have had a large expansion in the quantity of disordered sequence. The story for Archaea and Bacteria is less clear, where five methods out of nine show Archaea as having greater disordered content than Bacteria. The exception to this is that PV2, IUPred-L, Espritz-X and Espritz-N find Bacteria to have more disordered sequence than Archaea. This inversion in Bacterial and Archaeal disorder content between predictor variants such as seen with IUPred-S versus IUPred-L and Espritz-D versus other variants suggests that these two domains of life differ in the forms of disorder present if not the quantity. Looking at the interplay between structure and disorder over evolution, we see in Figure 5 all predictors register a pronounced switch to disorder between SCOP domains rather than within domains for Eukarya. Archaea and Bacteria being similar to each other in reduced coverage outside of domains and proportionally twice as much coverage within domains. This overlap of SCOP prediction and disorder prediction does not imply incorrect prediction for either category, as short-type disorder has specific function within structured domains as regions that undergo dynamic structural transitions (2).
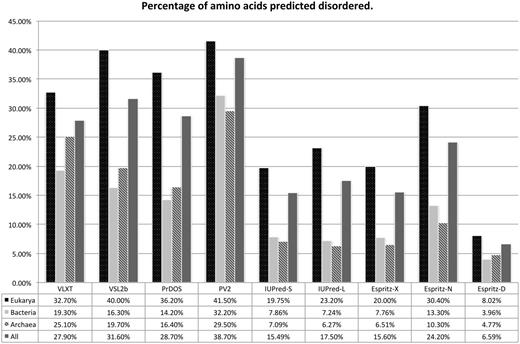
A bar chart grouped by prediction method of global percentage disorder predicted per domain of cellular life. The X axis shows results per domain grouped by predictor, the Y axis shows the percentage of all amino acid residues for a given domain of life predicted disordered by a given method.
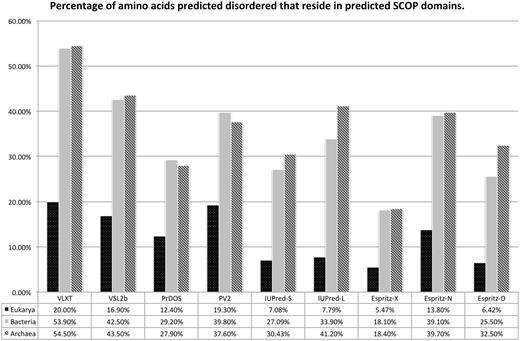
Amino acids which have been predicted to be disordered (Figure 4) were then sub-classified as either being inter- or intra-domain disorder. This figure shows a bar chart, with results grouped by predictor, of the percentage of disordered amino acids that reside within a predicted SCOP domain. The X axis shows results per domain grouped by predictor, the Y axis shows the percentage of all amino acid residues for a given domain of life predicted disordered by a given method.
DISCUSSION
The main content of this database is fairly straightforward yet of great value: comprehensive disorder prediction on genomes shown alongside structural domains. Similar work was done previously for the human proteome (40). This prior work included sequence conservation as a third feature, leading to three types of proteins for the human proteome: structured (52%), disordered (35%) and cryptic domains (18%), where cryptic domains were defined as sequences with high evolutionary conservation that failed to match any known structured domains and were thus assumed to be structured domains for which the structures had not yet been determined. This conclusion was based on the assumption that all disordered regions show high sequence variability. However, there are reports of regions of disorder that show high sequence conservation (26,41). Thus, an important use of D2P2 will be to determine which cryptic domains are predicted to be structured by multiple predictors and which cryptic domains are predicted to be disordered, thus partitioning these regions into likely globular domains of currently unknown structure and into likely regions of disorder with high sequence conservation. This work is in progress and will be reported when completed. Current disorder findings from D2P2 data for human (ENSEMBL release 63) using all predictors shows ∼37–50% of human amino acids predicted disordered with ∼29–39% of the amino acids being intra-domain disorder i.e. not found within SCOP domains. Structured domains cover ∼44% of amino acids leaving ∼17–27% of the amino acids unassigned to either SCOP domains or intra-domain disorder.
The data in D2P2 have been made as accessible as possible, and is provided interactively via a website including a graphical display with a consensus plot. We are anxious to communicate with users with regard to future developments, so users should not hesitate to suggest or provide additional tools or predictors to be added in the future.
Example use
D2P2 provides informative data for various types of biological investigation. A good example is the exploration of isoforms and their function. A recent study by Ellis et al. (42) showed that alternative splicing of proteins has rewired protein–protein interaction networks in neural tissue, and that these are important in tissue-specific function. Figure 1 shows the Bridging Integrator 1 (Bin1) gene from the study, and two of its most dimorphic isoforms (ENSP00000365281 and ENSP00000316779). It was change in disordered regions that were shown to alter Bin1 interaction with Dynamin 2 (Dnm2) facilitating endocytosis within neural-tissue. With D2P2 these forms of analyses can be automated with the addition of multiple sources of evidence for disordered regions. Additionally, the inclusion of PhosphoSitePlus curated PTM annotation lets us see that the disordered inserts between BIN1 isoforms also undergo posttranslational modifications as part of the regulatory process. The suggestion from the D2P2 data that Eukarya have a bias toward intra-domain disorder (Figure 5) suggests that this sort of study is likely to be increasingly important in characterizing the full complexity of protein interaction and regulation in Eukaryotes.
Further work
The principal future goal is to include more disorder predictors. Although the database has a substantial collection there are important predictors that need to be added, and furthermore important new predictors are likely to be developed over time. The other main future goal is to expand the sequences on which we have disorder predictions to include more genomes, e.g. thousands of viral genomes and other sequence sets that are already in SUPERFAMILY. We also intend to improve the interface and provide more tools for online analysis, e.g. tools to enable searches by Gene Ontology, tools for comparative genomics, analysis methods that take advantage of the domain-based sTOL (http://supfam.org/SUPERFAMILY/sTOL) and additional software that capitalizes on other tools attached to SUPERFAMILY.
FUNDING
Engineering and Physical Research Council (EPSRC) [EP/E501214/1 to M.E.O.]; Dissertation Fellowship awarded by the University of Alberta (to M.J.M.); Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery (to L.K.); Program of the Russian Academy of Sciences for “Molecular and Cellular Biology” (to V.N.U.); the US National Science Foundation [EF 0849803 to A.K.D. and V.N.U.]; Biotechnology and Biological Sciences Research Council (BBSRC) [BB/G022771/1 to J.G.]. Funding for open access charge: Engineering and Physical Research Council (EPSRC) [EP/E501214/1].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors acknowledge the efforts of all original authors for each disorder prediction method used in D2P2 and their indirect contribution to the data they present. Additionally, this resource would not have been possible without contributions from many open source developers our software is predicated on. The Bolyai Janos fellowship for Z.D. is gratefully acknowledged.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments