
Abstract
Cancer cells are often exposed to a metabolically challenging environment with scarce availability of oxygen and nutrients. This metabolic stress leads to changes in the balance between the endogenous synthesis and exogenous uptake of fatty acids, which are needed by cells for membrane biogenesis, energy production and protein modification. Alterations in lipid metabolism and, consequently, lipid composition have important therapeutic implications, as they affect the survival, membrane dynamics and therapy response of cancer cells. In this article, we provide an overview of recent insights into the regulation of lipid metabolism in cancer cells under metabolic stress and discuss how this metabolic adaptation helps cancer cells thrive in a harsh tumour microenvironment.
Similar content being viewed by others
Background
The development and progression of cancer is typically accompanied by marked changes in the tumour microenvironment. The rapid growth and expansion of tumour tissue often leads to a poor and aberrant blood supply, resulting in hypoxia and a limited supply of nutrients. To thrive under these changing and challenging conditions, cancer cells adapt their metabolism, including that of lipids.1,2,3 One of the key features of this metabolic adaptation is the elevated de novo synthesis of fatty acids (FAs), which is observed in many different cancer types4,5,6,7,8 and is generally believed to be required to provide rapidly proliferating cancer cells with a constant supply of FAs for membrane biogenesis, energy production and protein modification. Fatty acid synthase (FASN), the rate-limiting enzyme in the FA synthesis pathway, has been widely reported to promote cancer progression.3 However, studies have shown that cancer cells can also acquire exogenous FAs by upregulating various FA-uptake mechanisms.9,10,11
Several factors, including genetic mutations, play a significant role in determining the relative dependence of cancer cells on lipid uptake versus endogenous synthesis. For instance, cells transformed by oncogenic HRASG12V display elevated lipid uptake, whereas cells transformed by constitutively active (myristoylated) AKT undergo increased de novo synthesis.9 In addition, crosstalk between tumour cells and the microenvironment affects the cellular lipid acquisition mode. Oxygen and nutrient deprivation can each independently induce metabolic stress, which affects the balance between FA synthesis and uptake,3,9 and consequently the lipid composition of cancer cells.12,13,14 This metabolic flexibility is particularly important for cancer cells within tumour tissues that are often exposed to temporal fluctuations in oxygen and nutrient availability.
This review discusses the effects of oxygen and nutrient deprivation, as separate parameters and in combination, on various aspects of lipid metabolism in cancer cells. We have performed an extensive literature survey, and, through an in-depth analysis of the results disseminated throughout the published literature, tried to explain the inconsistencies in reports and to link these inconsistencies to different experimental/analytical methods, including differences in cell line models, cell culture conditions or data presentation strategies. Therefore, in this review we have focused not only on summarising the major findings but also on detailing the methodological variations in previous reports, which are provided as supplemental information. The findings discussed in this review reveal that oxygen and nutrient deprivation limit cancer cells’ metabolic flexibility that otherwise allows these cells to switch between different pathways of FA acquisition. Hence, under such conditions the pathway currently adopted by the cancer cell could be the preferred target of anti-neoplastic strategies. Otherwise, therapeutic strategies simultaneously targeting several routes of lipid provision might be required.
Lipid metabolism under hypoxic conditions
Hypoxia is a common feature of many human tumours and is a consequence of high cell proliferation rates, resulting in increased oxygen consumption, and aberrant blood vessel development. Hypoxia promotes aggressive malignancy and is associated with poor prognosis in a wide range of cancer types.15,16,17 Oxygen deprivation results in the expression of hypoxia-inducible factors (HIFs), which mediate multiple protective mechanisms that help in maintaining oxygen homoeostasis by reducing oxidative metabolism and oxygen consumption.18,19,20,21 One such inducible factor, HIF-1α, is a key regulator of multiple cancer-related metabolic pathways, including glycolysis, glycogenesis, the TCA cycle, nucleotide metabolism, amino acid metabolism, leptin metabolism, lipid metabolism and others.22,23,24,25
FA synthesis under hypoxic conditions
Several groups have studied the effects of hypoxic stress on FA synthesis in cancer cells, but the results of these studies appear to be largely inconsistent. For instance, FASN expression has been reported to be increased,26,27,28 decreased11,29 or unaffected30 in hypoxic cancer cells (Table 1, Fig. 1; FA Synthesis). The most obvious differences among these studies come from the selection of cancer type or cell line models (Table 1), suggesting the potential existence of a cell-type-specific regulation of FA synthesis under hypoxia. However, detailed examination of the previous reports revealed other subtle to substantial differences in cell culture methods (Supplementary Table 1), which might also induce variations in the results. For instance, in some studies, cells were serum-starved prior to hypoxia induction,31 hypoxia was applied in combination with nutrient deprivation,12 or full serum media was supplemented with exogenous lipids.32 Indeed, Lewis et al.11 reported that, in glial cancer cells, hypoxia alone led to the decreased expression of FA synthesis markers, while hypoxia in combination with low-serum conditions led to an increased expression of these markers. Interestingly, hypoxic regions within tumours growing in vivo display increased FASN expression (Supplementary Table 1).26 Hence, we can speculate that cells in these hypoxic cores are simultaneously deprived of oxygen and nutrients. Further studies are required to prove this hypothesis under in vivo settings. In addition, FASN expression under hypoxic conditions seems to be dependent on cell seeding density. FASN expression was downregulated in high-density HepG2 human hepatoblastoma cultures, while it remained unaffected in low-density cell cultures.29

