
Abstract
Malaria parasites have a complex life cycle that includes specialized stages for transmission between their mosquito and human hosts. These stages are an understudied part of the lifecycle yet targeting them is an essential component of the effort to shrink the malaria map. The human parasite Plasmodium falciparum is responsible for the majority of deaths due to malaria. Our goal was to generate transgenic P. falciparum lines that could complete the lifecycle and produce fluorescent transmission stages for more in-depth and high-throughput studies. Using zinc-finger nuclease technology to engineer an integration site, we generated three transgenic P. falciparum lines in which tdtomato or gfp were stably integrated into the genome. Expression was driven by either stage-specific peg4 and csp promoters or the constitutive ef1a promoter. Phenotypic characterization of these lines demonstrates that they complete the life cycle with high infection rates and give rise to fluorescent mosquito stages. The transmission stages are sufficiently bright for intra-vital imaging, flow cytometry and scalable screening of chemical inhibitors and inhibitory antibodies.
Similar content being viewed by others


Introduction
Malaria remains one of the most important infectious diseases in the world, impacting approximately a third of the world’s population and responsible for over 400,000 deaths annually1. The disease is caused by parasites of the genus Plasmodium, transmitted to their mammalian host by the infected Anopheline mosquitoes. Malaria parasites have two distinct stages in their mammalian hosts: an asymptomatic pre-erythrocytic stage, consisting of sporozoites and the liver stages into which they develop, and an erythrocytic stage when high parasite numbers lead to clinical disease. A small proportion of blood stage parasites differentiate to gametocytes which, when taken up in the blood meal, initiate infection in the mosquito, giving rise to oocysts on the midgut wall that produce sporozoites that migrate to the mosquito’s salivary glands. The transmission stages of Plasmodium, namely sporozoites and gametocytes, are bottlenecks in the parasite life cycle and their low numbers has limited investigations into their biology and chemotherapeutic interventions.
Rodent malaria models have enabled more in-depth study of the difficult to access transmission stages and have played a particularly important role in our understanding of pre-erythrocytic stages. Nonetheless, there are species-specific differences across the Plasmodium genus, such as the unique morphology and prolonged development of gametocytes of the Laverania, that make it critical to perform confirmatory studies and inhibitor screening with human malaria parasites. In order to facilitate these investigations, we generated transgenic Plasmodium falciparum lines expressing tdTomato or Green Fluorescent Protein (GFP) under the control of pre-erythrocytic or gametocyte stage promoters. Here we describe the generation of these parasites and their fluorescent properties throughout the development of the transmission stages.
Results and Discussion
Generation of fluorescent P. falciparum lines
Previous studies have generated fluorescent P. falciparum lines using the constitutively expressed elongation factor eEF1α (PF3D7_1357000) promoter and GFP variants2,3. While useful, the low intensity of fluorescence of these parasites during sporozoite and exoerythrocytic stages has limited their utility, particularly when imaging in mosquito and mammalian tissues where there is a high degree of autofluorescence in the green channel. We reasoned that using a red fluorescent protein in the place of GFP could surmount some of these challenges, as the red-shifted fluorescence is better suited for intravital tissue imaging. We chose tandem-dimer Tomato (tdTomato)4 as its intrinsic brightness is nearly three times higher than that of GFP and six times higher than the commonly-used red fluorescent protein mCherry5.
Additionally, we reasoned that strong, stage-specific promoters could be used to further boost the fluorescent signal during the sporozoite and exoerythrocytic stage. We tested two potential candidates, the promoter of the circumsporozoite protein (CSP) gene (PF3D7_0304600), which drives expression of the most abundant sporozoite surface protein, and the promoter of the Plasmodium Early Gametocyte 4 (PEG4, also known as eTRAMP10.3) (PF3D7_1016900) gene. PEG4 was first described as a protein expressed in the early stages of gametocytogenesis6, and its promoter region has been shown to be active at those times7. Indeed, the utility of this promoter to drive episomal transgene expression in P. falciparum gametocytes has been previously demonstrated7. However, fluorescence in mosquito stages was never assessed as these studies used a line that does not make infection-competent gametocytes. More recently, PEG4 was reported to be the homolog of the well-studied P. yoelii upregulated in sporozoites 4 (UIS4) gene8. The uis4 promoter has been a valuable tool for driving strong fluorescence in sporozoites of P. yoelii and P. berghei9. We hypothesized that the same would be true for P. falciparum, potentially providing a promoter that could drive expression in both sporozoites and gametocytes.
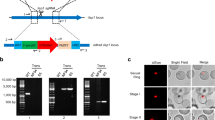
Typically, the production of genetically-modified P. falciparum parasites requires long in vitro selection and cloning times, which frequently results in the loss of the parasite’s ability to generate gametocytes or infect the mosquito. Beginning with a highly mosquito-infectious clone of the P. falciparum NF54 isolate10, we used zinc-finger nuclease (ZFN) technology to rapidly engineer a selectable marker-free Bxb1 phage integration site (attB) into the parasite genome (Fig. 1A). We selected the coding sequence of the pfs47 gene for attB introduction. Though recent work showed that Pfs47 is required for parasite evasion of the mosquito complement system in Anopheles gambiae mosquitoes11, it is not required for parasite survival in Anopheles stephensi12,13 and has been used by others as a locus for transgene expression in mosquito stages2,3. Once a stably-transfected parasite population was obtained, the line was cloned by limiting dilution, and the pfs47 locus was sequenced to verify the presence of the attB site (Fig. 1B). Clones were subsequently tested for gametocyte production and mosquito infectivity.

Introduction of the attB site into pfs47 locus and integration of tdTomato transgenes. (A) Schematic of the attB integration strategy. The expression of the pfs47-specific ZFN pair is under the control of the calmodulin promoter and the hsp86 3′UTR. The homologous donor sequence provided for double-strand break repair comprises a fragment of pfs47 stretching 0.68 kb upstream and 0.87 kb downstream of the ZFN target site (thunderbolt). The attB site is located between the two homologous regions and replaces the genomic pfs47 sequence containing the ZFNs binding site upon integration. The transfection plasmid pZFNpfs47-attB-hdhfr also contains a selectable marker for transient selection and propagation of the plasmid. (B) Sequence analysis of the unmodified endogenous pfs47 locus of the parental parasite line NF54 and the gene-edited recombinant locus NF54pf47-attB. (C) Schematic of the tdTomato plasmids used for transfection (PbDT = P. berghei DHFR-TS terminator, Cam pr = calmodulin promoter, BSD = Blasticidin S Deaminase selectable marker, hrp2T = HRP2 terminator) and of the NF54Pfs47attB attB locus pre- and post-integration of the tdTomato plasmids. Position of primers used to amplify the genomic locus (p13 & p14) and the integrated plasmid (p13 & p15) are shown. (D) PCR verification of plasmid integration. Primers p13 and p14 generate a 334 bp product from the unmodified pfs47attB locus (genomic), and primers p13 and p15 generate at 287 bp product upon tdTomato plasmid integration (integration). Shown is one gel with lines between lanes added to delineate the different Plasmodium clones. Primer sequences in Table S1.
A clone with high production of infectious gametocytes (NF54Pfs47attB C10) was then selected for secondary transfection. Using the Bxb1 integrase system14, a plasmid containing either Pf-ef1α-tdTomato, Pf-peg4-tdTomato, (Fig. 1C) or Pf-csp-GFP expression cassettes along with a Blasticidin S Deaminase (BSD) resistance cassette was introduced into the chromosomal pfs47 attB site. Stably transfected cultures were cloned by limiting dilution and tested for plasmid integration (Fig. 1D) and gametocyte production. Three clones of each of the tdTomato lines were passaged through mosquitoes to identify a clone producing robust sporozoite numbers. Pf-peg4-tdTomato Clone A and Pf-ef1α-tdTomato Clone 3D06 were chosen for further characterization. The Pf-csp-GFP line was not cloned and the uncloned line was studied.
Since these lines are the product of two sequential transfections and a limiting dilution cloning procedure, the tdTomato clones were further analyzed to determine whether they harbored the selectable marker cassettes used in each transfection. As expected, diagnostic PCRs performed with genomic DNA from both tdTomato lines had the BSD cassette (Supplementary Fig. 1). Because the Bxb1 attB site was introduced through marker-free homology directed repair, the episomal plasmid carrying the ZFNs and hDHFR marker was expected to have been lost after maintenance of the NF54Pfs47attB C10 line in the absence of WR99210 pressure. Unexpectedly, PCRs revealed that the Pf-ef1α-tdTomato line retained both the hDHFR cassette and the zinc finger components, whereas the Pf-peg4-tdTomato line did not (Supplementary Fig. 1). We attribute this to the initial NF54Pfs47attB C10 culture being comprised of two populations, one that had undergone ZFN-mediated recombination that integrated the attB locus without the remainder of the plasmid, and the other that underwent single-site crossover integration with the entire ZFN plasmid being integrated. The Pf-peg4-tdTomato clones were found by PCR to have been generated from parasites that had already lost the plasmid carrying the hDHFR cassette and the ZFNs, and thus carry only the BSD cassette. In contrast, the Pf-ef1α-tdTomato clones integrated into the parasites carrying the original ZFN plasmid and thus contain both the hDHFR and BSD cassettes (Supplementary Fig. 1).
To phenotypically confirm retention of the hDHFR selectable marker in the Pf-ef1α-tdTomato clone, we cultured blood-stage parasites of both tdTomato lines in the presence of 2.5 nM of the anti-folate antimalarial agent WR99210, which inhibits P. falciparum DHFR but allows hDHFR-expressing parasites to proliferate. In the absence of drug both lines grew well, were split on day 6, and continued their normal growth. In the presence of WR99210, the peg4-tdTomato clone began to die within the first 24 hr of drug pressure and no parasites were microscopically visible by day 3. We continued to follow the culture for an additional 9 days during which time no viable parasites were observed. In contrast, the Pf-ef1α-tdTomato parasites proliferated in the presence of 2.5 nM WR99210. While the retention of the hDHFR resistance cassette in Pf-ef1α-tdTomato clone will preclude its use for the discovery of novel anti-folate compounds, integration of the ZFN-expression plasmid should not interfere with other intended uses of this cell line.
Phenotypic characterization of P. falciparum fluorescent lines
To characterize the tdTomato clones and the uncloned GFP line in the mosquito host, Anopheles stephensi mosquitoes were fed with human blood containing gametocytes and subsequent parasite development in mosquitoes was followed. Oocyst numbers for the tdTomato lines were robust and as expected, both Pf-ef1α-tdTomato and Pf-peg4-tdTomato oocysts were fluorescent (Fig. 2A,B). Indeed at this stage, Pf-ef1α-tdTomato fluorescence is quite strong, likely because each oocyst consists of many genomes. Oocyst numbers in the GFP line were somewhat lower than what we observed in the tdTomato lines, though this level of infectivity is still within the range that can be useful for experimental analyses (Supplementary Fig. 2A,B).

Passage through the mosquito of P. falciparum-tdTomato lines is normal. (A) Mosquito midguts (whitelight) were dissected 10 days after the infectious blood meal and imaged using fluorescence microscopy to visualize associated Pf-peg4-tdTomato and Pf-ef1α-tdTomato oocysts (red). (B) Oocysts were counted based on their tdTomato fluorescence and data from two independent feeding experiments each are shown. Horizontal line shows median oocyst number. Groups were compared using an unpaired t-test with equal standard deviations. (C) Pf-peg4-tdTomato and Pf-ef1α-tdTomato lines produce normal number of salivary gland sporozoites. Shown is the mean +/− standard deviation of salivary gland loads from 20 mosquitoes.
In all three lines, oocyst sporozoites progressed to invade salivary glands with sporozoite numbers similar to those routinely observed in the NF54 parental line (Fig. 2C and Supplementary Fig. 2C). With both tdTomato lines we performed additional mosquito cycles, some in parallel with the NF54 parental line. The infectivity of these parasites for mosquitoes is consistent with that shown in Fig. 2: ef1α-tdTomato yielded a mean ± SEM of 48,934 ± 26,948 sporozoites per mosquito n = 3 cycles; peg4-tdTomato yielded 51,336 ± 6,581 sporozoites per mosquito n = 14 cycles; NF54 wild type yielded 33,116 ± 4,527 sporozoites per mosquito n = 6 cycles. As expected, salivary glands infected with Pf-ef1α-tdTomato sporozoites were dimly fluorescent, while Pf-peg4-tdTomato infected salivary glands were brightly fluorescent and could easily be visualized through the chitin exoskeleton of the mosquito (Fig. 3A), confirming that the peg4 promoter is highly active in salivary gland sporozoites. We then performed immunofluorescence assays on salivary gland sporozoites to compare the endogenous tdTomato or GFP fluorescence with the signal observed upon immunostaining of the major surface protein, the circumsporozoite protein (CSP). The tdTomato signal when under the control of the peg4 promoter gave the strongest fluorescent signal in sporozoites (Fig. 3B). GFP fluorescence under the csp promoter was robust but not as strong as that observed in the Pf-peg4-tdTomato sporozoites (Compare Fig. 3B and Supplementary Fig. 2D), while sporozoites expressing tdTomato under the ef1α promoter were only faintly fluorescent (Fig. 3B).

Immunofluorescence of sporozoites of P. falciparum-tdTomato lines. (A) The thorax of mosquitoes infected with the Pf-ef1α-tdTomato or Pf-peg4-tdTomato lines was visualized under a fluorescence stereoscope to detect the presence of the tdTomato sporozoites in the salivary glands. Note the red fluorescence observed in the thorax of mosquitoes infected with the P. falciparum-tdTomato lines compared to the absence of fluoresce in the uninfected mosquito. (B) Salivary gland sporozoites from mosquitoes infected with the Pf-ef1α-tdTomato or Pf-peg4-tdTomato were fixed and stained with an anti-PfCSP antibody (green) and analyzed by fluorescence microscopy to detect the cytoplasmic tdTomato (red) and the surface CSP (green). DNA was stained with DAPI (blue). Note the more intense signal detected from Pf-peg4-tdTomato sporozoites as compared to Pf-ef1α-tdTomato sporozoites, which match the fluorescence intensities from panel A.
We also investigated the fluorescence of the transgenic tdTomato lines in the liver stage of the parasite by performing in vitro infection assays using sporozoites and primary human hepatocytes. Immunofluorescence assays of liver stage parasites co-stained with an mAb to heat shock protein 70 (hsp 70) and DAPI to visualize the DNA, showed that both the Pf-ef1α-tdTomato and Pf-peg4-tdTomato lines were brightly fluorescent in this stage (Fig. 4A). Furthermore, live imaging confirmed that liver stage parasites were brightly fluorescent and could be clearly seen without immunostaining (Fig. 4B). These data indicate that both lines could be used to study and isolate P. falciparum liver stage parasites.

Immunofluorescene assays of late liver stage P. falciparum-tdTomato lines. (A) Primary human hepatocytes were infected with Pf-peg4-tdTomato and Pf-ef1α-tdTomato sporozoites. At 96 hours post infection, cells were fixed and stained for PfHsp70 (green) to visualize the parasite cytoplasm together with the endogenous tdTomato fluorescence (red). DNA was stained with DAPI (blue). Scale bars 5 μm. (B) Live confocal microscopy of Pf-peg4-tdTomato and Pf-ef1α-tdTomato EEFs at 96 hours post infection of primary human hepatocytes. Fluorescent images and overlays with the DIC images are shown. Scale bars 15 μm.
As mentioned above, PEG4 is likely the P. falciparum ortholog of the rodent malaria parasite protein UIS48, whose expression is highly upregulated in sporozoites and early liver stages15,16. In contrast to the rodent parasites, P. falciparum PEG4 is also expressed in gametocytes6. We therefore monitored the fluorescence of Pf-peg4-tdTomato gametocytes over the entire course of their development. We used flow cytometry to obtain fluorescence data from large numbers of gametocytes. Gametocytes could be clearly distinguished based on red fluorescence alone from stage III onward (day 5 of maturation). The fluorescence continued to increase as the gametocytes matured, reaching peak expression in mature stage V gametocytes (day 12) (Fig. 5A and Supplementary Fig. 3B). Images of gametocytes over the course of their development illustrate the expression of the tdTomato transgene in the different gametocyte stages of the Pf-peg4-tdTomato line (Fig. 5B), beginning as early as Stage I gametocytes (day 2 of maturation). Furthermore, exflagellation of male gametocytes could also be visualized (Fig. 5C). Thus this parasite line will be useful to laboratories studying the sexual stages of P. falciparum and could expedite the screening of drugs targeting gametocyte development.

tdTomato expression increases throughout Pf-peg4-tdTomato gametocyte development. (A) Mean fluorescence of >1000 gametocytes was measured on days 2-12 by flow cytometry. Values are normalized to the maximum mean tdTomato signal on day 12. Mean +/− standard deviation from two independent experiments is shown. (B) Representative phase and fluorescence images of Pf-PEG4-tdTomato stage I-V gametocytes with the nuclear stain Hoechst 33342. Scale bar = 1 micron. (C) Exflagellating Pf-peg4-tdTomato microgametes. Scale bar = 3 microns.
In conclusion, we have generated three fluorescent P. falciparum lines with chromosomally-integrated transgenes that are infective for the mosquito host. While the Pf-ef1a-tdTomato line produces a constitutive fluorescent signal throughout the cell cycle, the Pf-peg4-tdTomato line produces highly fluorescent gametocytes and sporozoites that are sufficiently bright to be used in flow cytometry and intravital imaging experiments. The Pf-peg4-tdTomato sporozoites could be used in combination with humanized mice17 to gain information on the behavior of human malaria sporozoites in vivo. To date, these types of studies have only been performed with rodent malaria parasites9,18. Furthermore, the Pf-peg4-tdTomato sporozoites and liver stages of both tdTomato lines are sufficiently bright to test inhibitors of sporozoite motility and liver stage development using high content imaging to score the results. The use of the peg4 promoter to drive tdTomato in gametocytes combines the best aspects of previous generations of fluorescent gametocyte reporter lines7,19,20, namely chromosomal integration, a strong promoter driving a bright red-fluorescent protein, and infectivity for the mosquito host. Additionally, the ability of flow cytometry to integrate stage-specific fluorescence reporters with measurements of DNA, RNA and hemozoin content, as well as mitochondrial membrane potential, will allow for multi-level cytological profiling of mixed stage cultures and more complex analyses of the impact of inhibitors on gametocyte maturation21. Overall, these lines will enable more rapid screening of both chemical inhibitors and potentially inhibitory antibodies on transmission stages of P. falciparum and aid in flow sorting transmission stages for downstream analyses such as transcriptomic and proteomic studies.
Methods
Asexual parasite culture and maintenance
Asexual blood stage parasites were propagated in 2% human erythrocytes in RPMI-1640 malaria culture medium containing 2 mM L-glutamine, 50 mg/L hypoxanthine, 25 mM HEPES, 0.225% NaHCO3, 10 mg/L gentamycin and 0.5% (w/v) Albumax II (Invitrogen). Parasites were maintained at 37 °C under an atmosphere of 5% O2, 5% CO2 and 90% N2.
Generation of the NF54Pfs47attB Plasmodium falciparum line
Zinc-finger nucleases (ZFN) directed against the P. falciparum 6-cysteine protein P47 (PlasmoDB ID PF3D7_1346800, also known as Pfs47) were purchased from Sigma Life Sciences. To introduce the attB site (from Mycobacterium smegmatis) into the pfs47 locus in P. falciparum, we generated a transfection plasmid with pfs47 specific ZFNs, a selectable human dhfr marker and a donor fragment consisting of two pfs47 homologous sequences flanking the attB site. The left and right ZFNs were 2A-linked and placed under the regulatory control of the 5′ calmodulin (PF3D7_1434200) promoter and the 3′ hsp86 (PF3D7_0708500) terminator22. The two homologous regions flanking the ZFN cut site were first amplified separately using the primer pairs p1 and p2 (0.68 kb) and p3 + p4 (0.87 kb). Primer p2 and p3 also contained the attB site and the two fragments were then linked by “splicing by overlap extension” PCR. The donor sequence was added to the ZFN-containing plasmid using the restriction enzymes AatII and BstAPI, thus yielding the final pZFNpfs47-attB-hdhfr transfection plasmid. Asexual NF54 blood-stage parasites were electroporated with purified circular plasmid pZFNpfs47-attB-hdhfr as described23. One day after electroporation, parasites were exposed to 2.5 nM WR99210 for 6 days to select for transformed parasites. Integration of the attB site was assessed by PCR analysis of bulk cultures, and cultures showing evidence of editing were selected for 96-well cloning by limiting dilution. Clones were identified by staining with 2× SYBR Green I and 165 nM MitoTracker Deep Red (Invitrogen) and assaying for growth by flow cytometry on an Accuri C6 cytometer24. To test for attB integration, P. falciparum genomic DNA was extracted and purified using DNeasy Blood kits (Qiagen). Pfs47 sequences were examined by PCR-amplifying the genomic locus with primers p5 + p6 flanking the pfs47 plasmid donor sequence. These products were amplified from bulk cultures or parasite clones. Sequencing was performed with the internal p7 and p8 primers. Four clones were expanded to test their ability to infect mosquitoes and of these, NF54Pfs47attB C10 gave the most robust mosquito infections and was therefore used for production of the fluorescent lines. Primer sequences in Table S1.
Generation of fluorescent P. falciparum lines
Plasmid construction was performed using standard molecular cloning techniques. The Pf-ef1α used was the same 806 bp sequence described and used by Talman et al.2, and was amplified by genomic DNA using primers p9 and p10. The 2227 bp Pf-peg4 upstream sequenced used was identical to that described by Buchholz et al.19, and was amplified from genomic DNA using primers p11 and p12. The Pf-csp sequence used consisted of a 1266 bp sequence from upstream of the csp coding sequence (PF3D7_0304600). The final plasmids were denoted pBattP-eEF1apr_tdTomato, pBattP_PEG4pr_tdTomato, and pLN_CSpr-GFP. Transfections were performed with 50 μg of the fluorescent protein plasmid along with 50 μg of the pINT integrase plasmid (genbank plasmid DQ813654.3)14 using the modified method described by Spalding et al.25. Transfections were selected with 2.5 μg/mL Blasticidin S until stable parasites were obtained. Transfected lines were cloned by limiting dilution as described above. Chromosomal integration of the plasmids was verified by PCR using primers p13 and p15, and compared to PCR of the unmodified pfs47attB locus with primers p13 and p14. Primer sequences in Table S1.
