
Abstract
FK506 binding protein 51 (FKBP51, also called FKBP5) belongs to a family of immunophilins, FK506 binding proteins (FKBPs). Members of this family are targets for drugs such as rapamycin and cyclosporine. Although FKBP5 shares characteristics with other FKBPs, it also has unique features, especially its role in the regulation of multiple signalling pathways and in tumourigenesis and chemoresistance. In this review, we will focus on the recently discovered role of FKBP5 in cancer aetiology and response to antineoplastic therapy.
Similar content being viewed by others

Main
FK506 binding protein 51 (FKBP51, also called FKBP5) is a 51-kDa FK506 binding protein that is a member of a family of immunophilins, FK506 binding proteins (FKBPs). Members of this family contain both FKBP domain(s) and tetratricopeptide repeat (TPR) domain(s) (Kang et al, 2008). The FKBP domain contains peptidylprolyl isomerase (PPIase) activity that catalyses the cis–trans conversion of peptidylprolyl bonds, a reaction that is important for protein folding (Fruman et al, 1994). The TPR domains at the C terminus are involved in protein–protein interactions (Scheufler et al, 2000). In general, TPR domains are all-helical structures consisting of 2–16 units of a consensus 34-aa motif. The domain is found in more than 800 different proteins from bacteria and humans. The first described and most well-known FKBP family member is FKBP12, a small protein with a single FKBP domain and a molecular size of 12 kDa. FK506 binding protein 12 is a major binding partner for FK506 and rapamycin (Siekierka et al, 1989). Additional members of this family have been identified on the basis of their ability to bind FK506, including FKBP13, FKBP25, FKBP38, FKBP52, and FKBP51 (named to reflect their molecular weights), all of which contain the FKBP domain that is responsible for drug ligand binding and PPIase activity (Kang et al, 2008).
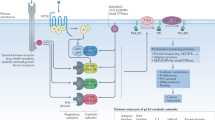
Human FKBP5 was first cloned from a HeLa cell cDNA library in 1995 (Baughman et al, 1995). It mediates FK506 inhibition of calcineurin in vitro and is highly expressed in murine T lymphocytes (Baughman et al, 1997). FK506 binding protein 5 contains two consecutive FKBP domains and a three-unit repeat of the TPR domain (Figure 1). The first FKBP domain (FK1) of FKBP5 shares 48% sequence identity to the FK domain of FKBP12 and has measurable PPIase activity, with a binding pocket for FK506 and rapamycin (Sinars et al, 2003). FK2 is also structurally similar to the FKBP domain of FKBP12, despite having only 26% sequence identity. However, FK2 lacks measurable PPIase activity. Presumably, FK2 resulted from an FK domain duplication event, but subsequently lost its PPIase activity, while it appeared to have gained protein interaction ability (Sinars et al, 2003). The FKBP5 structure study provides important initial insights into the relative orientations of the FK1, FK2, and TPR domains that are important for protein interaction and/or drug ligand binding (Sinars et al, 2003).

Structure of FKBP5 with its major domains that are critical for its function as well as with the list of its interactive proteins by domain. AKT=a serine/threonine protein kinase, also called PKB; AR=androgen receptor; FK=FKBP-type domain; GR=glucocorticoid receptor; Hsp90=heat shock protein 90; IKKα=IκB kinase α subunit; PHLPP=PH domain and leucine-rich repeat protein phosphatases; PR=progesterone receptor; TPR=tetratricopeptide repeat.
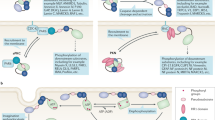
One of the major functions of FKBP5 is its involvement in the modulation of steroid receptor function, including progesterone, androgen and glucocorticoid receptors (GR), by forming a complex with the heat shock proteins Hsp90/Hsp70. Both FKBP5 and its family member FKBP4 (also called FKBP52) are involved in steroid receptor signalling, and their interchange served as the earliest known event in steroid receptor activation. Davies et al (2002) have used both in vitro and in vivo models to show the hormone-induced loss of FKBP5 and gain of FKBP4, providing evidence that the hormone-binding event causes an interchange of FKBP5 and FKBP4 immunophilins within the GR heterocomplex. The complex then dissociates, allowing the binding of GR to DNA-binding motifs in its target genes to regulate gene transcription (Figure 2A). Therefore, FKBP5 has been identified as a sensitive biomarker of corticosteroid responsiveness in vivo (Table 1) (Sinars et al, 2003; Rees-Unwin et al, 2007). In addition to a role in tumourigenesis (see below), numerous studies have also indicated that FKBP5 may have a role in psychiatric diseases such as depression and in response to the treatment of depression through modulation of hormone receptors (Schosser and Kasper, 2009). FK506 binding protein 5 and Hsp90 are also important for the clearance of tau, a microtubule-associated protein that accumulates in a group of neurodegenerative disorders such as Alzheimer's disease (Jinwal et al, 2010).

Schematic model of FKBP5 functions involved in several different signalling pathways, including glucocorticoid receptor (GR) signalling pathways (A), as well as NF-κB (B) and AKT–PHLPP (C) pathways. (A) In the absence of dexamethasone, FKBP5 is the primary immunophilin of the FKBP–Hsp90–GR complex in an inactive stable state. Upon dexamethasone binding, FKBP5 is displaced by FKBP4, and the complex can enter the nucleus. The complex then dissociates, allowing binding of GR to DNA-binding motifs of target genes, leading to apoptosis. (B) In the presence of doxorubicin or damage from radiation, FKBP5 might involve in the control of IKKα activity, which can induce IκBα degradation via its phosphorylation, nuclear translocation, and activation of NF-κB, and the expression of its target genes, consequently triggering cell apoptosis. (C) FK506 binding protein 5 interacts with PHLPP and AKT, acting as a scaffolding protein that promotes the interaction between AKT and PHLPP, thereby enhancing the dephosphorylation of AKT and inactivating AKT, which results in the blockade of AKT signalling for cell survival and leads to cell apoptosis or death.
Of equal or greater interest than these effects on steroid hormone signalling are recent studies that have indicated that FKBP5 could also be a biomarker for tumourigenesis and chemoresistance. Roles in those processes extend beyond its functions as a co-chaperone and a PPIase. Subsequent paragraphs will focus on the discovery of the role of FKBP5 in cancer aetiology and chemoresistance.
FK506 binding protein 51 expression and regulation in cancer
Human FKBP5 is highly expressed in multiple tissues, including kidney, skeletal muscle, liver, placenta, heart, and peripheral blood. However, its expression level is much lower in the pancreas, spleen, and stomach (Baughman et al, 1997). Although initial studies found no detectable FKBP5 in the brain, colon, or lung, subsequent studies have shown expression of FKBP5 in human brain tissues (Nair et al, 1997) and colon tissues (Mukaide et al, 2008). FK506 binding protein 5 expression can be induced through the activation of progesterone receptor, androgen receptor (AR), and GR (Ratajczak et al, 2003). Hormone response elements in the introns of FKBP5 regulate the induction of expression by glucocorticoids, progesterone, and androgens. For example, Febbo et al (2005) had identified FKBP5 as an androgen-inducible gene, and demonstrated a physical interaction between FKBP5 and AR. Recent studies have identified a long-range activation of FKBP5 transcription in prostate cancer cells by the AR via distal intronic enhancers (Makkonen et al, 2009). The FKBP5 locus harbours 13 in silico-predicted androgen response elements (AREs), most of which are located downstream from the transcription start site and are capable of binding AR in vitro. These results indicated that very distal AREs can act as robust enhancers, highlighting the potential importance of the long-range regulation of FKBP5 expression by the AR (Makkonen et al, 2009). FK506 binding protein 5 expression can also be induced by the GR in human lung cancer A549 cells, where FKBP5 mRNA are accumulated in response to dexamethasone exposure (Paakinaho et al, 2010). A major intronic enhancer within the FKBP5 locus was identified to bind to GR. There were also GREs identified in the proximal promoter of FKBP5. The GR is capable of activating FKBP5 transcription and also evoking changes in chromatin structure (Paakinaho et al, 2010). FK506 binding protein 5 can also function within an auto-regulatory negative feedback loop to modulate its own expression. Exogenous FKBP5 expression was able to downregulate the transcription of genomic FKBP5 in the presence of dexamethasone through this auto-regulation process (Park et al, 2007).
Both overexpression and downregulation of FKBP5 have been observed in human cancers. According to the Oncomine, FKBP5 is overexpressed in brain cancers, prostate cancer, lymphoma, head and neck cancer, and melanoma. On the other hand, FKBP5 has also been shown to be downregulated in pancreatic cancer (Pei et al, 2009), melanoma, colon cancer, and testicular cancer. Gene expression data for those studies are available through the Oncomine, and studies describing these observations are also referenced in this website (http://www.oncomine.org). Similar to its expression pattern, FKBP5 has been shown to either promote or suppress tumour growth through its regulation of different signalling pathways in a specific tissue environment.
Role of FKBP5 in tumourigenesis and response to antineoplastic therapy
FK506 binding protein 5 has been shown to be involved in several different signalling pathways, including steroid receptor signalling pathways, NF-κB, and AKT (a serine/threonine protein kinase, also called PKB)–PHLPP (leucine-rich repeat protein phosphatase) pathways, all of which have important roles in tumourigenesis and response to antineoplastic chemotherapy (Figure 2 and Table 1).
FK506 binding protein 5 is a key regulatory component of the androgen–receptor complex
Although FKBP5 knockout mice show normal androgen signalling (Yong et al, 2007), it has been linked to AR signalling and prostate cancer. As discussed above, FKBP5 itself is a target gene of AR. FK506 binding protein 5 can form a complex with AR to enhance AR transcriptional activity in prostate cancer (Febbo et al, 2005). Recently, Ni et al (2010) showed that FKBP5 stimulates the recruitment of cochaperone p23 to the ATP-bound form of Hsp90, forming an FKBP5–Hsp90–p23 superchaperone complex, which stimulates androgen-dependent transcription activation and cell growth. Therefore, FKBP5 might serve as a novel drug target to help disrupt AR-mediated signalling in prostate cancer.
FK506 binding protein 5 regulates the NF-κB pathway
Bouwmeester et al (2004) first identified a physical interaction between FKBP5 and nuclear factor IκB kinase α subunit (IKKα) during a proteomics study. In that study, FKBP5 was copurified with IKKα as well as IKKɛ, TGF beta activated kinase 1, and MAP kinase/ERK kinase kinase 1 (Bouwmeester et al, 2004). The interaction with IKKα was confirmed by co-immunoprecipitation. These results indicated that FKBP5 might have a role in NF-κB signalling.
Several studies by Romano et al have demonstrated that the downregulation of FKBP5 can sensitise cells to irradiation and anthracyclines through the regulation of NF-κB (Avellino et al, 2005). They showed that treatment of melanoma or acute lymphoblastic leukaemia cell lines with anthracycline would induce NF-κB activity. However, treatment with rapamycin significantly sensitised the cells to anthracycline by inhibition of the NF-κB activity. This effect was associated with FKBP5 activity, as downregulation of FKBP5 increased the rapamycin inhibition of NF-κB activity, and, in turn, increased apoptosis (Avellino et al, 2005). In addition, the same group also found that higher expression of FKBP5 might influence radioresistance through the activation of NF-κB in melanoma cells (Romano et al, 2010). They also found that FKBP5 was overexpressed in melanoma samples compared with normal skin tissue, and pretreatment of mice xenograft tumours with FKBP5-siRNA provoked massive apoptosis after irradiation (Romano et al, 2010). These experiments provided evidence that, both in vitro and in vivo, FKBP5 might be a promising target for radiosensitising agents against malignant melanoma.
FK506 binding protein 5 is a negative regulator of AKT activation in tumourigenesis and chemoresistance
Regulation of NF-κB by FKBP5 emphasised the potential importance of FKBP5 in tumourigenesis and response to therapy. However, it was not until very recently that another important function of FKBP5 was identified. Studies from our laboratory first demonstrated, through the use of genome-wide screening, that levels of FKBP5 were associated with response to two cytidine analogues, gemcitabine and cytosine arabinoside (AraC) (Li et al, 2008). Higher levels of expression of FKBP5 were associated with sensitivity and lower levels of expression were associated with resistance to gemcitabine and AraC. Furthermore, downregulation of FKBP5 desensitised pancreatic and breast cancer cell lines to several different classes of chemotherapeutic agents, including not only cytidine analogues but also taxanes, ironotecan, and etoposide (Li et al, 2008; Pei et al, 2009). In this case, the actions of FKBP5 on NF-κB activation or hormone induction could not explain the increased chemoresistance in cells with decreased FKBP5 expression, suggesting the existence of other mechanisms by which FKBP5 regulates cell survival.
We recently identified FKBP5 as a negative regulator of AKT (Pei et al, 2009). Full activation of AKT requires phosphorylation at Thr308 within the activation loop of the kinase domain and Ser473 at a hydrophobic motif just C-terminal to the kinase domain. Thr308 is phosphorylated by pyruvate dehydrogenase kinase isozyme 1, mitochondrial (Alessi et al, 1997), while Ser473 is phosphorylated by mTOR complex 2 (Sarbassov et al, 2005). AKT activity is also negatively regulated by lipid or protein phosphatases. Two protein phosphatases have been shown to dephosphorylate AKT and negatively regulate AKT acivity. Protein phosphatase 2 (PP2, also known as PP2A) holoenzymes dephosphorylate Ser308 (Padmanabhan et al, 2009), while PH domain and leucine-rich repeat protein phosphatase 1 and 2 (PHLPP1 and 2) dephosphorylate Ser473 (Brognard and Newton, 2008). Therefore, the balance between kinases and phosphatases determines AKT activity. We found that FKBP5 functions as a scaffolding protein that enhances the PHLPP–AKT interaction and facilitates PHLPP-mediated dephosphorylation of AKT-Ser473. Downregulation of FKBP5 results in decreased PHLPP–AKT interaction and increased AKT phosphorylation. Therefore, FKBP5 might also function as a tumour suppressor in the AKT signalling pathway, similar to phosphatase and tensin homologue (PTEN). Indeed, we found that FKBP5 levels were low or absent in pancreatic cancer cell lines and tissue samples from patients with pancreatic cancer, correlating with increased AKT Ser473 phosphorylation. We also observed downregulation of FKBP5 in breast cancer cell lines. High levels of FKBP5 led to decreased AKT phosphorylation and increased chemosensitivity, whereas low levels of FKBP5 resulted in increased AKT phosphorylation and decreased chemosensitivity. These observations make FKBP5 a potentially important biomarker for sensitivity to chemotherapy, and variation in FKBP5 levels might determine patient responses to chemotherapy (Figure 2C and Table 1) (Pei et al, 2009). In addition, determination of levels of FKBP5 might provide insights that could help to select patients for different combination therapies with inhibitors targeting the AKT pathway (Pei et al, 2009).
Conclusions
In summary, FKBP5 has altered expression levels in many different tumours. Through its influence on steroid receptor maturation, as well as NF-κB and AKT signalling pathways, FKBP5 plays an important role in tumourigenesis and response to antineoplastic therapy (Figure 2 and Table 1). All of these observations raise the possibility that FKBP5 might be a biomarker for chemoradiosensitivity. On the basis of studies using lymphoblastoid cell lines from 300 individuals, we have observed large variations in FKBP5 expression that correlate with cellular sensitivity to gemcitabine and AraC (Li et al, 2008). Future studies will be needed to confirm the role of FKBP5 in predicting chemosensitivity in clinical samples. In addition, it would be important to investigate factors that are responsible for variation in FKBP5 expression in individuals. Common single-nucleotide polymorphisms (SNPs) in the FKBP5 gene might contribute to variation in FKBP5 protein expression. Therefore, genotyping these SNPs and determining how they affect FKBP5 expression and, thus, drug sensitivity or tumourigenesis might be important next steps for this line of investigation. Additionally, the role of FKBP5 in tumourigenesis remains controversial. Whether FKBP5 functions as an oncogene or a tumour suppressor might depend on the tissue type and differences among pathways expressed in those tumours. For example, FKBP5 is found to be downregulated in pancreatic tumour tissue, while it is overexpressed in melanoma. These discrepancies need to be further clarified in the future using animal models.
Because of the role of FKBP5 in the regulation of multiple important biological pathways as discussed in this review, variation in FKBP5 could be of significance in many diseases beyond cancer, and in diseases such as diabetes (Tian, 2005). All of these possibilities will need to be investigated. Future studies will also be required to address questions such as how FKBP5 is regulated, and whether there are other pathways in which FKBP5 might be involved.
In summary, FKBP5 has multiple roles in the regulation of a variety of signalling pathways. Therefore, understanding the mechanisms underlying its regulation and its role in the development of cancer and in response to anti-neoplastic therapy could help make it possible to better individualise the treatment of human disease.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7: 261–269
Avellino R, Romano S, Parasole R, Bisogni R, Lamberti A, Poggi V, Venuta S, Romano MF (2005) Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood 106: 1400–1406
Baughman G, Wiederrecht GJ, Campbell NF, Martin MM, Bourgeois S (1995) FKBP51, a novel T-cell-specific immunophilin capable of calcineurin inhibition. Mol Cell Biol 15: 4395–4402
Baughman G, Wiederrecht GJ, Chang F, Martin MM, Bourgeois S (1997) Tissue distribution and abundance of human FKBP51, and FK506-binding protein that can mediate calcineurin inhibition. Biochem Biophys Res Commun 232: 437–443
Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Casari G, Drewes G, Gavin AC, Jackson DB, Joberty G, Neubauer G, Rick J, Kuster B, Superti-Furga G (2004) A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 6: 97–105
Brognard J, Newton AC (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol Metab 19: 223–230
Davies TH, Ning YM, Sanchez ER (2002) A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem 277: 4597–4600
Febbo PG, Lowenberg M, Thorner AR, Brown M, Loda M, Golub TR (2005) Androgen mediated regulation and functional implications of fkbp51 expression in prostate cancer. J Urol 173: 1772–1777
Fruman DA, Burakoff SJ, Bierer BE (1994) Immunophilins in protein folding and immunosuppression. FASEB J 8: 391–400
Jinwal UK, Koren III J, Borysov SI, Schmid AB, Abisambra JF, Blair LJ, Johnson AG, Jones JR, Shults CL, O’Leary III JC, Jin Y, Buchner J, Cox MB, Dickey CA (2010) The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J Neurosci 30: 591–599
Kang CB, Hong Y, Dhe-Paganon S, Yoon HS (2008) FKBP family proteins: immunophilins with versatile biological functions. Neurosignals 16: 318–325
Le Bihan S, Marsaud V, Mercier-Bodard C, Baulieu EE, Mader S, White JH, Renoir JM (1998) Calcium/calmodulin kinase inhibitors and immunosuppressant macrolides rapamycin and FK506 inhibit progestin-and glucocorticosteroid receptor-mediated transcription in human breast cancer T47D cells. Mol Endocrinol 12(7): 986–1001
Li L, Fridley B, Kalari K, Jenkins G, Batzler A, Safgren S, Hildebrandt M, Ames M, Schaid D, Wang L (2008) Gemcitabine and cytosine arabinoside cytotoxicity: association with lymphoblastoid cell expression. Cancer Res 68: 7050–7058
Makkonen H, Kauhanen M, Paakinaho V, Jaaskelainen T, Palvimo JJ (2009) Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Res 37: 4135–4148
Mukaide H, Adachi Y, Taketani S, Iwasaki M, Koike-Kiriyama N, Shigematsu A, Shi M, Yanai S, Yoshioka K, Kamiyama Y, Ikehara S (2008) FKBP51 expressed by both normal epithelial cells and adenocarcinoma of colon suppresses proliferation of colorectal adenocarcinoma. Cancer Invest 26: 385–390
Nair SC, Rimerman RA, Toran EJ, Chen S, Prapapanich V, Butts RN, Smith DF (1997) Molecular cloning of human FKBP51 and comparisons of immunophilin interactions with Hsp90 and progesterone receptor. Mol Cell Biol 17: 594–603
Ni L, Yang CS, Gioeli D, Frierson H, Toft DO, Paschal BM (2010) FKBP51 promotes assembly of the Hsp90 chaperone complex and regulates androgen receptor signaling in prostate cancer cells. Mol Cell Biol 30: 1243–1253
Paakinaho V, Makkonen H, Jaaskelainen T, Palvimo JJ (2010) Glucocorticoid receptor activates poised FKBP51 locus through long-distance interactions. Mol Endocrinol 24: 511–525
Padmanabhan S, Mukhopadhyay A, Narasimhan SD, Tesz G, Czech MP, Tissenbaum HA (2009) A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell 136: 939–951
Park J, Kim M, Na G, Jeon I, Kwon YK, Kim JH, Youn H, Koo Y (2007) Glucocorticoids modulate NF-kappaB-dependent gene expression by up-regulating FKBP51 expression in Newcastle disease virus-infected chickens. Mol Cell Endocrinol 278: 7–17
Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L (2009) FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16: 259–266
Ratajczak T, Ward BK, Minchin RF (2003) Immunophilin chaperones in steroid receptor signalling. Curr Top Med Chem 3: 1348–1357
Rees-Unwin KS, Craven RA, Davenport E, Hanrahan S, Totty NF, Dring AM, Banks RE, G JM, Davies FE (2007) Proteomic evaluation of pathways associated with dexamethasone-mediated apoptosis and resistance in multiple myeloma. Br J Haematol 139: 559–567
Romano MF, Avellino R, Petrella A, Bisogni R, Romano S, Venuta S (2004) Rapamycin inhibits doxorubicin-induced NF-kappaB/Rel nuclear activity and enhances the apoptosis of melanoma cells. Eur J Cancer 40(18): 2829–2836
Romano S, D’Angelillo A, Pacelli R, Staibano S, De Luna E, Bisogni R, Eskelinen EL, Mascolo M, Cali G, Arra C, Romano MF (2010) Role of FK506-binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ 17: 145–157
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307: 1098–1101
Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101: 199–210
Schosser A, Kasper S (2009) The role of pharmacogenetics in the treatment of depression and anxiety disorders. Int Clin Psychopharmacol 24: 277–288
Siekierka JJ, Hung SH, Poe M, Lin CS, Sigal NH (1989) A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 341: 755–757
Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J (2003) Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc Natl Acad Sci USA 100: 868–873
Tian R (2005) Another role for the celebrity: Akt and insulin resistance. Circ Res 96: 139–140
Yong W, Yang Z, Periyasamy S, Chen H, Yucel S, Li W, Lin LY, Wolf IM, Cohn MJ, Baskin LS, Sanchez ER, Shou W (2007) Essential role for co-chaperone Fkbp52 but not Fkbp51 in androgen receptor-mediated signaling and physiology. J Biol Chem 282: 5026–5036
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Li, L., Lou, Z. & Wang, L. The role of FKBP5 in cancer aetiology and chemoresistance. Br J Cancer 104, 19–23 (2011). https://doi.org/10.1038/sj.bjc.6606014
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6606014
Keywords
This article is cited by
-
Knocking out Fkbp51 decreases CCl4-induced liver injury through enhancement of mitochondrial function and Parkin activity
Cell & Bioscience (2024)
-
FKBP5 deficiency attenuates calcium oxalate kidney stone formation by suppressing cell–crystal adhesion, apoptosis and macrophage M1 polarization via inhibition of NF-κB signaling
Cellular and Molecular Life Sciences (2023)
-
Co-targeting HSP90 alpha and CDK7 overcomes resistance against HSP90 inhibitors in BCR-ABL1+ leukemia cells
Cell Death & Disease (2023)
-
Transcriptome sequencing reveals a lncRNA–mRNA interaction network in extramammary Paget’s disease
BMC Medical Genomics (2021)
-
Comparative analysis of FKBP family protein: evaluation, structure, and function in mammals and Drosophila melanogaster
BMC Developmental Biology (2018)