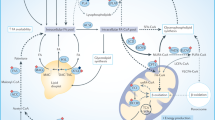
Overview of the major lipid metabolism pathways shown to be affected by metabolic stress. The figure highlights all the key lipid metabolism pathways activated in cancer cells. Major pathways are shown as boxes without outlines. The systematic names of these pathways are given at the bottom-right corner of each box. The numbers shown superscripted to each protein/metabolite indicate the reference number. For further details see the text and Supplementary Table 1. ACACA acetyl-CoA carboxylase 1, ACACB acetyl-CoA carboxylase 2, ACSS2 acyl Co-A synthetase-2, ADFP adipose differentiation protein, ATGL adipose triglyceride lipase, FA fatty acids, FABP3 fatty acid binding protein 3, FABP7 fatty acid binding protein 7, FFA free fatty acids, FASN fatty acid synthase, H hypoxia, HIF-1α hypoxia-inducible factor 1-α, HIF-2α hypoxia-inducible factor 2α, HIG2 hypoxia-inducible gene 2 protein, HMGCR 3-hydroxy-3-methylglutaryl-CoA reductase, LD lipid droplet, LCAD long-chain specific acyl-CoA dehydrogenase, MAG monoacylglycerol, MCAD medium-chain acyl-CoA dehydrogenase, MUFA monounsaturated fatty acids, PBMCs peripheral blood mononuclear cells, PC phosphatidylcholines, PE phosphatidylethanolamines, Pcho propargyl-choline, PI phosphatidylinositol, PS phosphatidylserine, PUFA polyunsaturated fatty acids, SCD stearoyl-CoA desaturase, SFA saturated fatty acids, SREBP sterol regulatory element-binding proteins, TG triglycerides
In hypoxic cancer cells, the expression of other enzymes that are involved in de novo lipid synthesis often show similar trends to those of FASN (Table 1). Most of these enzymes are under the transcriptional control of sterol regulatory-element binding protein-1 (SREBP-1). Hypoxia induces the activation of Akt and HIF-1, which is followed by the increased expression and activation of SREBP-1, which in turn induces the increased expression of FASN, among other biosynthetic enzymes.26 Conversely, the studies that reported a hypoxia-induced reduction in FASN expression showed that SREBP-1 expression is also decreased or unaffected under such conditions.11,29
In normoxic cells, glucose is the main source of acetyl-CoA for downstream lipid synthesis pathways (Fig. 1; FA synthesis and mevalonate pathway). However, hypoxia inhibits the entry of glucose-derived pyruvate into the TCA cycle,19,33 which consequently prevents glucose-based acetyl-CoA synthesis.19 To compensate, cancer cells adopt different metabolic mechanisms for FA synthesis—for instance, relying on glutamine or acetate as alternative substrates.34,35,36 Hypoxic breast, prostate, cervical, lung and colon cancer cells show increased acetate uptake, which has been shown to support tumour growth.12,34,37 Recent studies provide evidence that acetate is a fundamental nutrient that can fuel cancer growth under metabolic stress. Acetate can be endogenously generated through the removal of acetyl groups from histones by histone deacetylases38 and through hydrolysis of acetyl-CoA.39 It was recently shown that more than 80% of the intracellular acetate in cancer cells is generated de novo through glycolysis.40 While the expression of acyl-CoA synthetase short-chain family member 2 (ACSS2)—an enzyme that produces acetyl-CoA using acetate as a substrate—was found to be upregulated in hypoxic breast and prostate cancer cells,12,37 it was downregulated in hypoxic liver cancer cells;41 however, acetate supplementation increased ACSS2 and ACSS1 expression.41 These contrasting observations were attributed to the differences in cell culture methods. In addition, it has been shown that in hypoxic HepG2 cells acetate also functions as an epigenetic metabolite that enhances H3 acetylation levels at the promoter regions of FASN and acetyl-CoA carboxylase α (ACACA), resulting in increased expression of these genes, thus enhancing FA synthesis (Fig. 1; Nucleus).41 Interestingly, ACSS1 and ACSS2 were found to be involved in this acetate-induced epigenetic regulation of FA synthesis.41 Clinical samples of hepatocellular carcinoma with high ACSS1 and ACSS2 expression exhibit increased histone H3 acetylation and FASN expression.41 Thus, ACSS1 and ACSS2 were suggested to be potential therapeutic targets for cancer. It has been shown that co-silencing of FASN and ACSS2 induces cell death in biologically diverse cancer cell lines.13
Exogenous FA uptake under hypoxic conditions
In addition to adjusting for different substrates, hypoxic cancer cells might also use FA uptake pathways to compensate for reduced glucose-based de novo FA synthesis.3,9,10 Hypoxia increases FA uptake in breast cancer, ovarian cancer and glioblastoma cells by inducing the expression of FA-binding proteins (FABP3, FABP7 or FABP4), which are involved in the uptake and subcellular trafficking of FAs10,11,42 (Fig. 1, Table 1). Hypoxic cancer cells are known to accumulate lipid droplets.43 Bensaad et al.10 reported that this hypoxic accumulation of lipid droplets in breast cancer and glioblastoma cells is mediated by FABP-dependent FA uptake, while de novo FA synthesis is repressed. Gharpure et al.42 demonstrated that microRNA-409-3p regulates the expression of FABP4 in ovarian cancer cells. Hypoxia reduces the expression of microRNA-409-3p, thus potentially removing its inhibitory effect on FABP4, which subsequently results in increased FABP4 levels. These FABPs have been shown to be involved in tumour progression.10,42 In murine models, inhibition of FABP3,10 FABP710 and FABP442 impairs in vivo tumorigenesis.10 Clinical data also show that high FABP4 expression is significantly correlated with poor overall and progression-free survival.42,44
Expression of CD36, an FA uptake channel, is known to be upregulated under hypoxia in different non-transformed cells.45,46 CD36 has been previously implicated in tumour progression.7,8,47 Metastasis-initiating cells in human oral carcinomas display high levels of the CD36.48 Clinical data show that the presence of CD36+ metastasis-initiating cells correlates with a poor prognosis for numerous types of carcinomas, and inhibition of CD36 also impairs metastasis, at least in human melanoma- and breast cancer-derived tumours.48 Nevertheless, hypoxic regulation of CD36 in cancer cells is not studied in detail. Du et al.49 showed that at least in RCC4 clear cell renal cell carcinoma cells, hypoxic accumulation of lipid droplets was not mediated by CD36. Further studies are required to study this phenomenon in detail.