Gametocyte cultures and mosquito feeding
Gametocytes cultures were maintained as described above except with the following modifications: Culture medium contained 10% v/v human serum in the place of Albumax II, and parasites were propagated at 4% hematocrit. Instead of a gas incubator, cultures were maintained at 37 °C in a candle jar made from glass desiccators. Gametocyte cultures were initiated at 0.5% asynchronous asexual parasitemia from a low passage stock and maintained up to day 18 with daily media changes but without any addition of fresh erythrocytes. Day 15 to 18 cultures, containing largely mature gametocytes, were used for mosquito feeds: Cultures were centrifuged at 108 × g for 4 min and the parasite pellet was resuspended to final gametocytemia of 0.3% in a mixture of human O+ RBCs supplemented with 50% v/v human serum. Gametocytes were fed to Anopheles stephensi mosquitoes (Liston strain) that had been starved overnight, using glass membrane feeders. Unfed mosquitoes were removed after feeding. Fed mosquitoes were then maintained on a 10% sugar solution at 25 °C and 80% humidity with a 14:10 hr light:dark cycle including a 2 hr dawn/dusk transition. according to standard rearing methods. Human erythrocytes used to set up the cultures were collected weekly from healthy donors under the Johns Hopkins University Institutional Review Board approved protocol number NA_00019050. All experiments were performed in accordance with institutional guidelines and regulations and informed consent was obtained from all volunteers.
Oocyst and salivary gland sporozoite quantification
An. stephensi mosquitoes infected with Pf-ef1α-tdTomato, Pf-peg4-tdTomato or Pf-csp-GFP parasites were maintained at 25 °C, 80% humidity and on days 10 and 14 after the infective-blood meal, mosquitoes were dissected and midguts or salivary glands were harvested for sporozoite counts. On day 10 midguts were observed and photographed for oocyst counts by fluorescence and phase microscopy using an upright Nikon E600 microscope with a PlanApo 10× objective. On day 14, salivary glands from 30–50 mosquitoes were pooled, and released sporozoites were counted using a haemocytometer. For direct visualization of Pf-ef1α-tdTomato or Pf-peg4-tdTomato sporozoites in situ, infected mosquitoes were immobilized at 4 °C and transferred to a petri dish pre-chilled on ice. Mosquitoes were observed under an Olympus SZX7 fluorescence stereo microscope for detection of red fluorescence from transgenic sporozoites that had invaded the salivary glands inside the mosquito thoracic cavity. Images of the mosquitoes were obtained using the Q-Capture Pro 7 software. Uninfected mosquitoes were used as negative control.
Immunofluorescence assays (IFAs)
Sporozoites
Pf-ef1α-tdTomato, Pf-peg4-tdTomato or Pf-csp-GFP sporozoites were isolated from infected mosquitoes 16 days post-infection. Sporozoites were centrifuged onto a glass coverslip at 300 × g for 4 min to increase attachment to the coverslip. Sporozoites were fixed with 4% paraformaldehyde for 10 min, washed with PBS and then incubated with Alexa Fluor 488 or Alexa Fluor 594 labeled mAb 2A10, specific for the P. falciparum circumsporozoite protein26 diluted in PBS to a concentration of 1 μg/ml for 1 hour at room temperature. After three washes with PBS, sporozoites were mounted with ProLong Gold Antifade with DAPI (Thermofisher) and visualized on a Zeiss Axio Imager M2 fluorescence microscope for detection of tdTomato red fluorescence, PfCSP green fluorescence and DAPI. Sporozoite images were obtained and analyzed using the Volocity software.
Exoerythrocytic stages
Cryopreserved human primary hepatocytes, hepatocyte thawing, plating and maintenance medium were obtained from Triangle Research Labs (TRL, North Carolina). Growth and infection of primary hepatocytes was performed as previously outlined in27. Briefly, primary hepatocytes (donor 4051) were thawed and 300,000 hepatocytes were plated on collagen-coated coverslips in a 24 well plate. 48 hrs later, 3-5 × 105 Pf-ef1α-tdTomato or Pf-peg4-tdTomato sporozoites were added per well in Iscove’s Modified Dulbecco’s Medium (IMDM) with 2.5% FCS supplemented with penicillin, streptomycin and L-glutamine (complete growth medium), centrifuged at 380 × g for 5 min without break and then incubated at 37 °C for 4–6 hours. After incubation, cells were washed with PBS and maintained in complete growth medium supplemented with Fungizone (1.25 μg/ml, Gibco, Grand Island, NY). For IFAs, infected cells were fixed in 4% paraformaldehyde 96 hours post infection, washed in 1x PBS and permeabilized for 10 min at RT in 0.1% TritonX-100/PBS and incubated for 15 min in FX Signal Enhancer (Thermofisher #136933). Samples were blocked for 1 hour in 10% BSA/10% goat serum/0.1% TritonX100 and stained mAb 4C9 specific for Pf Hsp7028, followed by anti-mouse IgG-Alexa Fluor 488. Images were acquired using a LSM700 laser scanning confocal microscope (Zeiss AxioObserver, Carl Zeiss AG, Oberkochen, Germany) with a 63x PlanApo oil objective, and images were acquired using ZEN software (Carl Zeiss AG, Oberkochen, Germany). For live imaging, cells were directly imaged using an inverted Zeiss Axio Observer Z1 microscope with a Yokogawa CSU22 spinning disk using an EMCCD camera and 3i slidebook 5.0 software.
