




Abstract
Null mutations in the progranulin gene ( PGRN ) were recently reported to cause tau-negative frontotemporal dementia linked to chromosome 17. We assessed the genetic contribution of PGRN mutations in an extended population of patients with frontotemporal lobar degeneration (FTLD) ( N =378). Mutations were identified in 10% of the total FTLD population and 23% of patients with a positive family history. This mutation frequency dropped to 5% when analysis was restricted to an unbiased FTLD subpopulation ( N =167) derived from patients referred to Alzheimer's Disease Research Centers (ADRC). Among the ADRC patients, PGRN mutations were equally frequent as mutations in the tau gene ( MAPT ). We identified 23 different pathogenic PGRN mutations, including a total of 21 nonsense, frameshift and splice-site mutations that cause premature termination of the coding sequence and degradation of the mutant RNA by nonsense-mediated decay. We also observed an unusual splice-site mutation in the exon 1 5′ splice site, which leads to loss of the Kozac sequence, and a missense mutation in the hydrophobic core of the PGRN signal peptide. Both mutations revealed novel mechanisms that result in loss of functional PGRN. One mutation, c.1477C>T (p.Arg493X), was detected in eight independently ascertained familial FTLD patients who were shown to share a common extended haplotype over the PGRN genomic region. Clinical examination of patients with PGRN mutations revealed highly variable onset ages with language dysfunction as a common presenting symptom. Neuropathological examination showed FTLD with ubiquitin-positive cytoplasmic and intranuclear inclusions in all PGRN mutation carriers.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) is the overall term for a group of neurodegenerative diseases that account for 5–10% of all dementia patients and 10–20% of patients with onset before 65 years ( 1 , 2 ). FTLD includes several major clinical subtypes of which frontotemporal dementia (FTD) comprises the major group with early clinical symptoms consisting of behavior and personality dysfunctions followed by a cognitive decline eventually leading to dementia ( 3 ). FTLD patients with language dysfunction as the primary symptom, and with behavioral change appearing only later in the disease course, are further divided into semantic dementia (SD) and primary progressive aphasia (PPA) subtypes. Finally, in the FTD-motor neuron disease (MND) subtype, features of the amyotrophic form of MND are co-associated with FTD ( 4 ). In all subtypes of FTLD, a parkinsonian movement disorder can often develop during the course of the disease.
Neuropathologically, FTLD results from severe neurodegeneration of the frontal and/or temporal neocortices. On the basis of the profile of immunohistochemical staining and the pattern of intracellular inclusions, three major pathological subtypes have been defined: FTLD with tau-positive pathology (tau-positive FTLD), FTLD with tau-negative ubiquitin-positive inclusions (FTLD-U) and FTLD with no identifiable intracellular inclusions (dementia lacking distinctive histopathology, DLDH) ( 5 , 6 ). In recent years, the use of improved immunohistochemical analyses have emphasized the importance of the FTLD-U neuropathological subtype, resulting in a revised prevalence for FTLD-U of 40–65% and the recognition that DLDH is either extremely rare or non-existent ( 7 , 8 ). FTLD-U is characterized by ubiquitin-immunoreactive (ub-ir) neurites and neuronal cytoplasmic inclusions (NCI) in layer II of the frontal and temporal neocortex and in the dentate fascia of the hippocampus ( 8–12 ). In addition, familial FTLD-U patients frequently have ub-ir ‘cat-eye’ shaped or lentiform intranuclear inclusions (NII) with a similar distribution to the NCI ( 13 ).
FTLD is a genetically complex disorder with multiple genetic factors contributing to the disease. A positive family history of dementia is found in ~40% of FTLD patients, and in the majority of these patients, FTLD is inherited as an autosomal dominant trait. Genetic linkage studies have revealed FTLD loci and genes on chromosome 3p ( 14 ), chromosome 9q ( 15 ), chromosome 9p ( 16–18 ) (two loci) and chromosome 17q (two loci) ( 19–23 ). At a consensus conference in 1996, the autosomal dominant form of FTLD associated with loci on chromosome 17q21 was given the overall term FTD with parkinsonism linked to chromosome 17 (FTDP-17), reflecting the highly variable clinical phenotype in this group of families ( 24 ). In 1998, mutations in the microtubule-associated protein tau ( MAPT ) gene were first discovered in a proportion of these FTDP-17 families ( 21–23 ). To date, around 35 different confirmed pathogenic mutations in MAPT have been identified in over 100 families worldwide (FTD mutation database; http://www.molgen.ua.ac.be/FTDmutations/ ) ( 25 ). FTD patients with MAPT mutations consistently show tau-positive inclusion pathology at autopsy. However, in the past few years, an increasing number of FTLD-U families with significant evidence for linkage to the same chromosomal region as MAPT on 17q21 were reported, in which defined MAPT mutations could not be identified ( 10–12 , 26–28 ). These families all presented with ub-ir NCI and additionally with NII as a characteristic pathological feature ( 10–12 , 28 ). Recently, we showed that FTLD-U in these families is caused by null mutations in the gene encoding progranulin ( PGRN ), with the majority of these mutations causing premature termination of the coding sequence which leads to degradation of the mutant PGRN RNAs by nonsense-mediated decay (NMD) ( 19 , 20 ). We further demonstrated that this leads to a loss of functional PGRN protein, suggesting a haploinsufficiency mechanism.
PGRN is located 1.7 Mb centromeric of MAPT on chromosome 17q21 and consists of 12 coding and one non-coding exons covering a small genomic region of only 8 kb. It encodes a single biologically active precursor glycoprotein of 68.5 kDa that is composed of 7.5 tandem repeats of highly conserved motifs of 12 cysteines, which can be proteolytically cleaved by elastase-like activity to form a family of 6 kDa peptides called granulins ( 29 , 30 ). PGRN is a widely expressed secreted growth factor which plays a role in multiple processes including development, wound repair and inflammation by activating signaling cascades that control cell cycle progression and cell motility ( 29 , 31 ). PGRN has also been strongly linked to tumorogenesis ( 29 , 32 ). The normal function of PGRN and the individual granulins in neurons has yet to be determined; however, the identification of null PGRN mutations in FTLD-U patients suggests that reduced levels of this secreted mitogenic factor can lead to neurodegeneration.
In the present study, we report the first extensive mutation analyses of PGRN in 378 FTLD patients from a Mayo Clinic FTLD series, including 15 patients from a Minnesotan community-based (Olmsted County) dementia series and 167 patients from an Alzheimer's disease Research Centers (ADRC)-referral series. We thus estimated the genetic contribution of PGRN mutations to FTLD in general as well as to familial FTLD and pathologically confirmed FTLD-U subgroups. We also employed the community-based series to make an initial estimate of the frequency of PGRN mutations in all dementia patients. Our findings show that mutations in PGRN are a major cause of FTLD-U, expand the range of PGRN mutations identified to date and reveal novel mechanisms that lead to the loss of functional PGRN.
RESULTS
PGRN sequencing analyses in FTLD and ALS patient series
In a first phase, we performed systematic screening of PGRN in our FTLD and amyotrophic lateral sclerosis (ALS) patient series by direct sequencing of all 12 coding exons, the non-coding exon 0, the core promoter and the complete 3’ untranslated region (UTR). In the overall Mayo Clinic FTLD series ( N =378), we identified a total of 23 different pathogenic mutations, defined as mutations that would clearly lead to loss of functional PGRN protein consistent with previously reported mutations ( 1 ,2). Eighteen pathogenic mutations were found within the PGRN coding sequence and five intronic mutations were predicted to destroy exonic splice sites (Table 1 ). In addition, we identified 13 coding sequence variants (seven missense and six silent mutations) that likely represent non-disease-related polymorphisms (Table 2 ). No pathogenic mutations were observed in ALS patients. An additional eight non-pathogenic intronic sequence variants were identified (Supplementary Material, Table S1).
Clinicopathological findings in FTLD families with pathogenic PGRN mutations
| Patient | Origin | Disease presentation a | Mutation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at onset (years) | Age at death (years) | Pathological diagnosis [clinical] | Family history | Genomic b | Predicted cDNA c | Predicted protein d | Location | ||
| F161-1 | USA | 55 | 66 | FTLD-U (NII) | ND | g.100068T>C | c.2T>C | p.Met? | EX1 |
| 11696 | USA e | 56 | N/A | [PPA] | Y | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA99-175 | USA | 48 | 56 | FTLD-U (NII) | N | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA03-140 | USA | 63 | 65 | FTLD-U (NII) | ND | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| 8536 | USA | 65 | N/A | [FTD] | Y | g.100129insC | c.63insC | p.Asp22ArgfsX43 | EX1 |
| UBC17-68 | Canada | 57 | 61 | FTLD-U (NII) | Y | g.100156_100157insCTGC | c.90_91insCTGC | p.Cys31LeufsX35 | EX1 |
| F159-1 | USA e | 83 | N/A | [PPA] | Y | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| 0179-90 | Sweden | ND | ND | FTLD f | ND | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| F149-1 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.100205G>A | c.138+1G>A (IVS1+1G>A) | p.Met? | IVS1 |
| F142-1 | USA e | 69 | 76 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 367180 | USA g | 80 | 87 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 114209 | USA g | 61 | N/A | [PPA] | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 4504 | USA | 51 | 66 | FTLD-U (NII) | ND | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| B3485 | USA | 61 | 68 | FTLD f | Y | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| UBC11-1 | Canada | 66 | N/A | [FTD] | Y | g.101168_101171delCAGT | c.388_391delCAGT | p.Gln130SerfsX125 | EX4 |
| UBC14-9 | Canada | 55 | 60 | FTLD-U (NII) | Y | g.101343G>A | c.463-1G>A (IVS4-1G>A) | p.Ala155TrpfsX56 | IVS4 |
| A03-52 | USA | 56 | 61 | FTLD-U (NII) | N | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B4301 | USA | 66 | 72 | FTLD f | Y | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| 97-35 | USA | 51 | 53 | FTLD-U (NII) | ND | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B3802 | USA | 55 | 61 | FTLD f | Y | g.101703G>C | c.708+1G>C (IVS6+1G>C) | p.Val200GlyfsX18 | IVS6 |
| NP19870 | USA | 58 | 65 | FTLD-U | Y | g.101983_101984delTG | c.753_754delTG | p.Cys253X | EX7 |
| 12743 | USA | 56 | N/A | [CBS] | Y | g.102264G>C | c.836-1G>C (IVS7-1G>C) | p.Val279GlyfsX5 | IVS7 |
| F147-47 | USA e | 60 | 68 | FTLD-U (NII) | Y | g.102339_102340insTG | c.910_911insTG | p.Trp304LeufsX58 | EX8 |
| 4713 | USA | 56 | 65 | FTLD-U (NII) | Y | g.102340G>A | c.911G>A | p.Trp304X | EX8 |
| UBC19-1 | Canada | 55 | 61 | FTLD-U (NII) | Y | g.102363G>A | c.933+1G>A (IVS8+1G>A) | p.Val279GlyfsX5 | IVS8 |
| PPA1-1 | USA e | 65 | 73 | FTLD-U (NII) | Y | g.102516delG | c.998delG | p.Gly333ValfsX28 | EX9 |
| F129-2 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.102663delC | c.1145delC | p.Thr382SerfsX30 | EX9 |
| UBC4-1 | Canada | 62 | 71 | FTLD-U (NII) | Y | g.102675G>A | c.1157G>A | p.Trp386X | EX9 |
| 01-01 | USA | 49 | 54 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| UBC15-16 | Canada | 60 | 77 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| F153-1 | USA g | 52 | 56 | FTLD-U (NII) | Y | g.103132_103133insC | c.1395_1396insC | p.Cys466LeufsX46 | EX10 |
| NA01-249 | USA | 66 | 75 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| PPA3-1 | USA | 65 | N/A | [PPA] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NA02-297 | USA | 56 | 59 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| F144-1 | USA e | 54 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 9118 | USA | 48 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| A02-43 | USA | 57 | 61 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 05-44 | USA | 53 | 56 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NP12900 | USA | 69 | 72 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| Patient | Origin | Disease presentation a | Mutation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at onset (years) | Age at death (years) | Pathological diagnosis [clinical] | Family history | Genomic b | Predicted cDNA c | Predicted protein d | Location | ||