Hypoxia has been shown to increase FA uptake, mainly of monounsaturated FAs (MUFAs), in diverse cancer cell lines (Fig. 1).9 The reason for this specific MUFA uptake is not clearly understood, although it has been shown that cancer cells require MUFAs for survival.50 A specific balance between saturated FAs (SFAs) and unsaturated FAs is also critical for cancer cell progression. Alteration in the ratio of SFAs to MUFAs in cellular FA pools affects cell survival and proliferation via a number of interrelated mechanisms, including perturbations in mitochondrial function, heightened cellular reactive oxygen species (ROS), ER stress and apoptosis.51,52,53 FAs synthesised de novo are initially fully saturated; hence, a substantial fraction of de novo synthesised FAs will require desaturation by the activity of stearoyl-CoA desaturase-1 (SCD-1), a process that notably requires oxygen. Hypoxia therefore renders cells dependent on the uptake of exogenous unsaturated FAs. Indeed, limiting the supply of exogenous FAs to hypoxic cells leads to a critical deficiency in unsaturated FAs and results in cell death induced by endoplasmic reticulum stress.54 Thus, inhibiting FA uptake in hypoxic areas within tumours could represent a promising target for anticancer therapy. Indeed, a recent study has identified tamoxifen as a potential drug of interest that could inhibit FABP4 and subsequently affect migration of ovarian cancer cells.42
Lipid droplets and hypoxia
FAs acquired by cancer cells, either by endogenous synthesis or by exogenous uptake, are rapidly incorporated into cellular triglycerides (TGs), which form the core of lipid droplets in cells. Hypoxia-mediated accumulation of TGs and lipid droplets is accompanied by the increased expression of LIPIN1,43 an enzyme that catalyses the conversion of phosphatidic acid into diacylglycerol in the penultimate step of TG synthesis (Fig. 1; TG Synthesis). Hypoxia also promotes the storage of lipids in lipid droplets through the induction of perilipin 2 (PLIN2).10
Upon demand, fatty acyl moieties can be released from these stored TG deposits in lipid droplets via an intracellular lipolytic pathway (Fig. 1; Intracellular Lipolysis).55 Zechner et al.55 showed that monoglyceride lipase (MGLL) provides, by de-esterification, a stream of intracellular free FAs that can fuel cancer cell proliferation. Zhang et al.32 observed that intracellular lipolysis mediated by patatin-like phospholipase domain containing 2 (PNPLA2) [commonly known as adipose triglyceride lipase (ATGL)] is significantly reduced under hypoxic conditions.32 This lipolytic inhibition contributes to the accumulation of TGs and lipid droplets and cancer cell survival. The expression of PNPLA2 in hypoxic cancer cells remains unchanged; instead, PNPLA2 activity is inhibited by the protein encoded by hypoxia-inducible gene 2 (HIG2), a HIF-1 target.32 Experiments with murine xenograft models demonstrated that this inhibition of lipolysis by HIG2 is critical for tumour growth in vivo.32 Moreover, an abundance of HIG2 mRNA was observed in clinical samples collected from patients with renal clear cell carcinoma, colorectal adenocarcinoma, lung squamous cell carcinoma, bladder urothelial carcinoma, and uterine corpus endometrial carcinoma,32 indicating that PNPLA2 inhibition by HIG2 is a relevant mechanism for cancer pathophysiology in humans. As the accumulation of excess FAs can induce lipotoxicity, which might be especially important during the periods of hypoxia when FA uptake is upregulated, storage of excess FAs in lipid droplets through inhibition of PNPLA2-mediated lipolysis would constitute a conceivable strategy for cancer cells to evade lipotoxicity during hypoxic conditions.
FA oxidation under hypoxia
PNPLA2 also activates the FA oxidation pathway, leading to energy production, in normoxic cells.55 The de novo synthesis of FAs is considered an essential pathway for cancer cell survival and, according to the classical view, FA synthesis and FA oxidation cannot occur together. More recently, however, studies have challenged this notion and suggest that both metabolic pathways can be active simultaneously and independently of each other (reviewed in ref. 56). Under normal conditions, PNPLA2 channels excess FAs into the FA oxidation pathway by inducing the expression and activity of peroxisome proliferator-activated receptor-α (PPAR-α), a transcription factor and major regulator of lipid metabolism. However, sustained FA oxidation often results in increased generation of ROS, which may lead to oxidative stress and cell death.32 To counteract the damaging effects of ROS, PPARα also promotes the expression of various anti-oxidases, such as catalase and superoxide dismutase.57,58 In normoxia, mechanisms balancing ROS generation and degradation may maintain a steady-state redox environment. However, during hypoxia, a tilt towards excessive ROS production can occur as a result of increased electron leakage from the mitochondrial electron transport chain, leading to oxidative stress. Hence, inhibition of PNPLA2 activity by HIG2 under conditions of hypoxia also promotes cancer cell survival by reducing FA oxidation, ROS overproduction and oxidative damage. HIG2 is proposed to be a novel metabolic oncogenic factor that exerts its function by neutralizing the tumour suppressive role of PNPLA2. Development of drugs that disrupt HIG2-PNPLA2 interaction would liberate PNPLA2 and potentiate FA oxidation-driven ROS production to toxic levels, resulting in apoptotic death of hypoxic cancer cells.
A recent study49 showed that hypoxia also represses the expression of carnitine palmitoyltransferase 1A—a rate-limiting enzyme in long-chain FA oxidation—in clear-cell renal cell carcinoma. This reduces FA transport into the mitochondria and forces FA transport into lipid droplets for storage.
Huang et al.27 have reported another mechanism suppressing FA oxidation in hypoxia, by HIF-1α-mediated inhibition of acyl-CoA dehydrogenase medium-chain (ACADM) and acyl-CoA dehydrogenase long-chain (ACADL) expression. ACADM and ACADL catalyse the first step of FA oxidation in mitochondria (Fig. 1; β-Oxidation) but differ in the chain length of their FA substrates. Furthermore, ACADL is known to mediate unsaturated FA oxidation, whereas ACDM prefers SFAs as substrates. Interestingly, depletion of ACADL, but not ACADM, by HIF-1α promotes the progression of cancer cells, potentially by mediating the accumulation of unsaturated FAs. This observation was also found to be relevant in clinical settings. ACADL expression is downregulated in clinical hepatocellular carcinoma (HCC) samples in comparison to normal adjacent tissues.27 ACADL expression was shown to further decrease as HCC progressed to a higher clinical stage. ACADL expression in HCC patients was also linked to survival time, with patients expressing high levels of ACADL in their HCC lesions surviving much longer than those with low expression levels.
Extracellular lipolysis
Different types of cancer cells also express lipoprotein lipase (LPL), a lipolytic enzyme that is involved in the extracellular lipolysis of TG-rich lipoproteins (TGRL). LPL—bound to a heparin-like heparan sulfate proteoglycan motif either on the cancer cell surface or on the luminal surface of the vascular epithelium—has been speculated to release FAs from TGRLs (Fig. 1; Extracellular Lipolysis).7,8 These FAs can then enter the cancer cells via CD36, an FA uptake channel. In normal human preadipocytes, acute hypoxia strongly inhibits LPL-mediated lipolysis.59 LPL-regulated extracellular lipolysis in cancer cells may facilitate the hypoxia-induced FA uptake by rapidly releasing FAs from TGRLs in the tumour microenvironment. However, hypoxia-mediated regulation of LPL-regulated extracellular lipolysis in cancer cells has not been investigated in detail.
Lipid metabolism under conditions of nutrient deprivation
Poor blood perfusion in tumours reduces the availability of serum-derived factors and nutrients to the cancer cells, which might affect their lipid metabolism pathways. Regulation of the FA synthesis pathway in cancer cells cultivated in low-serum conditions has been studied (Table 1). Under such conditions, the availability of FAs, among several other nutrients, is restricted, making the cancer cells more reliant on endogenous synthesis for FA acquisition.12,60 As discussed above, the proliferation of various cancer cell types requires SCD1-mediated desaturation of endogenously synthesised SFAs into MUFAs, and SCD1, in turn, requires oxygen, so that, under oxygen-deprived conditions, cancer cells rely on an exogenous supply of MUFAs. However, in low-serum conditions, in which the exogenous supply of MUFAs is restricted, cancer cells instead upregulate their expression of SCD1 to facilitate increased endogenous FA desaturation. It has been reported that SCD1 inhibition induces cytotoxic effects on cancer cells in low-serum conditions, while it has little impact on cells cultured in full-serum conditions.50,60
This increased dependence of cancer cells on de novo FA synthesis and desaturation in low-serum conditions is thought to be mainly caused by the decreased availability of lipids/FAs. To further explore this hypothesis, we specifically investigated the effects of lipid deprivation on FA synthesis in cancer cells (Table 1) by cultivating cancer cells in media depleted of lipids or lipoproteins. We observed that in such conditions cancer cells differentially activated and thrived on endogenous lipid synthesis pathways.13 As expected, the expression and activity of SCD1 were also elevated.50 Therapeutic inhibition of SCD1 has been shown to efficiently reduce cancer cell proliferation by selectively depleting MUFAs.60,61,62 The inhibition of SCD1 increases the susceptibility of cancer cells towards chemotherapeutic drugs and metabolic inhibitors. Interestingly, it was shown that SCD inhibition is only detrimental to prostate cancer cell survival (in vitro) in the absence of exogenous lipids, particularly oleic acid.60 However, silencing of SCD reduces tumour growth in xenograft models of lung,63 gastric50 and liver cancer64, and also inhibits orthotopic growth of prostate cancer cells in vivo.60 Taken together, these studies suggest that unsaturated lipids are indeed limited within the tumour microenvironment. Further studies are required to demonstrate the effects of serum and lipid deprivation on other pathways involved in lipid metabolism.
Synergistic effects of hypoxia and nutrient deprivation on lipid metabolism
The combinatorial effects of hypoxia and nutrient deprivation, conditions that more closely mimic the in vivo tumour microenvironment, on de novo FA synthesis in cancer cells have been studied (Table 1). In such conditions, cancer cells upregulate de novo FA synthesis.11,12 As hypoxia also prevents glucose-based acetyl-CoA generation, cancer cells depend on glutamine or acetate as an alternative substrate for acetyl-CoA generation in conditions of hypoxia combined with low serum growth, to fuel their elevated FA synthesis pathway.12,35,65 Indeed, FA synthesis was shown to be decreased under hypoxic conditions, but increased when cancer cells were cultivated under hypoxia in combination with serum deprivation.11 As expected, expression of ACSS2 is elevated under conditions of oxygen deprivation and serum deprivation, but is mainly localised in the nucleus (Fig. 1; Acetate Metabolism).65 ACSS2 serves a dual function when oxygen and serum are limited: it facilitates the consumption of extracellular acetate as an alternative carbon source, but the increased nuclear localisation also enables cells to retain much of their endogenously produced acetate, which in turn allows cells to maintain sufficient acetylation of histones, to prevent initiation of apoptosis, and to maintain growth.65 The specific role of ACSS2 in cells that are hypoxic or metabolically stressed opens a therapeutic window in which ACSS2 could be rendered specifically toxic only to tumours. The therapeutic feasibility of pharmacologically targeting ACSS2 is currently being explored.
Effects of hypoxia and nutrient deprivation on lipid load and lipidomic profiles in cancer cells
As discussed above, hypoxic cancer cells modify the balance between FA synthesis and uptake, which leads to a significant accumulation of TGs and lipid droplets.43 Upon re-oxygenation, cells then use the lipids stored in droplets for energy production and antioxidant defence.10 However, the functional significance of the increased accumulation of lipid droplets during continued hypoxia is contentious. Hypoxic stress also promotes glycogen synthesis as a mechanism of storing glucose, while it is still available for later use as an anaerobic source of energy. However, as lipids can only produce energy through oxidative phosphorylation, a process drastically inhibited by hypoxia, a similar scenario may not arise in the case of lipids. Mylonis et al.43 proposed that TG storage by hypoxic cancer cells might help the cells to buffer the lipotoxicity caused by free FAs, which arises because of the suppressed respiratory-chain activity.66,67
Mammalian cells have a limited ability to synthesise polyunsaturated fatty acids (PUFAs) de novo, as they lack the Δ12 desaturase enzyme, and so cell membranes of lipogenic tumour tissues, with highly active de novo FA synthesis, are enriched in SFAs or MUFAs.68 As these FAs are less prone to lipid peroxidation than PUFAs, they make cancer cells more resistant to oxidative-stress-induced cell death68 and even to targeted therapies, including clinically used BRAF inhibitors. Furthermore, SFAs are packed more densely, and their increased levels are shown to alter membrane dynamics and limit drug uptake.68 Depletion of SFAs and MUFAs—by inhibiting FA synthesis—affects lateral and transversal membrane dynamics in cancer cells. It was shown that depletion of SFAs induces six-fold increase in the flip-flop rate of doxorubicin, accompanied by a significant increase in the intracellular accumulation of doxorubicin in prostate cancer cells.68 These findings suggest that tumour-associated FA synthesis protects the cancer cells against chemotherapeutic insults. They also highlight the significance of FA synthesis inhibitors as antineoplastic agents and chemotherapeutic sensitizers. Although several groups have studied the differential regulation of FA metabolism under metabolic stress, only a few have investigated lipidomic profiles10,28,30,31 (Table 1). It was reported that, under hypoxia and low serum conditions, the phosphatidylcholine and phosphatidylethanolamine profiles of breast cancer cells are altered, with an increase in the number of shorter and more saturated fatty acyl chains,12 indicating enhanced FA synthesis. HeLa cells, on the other hand, showed decreased levels of mono- and di-unsaturated, but increased levels of polyunsaturated, phospholipids (PLs).31
Schlaepfer et al.28 reported that hypoxic prostate cancer cells, and extracellular vesicles released by these cells, are significantly enriched in TGs due to the activation of the FA synthesis pathway. In addition, the TG sub-species profiles of these cells and the released extracellular vesicles were significantly altered. Cellular levels of myristic, palmitic and stearic FAs (all SFAs) were increased. The levels of linoleic acid and its derivative arachidonic acid, essential FAs implicated in tumour progression, were also significantly increased in the TGs of hypoxic cancer cells. Moreover, the levels of palmitic, stearic, linoleic and arachidonic acid were elevated in extracellular vesicles compared to those under normoxic conditions. Upon reoxygenation these cells break down the intracellularly stored lipids, and the generated energy induces increase in cell proliferation and the invasiveness potential of these cells. Interestingly, this increased proliferation and invasiveness were compromised by blocking the arachidonic acid pathway.28 Another study reported that, under hypoxic conditions, cellular levels of TGs with three double bonds significantly decreased in MCF7 breast cancer cells,10 but significantly increased in U87 glioblastoma cells.10 A recent study69 indicates that lipidomic profiles of leukaemia cells are predominantly affected by serum deprivation. Neutral lipid compositions are markedly modified under serum deprivation and, strikingly, the cellular levels of TG subspecies decreased with increasing number of double bonds in their fatty acyl chains. In contrast, cancer cells maintained lipid class homoeostasis under hypoxic stress. It was shown that, although the levels of individual lipid moieties alter under hypoxia, the robust averages of the broader lipid class remain unchanged. Another recent study70 also reported that low serum level affects TG composition in renal cancer cells, with significant decrease in the abundance of unsaturated TGs and a shift towards TG saturation. The authors also reported that hypoxia in combination with low serum induces decrease in levels of TGs harbouring unsaturated FAs and a shift towards increased TG saturation.
Hypoxia induces changes in the saturation index of membrane lipids in cancer cells that could affect cell membrane fluidity, dynamics and drug resistance. The role of hypoxia in drug resistance has been well known for at least 60 years now.71,72 Lipidomic analyses may also provide an explanation for the changes in membrane fluidity and dynamics observed in hypoxic cancer cells. The significance of alterations in TG subspecies profile in the context of tumour biology is being evaluated. Further studies, using broader lipidomic assays and larger panels of cell lines, are required to get a holistic view of lipidomic profiles in cancer cells under metabolic stress. These studies will not only help in understanding the role of lipids in cancer progression but also promote clinical applications of lipidomic profiling, including identification of potential therapeutic targets and diagnostic markers.
Conclusions
Dysregulation of lipid metabolism pathways in cancer cells has been widely reported. Multiple studies have indicated that such alterations are modulated by various cancer cell-intrinsic processes. However, emerging evidence suggests that these modifications are also mediated by crosstalk between the tumour microenvironment and metabolic circuits within cancer cells. The tumour microenvironment is mostly hypoxic. Although a few studies indicate that cancer cells might increase FA synthesis under hypoxia, most recent studies suggest that hypoxic cancer cells switch from endogenous FA synthesis to increased exogenous FA uptake because of the inhibition in glucose-based acetyl-CoA generation (Fig. 2). Alternatively, certain cancer cells, to compensate for this downregulation of glucose-based acetyl-CoA synthesis, switch to other carbon sources, such as glutamine or acetate. Nevertheless, FA desaturation by SCD1 requires oxygen and is therefore inhibited under hypoxic conditions, rendering cancer cells dependent on exogenous unsaturated FAs. In addition to the increased uptake of MUFAs, intracellular lipolysis and FA oxidation are downregulated under hypoxia, which results in the increased accumulation of TGs in lipid droplets. Increased TG synthesis and decreased lipolysis are both suggested to be strategies adopted by different cancer cells to evade lipotoxicity during hypoxia.

Effect of oxygen deprivation and/or nutrient deprivation on fatty acid (FA) metabolism in cancer cells. a Cancer cells with a sufficient supply of nutrients and oxygen mainly use glucose-derived acetyl-CoA for de novo FA synthesis to support rapid cell proliferation. They can also acquire FAs from the environment. b Different types of hypoxic cancer cells differentially regulate FA synthesis depending on various environmental factors—particularly, nutrient availability. Hypoxia inhibits the entry of glucose-derived pyruvate into the TCA cycle. Hence, cells either switch to alternative carbon sources (i.e. glutamine or acetate) for FA synthesis or increase their FA uptake. FA desaturation is impaired by oxygen deprivation; therefore, the uptake of unsaturated FAs is particularly enhanced. FAs are rapidly incorporated into cellular triglycerides (TGs) that enhance TG and lipid droplet (LD) accumulation. c Under nutrient and lipid restriction cancer cells mainly rely on endogenous FAs and desaturation. d When hypoxia is induced in combination with nutrient and lipid deprivation, cells cannot acquire FAs from the environment. Hence, they switch back to de novo FA synthesis, but fully depend on glutamine or acetate as an alternative substrate. Parts (b), (c) and (d) are drawn in comparison to the normoxic state depicted in (a). The line thickness represents the level of flux through the pathway. Dashed lines indicate biosynthetic pathways that are inactive due to lack of substrates or cofactors. The colours of the pathway boxes in (b), (c) and (d) represent upregulation (green), downregulation (grey) or differential regulation (green/grey) of the corresponding pathway in comparison to the normoxic state (with sufficient nutrient supply) in (a)
Nutrient and lipid deprivation renders cancer cells completely dependent on endogenous FA synthesis and desaturation. However, in conditions of hypoxia combined with nutrient and lipid deprivation, exogenous lipids are also in short supply. Hence, the cells switch back to de novo FA synthesis, but fully depend on glutamine or acetate as an alternative substrate. Different cancer cells may adopt different mechanisms to thrive under metabolic stress. Nevertheless, therapeutic targeting of de novo FA synthesis in cancer cells would be most effective under conditions that limit metabolic flexibility or, possibly, by concurrently targeting several routes of lipid acquisition. With the advent of better molecular techniques, including mass spectrometry imaging, to spatially visualise lipids in intact tissues, it will be possible to address several of the outstanding questions, to better correlate altered lipid profiles with changing microenvironments and to better explore the potential of lipid metabolism as an antineoplastic approach.
References
Beloribi-Djefaflia, S., Vasseur, S. & Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 5, e189 (2016).
Mullen, P. J., Yu, R., Longo, J., Archer, M. C. & Penn, L. Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718 (2016).
Röhrig, F. & Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16, 732 (2016).
Menendez, J. A. & Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763 (2007).
Brusselmans, K., Timmermans, L., Van de Sande, T., Van Veldhoven, P. P., Guan, G., Shechter, I. et al. Squalene synthase, a determinant of Raft-associated cholesterol and modulator of cancer cell proliferation. J. Biol. Chem. 282, 18777–18785 (2007).
Sebti, S. M. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell 7, 297–300 (2005).
Kuemmerle, N. B., Rysman, E., Lombardo, P. S., Flanagan, A. J., Lipe, B. C., Wells, W. A. et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 10, 427–436 (2011).
Zaidi, N., Lupien, L., Kuemmerle, N. B., Kinlaw, W. B., Swinnen, J. V. & Smans, K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog. Lipid. Res. 52, 585–589 (2013).
Kamphorst, J. J., Cross, J. R., Fan, J., de Stanchina, E., Mathew, R., White, E. P. et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl Acad. Sci. USA 110, 8882–8887 (2013).
Bensaad, K., Favaro, E., Lewis, C. A., Peck, B., Lord, S., Collins, J. M. et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 9, 349–365 (2014).
Lewis, C., Brault, C., Peck, B., Bensaad, K., Griffiths, B., Mitter, R. et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid-and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 34, 5128–5140 (2015).
Schug, Z. T., Peck, B., Jones, D. T., Zhang, Q., Grosskurth, S., Alam, I. S. et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 (2015).
Daniëls, V. W., Smans, K., Royaux, I., Chypre, M., Swinnen, J. V. & Zaidi, N. Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment. PLoS ONE 9, e106913 (2014).
Ackerman, D. & Simon, M. C. Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment. Trends Cell Biol. 24, 472–478 (2014).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Masson, N. & Ratcliffe, P. J. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab. 2, 3 (2014).
Rankin, E. B. & Giaccia, A. J. Hypoxic control of metastasis. Science 352, 175–180 (2016).
Gordan, J. D. & Simon, M. C. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr. Opin. Genetics Dev. 17, 71–77 (2007).
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L. & Denko, N. C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 (2006).
Rankin, E. & Giaccia, A. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 15, 678 (2008).
Semenza, G. L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29, 625 (2010).
Denko, N. C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705 (2008).
Harris, A. L. Hypoxia—a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38 (2002).
Semenza, G. L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721 (2003).
Semenza, G. L. HIF-1: upstream and downstream of cancer metabolism. Curr. Opin. Genetics Dev. 20, 51–56 (2010).
Furuta, E., Pai, S. K., Zhan, R., Bandyopadhyay, S., Watabe, M., Mo, Y.-Y. et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 68, 1003–1011 (2008).
Huang, D., Li, T., Li, X., Zhang, L., Sun, L., He, X. et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 8, 1930–1942 (2014).
Schlaepfer, I. R., Nambiar, D. K., Ramteke, A., Kumar, R., Dhar, D., Agarwal, C. et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 6, 22836 (2015).
Jung, S. Y., Jeon, H. K., Choi, J. S. & Kim, Y. J. Reduced expression of FASN through SREBP‐1 down‐regulation is responsible for hypoxic cell death in HepG2 cells. J. Cell. Biochem. 113, 3730–3739 (2012).
Valli, A., Rodriguez, M., Moutsianas, L., Fischer, R., Fedele, V., Huang, H.-L. et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1α-dependent and-independent pathways. Oncotarget 6, 1920 (2015).
Yu, Y., Vidalino, L., Anesi, A., Macchi, P. & Guella, G. A lipidomics investigation of the induced hypoxia stress on HeLa cells by using MS and NMR techniques. Mol. Biosyst. 10, 878–890 (2014).
Zhang X., Saarinen A. M., Hitosugi T., Wang Z., Wang L., Ho T. H. et al. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017; 6; https://doi.org/10.7554/eLife.31132.
Kim, J.-w, Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell. Metab. 3, 177–185 (2006).
Kamphorst, J. J., Chung, M. K., Fan, J. & Rabinowitz, J. D. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2, 23 (2014).
Metallo, C. M., Gameiro, P. A., Bell, E. L., Mattaini, K. R., Yang, J., Hiller, K. et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2012).
Wise, D. R., Ward, P. S., Shay, J. E., Cross, J. R., Gruber, J. J., Sachdeva, U. M. et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl Acad. Sci. USA 108, 19611–19616 (2011).
Yoshii, Y., Furukawa, T., Yoshii, H., Mori, T., Kiyono, Y., Waki, A. et al. Cytosolic acetyl‐CoA synthetase affected tumor cell survival under hypoxia: the possible function in tumor acetyl‐CoA/acetate metabolism. Cancer Sci. 100, 821–827 (2009).
Inoue, A. & Fujimoto, D. Enzymatic deacetylation of histone. Biochem. Biophys. Res. Commun. 36, 146–150 (1969).
Knowles, S. E., Jarrett, I. G., Filsell, O. H. & Ballard, F. J. Production and utilization of acetate in mammals. Biochem. J. 142, 401–411 (1974).
Liu, X., Cooper, D. E., Cluntun, A. A., Warmoes, M. O., Zhao, S., Reid, M. A. et al. Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175, 502–513. e513 (2018).
Gao, X., Lin, S.-H., Ren, F., Li, J.-T., Chen, J.-J., Yao, C.-B. et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960 (2016).
Gharpure, K. M., Pradeep, S., Sans, M., Rupaimoole, R., Ivan, C., Wu, S. Y. et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 9, 2923 (2018).
Mylonis, I., Sembongi, H., Befani, C., Liakos, P., Siniossoglou, S. & Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 125, 3485–3493 (2012).