Gametocyte assays
Pf-peg4-tdTomato gametocytes were induced synchronously according to previous methods29 and asexual stages were eliminated by treatment with 50 mM GlcNAc (Alfa Aesar, Haverhill, MA) and 20 U/ml heparin (Sigma) for 3 days following re-invasion. On days 2-12 of gametocytogenesis, cultures were stained with 16 μM Hoechst33342 (Invitrogen) and 50 nM DilC1(5) (Invitrogen) for 30 min at 37 °C. Mean tdTomato fluorescence was measured using a Cytek DxP12 flow cytometer for >1000 viable gametocytes, gated as infected (Hoechst 33342-high), hemozoin-containing (depolarized side scatter-high) RBCs with positive membrane potential (DilC1(5)-high). This gating strategy is illustrated in Supplementary Fig. 3A. Fluorescence images of stained gametocytes were acquired at 1000x magnification using a Leica DM-6000B. tdTomato and Hoechst 33342 Z-stacks were deconvoluted using the non-linear least square algorithm of the Deconvolution Lab2 plug-in of ImageJ and representative slices are shown along with DIC brightfield images. Image of tdTomato-positive microgametes was acquired at 600x magnification after treating cultures of Stage V (day 12) gametocytes with 20 µM Xanthurenic Acid (Sigma) at room temperature.
References
World Health Organization. World Malaria Report, http://www.who.int/iris/handle/10665/275867 (2018).
Talman, A. M., Blagborough, A. M. & Sinden, R. E. A Plasmodium falciparum strain expressing GFP throughout the parasite’s life-cycle. PLoS One 5, e9156 (2010).
Vaughan, A. M. et al. A transgenic Plasmodium falciparum NF54 strain that expresses GFP-luciferase throughout the parasite life cycle. Mol Biochem Parasitol 186, 143–7 (2012).
Shaner, N. C. et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22, 1567–72 (2004).
Shaner, N. C., Steinbach, P. A. & Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Methods 2, 905–9 (2005).
Silvestrini, F. et al. Genome-wide identification of genes upregulated at the onset of gametocytogenesis in Plasmodium falciparum. Mol Biochem Parasitol 143, 100–10 (2005).
Aingaran, M. et al. Host cell deformability is linked to transmission in the human malaria parasite Plasmodium falciparum. Cell Microbiol 14, 983–93 (2012).
Mackellar, D. C. et al. Plasmodium falciparum PF10_0164 (ETRAMP10.3) is an essential parasitophorous vacuole and exported protein in blood stages. Eukaryot Cell 9, 784–94 (2010).
Hopp, C. S. et al. Longitudinal analysis of Plasmodium sporozoite motility in the dermis reveals component of blood vessel recognition. Elife 4, https://doi.org/10.7554/eLife.07789 (2015).
Ponnudurai, T., Leeuwenberg, A. D. & Meuwissen, J. H. Chloroquine sensitivity of isolates of Plasmodium falciparum adapted to in vitro culture. Trop Geogr Med 33, 50–4 (1981).
Molina-Cruz, A. et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 340, 984–7 (2013).
van Schaijk, B. C. et al. Pfs47, paralog of the male fertility factor Pfs48/45, is a female specific surface protein in Plasmodium falciparum. Mol Biochem Parasitol 149, 216–22 (2006).
Canepa, G. E., Molina-Cruz, A. & Barillas-Mury, C. Molecular Analysis of Pfs47-Mediated Plasmodium Evasion of Mosquito Immunity. PLoS One 11, e0168279 (2016).
Nkrumah, L. J. et al. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat Methods 3, 615–21 (2006).
Kaiser, K., Matuschewski, K., Camargo, N., Ross, J. & Kappe, S. H. Differential transcriptome profiling identifies Plasmodium genes encoding pre-erythrocytic stage-specific proteins. Mol Microbiol 51, 1221–32 (2004).
