




Abstract
The pure hereditary spastic paraplegias (HSPs) are a group of conditions in which there is a progressive length-dependent degeneration of the distal ends of the corticospinal tract axons, resulting in spastic paralysis of the legs. Pure HSPs are most frequently inherited in an autosomal-dominant pattern and are commonly caused by mutations either in the SPG4 gene spastin or in the SPG3A gene atlastin. To identify binding partners for spastin, we carried out a yeast two-hybrid screen on a brain cDNA library, using spastin as bait. Remarkably, nearly all of the positive interacting prey clones coded for atlastin. We have verified the physiological relevance of this interaction using co-immunoprecipitation, glutathione S-transferase pull-down and intracellular co-localization experiments. We show that the spastin domain required for binding to atlastin lies within the N-terminal 80 residues of the protein, a region that is only present in the predominantly cytoplasmic, full-length spastin isoform. These data suggest that spastin and atlastin function in the same biochemical pathway and that it is the cytoplasmic function of spastin which is important for the pathogenesis of HSP. They also provide further evidence for a physiological and pathological role of spastin in membrane dynamics.
INTRODUCTION
The hereditary spastic paraplegias (HSPs) are a diverse group of genetically determined neurodegenerative conditions that all share the principal clinical feature of progressive spastic paralysis of the lower limbs. They are conventionally classified as pure or, when spastic paraplegia is accompanied by other prominent clinical manifestations, complex (1–3). Pure HSP is generally regarded as the most common type in Northern Europe and North America (4). In the pure HSPs, the most important cells involved are the corticospinal tract motor neurons (often termed ‘upper motor neurons’), and the clinical picture of progressive spastic paraplegia is reflected in pathological findings of progressive, length-dependent, distal ‘dying-back’ degeneration of the corticospinal tract axons (1). Identification of the genes involved in HSPs and elucidation of the function of the corresponding proteins is beginning to provide important insights into molecular pathways that are important for axonal maintenance and motor neuronal function.
Eight autosomal-dominant pure HSP loci have been mapped by genetic linkage studies (reviewed in 1,2,5). Rapid progress has been made in identifying the disease genes at these loci, with five, atlastin (SPG3A), spastin (SPG4), NIPA1 (SPG6), KIF5A (SPG10) and HSP60 (SPG13), having been isolated to date (6–10). Mutations in spastin and atlastin are the most frequent and are estimated to account for 40 and 10% of autosomal-dominant pure HSP families, respectively (5,6,11–13).
The spastin transcript is widely expressed within the nervous system and in non-neuronal tissues, but is most strongly expressed in subclasses of neurons (7,14,15). Immunolocalization studies in a variety of tissues and cell types have generally suggested that the protein has both a cytoplasmic and nuclear distribution (14–17). Recently, it has become clear that this is because of differential localization of two main isoforms of the protein: an N-terminal truncated form is nuclear and cytoplasmic, while a less abundant full-length form is actively exported from the nucleus to the cytoplasm (18). The cytoplasmic distribution was described as punctate and perinuclear in one study (16), whereas in another, spastin was found in cytoplasmic cellular regions containing dynamic microtubules, which in a neuronal cell line included the distal axon and axonal branch points (17). In human nervous tissues, spastin was located in the cytoplasm and synaptic terminals of several neuronal cell types, including motor neurons, although in other neuronal types, it was detected in the nucleus (15).
With regard to the cytoplasmic function of spastin, two main, perhaps related, cellular roles have been proposed. Spastin is an AAA ATPase closely related in sequence to p60 katanin, a microtubule severing protein, and a variety of data suggest that spastin is likely to have a role in severing microtubules (19,20). In cultured cells expressing epitope-tagged spastin, expression of the protein was associated with the appearance of broken microtubule bundles within the cytoplasm. In these experiments, the protein localized to discrete punctate cytoplasmic structures not corresponding to known organelles, but reminiscent of the punctate staining seen in some antibody studies of the endogenous protein (19). Further, expression of mutated ATPase-defective spastin resulted in striking cytoplasmic filaments that contained bundled microtubules (19,21). These results are compatible with the localization of endogenous spastin to cytoplasmic regions rich in dynamic microtubules. Data from Drosophila models also support a relationship between Drosophila spastin (D-spastin) and microtubule organization in neurons and other tissues (22,23,24).
Aside from its role in microtubule regulation, it has also been suggested that spastin may be involved in intracellular membrane traffic events. This suggestion was first made when it was recognized that spastin has an N-terminal microtubule interacting and trafficking (MIT) domain (25,26). Most, if not all, other proteins that contain this domain have roles in membrane traffic (26). Evidence for involvement of spastin in membrane traffic events was recently strengthened by our identification of the endosomal protein CHMP1B as a binding partner for spastin (27). CHMP1B localizes to endosomes and is associated with ESCRT-III (endosomal sorting complex required for transport-III), a protein complex which functions in the sorting of mono-ubiquinated proteins to the multivesicular body, a prelysosomal endocytic compartment (reviewed in 27). This study also identified a Golgi membrane protein, gp25L2, as a possible binding partner for spastin, although this interaction has not yet been characterized fully (27). As many membrane traffic events are linked to microtubule-based transport, it is not inconceivable that the membrane traffic and microtubule-regulating roles of spastin may be inter-related.
The atlastin gene consists of 14 exons, which code for a protein that is 558 amino acids long (6). The gene is expressed in many body tissues, with highest expression in brain and spinal cord (6). With rare exceptions, mutations in atlastin cause childhood onset HSP. Most atlastin mutations published to date have been missense mutations, with one frameshift mutation described (28–34).
The atlastin protein is a novel GTPase that is similar to members of the dynamin family of large GTPases (6). It has two putative transmembrane domains with membrane topology resulting in the N- and C-termini being exposed to the cytoplasm (35). The GTPase site is active and is formed from residues at three motifs, the P-loop, DxxG and RD motifs. The protein may oligomerize to form homotetramers (35).
The cellular distribution of atlastin has been studied in rat brain sections (35). Here, atlastin positive cells were most abundant in lamina V of the cerebral cortex, with both the neuronal soma and dendritic tree containing atlastin. At the subcellular level in cultured rat subcortical neurons, atlastin was predominantly located in the cis-Golgi apparatus, with punctate staining also seen in axons and dendrites. Some limited co-localization with endoplasmic reticulum (ER) markers was also seen (35). The precise functional role of atlastin at the Golgi membrane or in other membrane organelle sites remains to be clarified.
In this study, we set out to gain additional insights into the function of spastin, by identifying new binding partners for the protein. Our previous spastin yeast two-hybrid screen was directed against an erythroleukaemia cDNA library (27). In order to identify binding partners which might be important for spastin's function in the central nervous system, we repeated the yeast two-hybrid screen using a fetal brain cDNA library. Remarkably, nearly all of the positive interacting clones contained atlastin cDNA, strongly suggesting that these two HSP proteins interact with each other. We have confirmed this potential interaction by co-immunoprecipitation and pull-down experiments, along with co-localization studies using epitope-tagged wild-type and mutant proteins in neuronal and non-neuronal cell lines. Together, these data provide the first experimental evidence that two proteins involved in HSP physically interact. They suggest that spastin and atlastin participate in the same biochemical pathway and that the molecular pathology of spastin and atlastin HSP is related. They also provide further evidence that spastin is involved in membrane traffic events.
RESULTS
Yeast two-hybrid, glutathione S-transferase (GST) pull-down and co-immunoprecipitation experiments identify atlastin as a binding partner of spastin
We used full-length spastin protein as a ‘bait’ to screen a high-complexity human yeast two-hybrid fetal brain cDNA library (Clontech), using methods described previously (27). This screen generated 15 positive diploid clones that grew on selective media lacking adenine and were positive for β-galactosidase activity. When these primary interactions were retested in fresh yeast, eight were found to be both reproducible and bait specific, giving strong β-galactosidase activity and strong growth on selective medium lacking tryptophan, leucine and adenine (-W/L/A). Remarkably, all but two of the positive ‘prey’ clones contained atlastin coding sequence.
In order to verify the relevance of the putative interaction between spastin and atlastin, we first carried out GST pull-down experiments. GST–spastin successfully pulled down atlastin from a lysate of cells transiently transfected with a mammalian expression vector containing c-Myc-tagged atlastinΔTM (myc-atlastinΔTM; Fig. 1), indicating that the two proteins are capable of a biochemical interaction (Fig. 2A). We next characterized the spastin protein domains necessary for interaction with atlastin, by using yeast two-hybrid and GST pull-down experiments. We tested a prey atlastin vector against yeast two-hybrid bait vectors containing inserts coding for several truncated forms of spastin, spastinΔAAA, spastinΔN1 and spastinΔN2 (Fig. 1). We found that yeast two-hybrid interaction was maintained with the spastinΔAAA bait vector, but lost when the spastinΔN1 and spastinΔN2 baits were used (Fig. 2B). Pull-down assays using GST–atlastinΔTM and cell lysates from cells transfected with the mutant forms of spastin were also consistent with these results; GST–atlastinΔTM was able to pull down myc-spastinΔAAA and myc-spastinK388R (a form of spastin containing a disease-associated missense mutation), but not myc-spastinΔN1 or spastinΔN2-GFP, from lysates of cells transfected with the relevant construct (Fig. 2C). These data indicate that the region of spastin responsible for the interaction with atlastin lies at the N-terminal of the protein, from residues 1–80. Recent data have shown that spastin has two translation initiation sites, the first at codon 1 and the second at codon 87, resulting in the production of two main spastin isoforms, a rare, predominantly cytoplasmic, full-length form (∼68 kDa size) and a much more abundant shorter, nuclear and cytoplasmic isoform (∼60 kDa) (18). The situation is further complicated by the presence of alternative splicing events, which result in the presence of minor exon 4-deleted versions of the long and short isoforms (at ∼64 and 55 kDa, respectively). Our domain interaction data therefore suggest that only the long (i.e. 68 and 64 kDa) isoform versions of spastin are capable of interacting with atlastin.
We next attempted to verify the physiological relevance of the spastin–atlastin interaction by co-immunoprecipitation experiments. We co-immunoprecipited epitope-tagged myc-spastin and atlastin–GFP, by transiently expressing these proteins in HEK293T cells and immunoprecipitating with anti-GFP antibody. Subsequent immunobloting of the precipitate with anti-myc revealed a band of the size expected for myc-spastin (∼68 kDa; Fig. 2D). This band was not present when the GFP antibody was omitted from the immunoprecipitation reaction or when the immunoprecipitation was carried out using cells co-transfected with myc-spastin and GFP-empty vector (Fig. 2E).
Finally, we attempted to carry out co-immunoprecipitation experiments using antibodies to endogenous spastin and atlastin. We used two published antibodies in these experiments. The first was S51, a rabbit polyclonal anti-spastin antibody (a kind gift of Elena Rugarli) (17). This antibody can detect the main spastin isoforms in NSC34 cell lysates (Fig. 2F) and is capable of immunoprecipitating spastin. The second antibody was a rabbit polyclonal anti-atlastin antibody (5409—a kind gift of Craig Blackstone) (35). This antibody detects a single strong band at approximately the size predicted for atlastin (c. 55 kDa) on western blotting of NSC34 cell lysate (Fig. 2F). On immunoprecipitation of NSC34 cells under non-denaturing conditions using either the spastin S51 antibody or the atlastin antibody, we were unable to demonstrate any co-immunoprecipitation of the endogenous putative binding partner. As we were concerned that immunoprecipitation of atlastin by the atlastin antibody was weak under non-denaturing conditions, we investigated the capacity of this antibody to immunoprecipitate atlastin from NSC34 cells under denaturing conditions and found strong immunoprecipitation (Fig. 2G). We therefore carried out co-immunoprecipitation experiments under denaturing conditions, treating the cells with the chemical cross-linker dithiobis[succinnimidyl propionate] (DSP) before lysis, in order to preserve protein–protein interactions. However, we were still unable to demonstrate any co-immunoprecipitation of endogenous spastin (data not shown).
We hypothesized that the reason we could not demonstrate any interaction between the endogenous proteins was because of the very low abundance of full-length 68 kDa spastin (Fig. 2F), the form capable of interacting with atlastin. In view of this, we explored whether provision of additional full-length exogenous spastin might allow us to demonstrate co-immunoprecipitation with endogenous atlastin and we found that this was indeed the case. When we used the atlastin antibody under denaturing conditions to immunoprecipitate atlastin from NSC34 cells expressing myc-spastin, we found that myc-spastin was co-immunoprecipitated (Fig. 2H). No myc-spastin was co-immunoprecipitated when a spurious immunoprecipitating antibody was used (Fig. 2H).
In summary, we were able to identify an interaction between spastin and atlastin by yeast two-hybrid, GST pull-down and co-immunoprecipitation experiments. These results provide very strong evidence that spastin and atlastin are capable of binding to each other and that this interaction has physiological relevance.
Epitope-tagged atlastin and spastin co-localize in HeLa and NSC34 cells
Having established the presence of an interaction between spastin and atlastin, we next went on to determine whether there was any co-localization between the proteins in mammalian cells, and if so where the site of the interaction was, by examining the intracellular distribution of epitope-tagged proteins.
Before examining whether spastin and atlastin showed intracellular co-localization, we established the intracellular distribution in mammalian cells of epitope-tagged versions of atlastin, as this has not previously been described. To minimize the potential for tag-induced changes in distribution, we made constructs expressing either N-terminal c-Myc- or C-terminal GFP-tagged versions of full-length atlastin and examined the expression pattern of each construct by immunofluorescence using a confocal microscope. In each case, epitope-tagged atlastin appeared in a reticulate pattern throughout the cytoplasm of transiently transfected HeLa cells. This reticulate staining pattern strongly co-localized with calreticulin, a marker of the ER (Fig. 3A–F), and with antibodies to two other ER proteins, PDI (protein disulphide isomerase), a lumenal protein, and sec61α, an ER membrane protein (data not shown). In view of the interaction between spastin and atlastin, and the relationship between spastin and microtubules, we also examined whether atlastin co-localized with tubulin. We found partial co-localization between epitope-tagged atlastin and the anti-tubulin antibody YL1/2 (Fig. 3G–I), although there was no gross effect of atlastin expression on microtubule architecture. This co-localization between atlastin and microtubules is perhaps not surprising, as the ER is known to have a close association with the microtubule framework. We also tested for co-localization between epitope-tagged atlastin and a battery of other antibody markers, which included EEA1 (for early endosomes), M6PR (for late endosomes and trans-Golgi network), Lamp1 (for lysosomes), GM130 (for the Golgi apparatus) and TGN46 (for the trans-Golgi network). We found some limited co-localization between atlastin–GFP and M6PR (Fig. 3J–L) and GM130 (Fig. 3M–O), although we cannot be sure whether this might simply reflect the fact that the organelles these markers detect are located close to the microtubule organizing centre, where the expression of ER markers is also very intense.
In addition to experiments in HeLa cells, we examined the expression pattern of epitope-tagged atlastin in a mouse motor neuronal cell type, NSC34. This hybrid cell type is derived from a fusion between mouse spinal cord motor neurons and a neuroblastoma cell line and has many of the anatomical, pharmacological and electrical characteristics of neuronal cells (36). For c-Myc and -GFP-tagged atlastin, the expression pattern in NSC34 cells was very similar to that of the corresponding protein in HeLa cells. Atlastin expression was present both in the cell body and neurite extensions (data not shown).
Having established the intracellular distribution of epitope-tagged atlastin, we examined co-localization of epitope-tagged spastin and atlastin in HeLa cells (Fig. 4A–G) and NSC34 cells (Fig. 4H–J). We found partial but definite co-localization between the spastin and atlastin proteins and this did not depend on the nature or location of the epitope tag used. The co-localization in HeLa cells was typically in cytoplasmic puncta/small tubules, which also co-localized with the ER marker calreticulin (Fig. 4A–G). In NSC34 cells, co-localization occurred in the soma and in neurites (Fig. 4H–J).
As epitope-tagged spastin co-localized with atlastin on the ER, we next determined whether some epitope-tagged spastin is present on the ER in the absence of exogenous atlastin or whether expression of atlastin caused a redistribution of spastin to the ER. We found significant co-localization in puncta and tubules of a subset of myc-spastin with calreticulin (Fig. 4K–M), but this was not as strong as the ER localization when myc-spastin was co-expressed with atlastin-GFP (Fig. 4D–G), suggesting that co-expression with atlastin caused recruitment of spastin to the ER, perhaps because additional binding sites were provided there by atlastin expression. When GFP–spastin and spastin–GFP were expressed alone, co-localization with calreticulin was also present, but subjectively more limited than that with myc-spastin.
We next examined whether atlastin co-localized with ATPase-defective forms of spastin. We examined the effects of atlastin expression on a disease-causing spastin mutation, K338R. The spastin K388R mutation abrogates ATPase function and is associated with the development of an abnormal, filamentous phenotype when expressed in cultured cells. The abnormal filaments partially co-localize with markers of microtubules (19,27). We found that the spastin mutant retained a filamentous appearance when co-expressed with epitope-tagged atlastin and that the distribution of atlastin was dramatically changed so that the protein showed strong co-localization with the abnormal filaments (Fig. 4N–Q). In NSC34 cells, the abnormal filaments extended into neurite processes, and strong co-localization was seen here, as well as in the soma (data not shown). The redistribution of atlastin was accompanied by a redistribution of calreticulin, which also co-localized with the mutant spastin filaments (Fig. 4P). A similar redistribution of the ER was seen when myc-spastinK388R was expressed in HeLa cells in the absence of exogenous atlastin (Fig. 4R–T).
We have previously suggested that some spastin expression may be on endosomes, on the basis of data that show an interaction between spastin and the endosomal protein CHMP1B. We have also described colocalization between myc-spastin and a GFP-tagged endosomal marker RhoB (27). However, our previous co-localization experiments used a GFP-tagged RhoB construct (Clontech) that was inappropriate, as it contained a c-Myc epitope. We therefore reinvestigated the endosomal localization of transiently expressed spastin by examining HeLa cells co-transfected with myc-spastin and N-terminal GFP-tagged mammalian VPS4-E235Q (a kind gift of Paul Whitely) (37). The ATPase-defective GFP-VPS4-E235Q protein accumulates on swollen endosomes, which retain components of the ESCRT-III complex on their limiting membranes (37). We found significant partial co-localization between myc-spastin and GFP-VPS4-E235Q, strongly suggesting that a subpopulation of spastin is present on endosomal membranes (Fig. 4U–W). It therefore seems likely that spastin can be present on at least two membrane populations, endosomes and the ER.
In order to gain more information on the composition of the filaments associated with ATPase-defective spastin, we investigated them at the ultrastructural level. We transiently co-transfected HeLa cells with myc-spastinK388R and atlastin–GFP and examined the cells under the electron microscope. We found bundles of microtubules in the cytoplasm of co-transfected cells (Fig. 5A). Individual microtubules within these bundles had an abnormal appearance, having electron-dense striations. We also saw cytoplasmic structures with an abnormal honeycomb-like appearance in these cells (Fig. 5B) and these structures were sometimes continuous with the ER (Fig. 5B) or nuclear envelope (Fig. 5C). In some sections, the honeycomb structures were contiguous with the abnormal microtubule bundles (Fig. 5D).
DISCUSSION
In this study, we used a yeast two-hybrid approach to identify potential interacting partners for spastin, a protein mutated in autosomal-dominant pure HSP. Remarkably, this approach identified atlastin, another protein mutated in autosomal-dominant pure HSP, as a potential spastin-binding partner. We verified the relevance of this interaction in mammalian cells by a variety of approaches, which together provide compelling evidence that spastin and atlastin are true binding partners. This interaction is the first demonstration of a direct functional relationship between two proteins involved in HSP. Its presence strongly suggests that spastin and atlastin participate in the same biochemical pathway and that the molecular pathology of spastin and atlastin HSP is linked.
One issue that our study was not able to conclusively address was the subcellular site of the spastin–atlastin interaction. Our data with epitope-tagged proteins suggest that they interact on the ER, as we found that epitope-tagged atlastin strongly co-localized with ER markers (and to a lesser extent with the Golgi apparatus), that epitope-tagged full-length spastin showed partial, but definite, co-localization with the ER marker calreticulin and that when co-expressed, epitope-tagged full-length spastin and atlastin co-localized with each other and with calreticulin. It is possible that a subset of endogenous spastin might be located on the ER or the contiguous microtubule network—previous immunolocalization studies have not specifically examined whether a proportion of the cytoplasmic expression of spastin might be on or very close to the ER. Although a previous immunolocalization study found some endogenous atlastin on the ER, the main location of the endogenous protein was on the Golgi apparatus, a site which was less prominent with epitope-tagged atlastin (35). Explanations for this discrepancy could include an immunofluorescence cross-reaction of the atlastin antibody with another protein, the presence of ER and Golgi forms of atlastin with different sensitivity/available epitopes for the antibody or effects of the epitope tag on localization of the protein. However, the localization that we observed was independent of the site or nature of the epitope tag used. Transient expression of atlastin may have had an effect on its distribution, with over-expression saturating ER to Golgi transport pathways or affecting the balance between anterograde and retrograde ER/Golgi pathways, resulting in a relative increase in ER staining when compared with the endogenous pattern. Finally, the continuous distribution of atlastin that we saw throughout the ER was not typical of the aggregated clumps characteristic of over-expressed proteins that are misfolded in the ER. Further studies with additional spastin and atlastin antibodies will be required to conclusively resolve the subcellular location of the spastin–atlastin interaction.