Tothill, R. W., Tinker, A. V., George, J., Brown, R., Fox, S. B., Lade, S. et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 14, 5198–5208 (2008).
Mwaikambo, B. R., Yang, C., Chemtob, S. & Hardy, P. Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1- and phosphatidylinositol 3-kinase-dependent mechanisms. J. Biol. Chem. 284, 26695–26707 (2009).
Chabowski, A., Gorski, J., Calles-Escandon, J., Tandon, N. N. & Bonen, A. Hypoxia-induced fatty acid transporter translocation increases fatty acid transport and contributes to lipid accumulation in the heart. FEBS Lett. 580, 3617–3623 (2006)
Hale, J. S., Otvos, B., Sinyuk, M., Alvarado, A. G., Hitomi, M., Stoltz, K. et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 32, 1746–1758 (2014).
Pascual, G., Avgustinova, A., Mejetta, S., Martin, M., Castellanos, A., Attolini, C. S. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017).
Du, W., Zhang, L., Brett-Morris, A., Aguila, B., Kerner, J., Hoppel, C. L. et al. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat. Commun. 8, 1769 (2017).
Roongta, U. V., Pabalan, J. G., Wang, X., Ryseck, R.-P., Fargnoli, J., Henley, B. J. et al. Cancer cell dependence on unsaturated fatty acids implicates stearoyl-CoA desaturase as a target for cancer therapy. Mol. Cancer Res. 9, 1551–1561 (2011).
Inoguchi, T., Li, P., Umeda, F., Yu, H. Y., Kakimoto, M., Imamura, M. et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939–1945 (2000).
Alkhouri, N., Dixon, L. J. & Feldstein, A. E. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 3, 445–451 (2009).
van de Weijer, T., Schrauwen-Hinderling, V. B. & Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 92, 10–18 (2011).
Young, R. M., Ackerman, D., Quinn, Z. L., Mancuso, A., Gruber, M., Liu, L. et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress.. Genes Dev. 27, 1115–1131 (2013).
Zechner, R., Zimmermann, R., Eichmann, T. O., Kohlwein, S. D., Haemmerle, G., Lass, A. et al. FAT SIGNALS-lipases and lipolysis in lipid metabolism and signaling. Cell. Metab 15, 279–291 (2012).
Carracedo, A., Cantley, L. C. & Pandolfi, P. P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer 13, 227–232 (2013).
Khoo, N. K., Hebbar, S., Zhao, W., Moore, S. A., Domann, F. E. & Robbins, M. E. Differential activation of catalase expression and activity by PPAR agonists: implications for astrocyte protection in anti-glioma therapy. Redox Biol. 1, 70–79 (2013).
Liu, X., Jang, S. S., An, Z., Song, H., Kim, W.-D., Yu, J.-R. et al. Fenofibrate decreases radiation sensitivity via peroxisome proliferator-activated receptor α-mediated superoxide dismutase induction in HeLa cells. Radiat. Oncol. J. 30, 88 (2012).
Mahat, B., Chassé, É., Mauger, J.-F. & Imbeault, P. Effects of acute hypoxia on human adipose tissue lipoprotein lipase activity and lipolysis. J. Transl. Med. 14, 212 (2016).
Peck, B., Schug, Z. T., Zhang, Q., Dankworth, B., Jones, D. T., Smethurst, E. et al. Inhibition of fatty acid desaturation is detrimental to cancer cell survival in metabolically compromised environments. Cancer Metab. 4, 6 (2016).
Mason, P., Liang, B., Li, L., Fremgen, T., Murphy, E., Quinn, A. et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 7, e33823 (2012).
Hess, D., Chisholm, J. W. & Igal, R. A. Inhibition of stearoylCoA desaturase activity blocks cell cycle progression and induces programmed cell death in lung cancer cells. PLoS ONE 5, e11394 (2010).
Scaglia, N. & Igal, R. A. Inhibition of Stearoyl-CoA Desaturase 1 expression in human lung adenocarcinoma cells impairs tumorigenesis. Int. J. Oncol. 33, 839–850 (2008).
Budhu, A., Roessler, S., Zhao, X., Yu, Z., Forgues, M., Ji, J. et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 144, 1066–1075.e1061 (2013).
Bulusu, V., Tumanov, S., Michalopoulou, E., van den Broek, N. J., MacKay, G., Nixon, C. et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 18, 647–658 (2017).
Fakas, S., Qiu, Y., Dixon, J. L., Han, G.-S., Ruggles, K. V., Garbarino, J. et al. Phosphatidate phosphatase activity plays a key role in protection against fatty acid-induced toxicity in yeast. J. Biol. Chem. 286, 29074–29085 (2011).
Turkish, A., Sturley, S.L. Regulation of triglyceride metabolism. I. Eukaryotic neutral lipid synthesis: “Many ways to skin ACAT or a DGAT”. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G953–G95 (2007)
Rysman, E., Brusselmans, K., Scheys, K., Timmermans, L., Derua, R., Munck, S. et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 70, 8117–8126 (2010).
Lisec J., Jaeger C., Zaidi N. Cancer cell lipid class homeostasis is altered under nutrient-deprivation but stable under hypoxia. 2018: 382457; 10.1101/382457 %J bioRxiv.
Ackerman, D., Tumanov, S., Qiu, B., Michalopoulou, E., Spata, M., Azzam, A. et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 24, 2596–2605. e2595 (2018).
Gray, L. H., Conger, A. D., Ebert, M., Hornsey, S. & