
Abstract
The dynamics of transcription can be studied genome wide by high-throughput sequencing of nascent and newly synthesized RNA. 4-thiouridine (4SU) labeling in vivo enables the specific capture of such new transcripts, with 4SU residues being tagged by biotin linkers and captured using streptavidin beads before library production and high-throughput sequencing. To achieve high-resolution profiles of transcribed regions, an RNA fragmentation step before biotin tagging was introduced, in an approach known as transient transcriptome sequencing (TT-seq). We recently introduced a chemical approach for RNA fragmentation that we refer to as TTchem-seq. We describe how TTchem-seq can be used in combination with transient inhibition of early elongation using the reversible CDK9 inhibitor, 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB), to measure RNA polymerase II (RNAPII) elongation rates in vivo, a technique we call DRB/TTchem-seq. Here, we provide detailed protocols for carrying out TTchem-seq and DRB/TTchem-seq, including computational analysis. Experiments and data analysis can be performed over a period of 10–13 d and require molecular biology and bioinformatics skills.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
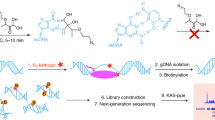
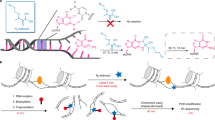

Data availability
All sequencing data are available under GEO no. GSE121826.
Code availability
All code used to analyze TTchem-seq and DRB/TTchem-seq is available at https://github.com/crickbabs/DRB_TT-seq/releases/tag/v1.2 and https://github.com/crickbabs/DRB_TT-seq.
References
Rabani, M. et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. 29, 436–442 (2011).
Windhager, L. et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 22, 2031–2042 (2012).
Dolken, L. et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972 (2008).
Duffy, E. E. et al. Tracking distinct RNA populations using efficient and reversible covalent chemistry. Mol. Cell 59, 858–866 (2015).
Fuchs, G. et al. 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol. 15, R69 (2014).
Gregersen, L. H. et al. MOV10 is a 5ʹ to 3ʹ RNA helicase contributing to UPF1 mRNA target degradation by translocation along 3ʹ UTRs. Mol. Cell 54, 573–585 (2014).
Herzog, V. A. et al. Thiol-linked alkylation of RNA to assess expression dynamics. Nat. Methods 14, 1198–1204 (2017).
Rabani, M. et al. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159, 1698–1710 (2014).
Schwalb, B. et al. TT-seq maps the human transient transcriptome. Science 352, 1225–1228 (2016).
Schwanhausser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Gregersen, L. H. et al. SCAF4 and SCAF8, mRNA anti-terminator proteins. Cell 177, 1797–1813 (2019).
Duffy, E. E., Canzio, D., Maniatis, T. & Simon, M. D. Solid phase chemistry to covalently and reversibly capture thiolated RNA. Nucleic Acids Res. 46, 6996–7005 (2018).
Duffy, E. E. & Simon, M. D. Enriching s4 U-RNA using methane thiosulfonate (MTS) chemistry. Curr. Protoc. Chem. Biol. 8, 234–250 (2016).
Michel, M. et al. TT-seq captures enhancer landscapes immediately after T-cell stimulation. Mol. Syst. Biol. 13, 920 (2017).
Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R. & Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol. 23, 232–237 (2005).
Saponaro, M. et al. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 157, 1037–1049 (2014).
Fuchs, G. et al. Simultaneous measurement of genome-wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB-seq. Nat. Protoc. 10, 605–618 (2015).
Chodosh, L. A., Fire, A., Samuels, M. & Sharp, P. A. 5,6-dichloro-1-beta-d-ribofuranosylbenzimidazole inhibits transcription elongation by RNA polymerase II in vitro. J. Biol. Chem. 264, 2250–2257 (1989).
Marshall, N. F. & Price, D. H. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 270, 12335–12338 (1995).
Churchman, L. S. & Weissman, J. S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373 (2011).
Mayer, A. et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161, 541–554 (2015).
Nojima, T. et al. Mammalian NET-Seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161, 526–540 (2015).
Milligan, L. et al. Strand-specific, high-resolution mapping of modified RNA polymerase II. Mol. Syst. Biol. 12, 874 (2016).
Schaughency, P., Merran, J. & Corden, J. L. Genome-wide mapping of yeast RNA polymerase II termination. PLoS Genet. 10, e1004632 (2014).
Core, L. J., Waterfall, J. J. & Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848 (2008).
Core, L. J. et al. Defining the status of RNA polymerase at promoters. Cell Rep. 2, 1025–1035 (2012).
Riml, C. et al. Osmium-mediated transformation of 4-thiouridine to cytidine as key to study RNA dynamics by sequencing. Angew. Chem. Int. Ed. Engl. 56, 13479–13483 (2017).
Schofield, J. A., Duffy, E. E., Kiefer, L., Sullivan, M. C. & Simon, M. D. TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat. Methods 15, 221–225 (2018).
Singh, J. & Padgett, R. A. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 16, 1128–1133 (2009).
Jonkers, I., Kwak, H. & Lis, J. T. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3, e02407 (2014).
Veloso, A. et al. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 24, 896–905 (2014).
Danko, C. G. et al. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol. Cell 50, 212–222 (2013).
Zylicz, J. J. et al. The implication of early chromatin changes in X chromosome inactivation. Cell 176, 182–197 e123 (2019).
Gressel, S. et al. CDK9-dependent RNA polymerase II pausing controls transcription initiation. Elife 6, e29736 (2017).
Miller, M. R., Robinson, K. J., Cleary, M. D. & Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods 6, 439–441 (2009).
Chen, T. & van Steensel, B. Comprehensive analysis of nucleocytoplasmic dynamics of mRNA in Drosophila cells. PLoS Genet. 13, e1006929 (2017).
Pai, A. A. et al. The kinetics of pre-mRNA splicing in the Drosophila genome and the influence of gene architecture. Elife 6, e32537 (2017).
Gay, L. et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes Dev. 27, 98–115 (2013).
Wachutka, L., Caizzi, L., Gagneur, J. & Cramer, P. Global donor and acceptor splicing site kinetics in human cells. Elife 8, e45056 (2019).
Mukherjee, N. et al. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat. Struct. Mol. Biol. 24, 86–96 (2017).
Burger, K. et al. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 10, 1623–1630 (2013).
Miller, C. et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
Munchel, S. E., Shultzaberger, R. K., Takizawa, N. & Weis, K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol. Biol. Cell 22, 2787–2795 (2011).
Sun, M. et al. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 22, 1350–1359 (2012).
Marzi, M. J. et al. Degradation dynamics of microRNAs revealed by a novel pulse-chase approach. Genome Res. 26, 554–565 (2016).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Hunt, S. E. et al. Ensembl variation resources. Database (Oxford) 2018, https://doi.org/10.1093/database/bay119 (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Lawrence, M. et al. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Mammana, A. H., Extract read count signals from bam files. R package v.1.12.11 https://doi.org/10.18129/B9.bioc.bamsignals (2016).
Acknowledgements
This work was supported by the Francis Crick Institute (which receives its core funding from Cancer Research UK (FC001166), the UK Medical Research Council (FC001166) and the Wellcome Trust (FC001166)) and by a grant from the European Research Council (agreement 693327 (TRANSDAM)). L.H.G. was supported by the EMBO-LTF program (EMBO ALTF 1026-2014). We thank A. Herlihy for helpful comments on the manuscript.
Author information
Authors and Affiliations
Contributions
L.H.G. and J.Q.S. conceived the experimental setup. L.H.G. performed all optimization steps and experiments. R.M. developed the bioinformatics pipeline for analysis of TTchem-seq and DRB/TTchem-seq. J.Q.S. supervised various aspects of the work. L.H.G. and J.Q.S wrote the manuscript with assistance from R.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Protocols thanks Didier Devys and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related link
Key reference using this protocol
Gregersen, L. H. et al. Cell 177, 1797–1813.e18 (2019): https://doi.org/10.1016/j.cell.2019.04.038
Integrated supplementary information
Supplementary Figure 1 Control for DRB inhibition and release, as well as proportion of yeast spike-ins in TTchem-seq.
(a) Slot blot of RNA from HEK293 cells treated with 100 μM DRB for 3.5 hrs either without release, or with release for the indicated time period together with 10 min 1 mM 4SU labelling. (b) Bioanalyzer result of fragmented purified 4SU-RNA from cells treated with 100 μM DRB for 3.5 hrs either with or without release for the indicated time. All samples were labelled with 1 mM 4SU for 10 min prior to harvest. Gel like images of the electropherogram are shown on the top. (c) Percent of sequencing reads mapping to the yeast genome when using 1% total yeast RNA as spike-ins.
Supplementary Figure 2 Protocol optimization.
(a) Increasing treatment time of total RNA with sodium hydroxide (0.167 M) results in decreasing RNA fragment size. 1.2% denaturing TBE gel. (b) Bioanalyzer results comparing Qiagen minElute RNA clean-up of 4SU-RNA after streptavidin pull-out using the recommended protocol which selects for RNA > 200 nt or the modified protocol using an increased volume of ethanol.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2.
Rights and permissions
About this article

Cite this article
Gregersen, L.H., Mitter, R. & Svejstrup, J.Q. Using TTchem-seq for profiling nascent transcription and measuring transcript elongation. Nat Protoc 15, 604–627 (2020). https://doi.org/10.1038/s41596-019-0262-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41596-019-0262-3
This article is cited by
-
Transcription-coupled repair of DNA–protein cross-links depends on CSA and CSB
Nature Cell Biology (2024)
-
High-sensitive nascent transcript sequencing reveals BRD4-specific control of widespread enhancer and target gene transcription
Nature Communications (2023)
-
Tox4 regulates transcriptional elongation and reinitiation during murine T cell development
Communications Biology (2023)
-
H3K4me3 regulates RNA polymerase II promoter-proximal pause-release
Nature (2023)
-
The SPOC domain is a phosphoserine binding module that bridges transcription machinery with co- and post-transcriptional regulators
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