Whether or not the localization of native atlastin is predominantly at the ER, at the Golgi, or at both, the fact that it interacts with spastin makes it very likely that spastin is active at more than one membrane site. We have previously identified the endosomal protein CHMP1B as a binding partner for spastin and we show here that full-length spastin partially co-localizes with GFP-VPS4-E235Q, an endosomal marker (27). A picture is therefore emerging of spastin as a microtubule-regulating protein that can be recruited by different adaptor proteins to several membrane sites, where it may play a role in local regulation of microtubules, perhaps coupled to the movement of membrane-bound organelles (especially in axons) and/or the remodelling of organelle architecture, or even have an entirely separate role unrelated to its microtubule severing function. The factors that influence recruitment of spastin to one site versus another are not yet known, but their elucidation will be important in further understanding the exact physiological role of the protein.
Spastin may also have other functions unrelated to membrane interactions. Several studies using antibodies to endogenous spastin have suggested that, in some cell types or at some stages of the cell cycle, spastin may have a nuclear localization (14,15,17). This nuclear spastin predominantly consists of the short N-terminal truncated isoform and its function is currently unknown (18). The scenario of an AAA family member having multiple sites of action is not unprecedented, with several documented examples of individual AAA proteins exerting a specific function, often involving regulation of substrate protein conformation, at multiple intracellular sites of action (38,39).
We examined the spastin domains that mediate the interaction with atlastin, and the region responsible resided within the first 80 residues of the protein (Fig. 6). Thus, only the predominantly cytoplasmic, full-length spastin isoform contains the region responsible for binding to atlastin, suggesting that it is the function of this isoform which is of direct importance for the pathogenesis of HSP. Interestingly, the full-length isoform is enriched in brain and spinal cord, the anatomical sites of HSP pathology (18). The atlastin-binding region overlaps the domain (residues 50–87) that is responsible for binding to microtubules and the centrosomal protein NA14, but is distinct from the region that interacts with CHMP1B (residues 81–194) (17,19,27). Thus, it appears that the different spastin locations and functions discussed earlier are reflected in the presence of binding sites for different adaptor proteins in its N-terminal region.
Our study also presents more information on the composition of the intracellular filaments seen on expression of ATPase-defective spastin mutants. Although it is unlikely that these filaments are relevant to those spastin mutations which cause haplo-insufficiency and it is possible that they represent an over-expression artefact, their composition may be relevant to HSP caused by spastin ATPase missense mutations. At the light-microscope level, epitope-tagged atlastin and the ER marker calreticulin were both redistributed to the abnormal microtubule bundles. On electron microscopy in cells transfected with myc-spastinK388R and atlastin–GFP, we observed bundled microtubules and abnormal honeycomb structures, which were sometimes seen in association with microtubule bundles. In some cases, it was apparent that the honeycomb structures were continuous with normal ER and with the nuclear envelope, suggesting that they may represent abnormal ER. It is not clear whether the reorganization of the ER that we have seen is a consequence of direct binding between mutant spastin and atlastin or whether it is an indirect effect via reorganization of the microtubule network associated with the ER, although it seems clear that the filaments are closely associated with membrane components.
In summary, we have identified two HSP proteins, spastin and atlastin, as binding partners. This suggests that the pathogenesis of HSP caused by mutation in these two proteins is fundamentally related and provides the first experimental evidence that HSP may be caused by abnormalities in proteins that are members of the same biochemical pathway. A more detailed understanding of the function of this pathway will not only give key insights into the causes of neurodegeneration in HSP and perhaps other neurological conditions but should also elucidate molecular mechanisms that are important for long-tract neuronal maintenance.
MATERIALS AND METHODS
Construction of yeast two-hybrid, GST and immunofluorescence vectors
Spastin yeast two-hybrid and mammalian expression constructs were constructed as described previously (27,40). Atlastin constructs were amplified by proof-reading Pfx polymerase (Invitrogen) PCR from IMAGE clone 3869877, using GatewayTM (Invitrogen) recombination cloning system-compatible primers designed from the reference cDNA sequence for each gene. For truncated cDNA constructs, Pfx PCR was carried out using appropriate Gateway (Invitrogen) recombination cloning system-compatible primers designed from within the reference cDNA. Gateway recombination cloning system pENTR201 and/or pENTR207 entry vectors containing the amplified cDNAs were constructed according to the manufacturer's instructions. Sequence of entry vector constructs was verified by sequencing on an ABI377 or 3700 sequencer, using BigDyeDT chemistry (Applied Biosystems).
The atlastin transcripts were then subcloned into Gateway-compatible vectors pcDNA3-C-Myc and/or pEGFP-N, which contained 5′-myc or 3′-GFP epitopes, respectively. The orientation of the atlastin sequences within the constructs was verified by direct sequencing on an ABI377 or 3700 sequencer.
For production of the GST-fusion cDNA constructs, full-length spastin cDNA and atlastinΔTM cDNA were amplified by PCR using the Pfu Turbo® polymerase enzyme (Stratagene). The primers used in the reaction were designed to produce a PCR product with 5′ and 3′ ends compatible with the GST gene fusion vector, pGEX-4T-3 (Amersham Biosciences). After restriction with NotI and SalI (New England Biolabs), the PCR product was ligated into the expression vector using T4 DNA ligase (New England Biolabs). The sequence of the constructs was verified as earlier.
Production of GST-fusion proteins
Transformation of the GST-fusion construct into BL21(DE3)pLysS cells was performed according to the manufacture's instructions (Invitrogen). Production of large-scale bacterial sonicates and subsequently GST-fusion protein purification were performed according to the Amersham Biosciences' GST gene fusion system instructions.
Yeast two-hybrid library screening, reporter assays and identification of interacting proteins
Yeast two-hybrid screening with the spastin bait clone was carried out as previously described, except the screen was carried out against a fetal brain cDNA Matchmaker Library (Clontech) (27). To identify interacting proteins, prey inserts were directly amplified from yeast using vector-specific primers, as previously described, and 3–5 µl of each PCR product was sequenced as described earlier (27).
Prey sequences were searched against locally held versions of the Homo sapiens Unigene and the EMBLminus databases using an automated BLAST (41) algorithm. Custom-built Perl modules and scripts were used to prefilter and format the raw BLAST output and determine whether the 5′ sequence read overlapped with the protein-coding region of a gene. Only matches to known protein-coding regions and to ESTs that have no defined open-reading frame were included as positives. Matches to 3′-UTRs and genomic DNA with no associated gene prediction were excluded. Singleton, unspliced ESTs that did not correspond to any gene predictions were also excluded.
Reconfirmation and specificity testing for yeast two-hybrid assays
To retest each interaction in fresh yeast, prey PCR product was cloned using Gateway recombination cloning system into the Gateway compatible pGAD-G vector, as described earlier. Competent MATα PJ69-4A yeast cells were transformed and mated as previously described. They were mated with the relevant bait, two different irrelevant baits or an empty bait vector, and then incubated for >5 h on YPAD plates. Diploids were selected on SD-Ura/Leu prior to testing for reporter activation by replicating onto SD-Ura/Leu/Ade.
