
Abstract
We used a cost-effective, non-invasive method to obtain high-quality DNA from buccal epithelial-cells (BEC) of premature infants for genomic analysis. DNAs from BEC were obtained from premature infants with gestational age ≤ 36 weeks. Short terminal repeats (STRs) were performed simultaneously on DNA obtained from the buccal swabs and blood from the same patient. The STR profiles demonstrated that the samples originated from the same individual and exclude any contamination by external DNAs. Whole exome sequencing was performed on DNAs obtained from BEC on premature infants with and without necrotizing enterocolitis and successfully provided a total number of reads and variants corroborating with those obtained from healthy blood donors. We provide a proof of concept that BEC is a reliable and preferable source of DNA for high-throughput sequencing in premature infants.
Similar content being viewed by others
Introduction
Understanding the genetic basis of a disease has vast potential benefit to healthcare. Obtaining genetic material for analysis is thus essential and has broad implications for understanding the pathogenesis of disease and for potentially designing individualized therapies. To this end, building repositories of genetic material may prove to be a useful tool. Several molecular genetic tests can be performed using dried blood spots, as is the case with statewide newborn screens. Other, more extensive testing, such as chromosome analysis, FISH (fluorescent in situ hybridization), microarray and PCR-based genotyping assays require whole blood samples. However, blood sampling is invasive, expensive and with limitations in preterm neonates. For these infants, every milliliter of blood is significant and relatively small volumes can constitute a large percentage of total blood volume. Additionally, obtaining blood for laboratory analysis may cause pain or discomfort and should only be collected when absolutely necessary.
The use of innovative and minimally invasive practices in pediatric and neonatal populations remains important. Buccal cells have previously been discredited as a source of reliable DNA in neonates, due to maternal epithelial cell contamination1,2,3,4. The purpose of this study is to evaluate the efficacy of already tried-and-tested buccal swab method to obtain high-quality DNA for high-throughput genomic analysis. This analysis includes: short tandem repeat (STR) analysis, Taqman Allelic Discrimination Assay, Single Nucleotide Polymorphisms (SNPs) genotyping by PCR-RFLP and more importantly whole exome sequencing (WES).
Results
Genomic DNA was successfully isolated from all samples (170 buccal brushes from 85 patients and 61 whole blood samples). Thirty-five (41%) premature neonates were extremely low birth weight, 33 (39%) were very low birth weight and 14 neonates (16.5%) were considered as low birth weight (Table 1). High quality DNA was obtained from buccal epithelial cells (BEC) with an average concentration 255.22 ng/μL (range: 89.5 to 421 ng/μl) and from whole blood (WB) (34.43 ng/μl; range 5.5 to 182.8 ng/μl). Interestingly, the DNA yield from BEC, per set of experiments, was significantly higher than WB (p ≤ 0.0001).
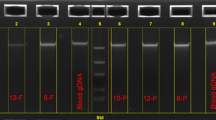
To confirm that the DNAs obtained from BEC are free of any external DNA contamination, we performed the STR (Short Terminal Repeat) on 12 DNA pairs (12 BEC and 12 WB) using AmpFlSTR® Plus and the results were analyzed by GeneMarker 2.4 (Softgenetics, PA). Full, single source profiles were obtained from all samples and the profile of each BEC sample matched at all 15 loci and Amelogenein with the WB sample from the same individual (Figure 1). These results confirmed that the DNA obtained from the buccal swabs was not contaminated by any external DNAs (Supplementary Table S1 online). Concomitantly, the same 12 DNA pairs were tested using six TaqMan Probe-based Allelic discrimination assays for detection of single nucleotide polymorphisms (SNPs). Data from genetic profiles obtained from BEC corroborate 100% with those obtained from WB cells (Supplementary Table S2 online). We then used these DNAs to amplify a 485 bp sequence in the regulatory region of the TRIM21 gene containing the polymorphic Bgl II site (C/T). PCR-RFLP reactions were successfully performed for all DNA from BEC samples (Supplementary Figure S1 online).

Electropherogram of four STR loci.
The electropherogram of STRs were obtained from the amplification of WB (A) and BEC (B) samples. Across the profile two or less alleles are present at each locus and peak height ratio, between sister alleles at heterozygous loci, is within the expected rage indicating that both are single source samples (i.e. absence of contamination). The two profiles are an exact match demonstrating that the samples originated from the same individual.