Matuschewski, K. et al. Infectivity-associated changes in the transcriptional repertoire of the malaria parasite sporozoite stage. J Biol Chem 277, 41948–53 (2002).
Sack, B. K. et al. Humoral protection against mosquito bite-transmitted Plasmodium falciparum infection in humanized mice. NPJ Vaccines 2, 27 (2017).
Amino, R. et al. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat Med 12, 220–4 (2006).
Buchholz, K. et al. A high-throughput screen targeting malaria transmission stages opens new avenues for drug development. J Infect Dis 203, 1445–53 (2011).
Adjalley, S. H. et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci USA 108, E1214–23, https://doi.org/10.1073/pnas.1112037108 (2011).
Shapiro, H. M. et al. Cytometry in malaria–a practical replacement for microscopy? Curr Protoc Cytom. Chapter 11: Unit 11.20, https://doi.org/10.1002/0471142956.cy1120s65 (2013).
Straimer, J. et al. Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat Methods 9, 993–8 (2012).
Fidock, D. A., Nomura, T. & Wellems, T. E. Cycloguanil and its parent compound proguanil demonstrate distinct activities against Plasmodium falciparum malaria parasites transformed with human dihydrofolate reductase. Mol Pharmacol 54, 1140–7 (1998).
Ekland, E. H., Schneider, J. & Fidock, D. A. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J 25, 3583–93, https://doi.org/10.1096/fj.11-187401 (2011).
Spalding, M. D., Allary, M., Gallagher, J. R. & Prigge, S. T. Validation of a modified method for Bxb1 mycobacteriophage integrase-mediated recombination in Plasmodium falciparum by localization of the H-protein of the glycine cleavage complex to the mitochondrion. Mol Biochem Parasitol 172, 156–60 (2010).
Nardin, E. H. et al. Circumsporozoite proteins of human malaria parasites Plasmodium falciparum and Plasmodium vivax. J Exp Med 156, 20–30 (1982).
Dumoulin, P. C. et al. Flow Cytometry Based Detection and Isolation of Plasmodium falciparum Liver Stages In Vitro. PLoS One 10, e0129623, https://doi.org/10.1371/journal.pone.0129623 (2015).
Tsuji, M., Mattei, D., Nussenzweig, R. S., Eichinger, D. & Zavala, F. Demonstration of heat-shock protein 70 in the sporozoite stage of malaria parasites. Parasitol Res. 80, 16–21 (1994).
Fivelman, Q.L. et al. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol Biochem Parasitol 154, 119–23 (2007).
Acknowledgements
The authors would like to thank the Bloomberg Family Foundation for their generous support of the Insectary and Parasitology core facilities at the Johns Hopkins Malaria Institute. We also thank Chris Kizito for expert mosquito rearing and the Johns Hopkins School of Medicine Microscopy Facility (MicFac). This work was supported by the National Institutes of Health (R01 AI132359 to P.S.; F31 pre-doctoral fellowship F31AI136405 to C.T.H.; Columbia Integrated Training Program in Infectious Diseases Research T32 AI100852 to J.L.S.), the Bill & Melinda Gates Foundation (OPP1040399 to D.A.F.), the Johns Hopkins Malaria Research Institute (post-doctoral fellowship to C.S.H. and pre-doctoral fellowship to P.C.D.), the Swedish Research Council (J.A.) and the Swedish Society of Medicine (J.A.).
Author information
Authors and Affiliations
Contributions
P.S. and D.A.F. conceived of and supervised the project. P.S., D.A.F., K.J.M., J.S., C.S.H., J.V.R., P.C.D., A.T., G.M. and B.F.C.K. designed the experiments. K.J.M., J.S., C.S.H., J.V.R., J.L.S.S., S.K., A.T., G.M., P.C.D., C.T.H., X.T., M.J.S., J.A. carried out the experiments. P.S. wrote the manuscript with support from K.J.M., J.S., B.F.C.K. and D.A.F. All authors provided critical feedback on the analysis and manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
McLean, K.J., Straimer, J., Hopp, C.S. et al. Generation of Transmission-Competent Human Malaria Parasites with Chromosomally-Integrated Fluorescent Reporters. Sci Rep 9, 13131 (2019). https://doi.org/10.1038/s41598-019-49348-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49348-x
This article is cited by
-
Sexual differentiation in human malaria parasites is regulated by competition between phospholipid metabolism and histone methylation
Nature Microbiology (2023)
-
A versatile Plasmodium falciparum reporter line expressing NanoLuc enables highly sensitive multi-stage drug assays
Communications Biology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