Antibodies
Monoclonal anti-myc primary antibody (mouse clone 4A6) was obtained from Upstate (New York). Monoclonal anti-microtubule antibody (rat clone YL1/2) and anti-GFP rabbit polyclonal antibodies were obtained from Abcam (Cambridge). Rabbit polyclonal anti-calreticulin antibody was obtained from Calbiochem (San Diego). Sheep polyclonal anti-TGN46 was obtained from Serotec (Oxford), and rabbit polyclonal anti-M6PR and mouse monoclonal anti-Lamp1 antibodies were available in-house. Mouse monoclonal anti-EEA1 and mouse monoclonal anti-GM130 were obtained from BD Transduction Laboratories (Oxford). Peroxidase-conjugated secondary antibodies for western blotting were obtained from Sigma. Alexafluor-labelled secondary antibodies for immunofluorescence were obtained from Molecular Probes (Oregon). The S51 rabbit anti-spastin polyclonal antibody was a kind gift of Elena Rugarli and the rabbit anti-atlastin 5409 antibody was a kind gift of Craig Blackstone.
Cell culture, transfection and immunofluorescence
HeLa cells were seeded onto sterile glass cover slips in six-well plates (∼0.5×105 cells/well). After 24 h, the cells were transfected with ∼400 ng of vector DNA, using the Effectene® transfection reagent (Qiagen), according to the manufacturer's instructions. Following transfection, cells were incubated for 24 or 48 h in RPMI-1640 medium (Sigma) containing 10% fetal bovine serum (FBS) (Sigma), 100 U/ml penicillin and 100 µg/ml streptomycin (Sigma) and 2 mml-glutamine (Sigma). NSC34 cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies), supplemented in the same way as the RPMI-1640 medium used for HeLa cells. NSC34 cells were seeded at 1×105 into six-well plates containing glass cover slips precoated with poly-d-lysine (Sigma). They were then transiently transfected with Effectene transfection reagent (Qiagen), according to the manufacturer's instructions, for 24–48 h before fixation.
Following transfection, cells were washed in PBS (Sigma) and fixed with 4% formaldehyde in PBS, at room temperature for 15 min. Fixed cells were washed in PBS before being permeabilized in PBS containing 0.1% Triton X-100 (Sigma) or 0.05% saponin (Sigma, used with M6PR and Lamp1 antibodies) at room temperature for 5–10 min. Permeabilized cells were then washed three times in PBS, incubated for 15 min in a blocking solution (PBS, 10% FBS, ±0.05% saponin) and then transferred to blocking solution containing the appropriate epitope-specific antibody at an appropriate dilution. After 60 min incubation, cover slips were washed three times in blocking solution and were then incubated for 60 min in blocking buffer containing the appropriate secondary antibodies, at a concentration of 1/300. Cover slips were then washed three times in PBS and once in distilled H2O, after which they were mounted in VectashieldTM (Vector Laboratories Inc.) medium on a glass slide. Stained samples were analyzed on a Zeiss 510 Meta confocal microscope. Images were recorded using LSM Image Analyzer software, and data were subsequently processed using Adobe Photoshop and Illustrator programs.
Electron microscopy
Fixation and processing of sections for microscopy was carried out according to the previously described methods (42).
Co-immunoprecipitation experiments
For the attempted co-immunoprecipitation of endogenous spastin and atlastin under non-denaturing conditions, untransfected NSC34 cells were plated into three 15 cm plates at 1×107. Twenty-four hours later, confluent cells were washed in PBS and lysed in 1 ml ice-cold NP-40 lysis buffer (see 27 for recipe). A 100 µl aliquot was then removed for use as a concentrated input fraction. The volume of NSC34 lysate was made up to 10 ml and this was split into two fractions. Fraction 1 represented total cell lysate, whereas fraction 2 was centrifuged at 13 000g for 5 min and supernatant removed for use as a soluble fraction. Both fractions were then divided further into 2.5 ml samples for positive/control co-immunoprecipitations. Anti-atlastin or anti-spastin polyclonal antiserum was then added to + fractions at 1/100, whereas anti-GFP (ab290 Abcam) antibody was added to control fractions. Fractions were then incubated on a rocking table at 4°C for 4 h. Following antibody incubations, 200 µl of a 50% slurry of protein-A Sepharose beads (Sigma) in NP-40 lysis buffer were added to all fractions and incubated overnight on a rocking table at 4°C. Following protein–antibody complex incubations, protein-A Sepharose beads were isolated by brief centrifugation and then washed five times for 20 min in 10 ml NP-40 lysis buffer. The final wash supernatant was then removed and beads were resuspended in 2× SDS–PAGE sample buffer. SDS–PAGE was then performed and immunoblotting carried out with appropriate antisera.
For transiently transfected HEK293T cells, cells were plated into two 15 cm plates at 1×107, and 24 h later, cells were co-transfected with atlastin-GFP and myc-spastin or pEGFP and myc-spastin. Twenty-four hours after transfection, cells were lysed in 1 ml ice-cold lysis buffer, and a 100 µl aliquot removed for use as a concentrated input fraction. Cells were then processed as described for NSC34 cells. An anti-GFP polyclonal antibody (ab290 Abcam) was added to + antibody fractions at 1/1000, and +/− antibody fractions were then incubated on a rocking table at 4°C for 4 h. Further processing was carried out as earlier.
A modified protocol was used for the co-immunoprecipitations carried out under denaturing conditions. These experiments were performed in NSC34 cells using denaturing conditions in the presence of a chemical cross-linker DSP (Pierce). NSC34 cells were harvested in PBS containing 1 mm DSP and incubated for 30 min at room temperature. 1 m Tris–HCl, pH7.5, was added to a final concentration of 10 mm to quench DSP activity and cells were incubated for a further 15 min at room temperature. Cells were resuspended in 100 µl per 1×107 cells denaturing lysis buffer containing 1% SDS, 50 mm Tris–HCl, pH 7.4, 5 mm EDTA, DNase I (Sigma) and protease inhibitors (Roche). Denatured lysate was then diluted into non-denaturing lysis buffer containing 1% Triton X-100, 50 mm Tris–HCl, pH 7.4, 300 mm NaCl, 0.5 mm EDTA and protease inhibitors to a final concentration of 0.1% SDS. Total cell lysate was cleared by centrifugation at 13 000 r.p.m. for 10 min and the soluble fraction was retained. Protein-A Sepharose beads were precoupled to 20 µg of the anti-atlastin (5409) polyclonal antibody and to a spurious control antibody (anti-GFP Abcam 6556), in the presence of DSP. DSP activity was quenched with 10 mm Tris–HCl, as earlier. Antibody-coupled protein-A Sepharose beads were then added to soluble cell lysates. Antigen–antibody complexes were allowed to form at 4°C for 2 h. Beads were then isolated and washed several times in non-denaturing lysis buffer. DSP cross-linking was cleaved by addition of SDS–PAGE sample buffer containing 5% β-mercaptoethanol at 100°C for 5 min and proteins were resolved by SDS–PAGE.
