
Abstract
Cell cycle progression is a tightly regulated process by which DNA replicates and cell reproduces. The major driving force underlying cell cycle progression is the sequential activation of cyclin-dependent kinases (CDKs), which is achieved in part by the ubiquitin-mediated proteolysis of their cyclin partners and kinase inhibitors (CKIs). In eukaryotic cells, two families of E3 ubiquitin ligases, anaphase-promoting complex/cyclosome and Skp1-Cul1-F-box protein complex, are responsible for ubiquitination and proteasomal degradation of many of these CDK regulators, ensuring cell cycle progresses in a timely and precisely regulated manner. In the past couple of decades, accumulating evidence have demonstrated that the dysregulated cell cycle transition caused by inefficient proteolytic control leads to uncontrolled cell proliferation and finally results in tumorigenesis. Based upon this notion, targeting the E3 ubiquitin ligases involved in cell cycle regulation is expected to provide novel therapeutic strategies for cancer treatment. Thus, a better understanding of the diversity and complexity of ubiquitin signaling in cell cycle regulation will shed new light on the precise control of the cell cycle progression and guide anticancer drug development.
Similar content being viewed by others


Facts
-
(1)
The cell cycle is a tightly orchestrated cellular process that governs the timely DNA replication and cell division events.
-
(2)
Sequential activation of cyclin-dependent kinases (CDKs) drives cell cycle progression in a timely and precisely regulated manner.
-
(3)
The activity of CDKs is modulated by cyclin partners and CDK inhibitors (CKIs), which are tightly controlled by the ubiquitin–proteasome system.
-
(4)
Two important types of E3 ligases, the anaphase-promoting complex or cyclosome (APC/C) and Skp1-Cul1-F-box (SCF) complexes, are dedicated to cell cycle control.
-
(5)
Targeting E3 ubiquitin ligases provides effective therapeutic strategies for cancer treatment.
Open questions
-
(1)
Unlike proteolytic signals, relatively little is known regarding the roles and mechanisms of non-proteolytic signals, such as the ones mediated by K6, K27, and K29 polyubiquitin chain, underlying the cell cycle control.
-
(2)
The specificity and diversity of deubiquitinating enzymes (DUBs) in regulating mitosis need to be further investigated.
-
(3)
How is the balance of ubiquitination–deubiquitination achieved to ensure accurate cell cycle progression remains elusive.
-
(4)
Unlike CDC20, the regulation of the enzymatic activity of CDH1 is not well defined yet.
-
(5)
The detailed mechanisms underlying spindle checkpoint imposed various E3 ligase activities of CDC20 toward different substrates need to be further investigated.
-
(6)
How those ubiquitination signaling events at the spindle checkpoint are integrated and orchestrated in a space–time-dependent manner remains not fully understood.
Introduction
The cell cycle is a series of tightly orchestrated molecular events that coordinately regulate DNA replication and chromosome segregation, eventually resulting in cell division and genetic material transmission. In eukaryotic cells, the cell cycle consists of four distinct phases, G1 phase (gap 1), S phase (DNA synthesis), G2 phase (gap 2), and M phase (mitotic) that proceed in a unidirectional manner (Fig. 1). The progression through each phase of the cell cycle is precisely regulated by a series of cyclin-dependent kinases (CDKs). The protein abundance of CDKs is constant, while their activities fluctuate throughout the cell cycle, which is mainly achieved by the periodic expression of cyclin coactivators and CDK inhibitors (CKIs). Briefly, in mid-to-late G1 phase, activation of Cyclin D-CDK4/6 complex mediates partial phosphorylation of the RB1 protein, releasing E2F transcription factors and thus allowing the expression of a set of genes that mediate cell cycle progression [1]. At the end of the G1 phase, the accumulation of Cyclin E activates CDK2 and promotes full phosphorylation of RB1 [2, 3], initiating cell cycle transition from G1 phase to S phase. As cell cycle enters S phase, Cyclin A, in replace of Cyclin E, associates with CDK2 to regulate the initiation of DNA replication and prevents the re-replication by phosphorylating particular DNA replication machinery components, such as CDC6 [4, 5]. Approaching late S phase, Cyclin A-CDK1 kinase activity is augmented, which coordinates with Cyclin A-CDK2 in G2 phase to promote mitotic entry [6,7,8]. The abundance of Cyclin B accumulates in M phase, resulting in Cyclin B-CDK1 complex activation and mitosis progression [9,10,11]. In addition to the fluctuating accumulation of cyclin activators, two families of CKIs, namely INK4 and CIP/KIP, also contribute to the periodic activation of CDKs over the course of the cell cycle. Briefly, the INK4 proteins (inhibitors of CDK4) specifically inhibit the catalytic subunit of CDK4 and CDK6, dephosphorylating RB1 and rendering its inhibitory effect on E2F transcription factors, while inhibitors of the CIP/KIP family have relatively more broad effects by modulating the kinase activities of Cyclin A-, B- and E-dependent kinases [12]. It is well characterized that the removal of cyclins is tightly regulated by the ubiquitin pathway and thus governs cell cycle progression in a time-efficient manner [13]. Moreover, the negative regulators of cyclin-CDK complex (CKIs), such as p21 and p27, have also been shown to be targeted for proteasomal degradation [14,15,16]. Altogether, these findings demonstrate that the cell cycle progression is predominantly regulated by the ubiquitin–proteasome system [17, 18].

The stages of the cell cycle are divided into four major phases: (1) G1 phase, also called the first gap phase. During the G1 phase, cells grow physically larger and duplicate cellular contents to prepare for the later steps; (2) S phase, cells synthesize a complete copy of DNA and duplicate the centrosome; (3) G2 phase, the second gap phase, cells grow more and prepare for mitosis; (4) M (mitotic) phase, during this phase, cells divide their copied DNA and cellular components, making two identical daughter cells. G0 phase is a quiescent stage that occurs outside of the cell cycle. During the G0 phase, cells are neither dividing nor preparing to divide. Sequential activation of Cyclin/CDKs drives cell cycle progression in a timely orchestrated manner. Briefly, Cyclin D1/CDK4 mainly functions in G1 phase to facilitate RB1 phosphorylation, releasing its suppression on E2F transcription factors; Cyclin E/CDK2 functions in S phase to control DNA replication; Cyclin A/CDK2 functions in later S phase to prepare the cell cycle entry into M phase; Cyclin B/CDK1 functions in M phase to be involved in regulation of chromatin separation. Additionally, three cell cycle checkpoints, G1/S checkpoint, G2-M DNA damage checkpoint, and spindle assembly checkpoint (SAC), are orchestrated to ensure the proper progression of the cell cycle. Protein structures of Cyclin/CDKs and RB1/E2F used here are as follows: RB1/E2F/DP (2AZE); Cyclin D1/CDK4 (2W9Z); Cyclin E/CDK2 (1W98); Cyclin A/CDK2 (6P3W); Cyclin B/CDK1 (4YC3).