| F161-1 | USA | 55 | 66 | FTLD-U (NII) | ND | g.100068T>C | c.2T>C | p.Met? | EX1 |
| 11696 | USA e | 56 | N/A | [PPA] | Y | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA99-175 | USA | 48 | 56 | FTLD-U (NII) | N | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA03-140 | USA | 63 | 65 | FTLD-U (NII) | ND | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| 8536 | USA | 65 | N/A | [FTD] | Y | g.100129insC | c.63insC | p.Asp22ArgfsX43 | EX1 |
| UBC17-68 | Canada | 57 | 61 | FTLD-U (NII) | Y | g.100156_100157insCTGC | c.90_91insCTGC | p.Cys31LeufsX35 | EX1 |
| F159-1 | USA e | 83 | N/A | [PPA] | Y | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| 0179-90 | Sweden | ND | ND | FTLD f | ND | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| F149-1 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.100205G>A | c.138+1G>A (IVS1+1G>A) | p.Met? | IVS1 |
| F142-1 | USA e | 69 | 76 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 367180 | USA g | 80 | 87 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 114209 | USA g | 61 | N/A | [PPA] | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 4504 | USA | 51 | 66 | FTLD-U (NII) | ND | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| B3485 | USA | 61 | 68 | FTLD f | Y | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| UBC11-1 | Canada | 66 | N/A | [FTD] | Y | g.101168_101171delCAGT | c.388_391delCAGT | p.Gln130SerfsX125 | EX4 |
| UBC14-9 | Canada | 55 | 60 | FTLD-U (NII) | Y | g.101343G>A | c.463-1G>A (IVS4-1G>A) | p.Ala155TrpfsX56 | IVS4 |
| A03-52 | USA | 56 | 61 | FTLD-U (NII) | N | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B4301 | USA | 66 | 72 | FTLD f | Y | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| 97-35 | USA | 51 | 53 | FTLD-U (NII) | ND | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B3802 | USA | 55 | 61 | FTLD f | Y | g.101703G>C | c.708+1G>C (IVS6+1G>C) | p.Val200GlyfsX18 | IVS6 |
| NP19870 | USA | 58 | 65 | FTLD-U | Y | g.101983_101984delTG | c.753_754delTG | p.Cys253X | EX7 |
| 12743 | USA | 56 | N/A | [CBS] | Y | g.102264G>C | c.836-1G>C (IVS7-1G>C) | p.Val279GlyfsX5 | IVS7 |
| F147-47 | USA e | 60 | 68 | FTLD-U (NII) | Y | g.102339_102340insTG | c.910_911insTG | p.Trp304LeufsX58 | EX8 |
| 4713 | USA | 56 | 65 | FTLD-U (NII) | Y | g.102340G>A | c.911G>A | p.Trp304X | EX8 |
| UBC19-1 | Canada | 55 | 61 | FTLD-U (NII) | Y | g.102363G>A | c.933+1G>A (IVS8+1G>A) | p.Val279GlyfsX5 | IVS8 |
| PPA1-1 | USA e | 65 | 73 | FTLD-U (NII) | Y | g.102516delG | c.998delG | p.Gly333ValfsX28 | EX9 |
| F129-2 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.102663delC | c.1145delC | p.Thr382SerfsX30 | EX9 |
| UBC4-1 | Canada | 62 | 71 | FTLD-U (NII) | Y | g.102675G>A | c.1157G>A | p.Trp386X | EX9 |
| 01-01 | USA | 49 | 54 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| UBC15-16 | Canada | 60 | 77 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| F153-1 | USA g | 52 | 56 | FTLD-U (NII) | Y | g.103132_103133insC | c.1395_1396insC | p.Cys466LeufsX46 | EX10 |
| NA01-249 | USA | 66 | 75 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| PPA3-1 | USA | 65 | N/A | [PPA] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NA02-297 | USA | 56 | 59 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| F144-1 | USA e | 54 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 9118 | USA | 48 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| A02-43 | USA | 57 | 61 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 05-44 | USA | 53 | 56 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NP12900 | USA | 69 | 72 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
a FTLD-U (NII), FTLD with ubiquitin-positive intranuclear inclusions; ND, not documented; N/A, not applicable.
b Numbering relative to the reverse complement of GenBank accession number AC003043 .1 and starting at nucleotide 1.
c Numbering according to GenBank accession number NM_002087 .2 starting at the translation initiation codon.
d Numbering according to GenPept accession number NP_002078 .1.
e FTLD-ADRC referral series.
f Ubiquitin staining was not performed.
g Olmsted-County community-based dementia series.
Clinicopathological findings in FTLD families with pathogenic PGRN mutations
| Patient | Origin | Disease presentation a | Mutation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at onset (years) | Age at death (years) | Pathological diagnosis [clinical] | Family history | Genomic b | Predicted cDNA c | Predicted protein d | Location | ||
| F161-1 | USA | 55 | 66 | FTLD-U (NII) | ND | g.100068T>C | c.2T>C | p.Met? | EX1 |
| 11696 | USA e | 56 | N/A | [PPA] | Y | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA99-175 | USA | 48 | 56 | FTLD-U (NII) | N | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA03-140 | USA | 63 | 65 | FTLD-U (NII) | ND | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| 8536 | USA | 65 | N/A | [FTD] | Y | g.100129insC | c.63insC | p.Asp22ArgfsX43 | EX1 |
| UBC17-68 | Canada | 57 | 61 | FTLD-U (NII) | Y | g.100156_100157insCTGC | c.90_91insCTGC | p.Cys31LeufsX35 | EX1 |
| F159-1 | USA e | 83 | N/A | [PPA] | Y | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| 0179-90 | Sweden | ND | ND | FTLD f | ND | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| F149-1 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.100205G>A | c.138+1G>A (IVS1+1G>A) | p.Met? | IVS1 |
| F142-1 | USA e | 69 | 76 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 367180 | USA g | 80 | 87 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 114209 | USA g | 61 | N/A | [PPA] | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 4504 | USA | 51 | 66 | FTLD-U (NII) | ND | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| B3485 | USA | 61 | 68 | FTLD f | Y | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| UBC11-1 | Canada | 66 | N/A | [FTD] | Y | g.101168_101171delCAGT | c.388_391delCAGT | p.Gln130SerfsX125 | EX4 |
| UBC14-9 | Canada | 55 | 60 | FTLD-U (NII) | Y | g.101343G>A | c.463-1G>A (IVS4-1G>A) | p.Ala155TrpfsX56 | IVS4 |
| A03-52 | USA | 56 | 61 | FTLD-U (NII) | N | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B4301 | USA | 66 | 72 | FTLD f | Y | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| 97-35 | USA | 51 | 53 | FTLD-U (NII) | ND | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B3802 | USA | 55 | 61 | FTLD f | Y | g.101703G>C | c.708+1G>C (IVS6+1G>C) | p.Val200GlyfsX18 | IVS6 |
| NP19870 | USA | 58 | 65 | FTLD-U | Y | g.101983_101984delTG | c.753_754delTG | p.Cys253X | EX7 |
| 12743 | USA | 56 | N/A | [CBS] | Y | g.102264G>C | c.836-1G>C (IVS7-1G>C) | p.Val279GlyfsX5 | IVS7 |
| F147-47 | USA e | 60 | 68 | FTLD-U (NII) | Y | g.102339_102340insTG | c.910_911insTG | p.Trp304LeufsX58 | EX8 |
| 4713 | USA | 56 | 65 | FTLD-U (NII) | Y | g.102340G>A | c.911G>A | p.Trp304X | EX8 |
| UBC19-1 | Canada | 55 | 61 | FTLD-U (NII) | Y | g.102363G>A | c.933+1G>A (IVS8+1G>A) | p.Val279GlyfsX5 | IVS8 |
| PPA1-1 | USA e | 65 | 73 | FTLD-U (NII) | Y | g.102516delG | c.998delG | p.Gly333ValfsX28 | EX9 |
| F129-2 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.102663delC | c.1145delC | p.Thr382SerfsX30 | EX9 |
| UBC4-1 | Canada | 62 | 71 | FTLD-U (NII) | Y | g.102675G>A | c.1157G>A | p.Trp386X | EX9 |
| 01-01 | USA | 49 | 54 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| UBC15-16 | Canada | 60 | 77 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| F153-1 | USA g | 52 | 56 | FTLD-U (NII) | Y | g.103132_103133insC | c.1395_1396insC | p.Cys466LeufsX46 | EX10 |
| NA01-249 | USA | 66 | 75 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| PPA3-1 | USA | 65 | N/A | [PPA] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NA02-297 | USA | 56 | 59 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| F144-1 | USA e | 54 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 9118 | USA | 48 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| A02-43 | USA | 57 | 61 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 05-44 | USA | 53 | 56 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NP12900 | USA | 69 | 72 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| Patient | Origin | Disease presentation a | Mutation | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Age at onset (years) | Age at death (years) | Pathological diagnosis [clinical] | Family history | Genomic b | Predicted cDNA c | Predicted protein d | Location | ||
| F161-1 | USA | 55 | 66 | FTLD-U (NII) | ND | g.100068T>C | c.2T>C | p.Met? | EX1 |
| 11696 | USA e | 56 | N/A | [PPA] | Y | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA99-175 | USA | 48 | 56 | FTLD-U (NII) | N | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| NA03-140 | USA | 63 | 65 | FTLD-U (NII) | ND | g.100092C>A | c.26C>A | p.Ala9Asp | EX1 |
| 8536 | USA | 65 | N/A | [FTD] | Y | g.100129insC | c.63insC | p.Asp22ArgfsX43 | EX1 |
| UBC17-68 | Canada | 57 | 61 | FTLD-U (NII) | Y | g.100156_100157insCTGC | c.90_91insCTGC | p.Cys31LeufsX35 | EX1 |
| F159-1 | USA e | 83 | N/A | [PPA] | Y | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| 0179-90 | Sweden | ND | ND | FTLD f | ND | g.100168delC | c.102delC | p.Gly35GlufsX19 | EX1 |
| F149-1 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.100205G>A | c.138+1G>A (IVS1+1G>A) | p.Met? | IVS1 |
| F142-1 | USA e | 69 | 76 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 367180 | USA g | 80 | 87 | FTLD-U (NII) | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 114209 | USA g | 61 | N/A | [PPA] | Y | g.100343delA | c.154delA | p.Thr52HisfsX2 | EX2 |
| 4504 | USA | 51 | 66 | FTLD-U (NII) | ND | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| B3485 | USA | 61 | 68 | FTLD f | Y | g.100423_100424delAG | c.234_235delAG | p.Gly79AspfsX39 | EX2 |
| UBC11-1 | Canada | 66 | N/A | [FTD] | Y | g.101168_101171delCAGT | c.388_391delCAGT | p.Gln130SerfsX125 | EX4 |
| UBC14-9 | Canada | 55 | 60 | FTLD-U (NII) | Y | g.101343G>A | c.463-1G>A (IVS4-1G>A) | p.Ala155TrpfsX56 | IVS4 |
| A03-52 | USA | 56 | 61 | FTLD-U (NII) | N | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B4301 | USA | 66 | 72 | FTLD f | Y | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| 97-35 | USA | 51 | 53 | FTLD-U (NII) | ND | g.101669_101670delCA | c.675_676delCA | p.Ser226TrpfsX28 | EX6 |
| B3802 | USA | 55 | 61 | FTLD f | Y | g.101703G>C | c.708+1G>C (IVS6+1G>C) | p.Val200GlyfsX18 | IVS6 |
| NP19870 | USA | 58 | 65 | FTLD-U | Y | g.101983_101984delTG | c.753_754delTG | p.Cys253X | EX7 |
| 12743 | USA | 56 | N/A | [CBS] | Y | g.102264G>C | c.836-1G>C (IVS7-1G>C) | p.Val279GlyfsX5 | IVS7 |
| F147-47 | USA e | 60 | 68 | FTLD-U (NII) | Y | g.102339_102340insTG | c.910_911insTG | p.Trp304LeufsX58 | EX8 |
| 4713 | USA | 56 | 65 | FTLD-U (NII) | Y | g.102340G>A | c.911G>A | p.Trp304X | EX8 |
| UBC19-1 | Canada | 55 | 61 | FTLD-U (NII) | Y | g.102363G>A | c.933+1G>A (IVS8+1G>A) | p.Val279GlyfsX5 | IVS8 |
| PPA1-1 | USA e | 65 | 73 | FTLD-U (NII) | Y | g.102516delG | c.998delG | p.Gly333ValfsX28 | EX9 |
| F129-2 | USA e | 56 | 63 | FTLD-U (NII) | Y | g.102663delC | c.1145delC | p.Thr382SerfsX30 | EX9 |
| UBC4-1 | Canada | 62 | 71 | FTLD-U (NII) | Y | g.102675G>A | c.1157G>A | p.Trp386X | EX9 |
| 01-01 | USA | 49 | 54 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| UBC15-16 | Canada | 60 | 77 | FTLD-U (NII) | Y | g.102989C>T | c.1252C>T | p.Arg418X | EX10 |
| F153-1 | USA g | 52 | 56 | FTLD-U (NII) | Y | g.103132_103133insC | c.1395_1396insC | p.Cys466LeufsX46 | EX10 |
| NA01-249 | USA | 66 | 75 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| PPA3-1 | USA | 65 | N/A | [PPA] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NA02-297 | USA | 56 | 59 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| F144-1 | USA e | 54 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 9118 | USA | 48 | N/A | [FTD] | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| A02-43 | USA | 57 | 61 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| 05-44 | USA | 53 | 56 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
| NP12900 | USA | 69 | 72 | FTLD-U (NII) | Y | g.103306C>T | c.1477C>T | p.Arg493X | EX11 |
a FTLD-U (NII), FTLD with ubiquitin-positive intranuclear inclusions; ND, not documented; N/A, not applicable.
b Numbering relative to the reverse complement of GenBank accession number AC003043 .1 and starting at nucleotide 1.
c Numbering according to GenBank accession number NM_002087 .2 starting at the translation initiation codon.
d Numbering according to GenPept accession number NP_002078 .1.
e FTLD-ADRC referral series.
f Ubiquitin staining was not performed.
g Olmsted-County community-based dementia series.
PGRN coding sequence variants with unknown significance
| Mutation | Frequency | Notes | |||||
|---|---|---|---|---|---|---|---|
| Genomic a | Predicted cDNA | Predicted protein | Location | rs number | Patients N (%) | Controls N (%) | |
| g.100121C>T | c.55C>T | p.Arg19Trp | EX1 | 1 (0.3) | — | ||
| g.100165C>T′ | c.99C>T | p.Asp33Asp | EX1 | 3 (0.8) | 1 (0.4) | p.Gly79AspfsX39 in one patient | |
| g.100453G>A | c.264G>A | p.Glu88Glu | EX2 | 1 (0.3) | — | ||
| g.100617T>C | c.313T>C | p.Cys105Arg | EX3 | 1 (0.3) | — | Not segregating with disease | |
| g.101164T>C | c.384T>C | p.Asp128Asp | EX4 | rs25646 | 17 (4.5) | 5 (2.2) | |
| g.101702C>T | c.708C>T | p.Asn236Asn | EX6 | 1 (0.3) | — | Mutant RNA not subject to NMD | |
| g.102290G>C | c.861G>C | p.Glu287Asp | EX8 | 1 (0.3) | — | ||
| g.102332G>A | c.903G>A | p.Ser301Ser | EX8 | 1 (0.3) | — | ||
| g.102488G>A | c.970G>A | p.Ala324Thr | EX9 | 1 (0.3) | — | ||
| g.102990G>A | c.1253G>A | p.Arg418Gln | EX10 | 1 (0.3) | 1 (0.4) | ||
| g.103034C>T | c.1297C>T | p.Arg433Trp | EX10 | 5 (1.3) | — | p.Ala155TrpfsX56 in one patient | |
| g.103251C>T | c.1422C>T | p.Cys474Cys | EX11 | 1 (0.3) | — | ||
| g.103373G>C | c.1544G>C | p.Gly515Ala | EX11 | rs25647 | 3 (0.8) | 1 (0.4) | p.Met1? in one patient |
| Mutation | Frequency | Notes | |||||
|---|---|---|---|---|---|---|---|
| Genomic a | Predicted cDNA | Predicted protein | Location | rs number | Patients N (%) | Controls N (%) | |
| g.100121C>T | c.55C>T | p.Arg19Trp | EX1 | 1 (0.3) | — | ||
| g.100165C>T′ | c.99C>T | p.Asp33Asp | EX1 | 3 (0.8) | 1 (0.4) | p.Gly79AspfsX39 in one patient | |
| g.100453G>A | c.264G>A | p.Glu88Glu | EX2 | 1 (0.3) | — | ||
| g.100617T>C | c.313T>C | p.Cys105Arg | EX3 | 1 (0.3) | — | Not segregating with disease | |
| g.101164T>C | c.384T>C | p.Asp128Asp | EX4 | rs25646 | 17 (4.5) | 5 (2.2) | |
| g.101702C>T | c.708C>T | p.Asn236Asn | EX6 | 1 (0.3) | — | Mutant RNA not subject to NMD | |
| g.102290G>C | c.861G>C | p.Glu287Asp | EX8 | 1 (0.3) | — | ||
| g.102332G>A | c.903G>A | p.Ser301Ser | EX8 | 1 (0.3) | — | ||
| g.102488G>A | c.970G>A | p.Ala324Thr | EX9 | 1 (0.3) | — | ||
| g.102990G>A | c.1253G>A | p.Arg418Gln | EX10 | 1 (0.3) | 1 (0.4) | ||
| g.103034C>T | c.1297C>T | p.Arg433Trp | EX10 | 5 (1.3) | — | p.Ala155TrpfsX56 in one patient | |
| g.103251C>T | c.1422C>T | p.Cys474Cys | EX11 | 1 (0.3) | — | ||
| g.103373G>C | c.1544G>C | p.Gly515Ala | EX11 | rs25647 | 3 (0.8) | 1 (0.4) | p.Met1? in one patient |
PGRN coding sequence variants with unknown significance
| Mutation | Frequency | Notes | |||||
|---|---|---|---|---|---|---|---|
| Genomic a | Predicted cDNA | Predicted protein | Location | rs number | Patients N (%) | Controls N (%) | |
| g.100121C>T | c.55C>T | p.Arg19Trp | EX1 | 1 (0.3) | — | ||
| g.100165C>T′ | c.99C>T | p.Asp33Asp | EX1 | 3 (0.8) | 1 (0.4) | p.Gly79AspfsX39 in one patient | |
| g.100453G>A | c.264G>A | p.Glu88Glu | EX2 | 1 (0.3) | — | ||
| g.100617T>C | c.313T>C | p.Cys105Arg | EX3 | 1 (0.3) | — | Not segregating with disease | |
| g.101164T>C | c.384T>C | p.Asp128Asp | EX4 | rs25646 | 17 (4.5) | 5 (2.2) | |
| g.101702C>T | c.708C>T | p.Asn236Asn | EX6 | 1 (0.3) | — | Mutant RNA not subject to NMD | |
| g.102290G>C | c.861G>C | p.Glu287Asp | EX8 | 1 (0.3) | — | ||
| g.102332G>A | c.903G>A | p.Ser301Ser | EX8 | 1 (0.3) | — | ||
| g.102488G>A | c.970G>A | p.Ala324Thr | EX9 | 1 (0.3) | — | ||
| g.102990G>A | c.1253G>A | p.Arg418Gln | EX10 | 1 (0.3) | 1 (0.4) | ||
| g.103034C>T | c.1297C>T | p.Arg433Trp | EX10 | 5 (1.3) | — | p.Ala155TrpfsX56 in one patient | |
| g.103251C>T | c.1422C>T | p.Cys474Cys | EX11 | 1 (0.3) | — | ||
| g.103373G>C | c.1544G>C | p.Gly515Ala | EX11 | rs25647 | 3 (0.8) | 1 (0.4) | p.Met1? in one patient |
| Mutation | Frequency | Notes | |||||
|---|---|---|---|---|---|---|---|
| Genomic a | Predicted cDNA | Predicted protein | Location | rs number | Patients N (%) | Controls N (%) | |
| g.100121C>T | c.55C>T | p.Arg19Trp | EX1 | 1 (0.3) | — | ||
| g.100165C>T′ | c.99C>T | p.Asp33Asp | EX1 | 3 (0.8) | 1 (0.4) | p.Gly79AspfsX39 in one patient | |
| g.100453G>A | c.264G>A | p.Glu88Glu | EX2 | 1 (0.3) | — | ||
| g.100617T>C | c.313T>C | p.Cys105Arg | EX3 | 1 (0.3) | — | Not segregating with disease | |
| g.101164T>C | c.384T>C | p.Asp128Asp | EX4 | rs25646 | 17 (4.5) | 5 (2.2) | |
| g.101702C>T | c.708C>T | p.Asn236Asn | EX6 | 1 (0.3) | — | Mutant RNA not subject to NMD | |
| g.102290G>C | c.861G>C | p.Glu287Asp | EX8 | 1 (0.3) | — | ||
| g.102332G>A | c.903G>A | p.Ser301Ser | EX8 | 1 (0.3) | — | ||
| g.102488G>A | c.970G>A | p.Ala324Thr | EX9 | 1 (0.3) | — | ||
| g.102990G>A | c.1253G>A | p.Arg418Gln | EX10 | 1 (0.3) | 1 (0.4) | ||
| g.103034C>T | c.1297C>T | p.Arg433Trp | EX10 | 5 (1.3) | — | p.Ala155TrpfsX56 in one patient | |
| g.103251C>T | c.1422C>T | p.Cys474Cys | EX11 | 1 (0.3) | — | ||
| g.103373G>C | c.1544G>C | p.Gly515Ala | EX11 | rs25647 | 3 (0.8) | 1 (0.4) | p.Met1? in one patient |
The 23 pathogenic mutations included a total of 20 mutations that are predicted to result in premature termination of the PGRN coding sequence. This group of mutations included nonsense ( N =4), frameshift ( N =12) and splice-site ( N =4) mutations. The truncating mutations were identified scattered over the PGRN gene in nine different exons but not in the 3′ end of exon 11 or in exon 12 (Fig. 1 ). Three additional mutations, two located in exon 1 and one located at the 5′ splice site of exon 1, were identified, which do not cause a simple truncation of the coding sequence but which are nonetheless almost certainly pathogenic. Mutation c.138+1G>A (IVS1+1G>A) is predicted to lead to the splicing out of exon 1, thereby removing the Met1 codon and all associated Kozac sequence, whereas c.2T>C (p.Met?) directly destroys the normal Kozac sequence by mutating the Met1 codon. The third mutation (c.26C>A; p.Ala9Asp) affects the hydrophobic core of the signal peptide sequence. None of the 23 pathogenic coding and splice-site mutations were observed by sequence analysis in 200 unrelated control individuals.
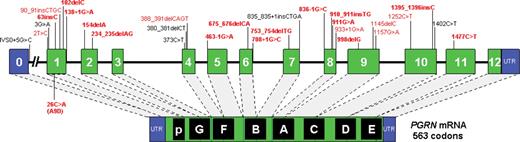
PGRN mutations identified in FTLD patients. Schematic representation of the PGRN gene and the mRNA encoding the PGRN protein, showing all PGRN mutations identified to date. Lettered boxes in the PGRN protein refer to the individual granulin repeats. Mutations are numbered relative to the largest PGRN transcript (GenBank accession number NM_002087 .2). Novel PGRN mutations identified in this study are shown in bold. All PGRN mutations that have been identified in the Mayo Clinic FTLD series are in red.
Segregation analysis for eight different mutations was performed in FTLD families, with at least one other affected family member available for genetic testing. This analysis showed segregation of the PGRN mutations with disease in all analyzed families (Supplementary Material, Fig. S1). Although all FTLD patients in these families carried the relevant PGRN mutation, we did observe five individuals from three different families who carried a pathogenic PGRN mutation but had not developed disease by the age of 60, including one individual without symptoms at the current age of 73.
In contrast to the pathogenic mutations, four of the 13 coding variants with unknown disease significance were observed in control individuals, including the silent mutation c.384T>C (p.Asp128Asp; rs25646) in exon 4 and the missense mutation c.1544G>C (p.Gly515Ala; rs25647) in exon 11, both previously reported in the NCBI dbSNP database ( www.ncbi.nih.gov/SNP ) (Table 2 ). Moreover, three variants were detected in patients already affected by another pathogenic PGRN mutation (Table 2 ). The missense mutation c.313T>C (p.Cys105Arg) was identified in the proband of FTLD family UBC20; however, sequence analyses of four affected and six unaffected relatives excluded segregation of this mutation, with the FTLD phenotype in this family indicating that it is likely a rare benign variant.
Detection of genomic rearrangements in PGRN region
To assess the possible contribution of large genomic insertion/deletion mutations to the overall PGRN mutation frequency in FTLD, we studied 100 patients from the ADRC-FTLD referral series and all FTLD patients ( N =15) from the Olmsted County community-based dementia series. In these patients, we applied long-range PCR analyses covering the complete PGRN coding region in either a single 4 kb fragment or in three 2 kb overlapping PCR fragments to detect large internal PGRN mutations. However, no evidence for large internal genomic rearrangements in PGRN , in either patient series, was found.
In addition, semi-quantitative PCR-based assays of exons 1 and 12 were performed in the complete FTLD population. These analyses have thus far shown no evidence of PGRN copy-number alterations consistent with genomic deletions or multiplications affecting the 5′ and 3′ ends of PGRN or the entire gene.
Frequency of PGRN mutations in the FTLD patient series
Pathogenic mutations in PGRN were detected in 39 patients or 10.5% of the Mayo Clinic FTLD series ( N =378) (Table 3 ). Within the subgroup of FTLD patients with a positive family history of a similar dementing disorder ( N =144), the PGRN mutation frequency was considerably higher (Table 3 ). More than one-fifth of the familial patients from our Mayo FTLD series (32 out of 144 analyzed patients or 22.2%) could be explained by mutations in PGRN . Family history was not documented for five PGRN mutation carriers, whereas the FTD phenotype was considered sporadic in two patients (Table 1 ). Patient NA99-175 carrying mutation c.26C>A (p.Ala9Asp) in the signal peptide showed first symptoms of dementia at the early age of 48 years, whereas his parents died at the ages of 66 and 70 years without signs of dementia. For patient A03-52, (c.675_676delCA; p.Ser226TrpfsX28) with an onset age of 56 years, one parent died at the age of 56 years from an unrelated illness, which may explain the lack of a positive family history. A pathological confirmation of the FTLD diagnosis was available for 30 PGRN mutation carriers. In all mutation carriers with immunohistochemical data available ( N =26), neuropathological findings were consistent with FTLD-U with NII, leading to an overall PGRN mutation frequency of 24.7% in the subpopulation of FTLD-U patients.
Type and frequency of PGRN mutations in Mayo FTLD patient series
| Mutation | Total FTLD population | FTLD community-based dementia series | FTLD-ADRC series | ||||||
|---|---|---|---|---|---|---|---|---|---|
| type | All ( N =378) | FH+ ( N =144) | Ub+ ( N =105) | All ( N =15) | FH+ ( N =10) | Ub+ ( N =5) | All ( N =167) | FH+ ( N =64) | Ub+ ( N =21) |
| Frameshift | 18 | 14 | 11 | 3 | 3 | 2 | 5 | 5 | 4 |
| Nonsense | 12 | 12 | 9 | — | — | — | 1 | 1 | — |
| Splice-site | 5 | 5 | 3 | — | — | — | 1 | 1 | 1 |
| Missense | 3 | 1 | 2 | — | — | — | 1 | 1 | — |
| Kozac | 1 | — | 1 | — | — | — | — | — | — |
| Total (%) | 39 (10.3) | 32 (22.2) | 26 (24.7) | 3 (20.0) | 3 (30.0) | 2 (40.0) | 8 (4.8) | 8 (12.5) | 5 (23.8) |
| Mutation | Total FTLD population | FTLD community-based dementia series | FTLD-ADRC series | ||||||
|---|---|---|---|---|---|---|---|---|---|
| type | All ( N =378) | FH+ ( N =144) | Ub+ ( N =105) | All ( N =15) | FH+ ( N =10) | Ub+ ( N =5) | All ( N =167) | FH+ ( N =64) | Ub+ ( N =21) |
| Frameshift | 18 | 14 | 11 | 3 | 3 | 2 | 5 | 5 | 4 |
| Nonsense | 12 | 12 | 9 | — | — | — | 1 | 1 | — |
| Splice-site | 5 | 5 | 3 | — | — | — | 1 | 1 | 1 |
| Missense | 3 | 1 | 2 | — | — | — | 1 | 1 | — |
| Kozac | 1 | — | 1 | — | — | — | — | — | — |
| Total (%) | 39 (10.3) | 32 (22.2) | 26 (24.7) | 3 (20.0) | 3 (30.0) | 2 (40.0) | 8 (4.8) | 8 (12.5) | 5 (23.8) |
FH+, positive family history of dementia; Ub+, pathological diagnosis of FTLD with ubiquitin-positive inclusions.
Type and frequency of PGRN mutations in Mayo FTLD patient series
| Mutation | Total FTLD population | FTLD community-based dementia series | FTLD-ADRC series | ||||||
|---|---|---|---|---|---|---|---|---|---|
| type | All ( N =378) | FH+ ( N =144) | Ub+ ( N =105) | All ( N =15) | FH+ ( N =10) | Ub+ ( N =5) | All ( N =167) | FH+ ( N =64) | Ub+ ( N =21) |
| Frameshift | 18 | 14 | 11 | 3 | 3 | 2 | 5 | 5 | 4 |
| Nonsense | 12 | 12 | 9 | — | — | — | 1 | 1 | — |
| Splice-site | 5 | 5 | 3 | — | — | — | 1 | 1 | 1 |
| Missense | 3 | 1 | 2 | — | — | — | 1 | 1 | — |
| Kozac | 1 | — | 1 | — | — | — | — | — | — |
| Total (%) | 39 (10.3) | 32 (22.2) | 26 (24.7) | 3 (20.0) | 3 (30.0) | 2 (40.0) | 8 (4.8) | 8 (12.5) | 5 (23.8) |
| Mutation | Total FTLD population | FTLD community-based dementia series | FTLD-ADRC series | ||||||
|---|---|---|---|---|---|---|---|---|---|
| type | All ( N =378) | FH+ ( N =144) | Ub+ ( N =105) | All ( N =15) | FH+ ( N =10) | Ub+ ( N =5) | All ( N =167) | FH+ ( N =64) | Ub+ ( N =21) |
| Frameshift | 18 | 14 | 11 | 3 | 3 | 2 | 5 | 5 | 4 |
| Nonsense | 12 | 12 | 9 | — | — | — | 1 | 1 | — |
| Splice-site | 5 | 5 | 3 | — | — | — | 1 | 1 | 1 |
| Missense | 3 | 1 | 2 | — | — | — | 1 | 1 | — |
| Kozac | 1 | — | 1 | — | — | — | — | — | — |
| Total (%) | 39 (10.3) | 32 (22.2) | 26 (24.7) | 3 (20.0) | 3 (30.0) | 2 (40.0) | 8 (4.8) | 8 (12.5) | 5 (23.8) |
FH+, positive family history of dementia; Ub+, pathological diagnosis of FTLD with ubiquitin-positive inclusions.
In the 15 FTLD patients derived from the community-based dementia population collected in Olmsted County (Minnesota, USA), two different PGRN mutations were detected in a total of three FTLD patients. The two patients carrying the same c.154delA (p.Thr52HisfsX2) mutation were independently ascertained; however, genealogical studies revealed that they were second-degree relatives from the large F142 family (Supplementary Material, Fig. S1). The data obtained in this small FTLD subgroup from Olmsted County can be extrapolated to the entire community-based dementia series of 649 patients, resulting in a PGRN mutation frequency of ∼0.5% in all types of dementia. In the ADRC-FTLD series, we identified mutations in eight FTLD patients (4.8% of 167), each carrying a different mutation (Table 3 ). Importantly, patients were not selected on the basis of family history or neuropathological subtype in this ADRC-FTLD series.
Founder effects of PGRN mutations
A total of 23 different pathogenic mutations were identified in 39 independently ascertained patients from our Mayo Clinic FTLD series. The most frequently observed mutation was c.1477C>T (p.Arg493X) located in exon 11, which was identified in eight independently ascertained FTLD patients. Five other mutations were observed more than once: mutations c.26C>A (p.Ala9Asp), c.154delA (p.Thr52HisfsX2) and c.675_676delCA (p.Ser226TrpfsX28) were identified in three patients, and c.102delC (p.Gly35GlufsX19), c.234_235delAG (p.Gly79AspfsX39) and c.1252C>T (p.Arg418X) were identified in two patients each (Table 1 ). To examine if patients carrying the same mutation could have had a common founder, we performed haplotype analyses with seven STR markers spanning a 7.5 Mb region around PGRN . The common c.1477C>T (p.Arg493X) mutation was identified in family PPA3, for which DNA of one additional affected and one unaffected individual was available, resulting in the unambiguous reconstruction of a disease haplotype in this family. When this haplotype was compared with individual genotype data of the seven additional patients carrying the c.1477C>T (p.Arg493X) mutation, we observed shared alleles between all patients for five consecutive STR markers spanning a 5.1 Mb region between D17S1299 and TAUPROM (Table 4 ). Shared haplotype analyses also supported a common genetic origin for each of the other six PGRN mutations that were observed in multiple independently ascertained FTLD patients (data not shown).
Shared haplotype analyses for PGRN p.Arg493X mutation in eight FTLD families
| Marker | Position | Frequency | PGRN p.Arg493X FTLD families | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (Mb) | (%) | PPA-3 | NA01-249 | NA02-297 | F144-1 | 9118 | A02-43 | NP12900 | 05-44 | |
| D17S1814 | 35.70 | 21.4 | 162 | 161-161 | 161-161 | 150- 162 | 166- 162 | 154- 162 | 164- 162 | 155-161 |
| D17S1299 | 36.20 | 19.3 | 200 | 196- 200 | 200- 200 | 208- 200 | 196- 200 | 200- 200 | 196- 200 | 196- 200 |
| D17S951 | 39.18 | 25.0 | 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 |
| c.1477C>T | 39.78 | T | C- T | C- T | C- T | C- T | C- T | C- T | C- T | |
| D17S934 | 40.41 | 16.1 | 180 | 180- 180 | 174- 180 | 180- 180 | 184- 180 | 182- 180 | 176- 180 | 174- 180 |
| D17S950 | 40.62 | 11.1 | 190 | 190- 190 | 184- 190 | 190- 190 | 180- 190 | 192- 190 | 178- 190 | 188- 190 |
| TAUPROM | 41.33 | 1.0 | 363 | 359- 363 | 359- 363 | 377- 363 | 345- 363 | 359- 363 | 361- 363 | 377- 363 |
| D17S806 | 43.17 | 1.9 | 181 | 169- 181 | 163- 181 | 173- 181 | 181- 181 | 173- 181 | 181- 181 | 173-175 |
| Marker | Position | Frequency | PGRN p.Arg493X FTLD families | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (Mb) | (%) | PPA-3 | NA01-249 | NA02-297 | F144-1 | 9118 | A02-43 | NP12900 | 05-44 | |
| D17S1814 | 35.70 | 21.4 | 162 | 161-161 | 161-161 | 150- 162 | 166- 162 | 154- 162 | 164- 162 | 155-161 |
| D17S1299 | 36.20 | 19.3 | 200 | 196- 200 | 200- 200 | 208- 200 | 196- 200 | 200- 200 | 196- 200 | 196- 200 |
| D17S951 | 39.18 | 25.0 | 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 |
| c.1477C>T | 39.78 | T | C- T | C- T | C- T | C- T | C- T | C- T | C- T | |
| D17S934 | 40.41 | 16.1 | 180 | 180- 180 | 174- 180 | 180- 180 | 184- 180 | 182- 180 | 176- 180 | 174- 180 |
| D17S950 | 40.62 | 11.1 | 190 | 190- 190 | 184- 190 | 190- 190 | 180- 190 | 192- 190 | 178- 190 | 188- 190 |
| TAUPROM | 41.33 | 1.0 | 363 | 359- 363 | 359- 363 | 377- 363 | 345- 363 | 359- 363 | 361- 363 | 377- 363 |
| D17S806 | 43.17 | 1.9 | 181 | 169- 181 | 163- 181 | 173- 181 | 181- 181 | 173- 181 | 181- 181 | 173-175 |
Shared haplotype analyses for PGRN p.Arg493X mutation in eight FTLD families
| Marker | Position | Frequency | PGRN p.Arg493X FTLD families | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (Mb) | (%) | PPA-3 | NA01-249 | NA02-297 | F144-1 | 9118 | A02-43 | NP12900 | 05-44 | |
| D17S1814 | 35.70 | 21.4 | 162 | 161-161 | 161-161 | 150- 162 | 166- 162 | 154- 162 | 164- 162 | 155-161 |
| D17S1299 | 36.20 | 19.3 | 200 | 196- 200 | 200- 200 | 208- 200 | 196- 200 | 200- 200 | 196- 200 | 196- 200 |
| D17S951 | 39.18 | 25.0 | 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 |
| c.1477C>T | 39.78 | T | C- T | C- T | C- T | C- T | C- T | C- T | C- T | |
| D17S934 | 40.41 | 16.1 | 180 | 180- 180 | 174- 180 | 180- 180 | 184- 180 | 182- 180 | 176- 180 | 174- 180 |
| D17S950 | 40.62 | 11.1 | 190 | 190- 190 | 184- 190 | 190- 190 | 180- 190 | 192- 190 | 178- 190 | 188- 190 |
| TAUPROM | 41.33 | 1.0 | 363 | 359- 363 | 359- 363 | 377- 363 | 345- 363 | 359- 363 | 361- 363 | 377- 363 |
| D17S806 | 43.17 | 1.9 | 181 | 169- 181 | 163- 181 | 173- 181 | 181- 181 | 173- 181 | 181- 181 | 173-175 |
| Marker | Position | Frequency | PGRN p.Arg493X FTLD families | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (Mb) | (%) | PPA-3 | NA01-249 | NA02-297 | F144-1 | 9118 | A02-43 | NP12900 | 05-44 | |
| D17S1814 | 35.70 | 21.4 | 162 | 161-161 | 161-161 | 150- 162 | 166- 162 | 154- 162 | 164- 162 | 155-161 |
| D17S1299 | 36.20 | 19.3 | 200 | 196- 200 | 200- 200 | 208- 200 | 196- 200 | 200- 200 | 196- 200 | 196- 200 |
| D17S951 | 39.18 | 25.0 | 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 | 170- 180 | 172- 180 |
| c.1477C>T | 39.78 | T | C- T | C- T | C- T | C- T | C- T | C- T | C- T | |
| D17S934 | 40.41 | 16.1 | 180 | 180- 180 | 174- 180 | 180- 180 | 184- 180 | 182- 180 | 176- 180 | 174- 180 |
| D17S950 | 40.62 | 11.1 | 190 | 190- 190 | 184- 190 | 190- 190 | 180- 190 | 192- 190 | 178- 190 | 188- 190 |
| TAUPROM | 41.33 | 1.0 | 363 | 359- 363 | 359- 363 | 377- 363 | 345- 363 | 359- 363 | 361- 363 | 377- 363 |
| D17S806 | 43.17 | 1.9 | 181 | 169- 181 | 163- 181 | 173- 181 | 181- 181 | 173- 181 | 181- 181 | 173-175 |
Phenotype of PGRN mutation carriers
In the FTLD patients with pathogenic mutations in PGRN , the mean age at onset of dementia was 59±7 years ( N =38), with a mean age at death of 65±8 years ( N =29) (Table 1 ). The clinical presentation was similar in the population of patients without PGRN mutations, although the age at death (70±12 years) was slightly later in non-carriers. However, as expected from the autosomal dominant FTLD-U families previously linked to chromosome 17 ( 10–12 ), a broad range in both the onset of dementia (48–83 years) and the age at death (53–87 years) was observed. No obvious correlation was noted between the onset of the first clinical symptoms and the location of each mutation in PGRN . In fact, variable onset ages were also observed for patients carrying identical PGRN mutations, with onset ages ranging from 48 to 69 years for carriers of the c.1477C>T (p.Arg493X) mutation and from 48 to 63 years for the c.26C>A (p.Ala9Asp) mutation. Using information on 68 affected and 16 asymptomatic PGRN mutation carriers, we generated a liability curve emphasizing the age-related disease penetrance for PGRN mutations and showed that only 50% of mutation carriers were affected by the age of 60, whereas >90% of carriers were affected at 70 years of age (Fig. 2 ). Clinically, FTD ( N =17) and PPA ( N =7) were the most frequently observed diagnoses, with language dysfunction as a prominent presenting symptom in 24% of the mutation carriers, compared with only 12% of the patients not carrying PGRN mutations. Notably, one patient (currently alive) was clinically diagnosed with corticobasal syndrome (CBS). Two patients received a clinical diagnosis of Alzheimer's disease (AD) with seizures and seven patients had a movement disorder (Parkinson disease, parkinsonism or FTD-MND). However, neuropathological autopsy findings for these nine patients were consistent with FTLD-U. Pathological confirmation of the clinical FTLD diagnoses was available for the majority of the mutation carriers (30/39, 77%) showing FTLD-U pathology with both cytoplasmic and NII in all patients for which exhaustive immunohistochemical data were available (26/30, 87%).
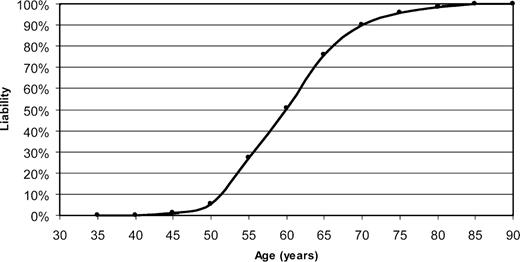
Liability curve for PGRN mutation carriers. The distribution of the age-related penetrance in PGRN mutation carriers identified in this study is shown. The disease penetrance was calculated in age groups of 5 years, starting at 30 years and ending at 85 years. A total of 69 affected and 16 non-affected PGRN mutation carriers were included in the analyses.
The variable onset age of dementia observed for many PGRN mutations and the potential incomplete penetrance associated with the PGRN IVS0+5G>C mutation, previously reported in a Belgian FTD population ( 20 ), emphasized the potential impact of modifying factors on the clinical presentation of FTLD in PGRN mutation carriers. Therefore, we analyzed the effect of the genotypes of the Apolipoprotein E ( APOE) gene and the extended H1 and H2 haplotypes of MAPT on the clinical presentation of FTLD in all mutation carriers identified in this study. No obvious effect on age of onset or age at death could be observed for the MAPT haplotypes, either within extended FTLD families or when all FTLD patients with null mutations in PGRN were included in the analysis (mean age at onset for H1/H1 carriers was 59±8 years and for H1/H2 carriers 59±7 years). Unexpectedly, patients carrying at least one APOE ε4 allele showed a significantly later disease onset (63±7 years; N =10) compared with APOE ε3ε3 carriers (57±7 years; N =29) ( P =0.01, unpaired t -test); however, this finding needs confirmation in larger patient series.
Mechanistic analyses of novel PGRN mutations
In five FTLD patients, we identified mutations affecting the splice sites of PGRN . These mutations are expected to lead to skipping of the affected exons, resulting in a frameshift and premature termination of the coding sequence. For the mutations affecting the 5′ splice site of exon 1 (c.138+1G>A; IVS1+1G>A) and the 3′ splice site of exon 5 (c.463-1G>A; IVS4-1G>A), frontal cortices of patients were available as a source of mRNA to study the effect of these mutations. RT–PCR transcript analyses in F149-1 carrying the c.138+1G>A mutation showed evidence for an aberrant product corresponding to the skipping of exon 1 (268 bp) in addition to the wild-type transcript (413 bp) (Fig. 3 ). The exclusion of exon 1 containing the start methionine codon from the PGRN mRNA is expected to block PGRN protein from being generated, creating a functional null allele. In contrast, no aberrant transcript was identified for patient UBC14-9 carrying c.463-1G>A (IVS4-1G>A) (Fig. 3 ). This mutation is likely to lead to skipping of exon 5 from the PGRN mRNA, resulting in a frameshift and a premature termination codon (PTC) in exon 6 (p.Ala155TrpfsX56) (Fig. 3 ). To determine whether the lack of an aberrant transcript for this mutation resulted from the specific degradation of mutant RNA (by NMD), we determined whether the brain RNA in this patient was derived from both PGRN alleles. To do this, we examined brain mRNA from patient UBC14-9 for the presence of the sequence variant c.1297C>T (p.Arg433Trp) in exon 10, which occurred on the opposite chromosome. Genomic DNA from patient UBC14-9 is heterozygous for this mutation, with the C-allele segregating on the disease haplotype. Comparison of sequence traces of PGRN exon 10 in gDNA and mRNA prepared from frontal cortex of patient UBC14-9 confirmed the absence of mutant RNA (C-allele) (Fig. 4 ). The three additional splice-site mutations identified in this study are all predicted to cause frameshifts and premature termination of the coding sequence and are therefore also likely to create null alleles through the degradation of the mutant RNAs; however, this could not be confirmed because a source of RNA was not available. In this group of splice-site mutations, c.708+1G>C (IVS6+1G>C) is expected to result in skipping of exon 6, resulting in a frameshift and a PTC in exon 7 (Val200GlyfsX18), whereas mutations c.836-1G>C (IVS7-1G>C) and c.933+1G>A (IVS8+1G>A) are both predicted to lead to skipping of exon 8, resulting in a frameshift and premature termination of translation in exon 9 (Val279GlyfsX5).
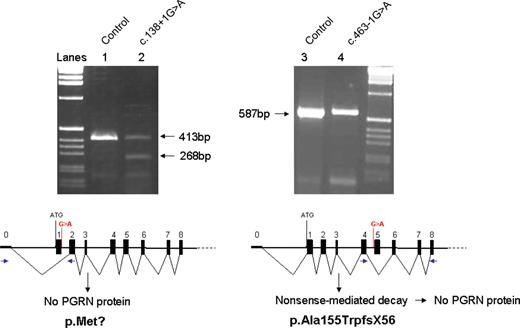
Transcript analyses of novel PGRN splice-site mutations. Agarose gel-electrophoresis of PGRN PCR amplicons obtained from first-strand cDNA prepared from frontal cortex of patients F149-1 (c.138+1G>A; lane 2) and UBC14-9 (c.463-1G>A; lane 4). The analysis of a control individual is included to show the expected transcript lengths for PGRN cDNA exons 0–2 (413 bp, lane 1) and PGRN cDNA exons 4–8 (587 bp, lane 3). For each mutation, a schematic presentation of the predicted splicing of the mutant PGRN transcript is shown. The positions of the mutations are indicated in red, and the locations of the PCR primers are in blue.
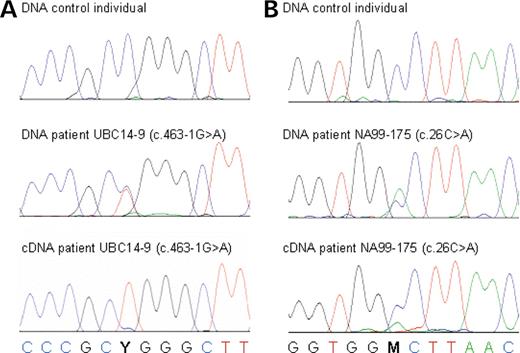
Nonsense-mediated decay of novel PGRN missense and splice-site mutation. Sequence traces of PGRN mutation c.1297C>T (p.Arg433Trp) located in exon 10, in gDNA and in cDNA prepared from frontal cortex of patient UBC14-9 carrying c.463-1G>A (IVS4-1G>A) ( A ) and of mutation c.26C>A (p.Ala9Asp) located in the signal peptide in gDNA and in cDNA prepared from frontal cortex of patient NA99-175 ( B ). Sequence traces from a control individual are included to show wild-type DNA sequence. For both mutations, a strong reduction in the levels of mutant cDNA is observed.
The missense mutation c.26C>A (p.Ala9Asp) was identified in three independently ascertained FTLD patients and is located in the hydrophobic core of the PGRN signal peptide. To determine whether the mutated PGRN signal peptide sequence resulted in a functional null allele, we compared gDNA and brain cDNA sequence traces of PGRN exon 1 of patient NA99-175 carrying p.Ala9Asp. Surprisingly, a substantial reduction in the amount of mutant RNA (A-allele) compared with wild-type RNA (C-allele) was detected (Fig. 4 ). Whether the mutant RNA is being reduced by a mechanism similar to NMD or by a different process will require additional mechanistic studies in lymphoblast cell lines derived from these patients.
DISCUSSION
The recent identification of mutations in PGRN , which cause ubiquitin-positive FTLD, finally established the presence of locus heterogeneity at chromosome 17q21 and explained why multiple large FTLD families with conclusive linkage to this region had previously failed to show MAPT mutations. To determine the frequency and the spectrum of mutations in PGRN in FTLD and dementia in general, we conducted the first systematic screen for PGRN mutations in an extended Mayo Clinic FTLD series comprising 378 patients. We identified 23 different PGRN mutations in 39 patients, together explaining the disease in 10.5% of this population and in 22.2% of FTLD patients with a positive family history of dementia. If the analysis was restricted to patients with a neuropathological diagnosis of FTLD-U, the frequency of PGRN mutations was 24.7%.
The estimate for the PGRN mutation frequency (10.5%) in the 378 FTLD patients in this entire series is subject to considerable ascertainment bias. As a consequence of the inclusion of a significant number of patients from tertiary referral centers ( N =196), our population was enriched for familial FTLD patients ( N =144) and for patients with a pathological diagnosis of FTLD-U ( N =105). Indeed, within the tertiary-center referred subgroup of patients, the frequency of PGRN mutations was considerably higher, with 28 out of 196 (14.3%) mutation carriers or 21 out of 71 (29.6%) in familial patients. To obtain a more representative estimate of the PGRN mutation frequency, we separately analyzed a subgroup of 167 patients from an unselected population of FTLD patients that had been ascertained by five NIH ADRC-funded centers (Mayo Clinics in Rochester, Jacksonville and Scottsdale, UCSF, UCLA) between 1990 and 2006. In this ADRC-FTLD series, the PGRN mutation frequency was significantly lower: eight PGRN mutation carriers accounting for 4.8% of the population were identified. An overall PGRN mutation frequency of around ∼5% is therefore likely to be closer to the actual frequency of PGRN mutations that will be encountered in Neurology Clinics among patients with all forms of FTLD. This estimate for the PGRN mutation frequency in unselected FTLD patients is lower than the figure of 10.7% previously obtained from a similar series of 105 FTD patients from Belgium ( 20 ). However, in the Belgium population, eight out of 11 patients carried the same IVS0+5G>C PGRN mutation, reflecting a strong founder effect in this population. Not surprisingly, we did not observe a founder effect on a similar scale in our US patient population, presumably explaining the lower frequency estimate obtained from our ADRC-FTLD referral series. Previously, MAPT mutation analyses in the ADRC-FTLD referral subgroup had identified seven patients with pathogenic MAPT mutations accounting for 4.4% of the population ( MAPT mutations in 7/160 analyzed patients). Together, these data suggest that PGRN and MAPT mutations will have similar frequencies in the general FTLD patient population.
To further assess the contribution of PGRN mutations to dementia in general, we analyzed FTLD patients ascertained as part of the Olmsted County (Minnesota) community-based dementia study. Out of 649 dementia patients (all diagnoses) collected between 1987 and 2006, 15 patients (2.3%) were diagnosed with FTLD. In total, we identified three PGRN mutation carriers in this population, leading to a mutation frequency of 20% within this small group of FTLD patients and 0.5% for all types of dementia. The actual frequency of PGRN mutations in the general dementia population may be even higher, as PGRN mutations were not excluded in patients with other types of dementia.
As expected from our previous study ( 19 ), the pathogenic mutations identified in the current Mayo Clinic FTLD patient series predominantly included nonsense ( N =4) and frameshift ( N =12) mutations ( 19 ). We previously showed that mutations of this type introduce PTCs that induce the degradation of the mutant RNA by NMD ( 19 ). This results in functional null alleles with the accompanying reduction in PGRN protein. NMD is a surveillance mechanism designed to protect the organism against the deleterious effects of truncated proteins that would be produced if the nonsense transcripts were stable ( 33 ). The general requirements for the induction of NMD are the presence of at least one intron downstream of the PTC and the location of the PTC at least 50–55 nucleotides upstream of the 3′ most exon–exon junction ( 34 , 35 ). All the nonsense and frameshift mutations identified in this study introduce PTCs that fulfill these criteria, with the most C-terminal PTC introduced into exon 11 by the c.1395_1396insC (p.Cys466LeufsX46) mutation at 112–115 nucleotides upstream of the 3′ splice site of exon 11. Significantly, PGRN mutations were not identified in exon 12, presumably reflecting the fact that nonsense or frameshift mutations occurring in the most 3′ exon would not activate NMD and therefore would likely result in the production of functional or partially functional PGRN.
In addition to nonsense and frameshift mutations, we identified five splice-site mutations that are expected to result in skipping of the affected exons (exons 1, 5, 6 and 8). The exonic structure of PGRN is organized such that skipping of either one of exons 3, 4, 5, 6, 7 or 8 will result in a frameshift of the coding region and the introduction of a PTC, again initiating NMD leading to the loss of the mutant RNA. In this study, we were able to perform transcript analysis on cDNA isolated from frontal cortices of patients carrying the c.138+1G>A (IVS1+1G>A) and c.463-1G>A (IVS4-1G>A) mutations. In a patient with the 3′ splice-site mutation in exon 5 (c.463-1G>A, IVS4-1G>A), we demonstrated that the PGRN RNA was derived only from the normal allele, consistent with degradation of the mutant RNA by NMD. This result confirmed that this type of splice-site mutation generates a null allele. In contrast, the c.138+1G>A (IVS1+1G>A) mutation is unusual, as it is predicted to result in the skipping of exon 1, leading to the removal of the start methionine codon and the complete block of normal PGRN protein production. RT–PCR transcript analysis in a patient carrying this mutation detected mutant mRNA with exon 1 aberrantly spliced out without any obvious reduction in the level of mutant compared to wild-type RNA (Fig. 3 ). This suggests that the c.138+1G>A (IVS1+1G>A) mutant RNA is not subject to NMD, presumably because of the loss of the normal Kozac sequence. Initiation of NMD requires an initial round of translation, thus absence of PGRN protein production from the mutant c.138+1G>A (IVS1+1G>A) transcript, likely prevents the mutant RNA from being degraded by NMD ( 33 ).
In three FTLD patients, we identified a missense mutation at position −9 in the hydrophobic core region of the signal peptide sequence of PGRN (c.26C>A; p.Ala9Asp), introducing a charged residue into the mutant signal peptide (residues are numbered negatively starting from the site of cleavage toward the N-terminus). Signal peptides fulfill a critical role in the targeting of proteins to the ER and translocation of proteins across the ER membrane ( 36 ). Cotranslational targeting of a protein to the ER is initiated when the signal recognition particle (SRP) interacts with the hydrophobic region present at the N-terminus of the nascent chain. It has been shown that introduction of only a single charged amino acid into this hydrophobic core renders that sequence non-functional for SRP-mediated targeting, thereby blocking the targeting and translocation of protein across the ER membrane. The importance of the signal peptide for proper protein localization led us to suggest that unlike all other PGRN missense mutations identified in this study, c.26C>A (p.Ala9Asp) is likely pathogenic. This hypothesis was supported by analyses that showed that all three FTLD patients carrying the c.26C>A (p.Ala9Asp) mutation shared a haplotype consistent with a common genetic founder. Moreover, RT–PCR transcript analyses showed strongly reduced levels of mutant RNA in the frontal cortex of a patient carrying the c.26C>A (p.Ala9Asp) mutation. The selective loss of mutant RNA by a mutation that causes a disruption of the signal peptide and presumably mislocalization of the PGRN protein is somewhat surprising. Currently, the mechanism that leads to the loss of mutant c.26C>A RNA is unclear; however, it is possible that the close link between mRNA translation and signal peptide recognition allows the induction of a process similar to NMD. Alternatively, selective silencing of the mutant allele may be induced. Regardless, c.26C>A (p.Ala9Asp) should be considered pathogenic, as this mutation will cause loss of functional PGRN and/or mislocalization of the PGRN protein.
Our PGRN mutation analyses further identified 13 rare missense and silent sequence variants with unknown pathogenic significance in the FTLD patient population and four additional missense variants (p.Thr18Met, p.Pro134Leu, p.Met286Thr and p.Ser301Pro) in control individuals. On the basis of their presence in controls (p.Asp33Asp, p.Asp128Asp, Arg418Gln and p.Gly515Ala), their non-segregation with FTLD in a family (p.Cys105Arg) or the concomitant occurrence of another pathogenic PGRN mutation in the same patient (p.Asp33Asp and p.Arg433Trp), these variants likely represent rare benign sequence variants unrelated to FTLD. Interestingly, one of the missense mutations c.1297C>T (p.Arg433Trp) was identified in five unrelated FTLD patients, whereas it was absent in control individuals. Although one of these patients also carried the c.463-1G>A (IVS4-1G>A) splice-site mutation in PGRN , which results in the loss of PGRN mRNA, we cannot exclude that this patient may be a compound heterozygote, carrying two pathogenic mutations. However, the location of the Arg433 residue outside of the functional granulin repeat units supports the idea that c.1297C>T (p.Arg433Trp) is likely to be non-pathogenic.
It is interesting that no missense mutations outside of the signal peptide have thus far been unambiguously shown to be pathogenic by causing loss of PGRN function. This notable absence might be explained by the presence of 7.5 highly conserved cysteinyl repeats in PGRN. This repeat structure may introduce considerable functional redundancy into the protein such that if one repeat unit is disrupted by a missense change, the other repeats maintain normal or near normal levels of PGRN function. The fact that these PGRN repeat units can be proteolytically cleaved to multiple independent, biologically active, granulin peptides is consistent with this hypothesis.
In two patients, silent sequence variants were identified, which affect the last nucleotide of exon 2 (c.264G>A; p.Glu88Glu) and exon 6 (c.708C>T; p.Asn236Asn), respectively, and therefore potentially affect exonic splicing. However, RT–PCR transcript analyses performed on patient A02-83 carrying c.708C>T could not detect evidence of exon 6 skipping and showed normal expression levels of mutant and wild-type RNA. This implied that this mutation does not significantly alter exonic splicing and is therefore also unlikely to be disease-related.
A comparison of the clinical phenotype in all patients carrying pathogenic PGRN mutations that were identified in this study (Table 1 , Supplementary Material, Fig. S1) showed highly variable ages of disease onset and death. However, the clinical presentation did not correlate with the actual position or type of mutation in PGRN . This presumably reflects the fact that all PGRN mutations have a similar effect on PGRN production—the creation of a functional null allele. Analyses of APOE genotypes and extended MAPT haplotypes in PGRN mutation carriers indicated that the APOE ε4 allele might explain a proportion of the variability, although this will require confirmation in a larger group of PGRN mutation carriers, whereas MAPT haplotypes showed no effect on age of onset or age at death. Using the combined data of more than 80 mutation carriers identified in this study, we constructed a liability curve for PGRN mutation carriers. This curve emphasizes the highly variable onset ages of FTLD in mutation carriers and demonstrates that a significant proportion of patients remain unaffected until old age (only 50% of the carriers are affected by the age of 60). These findings are consistent with the previously published variable disease onset in 17-linked FTLD-U families [UBC17 ( 10 ), 1083 ( 11 ), DR8 ( 12 )] that are now all explained by PGRN mutations ( 19 , 20 ). Although we are aware of the potential caviat that sampling of individual FTLD patients and their relatives in this study has not been performed in a rigorous epidemiological context, the liability curve presented here will have important implications for the design of future diagnostic PGRN screenings. The identification of a PGRN mutation in a patient with dementia will be extremely helpful to establish an accurate differential diagnosis; however, asymptomatic individuals should only consider testing in the context of strict genetic counseling, with ongoing follow-up and management. In addition, the relatively frequent occurrence of rare missense mutations with unknown pathological significance, as detected in both FTLD patients and control individuals in this study, further emphasizes the complexity associated with the clinical validity of PGRN mutations.
In patients with PGRN mutations, language dysfunctions were a common presenting symptom affecting 24% of the carriers compared with only 12% of the non- PGRN mutation patients. An additional feature of the clinical and neuropathological phenotypes in these patients was the absence or near complete absence of MND. Consistent with the rare appearance of MND in PGRN mutation carriers, we also did not observe any pathogenic PGRN mutations in a series of 48 ALS patients. Neuropathological findings showed that all PGRN mutation carriers for which ubiquitin staining was available ( N =26) developed ubiquitin-immunoreactive NCI and NII. The presence of NII in all these patients confirmed that NIIs are a consistent feature of the neuropathology in the PGRN -positive patients; however, NIIs are not specific to PGRN mutation carriers in this FTLD patient series.
The various PGRN mutations identified in this study predict a uniform disease mechanism for all mutations—the loss of functional PGRN (haploinsufficiency). This led us to hypothesize that genomic rearrangements, such as deletions and/or duplications within the PGRN genomic region, could also lead to an FTLD phenotype. However, extensive mutation analyses within the FTLD patient series by both long-range PCR and semi-quantitative fragment-length analysis have thus far not identified evidence of genomic PGRN deletion/multiplication mutations. Although these methods need to be applied to additional patients to determine whether genomic deletion/insertion mutations are a rare cause of PGRN -associated FTLD, our current data do suggest that large deletions and duplications are unlikely to significantly affect the frequency of PGRN mutations estimated in our FTLD patient series. The absence of genomic deletions in this series means that, as yet, no pathogenic PGRN mutations have been identified, which block the transcription of mutant PGRN RNA. This leaves open the possibility that the production of mutant RNA is somehow required for initiation of the neurodegenerative cascade by these mutations. The identification of a large deletion mutation affecting the PGRN genomic region in an FTLD family would thus have important mechanistic implications.
The most frequent mutation identified in this study was c.1477C>T (p.Arg493X), located in exon 11, which was present in eight apparently unrelated FTLD patients, accounting for the disease in 2% of our complete FTLD population. Using haplotype-sharing studies, we demonstrated that this mutation represents a founder mutation with a haplotype of ~5 cM, which is shared between all p.Arg493X FTLD patients. By means of genealogical studies, a common ancestry could further be established for two of these patients (F144-1 and NP12900) who reside in the Southern USA. For all additional PGRN mutations that were identified in multiple apparently unrelated FTLD patients in this study ( N =6), shared haplotypes could be reconstructed, suggesting common genetic origins for each of these mutations. Previously, PGRN mutation IVS0+5G>C was already reported as a founder mutation, explaining the dementia in ~8% of patients from a Belgium FTD population ( 20 ).
The identification of null mutations in PGRN as a cause of FTLD-U represents a landmark research finding that will likely prove critical to our understanding of the disease in this group of patients. In this study, we have demonstrated that PGRN mutations account for ∼20% of familial FTLD patients and for a significant proportion (∼5%) of all FTLD. Moreover, the large number of mutation carriers identified among our patients provided considerable information on the range of different PGRN mutations that are associated with FTLD-U and on the clinical phenotype of these patients. Future studies will need to focus on the mechanism by which PGRN mutations lead to neurodegeneration and, in particular, the relationship between PGRN haploinsufficiency and the formation of the ubiquitin-positive neuronal inclusions that characterize the neuropathology of PGRN mutation carriers and patients with FTLD-U in general.
MATERIALS AND METHODS
FTLD patients and control series for PGRN mutation screening
The Mayo Clinic FTLD series comprised 378 patients, 210 clinically diagnosed FTLD patients and 168 pathologically confirmed FTLD patients. The mean onset age of dementia was 60±11 years (range 32–83). Among the 168 deceased patients, their mean age at death was 70±12 years (range 39–97). The main syndromic clinical diagnoses were FTD and PPA, with rare occurrences of SD, CBS and FTD-MND. Among autopsied patients, FTLD-U was the major neuropathological subtype ( N =105, 62.5%).
The overall Mayo Clinic FTLD series included three patient subgroups: 15 FTLD patients from the Olmsted County community-based dementia series, 167 FTLD patients referred to NIH-funded ADRCs (ADRC-FTLD referral series) and 196 patients ascertained from multiple tertiary referral centers.
The Olmsted County community-based dementia series.
This series included 15 FTLD patients (seven clinical; eight pathological) among 649 patients with clinical dementia collected between 1987 and June 2006 in Olmsted County, Minnesota, USA. This series also contained 536 patients with possible or probable AD, 10 patients with vascular dementia, 36 patients with Lewy-body dementia (LBD) and 52 patients with other neurodegenerative forms of dementia. All FTLD patients (13 FTD and two PPA) were included in the mutation screening. The mean age at onset in the FTLD patients was 65±11 years (range 50–81), mean age at death was 79±12 years (range 54–96) and 67% (10/15) had a positive family history of dementia. The diagnoses were based on clinical findings and imaging.
The ADRC-FTLD patient referral series.
This series comprised 167 FTLD patients ascertained by referral to five NIH ADRC-funded centers: Mayo Clinic ADRC Rochester, Minnesota ( N =66), Mayo Clinic ADC Jacksonville, Florida ( N =51), Arizona ADC Scottsdale, Arizona ( N =11), University of California, San Francisco (UCSF) ADC ( N =21) and University of California, Los Angeles (UCLA) ADC ( N =18). The mean age at onset of dementia in this series was 59±10 years (range 32–83), and 38% had a positive family history of dementia. Primary clinical diagnoses included FTD, PPA, AD, CBS, FTD-MND, posterior cortical atrophy and unspecified dementia. Pathological examination was performed in 28 of the 167 patients and the mean age at death in this group was 71±9 years (range 39–84). The FTLD-U pathological subtype was observed in 21 patients (75%). In addition, three patients were subsequently pathologically diagnosed with CBS, two with AD and one with atypical progressive supranuclear palsy/LBD. All 167 patients included in the PGRN mutation screening received a clinical diagnosis of FTLD.
The tertiary referral series.
A total of 196 FTLD (64 clinical, 132 pathological) patients were ascertained by multiple tertiary referral centers. The majority ( N =112) of patients were obtained through nine brain banks within the USA: the State of Florida Alzheimer's disease Initiative ( N =29), Albert Einstein College of Medicine ( N =7), Northwestern University Feinberg School of Medicine ( N =8), Sun Health Research Institute ( N =18), University of Texas Southwestern Medical Center ( N =14), the Society of Progressive Supranuclear Palsy ( N =5), Boston University ( N =7), Duke University ( N =17) and Harvard Brain Bank ( N =7). The remaining 84 FTLD patients were ascertained through international collaborations with the University of British Columbia (Canada), University of Helsinki (Finland), Lund University Hospital, Lund (Sweden), the National Institutes of Health, Cornell University Medical School and Tronderbrain project from Trondheim, Norway.
Control individuals.
A total of 200 control individuals (mean age 76±10 years) were ascertained through the Mayo Clinics in Jacksonville, Florida and Scottsdale, Arizona.
ALS patient series for PGRN mutation screening
The ALS patient series comprised 48 patients, of which 27 were pathologically confirmed. ALS patients were recruited through the Neuromuscular Clinic, Mayo Clinic Jacksonville ( N =17) and through international collaborations ( N =4). Pathologically confirmed ALS patients were obtained from the Neuropathological Core, Mayo Clinic Jacksonville ( N =17), Harvard Brain Bank ( N =6), Northwestern University Feinberg School of Medicine ( N =1) and Sun Health Research Institute ( N =3). The mean age at onset of ALS was 57 years (range 30–75) and a family history of ALS was present in 40% of the patients.
PGRN gene sequencing
The 12 coding exons of PGRN were amplified by PCR using our previously published primers and protocol ( 19 ). In addition, we developed novel PGRN PCR and sequencing primers designed to amplify up to three PGRN exons in a single fragment, allowing for higher throughput analyses (Supplementary Material, Table S2). PCR primers flanking the non-coding exon 0 and the 3′-UTR of PGRN were also developed (Supplementary Material, Table S2). Standard 25 µl PCR reactions were performed using Qiagen PCR products, with addition of Q-solution for PGRN exons 1–3 and 7–9 (Qiagen, Valencia, CA, USA) and a final concentration of 0.8 µ m for each primer. The annealing temperature for PGRN exons 1–3 and 7–9 was 66°C and for PGRN exons 4–6 and 10–12 was 64°C. The PGRN exon 0 and 3′-UTR fragments were cycled using touchdown protocols of 70–55 and 58–48°C, respectively. PCR products were purified with Multiscreen plates (Millipore, Billerica, MA, USA) and sequenced in both directions on an ABI 3730 with the Big Dye chemistry following manufacturer's protocol (Applied Biosystems, Foster City, CA, USA).
Genomic characterization of PGRN
The presence of internal PGRN genomic insertions/deletions or rearrangements was analyzed by long-range PCR of the complete 4 kb PGRN coding sequence in a single fragment using the Expand Long Template PCR System kit (Roche, IN, USA) and, alternatively, by PCR of three overlapping 2 kb PGRN coding sequence fragments. Long-range PCR reactions were performed with primers GRN1-3F and GRN10-12R (Supplementary Material, Table S2) using the standard PCR protocol (buffer 1) and cycling conditions recommended by the Expand Long Template PCR kit. Pvu II restriction enzyme digest was performed on the 4 kb PCR product, resulting in nine fragments (1463, 608, 474, 445, 275, 271, 182, 142 and 63 bp) that could be readily sized. The smaller 2 kb PGRN coding sequence fragments that spanned exons 1–6, exons 4–9 and exons 7–12 were amplified using the GRN PCR primers developed for high-throughput sequencing (GRN1-3F/GRN4-6R, GRN7-9F/GRN10-12R, GRN4-6F/GRN7-9R) (Supplementary Material, Table S2). Standard 25 µl PCR reactions were performed using Qiagen PCR products with Q-solution using a 66–61°C touchdown protocol for all primer sets and subsequent restriction enzyme digestion with Rsa I (exons 1–6: 727, 588, 467, 79 and 66 bp; exons 4–9: 823, 316, 233, 207, 153 and 90 bp; exons 7–12: 1108, 266, 207, 153, 124, 90 and 83 bp). Agarose gel electrophoresis was performed on Pvu II and Rsa I digested fragments (2% gel) as well as on the undigested (1% gel) PCR products to detect aberrant banding patterns that might indicate the presence of a genomic abnormality. If an aberrant pattern was detected, the PCR reaction was repeated for confirmation and sequencing analyses with internal PGRN primers were performed as described earlier.
PGRN copy-number analyses
To detect duplication or deletion mutations affecting the 5′ or 3′ end of PGRN or the entire PGRN gene, semi-quantitative assessment of genomic copy number for exons 1 and 12 of PGRN was made relative to two endogenous genes: glyceraldehyde-3-phosphate dehydrogenase ( GAPDH ) and β-2-microglobulin (β 2M ). Multiplexed PCRs contained fluorescently labeled primers (Supplementary Material, Table S3) designed to amplify the four products in a single reaction. This allowed relative peak heights to be measured in a linear phase PCR. Comparisons of peak height of the two endogenous amplicons to each other acted as a quality control, and ratios of exons 1 and 12 to both GAPDH and β2M were used to determine relative copy number of PGRN . PCR reactions were performed using 50 ng of DNA in 25 µl for 25 cycles at a 65°C annealing temperature using Qiagen reagents (Qiagen, Valencia, CA, USA) with Q-solution and at a final primer concentration of 0.4 µ m . Each sample was independently assessed in two separate PCRs, one using a 30 s extension time per cycle and the other using 2 min per cycle. Fluorescent amplicons were analyzed twice, on an ABI 3100 and an ABI3730 genetic analyzer, using GENEMAPPER and GENESCAN/GENOTYPER software (Applied Biosystems, CA, USA).
Haplotype sharing studies
Seven different PGRN mutations [c.26C>A (p.Ala9Asp), c.102delC (p.Gly35GlufsX19), c.154delA (p.Thr52HisfsX2), c.234_235delAG (p.Gly79AspfsX39), c.675_676delCA (p.Ser226TrpfsX28), c.1252C>T (p.Arg418X) and c.1477C>T (p.Arg493X)] were identified in multiple independently ascertained patients. To examine whether the FTLD patients with the same PGRN mutation could have shared a common founder, we typed seven STR markers spanning a region of 7.5 Mb flanking PGRN at chromosome 17q21. All markers were amplified with one fluorescently labeled primer, and PCR fragments were analyzed on an automated ABI3100 DNA-analyzer. Alleles were scored using the GENOTYPER software (Applied Biosystems). For six markers (D17S1814, D17S1299, D17S951, D17S934, D17S950 and D17S806), CEPH allele frequencies were used to estimate the allele frequency of the shared alleles in control individuals (CEPH genotype database; http://www.cephb.fr/cephdb/ ). The novel marker TAUPROM was PCR amplified using TAUPROM-F: FAM-ACCGCGGCCAGCCATAACTCT and TAUPROM-R: ATCAAGGCACCTCAACATAATAAT, and allele frequencies were estimated in a population of 180 unrelated control individuals.
APOE and MAPT genotyping in PGRN mutation carriers
PGRN mutation carriers were genotyped for the extended H1 and H2 MAPT haplotypes based on the previously described H2 variant rs1052553, using a Taqman SNP genotyping assay on the 7900HT Fast Real Time PCR system ( 37 ). Genotype calls were made using the SDS v2.2 software (Applied Biosystems). APOE alleles were determined as described previously ( 38 ).
RT–PCR analysis of PGRN RNA
To determine whether specific novel mutations in PGRN caused loss of the mutant RNA by NMD or similar mechanism, PGRN RNA was analyzed where frozen brain tissue was available. Frontal brain tissue from patients F149-1 (c.138+1G>A; IVS1+1G>A), NA99-175 (c.26C>A; p.Ala9Asp), UBC14-9 (c.463-1G>A; IVS4-1G>A) and A02-83 (c.708C>T; p.Asn236Asn) was homogenized and total RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized starting from total RNA with random hexamer and oligo-dT primers using the Superscript II First-Strand Synthesis System for RT–PCR kit (Invitrogen). PCR was performed on brain cDNA, using primers spanning exon 1 in patients carrying c.138+1G>A (IVS1+1G>A) and c.26C>A (p.Ala9Asp) (cEX0F: CAGGGAGGAGAGTGATTTG; cEX2R: GCAGAGCAGTGGGCATCAAC) and using primers spanning exon 6 in patient A02-83 carrying the silent c.708C>T (p.Asn236Asn) mutation (cEX4F: TGCTGTGTTATGGTCGATG; cEX8R: GTACCCTTCTGCGTGTCAC). The same cEX4F and cEX8R primers were used for cDNA analysis of patient UBC14-9 carrying the c.463-1G>A (IVS4-1G>A) mutation in intron 4. In addition, we performed PCR on brain cDNA of patient UBC14-9 with primers spanning exon 10 to determine the number of transcribed alleles based on the presence of the c.1297C>T (p.Arg433Trp) missense mutation (cEX8F: ATACCTGCTGCCGTCTAC; cEX11R: ACGTTGCAGGTGTAGCCAG). RT–PCR products were analyzed on a 2% agarose gel and sequenced to determine the relative amounts of wild-type and mutant mRNA.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
ACKNOWLEDGEMENTS
The authors wish to acknowledge and thank the families who contributed samples that were critically important to the completion of this study; Bruce Miller (UCSF), Joel Kramer (UCSF) and Mario Mendez (UCLA) for provision of clinical FTLD samples for the ADRC referral patient series as part of NIA funded project RO1 AG023195 (D. Knopman, PI); Dr Kevin Boylan (Mayo Clinic) for providing the ALS patients; Francine Parfitt, Nancy Haukom, Rita Fletcher, Josie Pagdangannon, Margaret Turk and Audrey Strongosky (all Mayo Clinic) for collection and sampling of FTLD patient series; Manjari Mishra, PhD (Northwestern University Brain Bank) and The Harvard Brain Tissue Resource Center (supported by MH/NS31862) for FTLD brain tissue samples; Dr Jan Aalsy and the Tronderbrain project staff, The FTD research team at Vancouver Coastal Health and the University of British Columbia all for ascertainment and follow-up of specific FTLD familial patients. The work was supported by NIH grants P01 AG017216 and R01 AG026251 (to M.H.), P50-NS40256-06 (to D.D. and Z.W.), the Mayo Clinic ADRC (P50 AG16574, R. Petersen, PI; to M.H., B.B. and R.P.), The Mayo Clinic Research Foundation (to M.H. and D.D.) and the Robert and Clarice Smith Fellowship program (to S.M.). In addition, M.M. was funded by the Alzheimer's Disease Core Center grant to Northwestern University (P30 AG013854, M. Mesulam, PI), S.W. by a National Alzheimer's Disease Coordinating Center Grant and E.B. by a grant from the Association for Frontotemporal Dementia. I.R.M. and H.F. were funded by the Canadian Institutes of Health Research Operating Grant no. 74580. C.L.W. was funded by NIH ADC grant AG12300 and by the Winspear Family Center for Research on the Neuropathology of Alzheimer's Disease. The Sun Health Research Institute Brain Donation Program (T.G.B.) is supported by the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center) and the Arizona Biomedical Research Commission (contract 0011, Arizona Parkinson's Disease Center). S.M. is a Ruth L. Kirschstein NRSA postdoctoral fellow (AG024030). S.M.P.-B. received grants from the Medical Research Council (UK) and Motor Neuron Disease Association. R.R. is a postdoctoral fellow of the Fund for Scientific Research Flanders (FWO-F) and a visiting scientist from the Neurodegenerative Brain Diseases Group of the Department of Molecular Genetics, VIB, University of Antwerp, Belgium.
Conflict of Interest statement. The authors declare that they have no competing financial interests.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}