GST-fusion protein pull-down experiments
Glutathione–Sepharose beads (Amersham Biosciences) were washed in a pull-down buffer that consisted of 1% NP-40 and PBS solution with protease inhibitors (Complete Mini protease inhibitor cocktail tablets, Roche Diagnostic) and made up to a 50% slurry in this buffer. HeLa cells were transiently transfected with expression vector constructs of epitope-tagged spastin or atlastin, using the Effectene Transfection kit (Qiagen), as described earlier. Cells were harvested 24–48 h after transfection into the wash buffer and freeze/thawed to −20°C.
The pull-down reaction consisted of 50 µl of 50% glutathione–Sepharose beads, 500 µg cell cytosol and 100 µg GST-fusion protein. The total volume was made up to 0.5 ml in a 1.5 ml tube with pull-down buffer (1% NP-40, PBS, 5 mm MgCl2 and 5 mm ATP). The GST negative control used an equi-molar amount of GST protein. Tubes were incubated at 4°C on a rotating wheel overnight. The beads were spun at 13 000 for 15 s in a bench top centrifuge and then washed with 1 ml of the buffer. This cycle was repeated four times. Prior to the fourth wash, the pull-down mix was transferred to clean 1.5 ml tubes to eliminate any possible binding of protein to the interior of the plastic tube. Protein bound to the beads was eluted after four wash/spin cycles with 55 µl of SDS sample buffer (188 mm TRIS, 6% SDS, 30% glycerol, 0.03% Bromophenol blue and 10% β-mercaptoethanol, pH 6.8). The samples were heated to 95°C for 5 min and then spun at 13 000 speed in a bench top centrifuge for 15 s. The supernatants were loaded on a 10% SDS–polyacrylamide gel and immunoblotted with anti-myc or anti-GFP primary antibody as appropriate.
ACKNOWLEDGEMENTS
The pGBDU-C series of vectors and the PJ69-4A MATa and MATα yeast strains were kindly provided by Dr Philip James (Department of Biomolecular Chemistry, University of Wisconsin, Madison, WI 53706-1532, USA). The S51 spastin antibody was a kind gift of Dr Elena Rugarli (Telethon Institute, Naples, Italy), the atlastin antibody was a kind gift of Craig Blackstone (NIH, MD, USA), the GFP-VPS4EQ construct was a kind gift of Paul Whitely (Univeristy of Bath, UK) and NSC34 cells were a kind gift of Dr Neil Cashman (University of Toronto, Canada). This work has been supported by grants from the Wellcome Trust and Medical Research Council. E.R. is a Wellcome Trust Advanced Clinical Fellow. T.L.E. is supported by a Raymond and Beverly Sackler Fellowship and an award from the Cambridge Commonwealth Trust.
Conflict of Interest statement. None declared.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.
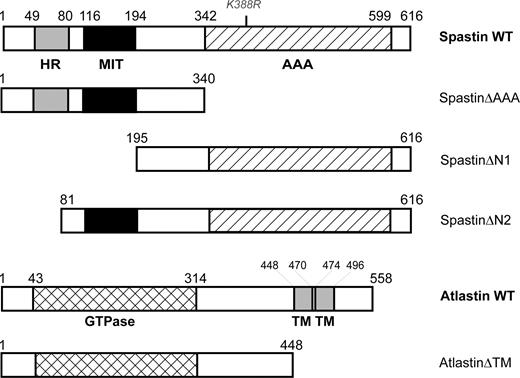
Figure 1. Constructs used in this study. Schematic diagram showing the domain structure of wild-type (WT) spastin and atlastin, along with the structure of other inserts used in constructs. Amino acid numbering is given above each construct, whereas the position of the K388R point mutation examined in the study is shown in grey font above the WT spastin illustration. HR, hydrophobic region; AAA, AAA ATPase cassette; GTPase, GTPase cassette; TM, transmembrane domain.
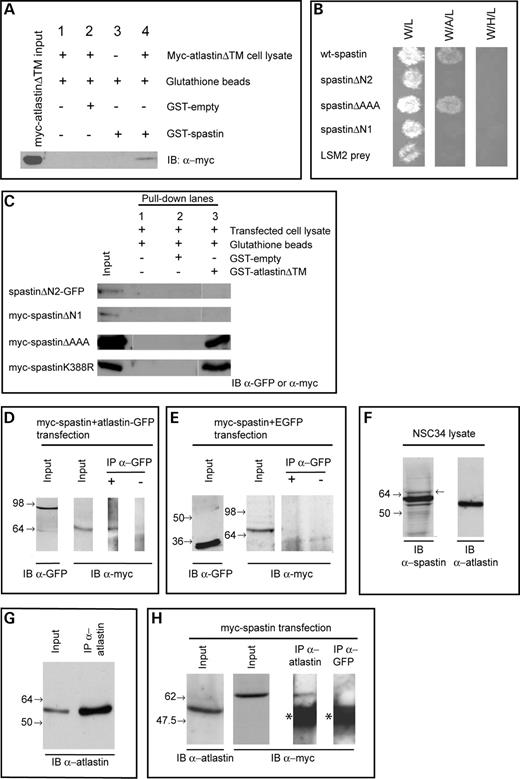
Figure 2. Spastin and atlastin interact biochemically. > (A) GST–spastin was used in a pull-down experiment using cell lysate from HeLa cells transfected with myc-atlastinΔTM. A band of the correct size for myc-atlastinΔTM was seen in the pull-down lane (lane 4), but not in lanes from experiments lacking GST protein (lane 1), experiments using empty-GST instead of GST–spastin (lane 2), or experiments in which no cell lysate was added (lane 3). (B) Investigation of spastin domains responsible for the interaction with atlastin was carried out using yeast two-hybrid. Control column 1 shows the growth of diploid colonies on media lacking tryptophan and leucine (-W/L) to select for the presence of both bait and prey vectors. Column 2 shows the growth of diploid colonies on media lacking tryptophan, leucine and adenine (-W/A/L), which requires the activation of the Ade reporter gene, whereas control column 3 shows the growth of diploid colonies on media lacking tryptophan, leucine and histidine (-W/H/L), which requires the activation of the His reporter gene. In each case, spastin or a spastin mutant was used as the ‘bait’ protein, whereas atlastin was used as ‘prey’. Full-length spastin and spastinΔAAA interact with atlastin, but spastinΔN1 and spastinΔN2 do not. No interaction was present with a non-specific prey (LSM2). (C) GST pull-down experiments using GST–atlastinΔTM versus cell lysates from cells transfected with the stated constructs confirmed the yeast two-hybrid results. The GST-fusion protein was able to pull down myc-spastinΔAAA and myc-spastinK388R, but not myc-spastinΔN1 or spastinΔN2-GFP. (D) Co-immunoprecipitation of transiently transfected myc-spastin and atlastin-GFP in HEK293T cells. Twenty-four hours after transfection, tagged proteins were immunoprecipitated (IP) using an anti-GFP polyclonal antibody and immunoblotted (IB) with an anti-myc monoclonal antibody. A band of the expected size for transfected full-length myc-spastin was co-immunoprecipitated with atlastin-GFP. No band was present when the anti-GFP antibody was omitted from the immunoprecipitation reaction. (E) Attempted co-immunoprecipitation of transiently transfected myc-spastin and vector-only EGFP as a negative control for (D). Myc-spastin was not co-immunoprecipitated with GFP. For parts (D) and (E), input lanes show α-myc and α-GFP immunoblots of input fractions, confirming expression of the relevant proteins in the cell lysates used for the immunoprecipitations. (F) Endogenous spastin and atlastin are present in NSC34 cells. Spastin was identified using the S51 spastin antibody. This antibody reveals three bands in this blot, corresponding to different isoforms of spastin—full-length (68 kDa, arrow to right of lane marks position), short form (60 kDa) and short form variant lacking exon 4 (55 kDa). The full-length isoform lacking exon 4 (64 kDa) is of such low abundance that it is not seen on this blot. Using the 5409 anti-atlastin antibody, a single band of ∼55 kDa was seen. (G) Under denaturing conditions, atlastin is strongly immunoprecipitated from NSC34 cells by the 5409 atlastin antibody. The left lane shows the input cell lysate, whereas the right lane shows the immunoprecipitate. Both were blotted with the atlastin antibody. (H) Endogenous atlastin and myc-spastin are co-immunoprecipitated from NSC34 cells. Anti-atlastin 5409 antibody was used to immunoprecipitate under denaturing conditions and immunoblotting was performed using anti-myc. A band of the size expected for myc-spastin is present in the input lysate and in the immunoprecipitate. This band was absent when a spurious control antibody (anti-GFP) was used to carry out the immunoprecipitation. The asterisks mark the position of immunoglobulin bands—these were also present when the primary anti-myc antibody was omitted from the immunoblotting procedure (data not shown). The position of relevant marker bands is shown to the left of each blot.
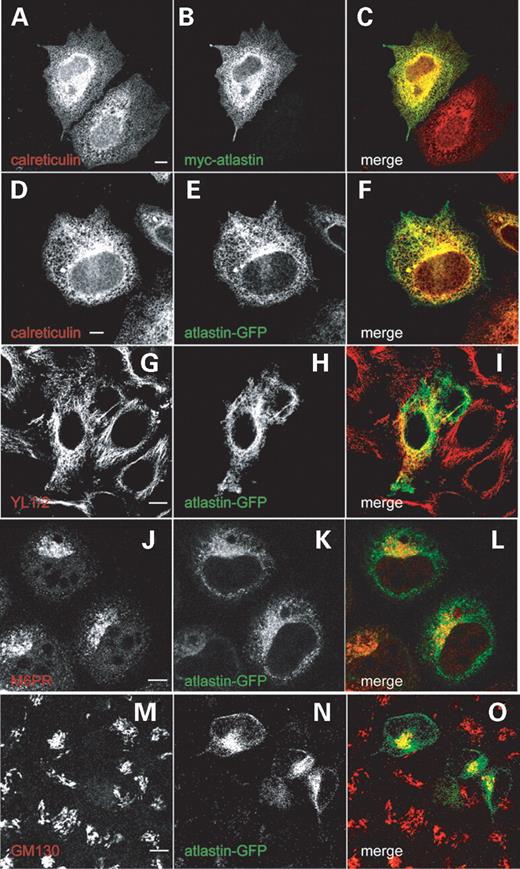
Figure 3. Epitope-tagged wild-type and mutant atlastin co-localize with ER and microtubule markers. The images show Hela cells labelled with the antibody marker shown in (A, D, G, J, M). (B, E, H, K, N) show the expression pattern in the same cells, transiently transfected with the construct shown in each panel. The colour of text in each of these panels represents the image colour in the corresponding merged panels (C, F, I, L, O). Both N-terminal myc-tagged (A–C) and C-terminal GFP-tagged (D–F) atlastin co-localize tightly with calreticulin. Wild-type atlastin also partially co-localizes with the microtubule marker YL1/2 (G–I). This co-localization was strongest in the perinuclear region. There is also partial co-localization with the late endosomal mannose-6-phosphate receptor (M6PR) (J–L) and the Golgi marker GM130 (M–O), particularly in a focal perinuclear region where atlastin showed strong expression. Cells were fixed 24 h after transfection. Size bars indicate 10 µm.
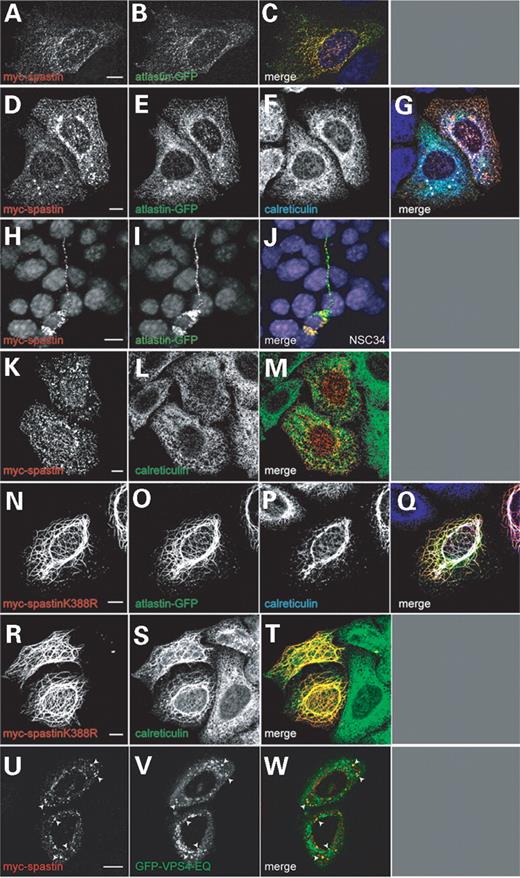
Figure 4. Epitope-tagged atlastin and spastin co-localize in HeLa and NSC34 cells. Hela cells (A–G and K–W) or NSC34 cells (H–J) were co-transfected with myc-spastin and atlastin-GFP (A–J), myc-spastin alone (K–M), myc-spastinK388R and atlastin-GFP (N–Q), myc-spastinK388R alone (R–T) or myc-spastin and GFP-VPS4-EQ (U–W). The cells shown in (D–G) and (K–T) were also labelled with the ER marker anti-calreticulin (F, L, P, S). (C, G, J, M, Q, T, W) show the merged images of the cells shown in (A and B), (D–F), (H and I), (K and L), (N–P), (R and S) and (U and V), respectively, with the colour of the text in each panel indicating the image colour in the merged picture. In each of the cells shown in (A–J), there is definite co-localization between the spastin and atlastin proteins. Both the co-localized proteins (D–G) and myc-spastin expressed alone (K–M) also co-localize with calreticulin. In NSC34 cells (H–J), co-localization was seen in both the soma and neurite extensions. In cells expressing mutant spastin and wild-type atlastin (N–Q), atlastin and calreticulin are redistributed to co-localize with the mutant spastin in filaments. Exogenous atlastin expression is not necessary for calreticulin to be redistributed in this way (R–T). A proportion of myc-spastin expression was also on endosomes, as it partially co-localized with GFP-VPS4-EQ (U–W, arrowheads). Cells were fixed 24 h after transfection. Size bars indicate 10 µm.
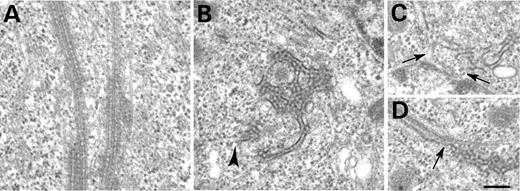
Figure 5. The ultrastructure of the filaments associated with ATPase-defective spastin. >At the electron microscope level, cells co-transfected with myc-spastinK388R and atlastin-GFP had abnormal bundles of microtubules in the cytoplasm (A). The microtubules had an unusual electron dense coating. Dense honeycomb structures were also seen, and in some places, these were continuous with the ER (B, arrowhead) or with the nuclear membrane (C, arrows). In some cases, they were contiguous with the abnormal microtubule bundles (D, arrow). All panels show HeLa cells that were fixed 24 h after transfection. Size bar represents 200 nm; all pictures are at the same magnification.
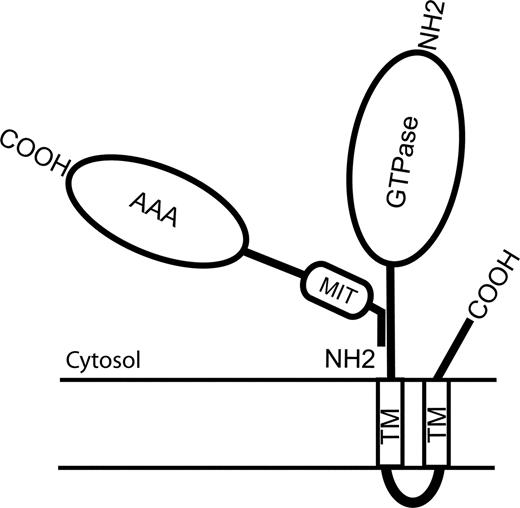
Figure 6. Cartoon diagram of interaction between spastin and atlastin. The region of atlastin responsible for binding to spastin lies N-terminal to the first TM. For spastin, the region responsible for binding to atlastin resides within the first 80 residues of the protein. This overlaps the region of the protein (residues 50–87) that is responsible for binding to microtubules, but is distinct from the MIT and AAA ATPase domains.
References
Harding, A.E.(
Fink, J.K. (
Reid, E. (
Polo, J.M., Calleja, J., Combarros, O. and Berciano, J. (
Fink, J.K. (
Zhao, X., Alvarado, D., Rainier, S., Lemons, R., Hedera, P., Weber, C.H., Tukel, T., Apak, M., Heiman-Patterson, T., Ming, L. et al. (
Hazan, J., Fonknechten, N., Mavel, D., Paternotte, C., Samson, D., Artiguenave, F., Davoine, C.S., Cruaud, C., Dürr, A., Wincker, P. et al. (
Rainier, S., Chai, J.H., Tokarz, D., Nicholls, R.D. and Fink, J.K. (
Reid, E., Kloos, M., Ashley-Koch, A., Hughes, L., Bevan, S., Svenson, I.K., Lennon Graham, F., Gaskell, P.C., Dearlove, A., Pericak-Vance, M.A. et al. (
Hansen, J.J., Durr, A., Cournu-Rebeix, I., Georgopoulos, C., Ang, D., Nielsen, M.N., Davoine, C.S., Brice, A., Fontaine, B., Gregersen, N. and Bross, P. (
Fonknechten, N., Mavel, D., Byrne, P., Davoine, C.S., Cruaud, C., Boentsch, D., Samson, D., Coutinho, P., Hutchinson, M., McMonagle, P. et al. (
Meijer, I.A., Hand, C.K., Cossette, P., Figlewicz, D.A. and Rouleau, G.A. (
Sauter, S., Miterski, B., Klimpe, S., Bonsch, D., Schols, L., Visbeck, A., Papke, T., Hopf, H.C., Engel, W., Deufel, T. et al. (
Charvin, D., Cifuentes-Diaz, C., Fonknechten, N., Joshi, V., Hazan, J., Melki, J. and Betuing, S. (
Wharton, S.B., McDermott, C.J., Grierson, A.J., Wood, J.D., Gelsthorpe, C., Ince, P.G. and Shaw, P.J. (
Beetz, C., Brodhun, M., Moutzouris, K., Kiehntopf, M., Berndt, A., Lehnert, D., Deufel, T., Bastmeyer, M. and Schickel, J. (
Errico, A., Claudiani, P., D'Addio, M. and Rugarli, E.I. (
Claudiani, P., Riano, E., Errico, A., Andolfi, G. and Rugarli, E.I. (
Errico, A., Ballabio, A. and Rugarli, E.I. (
Evans, K.J., Gomes, E.R., Reisenweber, S.M., Gundersen, G.G. and Lauring, B.P. (
McDermott, C.J., Grierson, A.J., Wood, J.D., Bingley, M., Wharton, S.B., Bushby, K.M. and Shaw, P.J. (
Trotta, N., Orso, G., Rossetto, M.G., Daga, A. and Broadie, K. (
Sherwood, N.T., Sun, Q., Xue, M., Zhang, B. and Zinn, K. (
Roll-Mecak, A. and Vale, R.D. (
Patel, H., Cross, H., Proukakis, C., Hershberger, R., Bork, P., Ciccarelli, F.D., Patton, M.A., McKusick, V.A. and Crosby, A.H. (
Ciccarelli, F.D., Proukakis, C., Patel, H., Cross, H., Azam, S., Patton, M.A., Bork, P. and Crosby, A.H. (
Reid, E., Connell, J., Edwards, T.L., Duley, S., Brown, S.E. and Sanderson, C.M. (
Muglia, M., Magariello, A., Nicoletti, G., Patitucci, A., Gabriele, A.L., Conforti, F.L., Mazzei, R., Caracciolo, M., Ardito, B., Lastilla, M. et al. (
Tessa, A., Casali, C., Damiano, M., Bruno, C., Fortini, D., Patrono, C., Cricchi, F., Valoppi, M., Nappi, G., Amabile, G.A. et al. (
Dalpozzo, F., Rossetto, M.G., Boaretto, F., Sartori, E., Mostacciuolo, M.L., Daga, A., Bassi, M.T. and Martinuzzi, A. (
Wilkinson, P.A., Hart, P.E., Patel, H., Warner, T.T. and Crosby, A.H. (
Sauter, S.M., Engel, W., Neumann, L.M., Kunze, J. and Neesen, J. (
D'Amico, A., Tessa, A., Sabino, A., Bertini, E., Santorelli, F.M. and Servidei, S. (
Hedera, P., Fenichel, G.M., Blair, M. and Haines, J.L. (
Zhu, P.P., Patterson, A., Lavoie, B., Stadler, J., Shoeb, M., Patel, R. and Blackstone, C. (
Cashman, N.R., Durham, H.D., Blusztajn, J.K., Oda, K., Tabira, T., Shaw, I.T., Dahrouge, S. and Antel, J.P. (
Whitley, P., Reaves, B.J., Hashimoto, M., Riley, A.M., Potter, B.V. and Holman, G.D. (
Dreveny, I., Pye, V.E., Beuron, F., Briggs, L.C., Isaacson, R.L., Matthews, S.J., McKeown, C., Yuan, X., Zhang, X. and Freemont, P.S. (
Whiteheart, S.W. and Matveeva, E.A. (
Horton, R.M., Hunt, H.D., Ho, S.N., Pullen, J.K. and Pease, L.R. (
Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}