Ubiquitin is an ubiquitously expressed small regulatory protein in living cells [19]. The addition of ubiquitin to a substrate protein is called ubiquitination, which is catalyzed by three types of enzymes, ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s), involving three major steps [20]. Briefly, the ubiquitin protein is first activated by E1-mediated catalysis of the acyl-adenylation of the C-terminus of the ubiquitin protein, followed by transferring ubiquitin to an active cysteine residue in the context of ATP providing energy. Then, E2 ubiquitin-conjugating enzymes catalyze the transfer of the ubiquitin from E1 to the active site cysteine of E2. Finally, an E3 ubiquitin ligase brings the substrate and ubiquitin-loaded E2 together, catalyzing the transfer of the ubiquitin from E2 to the substrate (Fig. 2). The ubiquitination process can involve either a single ubiquitin protein (monoubiquitination) or a chain of ubiquitin linked via different lysine residues of the ubiquitin molecule (termed as polyubiquitination). As ubiquitin possesses seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) and one N-terminal methionine (M1) that can serve as docking points of additional ubiquitin chain formation, polyubiquitination of target protein exhibits distinct functional consequences depending on the lysine residue of the ubiquitin that is linked (Fig. 3).

Ubiquitination is an enzyme-mediated posttranslational modification by which the ubiquitin protein is attached to a substrate protein. This process involves three main steps: (1) activation step, the ubiquitin protein is activated by an E1 ubiquitin-activating enzyme, with ATP providing energy; (2) conjugation step, the ubiquitin protein is transferred from E1 to the active site of an E2 ubiquitin-conjugating enzyme; (3) ligation step, the ubiquitin protein is attached to the substrate (sub) with the catalyzation of an E3 ubiquitin ligase. Protein structures used here are as follows: Ub (1UBQ); E1–Ub complex (6DC6); E2–E3–Ub complex (4AP4).

Ubiquitin is a small protein (8.6 kDa) that is expressed in all eukaryotic cells. There are eight amino acids (the N-terminal methionine M1 and seven lysine residues: K6, K11, K27, K29, K33, K48, and K63) that can serve as docking points for additional ubiquitin addition. The ubiquitination can be either a single ubiquitin protein (monoubiquitination) or a chain of ubiquitin (polyubiquitination). The variety of different modifications confers the diversity of linkage-dependent function of ubiquitination. Structure of ubiquitin is 1UBQ.
Briefly, K11-linked ubiquitin chain was found to regulate the substrates of the anaphase-promoting complex/cyclosome (APC/C) complex and control progression through mitosis, while Skp1-Cul1-F-box (SCF) ubiquitin ligase complex catalyzed K48-linked polyubiquitination and subsequent proteasomal degradation of substrates to modulate cell cycle progression [21,22,23]. To gain more insights into the functional diversity and specificity of linear-, mono- and linkage-dependent polyubiquitination modification, readers are encouraged to refer to the extensive literature which has been summarized previously [24,25,26]. Here, we will mainly focus on summarizing the physiological role of the ubiquitin signaling in cell cycle control and tumorigenesis, with primary purpose to provide a better understanding of ubiquitination-mediated cell cycle regulation and ubiquitin ligase targeted anticancer therapies.
Overview of the function of APC/C and SCF E3 ligases in modulating cell cycle progression
Progression through the cell cycle is determined by phosphorylation of CDK substrates [27, 28]. To ensure the cell cycle progression occurs in an ordered manner, the oscillating activity of CDKs is established and tightly orchestrated by multiple mechanisms including transcription, phosphorylation, as well as periodic degradation of their cyclin coactivators and CKIs as mentioned above [13,14,15,16, 29]. Of note, the proteolytic degradation of regulators of CDKs is primarily controlled by two families of E3 ubiquitin ligases in mammalian cells, APC/C, and SCF protein complex [30].
The APC/C is a multi-subunit cullin-RING E3 ubiquitin ligase that functions in mitotic phase and G1 phase, regulating cell cycle progression through M phase and entry into S phase [31, 32]. The temporal regulation of APC/C activity is prominently achieved through combination of two structurally relevant coactivators, CDC20 and CDH1, which are sequentially activated to regulate mitotic progress and G1 stabilization. Briefly, mitotic phosphorylation of APC1 relieves its auto-inhibition and promotes APC/C activation by facilitating CDC20 engagement [33, 34]. Activation of APC/CCDC20 then mediates the proteasomal degradation of Cyclin B1 and Securin, facilitating chromosome segregation and anaphase onset [31, 35, 36]. In addition, degradation of Cyclin B1 inactivates CDK1, preventing APC/C-CDC20 combination while releasing its inhibitory phosphorylation of CDH1 [37]. Simultaneously, CDC14 is released and activated with the onset of anaphase, dephosphorylating and activating the APC/CCDH1 E3 ligase [38, 39]. Together, suppression of CDK1 and activation of CDC14 build up a swift transition from APC/CCDC20 to APC/CCDH1 during anaphase. Activation of CDH1 then mediates a large number of mitotic and G1 regulators for ubiquitination and proteasomal degradation, such as Cyclin B1, PLK1, CDC20, FOXM1, and SKP2, facilitating irreversible mitotic exit and G1 maintenance [40]. As cells reach late G1 phase, multiple mechanisms are then employed, such as CDK-mediated phosphorylation, degradation of the E2 enzyme UBE2C, and accumulation of pseudo-substrate EMI1, to inactivate CDH1 to facilitate G1/S transition [40]. Collectively, our current knowledge suggests that the APC/C is mainly active from mitosis through late G1 phase over the course of the cycle (Fig. 4).

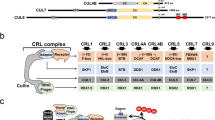
APC/C and SCF E3 ligases serve as the two important types of E3 enzymes to regulate the cell cycle progression. Briefly, APC/C-CDC20 functions in prophase to metaphase to mediate the ubiquitination and proteasomal destruction of Cyclin A/B, Securin; APC/C-CDH1 functions from anaphase to G1 phase to modulate the protein stability of CDC6, PLK1, FOXM1, Cyclin A/B, and Aurora A/B, ensuring M phase progression and G1 phase maintenance. Four F-box proteins of SCF E3 ligase complex have been well-documented to function in regulation of the cell cycle progression: FBXW7, βTrCP, SKP2, and Cyclin F. FBXW7 functions largely as a tumor suppressor to mediate the protein destruction of MYC and Cyclin E. By contrast, SKP2 is believed to serve as an oncogene, which mediates the ubiquitination and degradation of CDK inhibitors, such as p21, p27, and p57. βTrCP is found to play a dual role in controlling CDK1 activity, turning it on by inducing Claspin and WEE1 degradation in G2 phase, and turning it off by inducing the degradation of CDC25A, EMI1, and REST. Cyclin F functions in G2 phase to restrict the activity of E2F, the synthesis of replicative histones (SLBP), and the levels of ribonucleotides (RRM2), as well as regulate centrosomal duplication (CP110). In addition, the APC/C and SCF E3 ligases control each other. For example, CDH1 mediates the degradation of SKP2 in G1 phase, while βTrCP and Cyclin F were reported to control the destruction of CDH1. Moreover, CDH1 is responsible for mediating CDC20 for proteasomal degradation, while the protein stability of Cyclin F was shown to be controlled by βTrCP.
The SCF complex contains three core subunits Cullin, SKP1, and RBX1, as well as a variable F-box protein. In comparison to APC/C, the number of substrates of SCF complex is enormous due to the variety of F-box proteins. Although almost 70 F-box proteins have been reported in mammals [41], only four of them, SKP2, FBXW7, βTrCP, and Cyclin F, have been well characterized in the cell cycle regulation [42,43,44].
In the early G1 phase, the E3 ligase activity of SKP2 is suppressed due to the active presence of APC/CCDH1 [45, 46]. However, when cells approach late G1 phase, the enzymatic activity of CDH1 is diminished and phosphorylation of SKP2 by Cyclin E-CDK2 protects it from APC/CCDH1-mediated proteasomal degradation, conferring activation of the SCFSKP2 E3 ligase complex [40, 47]. Meanwhile, the activated Cyclin E-CDK2 complex mediates phosphorylation and ubiquitination of p27 [48, 49]. Subsequently, the phosphorylated p27 is recognized and ubiquitinated by SKP2, leading to its proteasomal degradation [50]. Consequently, degradation of p27 relieves its suppression on Cyclin E-CDK2, leading to a positive feedback loop which contributes to RB1 full phosphorylation and the G1/S transition [2, 3]. In addition to p27, the proteolytic degradation of the other two CIP/KIP members, p21 and p57, is also controlled by SKP2 [51, 52]. Given the importance of the CIP/KIP family of CKIs in regulating cell cycle transition [12, 53], it is conceivable that the disruption of SKP2 E3 ligase activity would cause dysregulation of cell cycle progression. As mentioned above, Cyclin A association with CDK2, in replace of Cyclin E, is involved in the regulation of the initiation of DNA synthesis when cells enter S phase [4, 5]. Thus, the timely removal of free Cyclin E is necessary to ensure cell progresses forward through the cell cycle. In support of this notion, SKP2 was found to be capable of ubiquitinating free Cyclin E for proteasomal degradation [54].
When cells entry into G2 phase, Cyclin F ubiquitinates and restricts the activity of E2Fs, the main and most critical transcriptional engines of the cell cycle [55, 56]; mediates degradation of SLBP to limit H2A.X accumulation and apoptosis upon genotoxic stress [57]; controls genome integrity and centrosome homeostasis by degrading Ribonucleotide Reductase M2 (RRM2) and CP110, respectively [58, 59]. Interestingly, the protein stability of Cyclin F is modulated by βTrCP to control timely mitotic progression [60].
During the early stage of mitosis, Cyclin A associates and activates CDK1, driving the initiation of chromosome condensation [61,62,63]. Once the activity of APC/CCDC20 is turned on in prometaphase, Cyclin A is ubiquitinated and degraded by the proteasome [64, 65]. Of note, destruction of Cyclin B, another crucial mitotic cyclin that can be targeted by the APC/CCDC20 for proteasomal degradation, is initiated during the metaphase, and occurs significantly later than the destruction of Cyclin A [66, 67]. Investigation of this difference of temporal degradation between Cyclin A and Cyclin B suggests that destruction of Cyclin A is likely spindle checkpoint independent, while the proteolytic degradation of Cyclin B1 is largely sensitive to the spindle assembly checkpoint (SAC) [68, 69]. Therefore, degradation of Cyclin B1 by APC/CCDC20 is blocked by MAD2 in prometaphase when chromosomes are not fully attached to the mitotic spindles [70, 71]. Moreover, protein stability of CDT2 was found to be regulated by FBXO11 ubiquitin ligase [72]. Collectively, the tightly orchestrated sequential destruction of mitotic regulators contributes to dictate the timing of events during mitotic exit.
The protein abundance of CDH1 diminished during late G1 phase (Fig. 5), which is partially mediated by βTrCP- and Cyclin F-directed proteasomal degradation [73, 74]. Moreover, βTrCP has been found to play dual roles in controlling CDK1 activity, turning it on by inducing WEE1 and Claspin degradation during G2 phase [75, 76], and turning it off by inducing the degradation of EMI1 and CDC25A in M and S phase, respectively [77, 78]. Activation of CDK1 in G2 phase phosphorylates EMI1, priming it for recognition and degradation by βTrCP, ensuring the timely activation of APC/C [77, 79]. In addition, βTrCP also controls APC/C E3 ligase activity in part by mediating the degradation of REST, a repressor of MAD2 transcription [80]. In addition to the well-established SKP2, βTrCP, FBXW7, and Cyclin F, roles of other F-box proteins involved in regulating cell cycle progression have been summarized elsewhere [81].

A Western blot showing the protein abundance of APC/C and SCF adapter proteins over the course of the cell cycle. B Quantification of protein density showing the accumulation of protein levels of APC/C and SCF adapter proteins throughout the cell cycle.
Role of ubiquitination signaling in cell cycle checkpoints
It is well established that three checkpoints operate in eukaryotic cells to ensure ordered and accurate cell cycle progression (Fig. 1). In addition to the roles of ubiquitination in cell cycle regulation mentioned above, ubiquitin signaling is also involved in mediating cell cycle checkpoint response. In the G1/S checkpoint, phosphorylation and degradation of CDH1 are required to release its inhibition on SKP2, allowing p27 destruction and consequent Cyclin E-CDK2 activation. The G2/M checkpoint prevents cells initiating mitosis in the context of damaged or incompletely replicated DNA. Upon DNA damage, activation of ATR phosphorylates and activates CHK1 protein kinase, which then mediates phosphorylation and proteasomal degradation of CDC25A in a SCFβTrCP-dependent manner [82, 83]. Suppression of CDC25A prevents CDK1 from dephosphorylation and activation, arresting cells in G2 phase for sufficient DNA damage repair [83, 84]. Moreover, CHK1-mediated phosphorylation of RAD51 counteracts EMI1-dependent degradation, thereby restoring RAD51-dependent homologous recombination (HR) repair [85]. Of note, recent findings showed that DNA damage-induced activation of ATM phosphorylates p53 and facilitates its binding with FBXW7, leading to subsequent p53 ubiquitination and proteasomal degradation [86]. In addition to the ubiquitination of key cell cycle regulators, histone ubiquitination also plays crucial roles in DNA damage response and cell cycle advance. For example, site-specific ubiquitination of H2A organizes the spatio-temporal recruitment of DNA repair factors to contribute to DNA repair pathway choice between homologous recombination (HR) and non-homologous end joining (NHEJ) [87], while deubiquitination of H2A is required for chromosome segregation when cells enter mitosis [88]. The M checkpoint is also known as spindle assembly checkpoint (SAC), by which cells assess whether all chromosomes are properly attached to the spindle. In the context of chromatids being misplaced, kinetochores activate the SAC, which then inhibits the E3 ligase activity of APCCDC20 and delays cell division until accurate chromosome segregation can be guaranteed [89]. By contrast, once all chromosomes are correctly attached to the microtubule spindle apparatus, APCCDC20 mediates Cyclin B1 and Securin for ubiquitination and proteasomal degradation, allowing for chromosome segregation and metaphase-to-anaphase transition [31, 35, 36].
Substrate recognition by APC/C and SCF E3 ligase
Recognition of the substrates by corresponding E3 ligases is achieved by short destruction-mediating sequence elements, which is named degron [90]. The best-studied degron in targets of APC/C are the nine-amino acid destruction box (D-box: RxxLxxxxN) and the KEN box (KENxxxN), which are preferred by CDH1 and CDC20 or CDH1, respectively [13, 91] (Table 1). Nonetheless, a spectrum of other amino acid sequences has also been found to be recognized by the APC/C complex, such as the ABBA motif ([ILVF]x[ILMVP][FHY]x[DE]) which was identified in Cyclin A, BUBR1, BUB1, and Acm1 [92]. In comparison to APC/C, F-box proteins recognize their substrates in multiple ways, among which the best-characterized F-box proteins bind to phosphodegrons in their substrates [93]. Thus, phosphorylation of the substrates plays an important role for F-box protein-mediated recognition and ubiquitination. βTrCP recognizes the DSGxxS/T degron in which the serine residues or serine and threonine residues are phosphorylated [93, 94] (Table 1). For example, CDK1 phosphorylation of the DSG degron of EMI1 primes its recognition and destruction by βTrCP to activate APC/C complex [77, 95]. Substrates of FBXW7 usually contain a canonical degron S/TPPxS/T [93, 96] (Table 1). Serving as an example, CDK2 phosphorylation of the TPPxS of Cyclin E determines its recognition and ubiquitination by FBXW7 [97, 98]. Unlike βTrCP and FBXW7, SKP2-dependent ubiquitination and degradation of CKIs, such as p27, requires not only the CDK-mediated phosphorylation, but also an accessory protein, CKS1, representing a cofactor-dependent substrate recognition [48,49,50, 99]. Cyclin F contains three separate modules, the pseudo-catalytic, substrate recruitment, and regulatory modules. It was reported to utilize the hydrophobic patch in the cyclin domain to bind the CY-containing substrates [44]. Mechanisms and functions of substrate recognition by F-box protein have been extensively summarized in [44, 93].
Role of APC/C and SCF E3 ligases in tumorigenesis
Accumulating evidence have shown that CDC20 is frequently overexpressed in a wide range of cancers, indicating that it might function as an oncoprotein [32, 100]. From the perspective of cell cycle, degradation of Cyclin B and Securin is required for the onset of anaphase [35, 101]. It is thus conceivable that the loss of CDC20 causes metaphase arrest in mouse embryos [102]. In support of the oncogenic role, genetic ablation of CDC20 results in efficient tumor regression [103], while the loss of CDC20 inhibition promotes tumorigenesis [104], advocating CDC20 as a potential therapeutic target for cancer treatment [105]. In contrast, CDH1 has been found to be downregulated in a large variety of human cancers [32, 100]. CDH1-deficient cells proliferate inefficiently and CDH1 heterozygous animals show increased susceptibility to spontaneous tumors, largely conferring CDH1 a tumor suppressor role [106]. In addition, accumulation of SKP2 due to the loss of CDH1 is considered to promote proteasomal degradation of CIP/KIP family of CKIs and thus facilitate tumorigenesis.
Regarding the role of SCF E3 complex in cancer development, emerging evidence suggest that it acts in a F-box protein- and context-dependent manner [107,108,109]. Specifically, SKP2 is a well-defined oncoprotein and was found to be overexpressed in various human cancers [109, 110]. Targets of SKP2 are mainly tumor suppressor proteins including p21, p27, p57, p130, and CDT1 [50,51,52, 111, 112]. Therefore, SKP2 exerts its oncogenic function mainly through degradation of its tumor suppressive targets. In support of the oncogenic role of SKP2, pharmacological inhibition of SKP2 was found to be able to restrict cancer progression [113]. In contrast to SKP2, FBXW7 is believed to function mainly as a tumor suppressor by targeting various oncogenic proteins for degradation [96, 107,108,109]. For example, proteasomal destruction of Cyclin E through FBXW7-mediated ubiquitination blocks CDK2 activation in late G1 phase and thus delays G1/S transition, arresting cells in G1 phase [97, 98, 114]. Another well-established oncogenic substrate of FBXW7 is MYC [115], which serves as a transcription factor involved in the genesis of many human cancers [116]. Regarding Cyclin F, it is believed to function as a tumor suppressor by controlling genome integrity and centrosome duplication by regulating the protein stability of RRM2 and CP110, respectively [44, 58, 59]. Looking at the substrate list of βTrCP, it is obvious that βTrCP plays a dual role in regulating CDK1 activity, turning it on by inducing WEE1 and Claspin destruction [75, 76], while turning it off by targeting EMI1 and CDC25A for proteasomal degradation [77, 78]. Importantly, preclinical studies have validated WEE1 inhibition as a viable therapeutic target in treating cancer [117], and CDC25A is also deemed as a suitable therapeutic target for cancer treatment [118], establishing βTrCP as a tumor suppressor. On the other hand, βTrCP was found to be involved in mediating the proteasomal degradation of tumor suppressors, such as FOXO3 and DEPTOR [119, 120]. Taking these results into consideration, βTrCP might be expected to be oncogenic and exert a tumor suppressive role in a context-dependent manner [107, 109].
Conclusion and perspective
In the present review, we mainly summarized the proteolytic signals involved in the cell cycle control. Moreover, non-proteolytic ubiquitination of the cell cycle regulators also plays crucial roles in controlling cell cycle progression. For example, the endoplasmic reticulum lipid associated protein 2 was found to interact and facilitate K63-linked ubiquitination and stabilization of Cyclin B1, facilitating mitosis exit [121]. Di-ubiquitination of the minichromosome maintenance protein 10 is required for its interaction with PCNA to facilitate DNA elongation in S phase [122]. Although accumulating evidence supporting critical roles of non-proteolytic ubiquitin signals in regulating cell cycle progression, unlike the functions of proteolytic ubiquitin signals which have been studied extensively, relatively little is known regarding the roles and mechanisms underlying cell cycle control that go beyond proteasomal degradation. In addition, it is well characterized that ubiquitination is a reversible process as the ubiquitin can be removed from the modified proteins by an array of deubiquitinating enzymes (DUBs) [123]. Of note, DUBs have been found to play critical roles in regulation of mitosis [124], and small molecular inhibitors against DUBs are expected to offer novel therapeutic opportunities for cancer treatment [125]. However, the roles and substrates of DUBs in regulating cell cycle events remain not well understood. In particular, how is the balance of ubiquitination–deubiquitination achieved to ensure accurate cell cycle progression remains elusive and needs additional in-depth investigations.
With respect to the well-established APC/C E3 ligase complex, activation of CDC20 is required for anaphase onset, while CDH1 plays a central role in mediating mitosis exit and G1 maintenance. The ordered activation of CDC20 and CDH1 is essential for accurate mitosis progression. Mitotic phosphorylation of APC/C relieves its auto-inhibition and facilitates CDC20 engagement [33, 34], while BUB1-mediated phosphorylation of CDC20 upon spindle checkpoint activation inhibits the ubiquitin ligase activity of APCCDC20, ensuring the fidelity of chromosome segregation [126]. Although the protein stability of CDH1 has been reported to be modulated by βTrCP and Cyclin F [73, 74], regulation of CDH1 E3 ligase activity is not well known yet. A previous study has shown that there are 19 serine and threonine residues on CDH1 that can be phosphorylated by multi-kinases in vivo, indicating that the phosphoregulation of CDH1 is much more complex [127]. Another intriguing phenomenon is about the timing of degradation of proteins controlled by the same substrate adapters. We know that CDC20 functions as an upstream adapter protein for Cyclin A, Cyclin B, and Securin, mediating their ubiquitination and proteasomal degradation during mitosis. Interestingly, degradation of Cyclin A proceeds before that of Cyclin B and Securin, which is governed by the presence of spindle checkpoint signaling [128]. However, the detailed mechanisms underlying the spindle checkpoint imposed various E3 ligase activities of CDC20 toward different substrates need to be further investigated.
Overall, the cell cycle progression is tightly regulated to ensure the genomic integrity and identity in daughter cells, and ubiquitin signaling involves almost each step of the cell cycle. Dysregulation of the ubiquitination modification led to uncontrolled cell cycle progression and eventually resulted in tumorigenesis [18]. Based upon this notion, targeting the ubiquitin system has provided effective therapeutic strategies for cancer treatment [129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146]. At the moment, how these signaling events are integrated and orchestrated in a time–space-dependent manner remains not fully understood. In addition, only a handful of drugs targeting the ubiquitin system have been approved by the FDA. Therefore, a better understanding of the ubiquitin signaling in cell cycle control will expand and diversify the range of anticancer strategies and benefit the clinical treatment of cancer patients in the future.
References
Weinberg RA. The Retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30.
Zarkowska T, Mittnach S. Differential phosphorylation of the Retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem. 1997;272:12738–46.
Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006.
Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–71.
Petersen BO, Lukas J, Sørensen CS, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999;18:396–410.
Furuno N, Elzen ND, Pines J. Human cyclin A is required for mitosis until mid prophase. J Cell Biol. 1999;147:295–306.
Boer LD, Oakes V, Beamish H, Giles N, Stevens F, Torres MS, et al. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene. 2008;27:4261–8.
Vigneron S, Sundermann L, LabbéJC, Pintard L, Radulescu O, Castro A, et al. Cyclin A-cdk1-dependent phosphorylation of Bora is the triggering factor promoting mitotic entry. Dev Cell. 2018;45:637–50.
Lindqvist A, Zon WV, Rosenthal CK, Wolthuls RM. Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 2007;5:e123. https://doi.org/10.1371/journal.pbio.0050123.
Ferrero M, Ferragud J, Orlando L, Valero L, Pino MS, Farràs R, et al. Phosphorylation of AIB1 at mitosis is regulated by CDK1/Cyclin B. PLoS ONE. 2011;6:e28602. https://doi.org/10.1371/journal.pone.0028602.
Guo L, Mohd KS, Ren H, Xin G, Jiang Q, Clarke PR, et al. Phosphorylation of importin-alpha1 by CDK1-Cyclin B1 controls mitotic spindle assembly. J Cell Sci. 2019;132:jcs232314. https://doi.org/10.1242/jcs.232314.
Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12.
Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–8.
Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–72.
Tomoda K, Kubota Y, Kato J. Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature. 1999;398:160–5.
Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115:71–82.
Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol. 1997;9:788–99.
Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–81.
Goldstein G, Scheid M, Hammerling U, Schlesinger DH, Niall HD, Boyse EA. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc Natl Acad Sci USA. 1975;72:11–5.
Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79.
Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D. K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell. 2010;39:477–84.
Wickliffe KE, Williamson A, Meyer HJ, Kelly A, Rape M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol. 2011;21:656–63.
Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73:3497–506.
Emmerich CH, Schmukle AC, Walczak H. The emerging role of linear ubiquitination in cell signaling. Sci Signal. 2011;4:re5. https://doi.org/10.1126/scisignal.2002187.
Akutsu M, Dikic I, Bremm A. Ubiquitin chain diversity at a glance. J Cell Sci. 2016;129:875–80.
Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422.
Suryadinata R, Sadowski M, Sarcevic B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci Rep. 2010;30:243–55.
Swaffer MP, Jones AW, Flynn HR, Snijders A, Nurse P. CDK substrate phosphorylation and ordering the cell cycle. Cell. 2016;167:1750–61.
Obaya AJ, Sedivy JM. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol Life Sci. 2002;59:126–42.
Vodermaier HC. APC/C and SCF: controlling each other and the cell cycle. Curr Biol. 2004;14:R787–96.
Barford D. Structure, function and mechanism of the anaphase promoting complex (APC/C). Q Rev Biophys. 2011;44:153–90.
Zhang J, Wan L, Dai X, Sun Y, Wei W. Functional characterization of anaphase promoting complex/cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim Biophys Acta. 2014;1845:277–93.
Qiao R, Weissmann F, Yamaguchi M, Brown NG, VanderLinden R, Imre R, et al. Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc Natl Acad Sci USA. 2016;113:E2570–8.
Zhang S, Chang L, Alfieri C, Zhang Z, Yang J, Maslen S, et al. Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature. 2016;533:260–4.
Chang DC, Xu N, Luo KQ. Degradation of cyclin B is required for the onset of anaphase in mammalian cells. J Biol Chem. 2003;278:37865–73.
Singleton MR, Uhlmann F. Separase-securin complex: a cunning way to control chromosome segregation. Nat Struct Mol Biol. 2017;24:337–9.
Crasta K, Lim HH, Giddings TH Jr, Winey M, Surana U. Inactivation of Cdh1 by synergistic action of Cdk1 and polo kinase is necessary for proper assembly of the mitotic spindle. Nat Cell Biol. 2008;10:665–75.
Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol Cell. 1998;2:709–18.
Sullivan M, Uhlmann F. A non-proteolytic function of separase links the onset of anaphase to mitotic exit. Nat Cell Biol. 2003;5:249–54.
Li M, Zhang P. The function of APC/CCdh1 in cell cycle and beyond. Cell Div. 2009;4. https://doi.org/10.1186/1747-1028-4-2.
Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 2004;18:2573–80.
Ang XL, Harper JW. SCF-mediated protein degradation and cell cycle control. Oncogene. 2005;24:2860–70.
Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol. 2005;16:323–33.
D’Angiolella V, Esencay M, Pagano M. A cyclin without cyclin-dependent kinases: cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 2013;23:135–40.
Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCFSkp2–Cks1 ubiquitin ligase by the APC/CCdh1 ubiquitin ligase. Nature. 2004;428:190–3.
Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–8.
Geneviève R, Coulombe P, Tanguay PL, Boutonnet C, Meloche S. Phosphorylation of Skp2 regulated by CDK2 and Cdc14B protects it from degradation by APCCdh1 in G1 phase. EMBO J. 2008;27:679–91.
Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997;11:1464–78.
Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181–9.
Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1:193–9.
Bornstein G, Bloom J, Sitry-Shevah S, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278:25752–7.
Kamura T, Hara T, Kotoshiba S, Yada M, Ishida N, Imaki H, et al. Degradation of p57Kip2 mediated by SCFSkp2- dependent ubiquitylation. Proc Natl Acad Sci USA. 2003;100:10231–6.
Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev. 2004;18:851–5.
Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27Kip1, polyploidy and centrosome overduplication. EMBO J. 2000;19:2069–81.
Clijsters L, Hoencamp C, Calis JJ, Marzio A, Handgraaf SM, Cuitino MC, et al. Cyclin F controls cell-cycle transcriptional outputs by directing the degradation of the three activator E2Fs. Mol Cell. 2019;74:1264–77.
Burdova K, Yang H, Faedda R, Hume S, Chauhan J, Ebner D, et al. E2F1 proteolysis via SCF-cyclin F underlies synthetic lethality between cyclin F loss and Chk1 inhibition. EMBO J. 2019;38:e101443. https://doi.org/10.15252/embj.2018101443.
Dankert JF, Rona G, Clijsters L, Geter P, Skaar JR, Bermudez-Hernandez K, et al. Cyclin F-mediated degradation of SLBP limits H2A.X accumulation and apoptosis upon genotoxic stress in G2. Mol Cell. 2016;64:507–19.
D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, et al. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–34.
D’Angiolella V, Donato V, Vijayakumar S, Saraf A, Florens L, Washburn MP, et al. SCFCyclin F controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature. 2010;466:138–42.
Mavrommati I, Faedda R, Galasso G, Li J, Burdova K, Fischer R, et al. β-TrCP- and casein kinase II-mediated degradation of Cyclin F controls timely mitotic progression. Cell Rep. 2018;24:3404–12.
Li C, Vassilev A, DePamphilis ML. Role for Cdk1(Cdc2)/cyclin A in preventing the mammalian origin recognition complex’s largest subunit (Orc1) from binding to chromatin during mitosis. Mol Cell Biol. 2004;24:5875–86.
Gong D, Ferrell JE Jr. The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol Biol Cell. 2010;21:3149–61.
Abe S, Nagasaka K, Hirayama Y, Kozuka-Hata H, Oyama M, Aoyagi Y, et al. The initial phase of chromosome condensation requires Cdk1-mediated phosphorylation of the CAP-D3 subunit of condensin II. Genes Dev. 2011;25:863–74.
Elzen N, Pines J. Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol. 2001;153:121–36.
Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, Hunt T. Anaphase-promoting complex/cyclosome–dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol. 2001;153:137–48.
Hershko A. Mechanisms and regulation of the degradation of cyclin B. Philos Trans R Soc Lond B Biol Sci. 1999;354:1571–75.
Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–7.
Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell. 2002;9:931–43.
Wolthuis R, Clay-Farrace L, Zon W, Yekezare M, Koop L, Ogink J, et al. Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell. 2008;30:290–302.
Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–83.
Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty V, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci USA. 2004;101:4459–64.
Rossi M, Duan S, Jeong YT, Horn M, Saraf A, Florens L, et al. Regulation of the CRL4Cdt2 ubiquitin ligase and cell-cycle exit by the SCFFbxo11 ubiquitin ligase. Mol Cell. 2013;49:1159–66.
Fukushima H, Ogura K, Wan L, Lu Y, Li V, Gao D, et al. SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep. 2013;4:803–16.
Choudhury R, Bonacci T, Arceci A, Lahiri D, Mills CA, Kernan JL, et al. APC/C and SCFCyclin F constitute a reciprocal feedback circuit controlling S-phase entry. Cell Rep. 2016;16:3359–72.
Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TrCP. Proc Natl Acad Sci USA. 2004;101:4419–24.
Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, et al. SCFβTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–29.
Margottin-Goguet F, Hsu JY, Loktev A, Hsieh HM, Reimann J, Jackson PK. Prophase destruction of Emi1 by the SCFβTrCP/Slimb ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev Cell. 2003;4:813–26.
Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, et al. Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91.
Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, et al. Control of meiotic and mitotic progression by the F box protein β-Trcp1 in vivo. Dev Cell. 2003;4:799–812.
Guardavaccaro D, Frescas D, Dorrello NV, Peschiaroli A, Multani AS, Cardozo T, et al. Control of chromosome stability by the β-TrCP-REST-Mad2 axis. Nature. 2008;452:365–9.
Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–51.
Jin J, Shirogane T, Xu L, Nalepa G, Qin J, Elledge SJ, et al. SCFβ-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003;17:3062–74.
Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–73.
Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase-combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23:2050–6.
Marzio A, Puccini J, Kwon Y, Maverakis NK, Arbini A, Sung P, et al. The F-box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol Cell. 2019;73:224–37.
Cui D, Xiong X, Shu J, Dai X, Sun Y, Zhao Y. FBXW7 confers radiation survival by targeting p53 for degradation. Cell Rep. 2020;30:497–509.
Uckelmann M, Sixma TK. Histone ubiquitination in the DNA damage response. DNA Repair. 2017;56:92–101.
Joo HY, Zhai L, Yang C, Nie S, Erdjument-Bromage H, Tempst P, et al. Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature. 2007;449:1068–72.
Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966–80.
Varshavsky A. Naming a targeting signal. Cell. 1991;64:13–5.
Pfleger CM, Kirschner MW. The KEN box- an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14:655–65.
Fiore BD, Davey NE, Hagting A, Izawa D, Mansfeld J, Gibson TJ, et al. The ABBA motif binds APC/C activators and is shared by APC/C substrates and regulators. Dev Cell. 2015;32:358–72.
Skaar JR, Pagan JK, Pagano M. Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol. 2013;14:369–81.
Fuchs SY, Spiegelman VS, Kumar KG. The many faces of β-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene. 2004;23:2028–36.
Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 2001;105:645–55.
Cheng Y, Li G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev. 2012;31:75–87.
Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–7.
Ye X, Nalepa G, Welcker M, Kessler BM, Spooner E, Qin J, et al. Recognition of phosphodegron motifs in human cyclin E by the SCFFbw7 ubiquitin ligase. J Biol Chem. 2004;279:50110–9.
Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, et al. Structural basis of the Cks1-dependent recognition of p27Kip1 by the SCFSkp2 ubiquitin ligase. Mol Cell. 2005;20:9–19.
Smolders L, Teodoro JG. Targeting the anaphase promoting complex: common pathways for viral infection and cancer therapy. Expert Opin Ther Targets. 2011;15:767–80.
Hornig N, Knowles PP, McDonald NQ, Uhlmann F. The dual mechanism of separase regulation by securin. Curr Biol. 2002;12:973–82.
Li M, York JP, Zhang P. Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Mol Cell Biol. 2007;27:3481–8.
Manchado E, Guillamot M, Càrcer G, Eguren M, Trickey M, García-Higuera I, et al. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55α, δ phosphatase. Cancer Cell. 2010;18:641–54.
Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol. 2009;185:983–94.
Wang Z, Wan L, Zhong J, Inuzuka H, Liu P, Sarkar FH, et al. Cdc20: a potential novel therapeutic target for cancer treatment. Curr Pharm Des. 2013;19:3210–4.
García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol. 2008;10:802–11.
Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8:438–49.
Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93.
Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–47.
Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, et al. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci USA. 2001;98:5043–8.
Bhattacharya S, Garriga J, Calbó J, Yong T, Haines DS, Graña X. SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene. 2003;22:2443–51.
Li X, Zhao Q, Liao R, Sun P, Wu X. The SCFSkp2 ubiquitin ligase complex interacts with the human replication licensing factor Cdt1 and regulates Cdt1 degradation. J Biol Chem. 2003;278:30854–8.
Chan C, Morrow JK, Li C, Gao Y, Jin G, Moten A, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell. 2013;154:556–68.
Minella AC, Welcker M, Clurman BE. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proc Natl Acad Sci USA. 2005;102:9649–54.
Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA. 2004;101:9085–90.
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
Do K, Doroshow JH, Kummar S. Wee1 kinase as a target for cancer therapy. Cell Cycle. 2013;12:3159–64.
Cangi MG, Cukor B, Soung P, Signoretti S, Moreira G Jr, Ranashinge M, et al. Role of the Cdc25A phosphatase in human breast cancer. J Clin Invest. 2000;106:753–61.
Tsai W, Chung YM, Zou Y, Park S, Xu Z, Nakayama K, et al. Inhibition of FOXO3 tumor suppressor function by βTrCP1 through ubiquitin-mediated degradation in a tumor mouse model. PLoS ONE. 2010;5:e11171. https://doi.org/10.1371/journal.pone.0011171.
Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCFβTrCP E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44:304–16.
Zhang X, Cai J, Zheng Z, Polin L, Lin Z, Dandekar A, et al. A novel ER-microtubule-binding protein, ERLIN2, stabilizes Cyclin B1 and regulates cell cycle progression. Cell Disco. 2015;1:15024. https://doi.org/10.1038/celldisc.2015.24.
Das-Bradoo S, Ricke RM, Bielinsky AK. Interaction between PCNA and diubiquitinated Mcm10 is essential for cell growth in budding yeast. Mol Cell Biol. 2006;26:4806–17.
Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta. 2004;1695:189–207.
Park J, Cho J, Kim EE, Song EJ. Deubiquitinating enzymes: a critical regulator of mitosis. Int J Mol Sci. 2019;20:5997. https://doi.org/10.3390/ijms20235997.
Pfoh R, Lacdao IK, Saridakis V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr Relat Cancer. 2015;22:T35–54.
Tang Z, Shu H, Oncel D, Chen S, Yu H. Phosphorylation of Cdc20 by Bub1 provides a catalytic mechanism for APC/C inhibition by the spindle checkpoint. Mol Cell. 2004;16:387–97.
Hall MC, Warren EN, Borchers CH. Multi kinase phosphorylation of the APC/C activator Cdh1 revealed by mass spectrometry. Cell Cycle. 2004;3:1278–84.
Clijsters L, Ogink J, Wolthuis R. The spindle checkpoint, APC/CCdc20, and APC/CCdh1 play distinct roles in connecting mitosis to S phase. J Cell Biol. 2013;201:1013–26.
Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–44.
Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26:484–98.
Walczak H, Iwai K, Dikic I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012;10:23. https://doi.org/10.1186/1741-7007-10-23.
Wu-Baer F, Ludwig T, Baer R. The UBXN1 protein associates with autoubiquitinated forms of the BRCA1 tumor suppressor and inhibits its enzymatic function. Mol Cell Biol. 2010;30:2787–98.
Durcan TM, Tang MY, Pérusse JR, Dashti EA, Aguileta MA, McLelland GL, et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014;33:2473–91.
Locke M, Toth JI, Petroski MD. Lys11- and Lys48-linked ubiquitin chains interact with p97 during endoplasmic-reticulum-associated degradation. Biochem J. 2014;459:205–16.
Nucifora FC Jr, Nucifora LG, Ng CH, Arbez N, Guo Y, Roby E, et al. Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat Commun. 2016;7:11792. https://doi.org/10.1038/ncomms11792.
Palicharla VR, Maddika S. HACE1 mediated K27 ubiquitin linkage leads to YB-1 protein secretion. Cell Signal. 2015;27:2355–62.
Chastagner P, Israël A, Brou C. Itch/AIP4 mediates Deltex degradation through the formation of K29-linked polyubiquitin chains. EMBO Rep. 2006;7:1147–53.
Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J. 2008;411:249–60.
Yuan WC, Li YR, Lin SY, Chang LY, Tan YP, Hung CC, et al. K33-linked polyubiquitination of coronin 7 by Cul3-KLHL20 ubiquitin E3 ligase regulates protein trafficking. Mol Cell. 2014;54:596–600.
Huang H, Jeon MS, Liao L, Yang C, Elly C, Yates JR 3rd, et al. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity. 2010;33:60–70.
Liu Z, Dong X, Yi HW, Yang J, Gong Z, Wang Y, et al. Structural basis for the recognition of K48-linked Ub chain by proteasomal receptor Rpn13. Cell Discov. 2019;5. https://doi.org/10.1038/s41421-019-0089-7.
Zhang L, Xu M, Scotti E, Chen ZJ, Tontonoz P. Both K63 and K48 ubiquitin linkages signal lysosomal degradation of the LDL receptor. J Lipid Res. 2013;54:1410–20.
Ohtake F, Tsuchiya H, Saeki Y, Tanaka K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci USA. 2018;115:E1401–8.
Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene. 2010;29:4493–503.
Erpapazoglou Z, Walker O, Haguenauer-Tsapis R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells. 2014;3:1027–88.
Lauwers E, Jacob C, André B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J Cell Biol. 2009;185:493–502.
Taguchi SI, Honda K, Sugiura K, Yamaguchi A, Furukawa K, Urano T. Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS Lett. 2002;519:59–65.
Lindon C, Pines J. Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. J Cell Biol. 2004;164:233–41.
Laoukili J, Alvarez-Fernandez M, Stahl M, Medema RH. FoxM1 is degraded at mitotic exit in a Cdh1-dependent manner. Cell Cycle. 2008;7:2720–6.
Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell. 2005;8:25–33.
Acknowledgements
We apologize for not citing all relevant reports owing to space limitations. This work was supported in part by funding from NIH (R01CA200651 to WW).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
WW is a co-founder and consultant for the ReKindle Therapeutics. Other authors declare no competing financial interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dang, F., Nie, L. & Wei, W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ 28, 427–438 (2021). https://doi.org/10.1038/s41418-020-00648-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00648-0
This article is cited by
-
O-GlcNAcylation of E3 ubiquitin ligase SKP2 promotes hepatocellular carcinoma proliferation
Oncogene (2024)
-
Uncovering Cell Cycle Dysregulations and Associated Mechanisms in Cancer and Neurodegenerative Disorders: A Glimpse of Hope for Repurposed Drugs
Molecular Neurobiology (2024)
-
The cross talk of ubiquitination and chemotherapy tolerance in colorectal cancer
Journal of Cancer Research and Clinical Oncology (2024)
-
Leveraging a disulfidptosis/ferroptosis-based signature to predict the prognosis of lung adenocarcinoma
Cancer Cell International (2023)
-
High expression of ABCF1 is an independent predictor of poor prognosis in bladder cancer
BMC Urology (2023)