Whole exome sequencing is the state-of-art means of genomic analysis. In an effort to evaluate whether the quality of the buccal epithelial cell DNA in healthy and pathological cases were adequate for next generation DNA sequencing technologies, we performed whole exome sequencing on four samples: two healthy premature infants and two infants with necrotizing enterocolitis (Bell's Stage III). The total number of reads for the controls #1, #2 and patients #1 and #2 were respectively 18,448,882, 24,206,718, 16,874,844 and 31,507,076. The average coverage was evaluated at 17.1×. The total number of coding variants discovered that passed analysis parameters was 18,649 ± 1,781 (Table 2). Our data corroborate with laboratory results obtained from the whole blood of healthy donors (unpublished data) and other previously published studies5,6
Our data provides the proof of concept that an already tried-and-tested buccal swab method is a reliable, inexpensive, non-invasive and suitable for biobanking of genomic materials. The DNA from BEC meets quantitative and qualitative requirements for high-throughput screening and next generation sequencing technologies.
Discussion
Our cohort of eighty-five premature infants is larger than any previously published studies on the use of BEC for DNA extraction in this population and is the only one focused exclusively on premature infants7,8,9,10,11,12.
Although whole blood samples provide generous amounts of good quality DNA, its collection remains invasive, expensive and technical difficulties associated with phlebotomy in small, sick preterm neonates often limit the volume of blood obtained and therefore, reduce the possibility of genomic testing. Phlebotomy from a neonate requires a skilled practitioner and the use of a large number of DNA purification columns (7 to 8 purification columns for 750 μl of blood), which significantly increases the cost and the extraction time. In addition, blood drawing, placement of peripheral and central vascular catheters, can cause pain and discomfort, compromise the skin integrity and increase the risk of infection in premature neonates. Alternatively, BEC can be collected by any trained member of a clinical care or research team and does not require the use of a particular extraction kit, reducing the overall cost and does not increase the odds of the infection associated with venipuncture. Research dedicated to advancing the care of premature neonates has necessitated investigation into reliable sources of genomic DNA. This study successfully validated the use of BEC as a non-invasive and reliable source of genomic DNA for use in a variety of genetic assays.
The issue of possible contamination always remains paramount. STR is a reliable method to determine whether or not any contamination with external DNA exists. To confirm the BEC and blood samples were from the sample individual, we utilized STR analysis of matched BEC and blood samples from 12 patients. Historically, the STR analysis has been used to ensure that a prenatal fetal sample is not contaminated with maternal cells prior to assaying the prenatal fetal sample13. Therefore, it constitutes a very sensitive method able to detect the DNA from single cell. All 15 STR loci and amelognein showed similar profile between the matched BEC and blood samples. This demonstrates there was not any contamination of the BEC and blood samples by maternal or any other DNA.
While the use of BEC for genomic DNA is not a novel method, we successfully showed that the improved methodology can be used for genetic analysis and the state-of-art genomic technology such as whole exome sequencing.
The collection of DNA from BEC provides high quality and quantity DNA for genomic studies. Furthermore, it will allow for easy re-sampling in premature or newborn infants if an assay fails.
Methods and Patients
Patient selection
Following Institutional Review Board approval protocol at Children's National Health system, parental consent were obtained from all patients with a gestational age of less than 36 weeks included in this study. Preterm infants were recruited at the Neonatal Intensive Care Unit (NICU) at Children's National Health System, a 54 bed, level IV NICU. All enrolled patients were Nil Per Os (NPO) at the time of buccal swab and blood collection. Patient demographics are represented in Table 1.
Sample collection and DNA extraction
Buccal swabs were collected from 85 patients with a gestational age ranging from 24–36 weeks using cytology brushes. Briefly, 2 brushes were twirled on the inside of each cheek for less than 10 seconds. 0.75 mL of blood was also collected from 61 patients. DNA was extracted from blood or buccal swab specimens using Qiagen Buccal Cell and DNAeasy Kit (Qiagen Sciences, MD) respectively. We modified the buccal cell extraction kit for each experiment set to accommodate a three-fold increase in sample processing for a cell lysate volume of 900 μL. 100 μL of whole blood in EDTA was used for DNA extraction, corresponding to the recommended upper volume limit by the Qiagen DNA extraction kit for one set of experiments.
Short tandem repeat (STR) analysis
DNAs were diluted to a final concentration of 0.5 ng/μL. One μL of each samples was amplified with AmpFlSTR® Identifiler® Plus (Applied Biosystems) with 2 μL of reaction mix, 1 μL of primer mix, 1 μL dH2O in a 5 μL final volume. The amplification cycle was 11 min. at 95°C, 28×(20 sec. at 94°C, 3 min. at 59°C), 10 min at 60°C, ∞ at 4°C. To prepare samples for electrophoresis, 10 μL of LIZ 120 size standard was added to 400 mL of Hi-Di formamide (Applied Biosystems) and 1 mL of sample was added to 10 mL of the Formamide/ILS mixture. The AmpFlSTR® Identifiler® Plus kit contains 15 STR systems (D8S1179, D21S11, D7S820, CSF1PO, D3S1358, TH01, D13S317, D16S539, D2S1338, D19S433, VWA, TPOX, D18S51, D5S818 and FGA). Samples were electrophoresed on the 3130 Genetic Analyzer (Applied Biosystems), using a 36 cm capillary and POP-7 polymer with injection parameters of 1.2 kV for 16 s. STR fragment analysis was GeneMarker 2.4 (Softgenetics, PA).
Taqman Probe-based assay
10 ng of DNA obtained from the buccal swabs and whole blood were analyzed for six SNPs (rs1799983 [C___3219460_20], rs854560 [C___2259750_20], rs1137101 [C___8722581_10], rs1815739 [C____590093_1_], rs1046502 [C___7577769_10] and rs4871385 [C__12060045_20) according to the manufacturer's protocol (Life Technologies, CA). Briefly, this method employs the 5' nuclease activity of Taq polymerase to detect a fluorescent reporter signal generated during PCR reactions. Data were collected on a Life Technologies 7900HT Sequence Detection System and analyzed using the SDS 2.4 software.
PCR-restriction fragment length polymorphism (PCR- RFLP) analysis
200 ng of BEC DNA from 85 premature infants were subject to PCR using 5′ CTG TAC ATC CAC AGT GAG C 3′ (Forward primer) and 5′ CAT CCC TTG TCA GAT GGA TAG 3′ (Reverse primer). The PCR products were then digested with the restriction enzyme Bgl II (New England BioLabs, MA) to determine a polymorphism in the TRIM21 gene according to previously published data14,15.
Genomic DNA quantification and quality assessment
Quality of genomic DNA was assessed by 1% agarose gel. Samples that pass the gel check proceed to quantification using Qubit 2.0 Fluorometer using Qubit® dsDNA BR Assay Kits (Invitrogen, CA).
Illumina DNA library preparation
DNA library preparation was completed using Illumina's TruSeq DNA Sample Prep v2 kit protocol (Illumina, CA). The DNA (1 μg) was randomly fragmented by the Covaris S220 (Covaris, MA) using insert sizes of 100 to 900 bp. DNA quality was checked by analysis of samples on the Agilent 2100 Bioanalyzer using a DNA High Sensitivity chip followed by quantification on the Qubit 2.0 Fluorometer (Life Technologies).
Exome enrichment was carried out using the standard protocol for the Illumina TruSeq Exome Enrichment Kit (Illumina, CA). Combining 500 ng from each sample creates library pools (only samples with different index adapters were pooled together). Following the Illumina Trueseq Exome Enrichment protocol, the quality of the final libraries is checked using an Agilent High Sensitivity DNA Bioanalyzer chip (Agilent, CA). The libraries are then quantified using the Qubit 2.0 Fluorometer and normalized to 1 ng/ul.
Quantative PCR (qPCR)
The Kapa Biosystems Library Quantification Kit-Illumina/ABI Prism kit is used for the qPCR (Kapa Biosystems, MA). The quality of the final libraries are checked using an Agilent High Sensitivity DNA Bioanalyzer chip (Agilent, CA). The libraries are quantified using the Qubit 2.0 Fluorometer and the libraries are normalized to 1 ng/ul. The qPCR is performed on the normalized library with a Life Technologies 7900HT Real Time PCR System to determine the concentration. All of the libraries are pooled together and normalized to single concentration (4 nM). For the HiscanSQ analysis seven pools were created has and each pool has four samples for a total of 28 samples. qPCR is performed on the library pools using the ABI 7900HT Fast Real Time PCR System to validate the final concentration. Thermal cycling parameters were as follows: with the following conditions: 95°C for 5 minutes followed by 35 cycles of 95°C for 30 seconds, 60°C for 30 seconds.
Illumina cluster generation and sequencing
The Illumina cBot was used to hybridize the libraries to the flowcell and generate clusters. The flowcell was then loaded onto the Illumina HiScan along with the TruSeq SBS v3 200 cycle kit (Illumina, CA) and ran on a 101 × 7 × 101 paired end single multiplexed program. The exome sequencing analysis took approximately 10 days to finish.
Genome analysis tool kit for exome analysis
We sequenced four samples on one lane on an Illumina HiScanSQ system, aligned the resulting reads to the hg19 reference genome with Burrows-Wheeler alignment (BWA)16, applied the genome analysis tool kit (GATK)17 base quality score recalibration, insertion/deletion realignment, duplicate removal and performed SNP and insertion-deletion18 discovery and genotyping across all four samples simultaneously using standard hard filtering parameters or variant quality score recalibration19.
Statistical analysis
A student's two-tailed t-test was used to compare yields between whole blood and buccal cells.
Additional information
Statement of financial support The project described was supported by Award Number UL1RR031988 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
References
Babovic-Vuksanovic, D. et al. Guidelines for buccal smear collection in breast-fed infants. Am J Med Genet 84, 357–360, 10.1002/(SICI)1096-8628 (1999).
Babovic-Vuksanovic, D., Michels, V. V., Law, M. E., Lindor, N. M. & Jalal, S. M. Maternal cell contamination of buccal smear samples in nursing neonates. Clin Genet 53, 114–118, 10.1111/j.1399-0004 (1998).
Harris, C., Wilkerson, C., Clark, K., Lazarski, K. & Meisner, L. Potential use of buccal smears for rapid diagnosis of autosomal trisomy or chromosomal sex in newborn infants using DNA probes. Am J Med Genet 53, 355–358, 10.1002/ajmg.1320530410 (1994).
Schad, C. R. et al. Application of fluorescent in situ hybridization with X and Y chromosome specific probes to buccal smear analysis. Am J Med Genet 66, 187–192, 10.1002/(SICI)1096-8628 (1996).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12, 745–755, 10.1038/nrg3031 (2011).
Wang, L. et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res 22, 208–219, 10.1101/gr.123109.111 (2012).
Bennett, L. C., Kraemer, R. & Liechti-Gallati, S. Buccal cell DNA analysis in premature and term neonates: screening for mutations of the complete coding region for the cystic fibrosis transmembrane conductance regulator. Eur J Pediatr 159, 99–102 (2000).
Gavriel, G., Modi, N., Stanier, P. & Moore, G. E. Neonatal buccal cell collection for DNA analysis. Arch Dis Child Fetal Neonatal Ed 90, F187, 10.1136/adc.2004.062661 (2005).
Lench, N., Stanier, P. & Williamson, R. Simple non-invasive method to obtain DNA for gene analysis. Lancet 1, 1356–1358, 10.1016/S0140-6736(88)92178-2 (1988).
Parad, R. B. Buccal cell DNA mutation analysis for diagnosis of cystic fibrosis in newborns and infants inaccessible to sweat chloride measurement. Pediatrics 101, 851–855, 10.1542/peds.101.5.851 (1998).
Richards, B. et al. Multiplex PCR amplification from the CFTR gene using DNA prepared from buccal brushes/swabs. Hum Mol Genet 2, 159–163, 10.1093/hmg/2.2.159 (1993).
Zheng, S., Ma, X., Buffler, P. A., Smith, M. T. & Wiencke, J. K. Whole genome amplification increases the efficiency and validity of buccal cell genotyping in pediatric populations. Cancer Epidemiol Biomarkers Prev 10, 697–700 (2001).
Kroneis, T. et al. Combined molecular genetic and cytogenetic analysis from single cells after isothermal whole-genome amplification. Clinical chemistry 57, 1032–1041, 10.1373/clinchem.2011.162131, 10.1373/clinchem.2011.162131 (2011).
Tatari-Calderone, Z. et al. rs660 polymorphism in Ro52 (SSA1; TRIM21) is a marker for age-dependent tolerance induction and efficiency of alloimmunization in sickle cell disease. Mol Immunol 47, 64–70, 10.1016/j.molimm.2008.12.027 (2009).
Tsugu, H., Horowitz, R., Gibson, N. & Frank, M. B. The location of a disease-associated polymorphism and genomic structure of the human 52-kDa Ro/SSA locus (SSA1). Genomics 24, 541–548, 10.1006/geno.1994.1664 (1994).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, 10.1093/bioinformatics/btp698 (2010).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, 10.1101/gr.107524.110 (2010).
Kugathasan, S. et al. Epidemiologic and clinical characteristics of children with newly diagnosed inflammatory bowel disease in Wisconsin: a statewide population-based study. J Pediatr 143, 525–53 (2003).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498, 10.1038/ng.806 (2011).
Author information
Authors and Affiliations
Contributions
M.S. enrolled patients, medical data analysis and contributed to writing the manuscript; C.C. performed the research, contributed to data analysis and writing the manuscript; J.M.D. contributed to genomic data generation and analysis; D.P. contributed to STRs data generation and analysis; A.L.B. contributed with the institutional review board protocol writing and submission; S.V. performed statistical analysis, contributed to data analysis; K.R.B. contributed to patients enrollment and data analysis; N.C.L. contributed to writing the manuscript and data analysis; and A.D.S. contributed data analysis and review the surgical data; Z.T.C. participated in data analysis, study design and prepared the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Said, M., Cappiello, C., Devaney, J. et al. Genomics In Premature Infants: A Non-Invasive Strategy To Obtain High-Quality DNA. Sci Rep 4, 4286 (2014). https://doi.org/10.1038/srep04286
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04286
This article is cited by
-
Are Immune Modulating Single Nucleotide Polymorphisms Associated with Necrotizing Enterocolitis?
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
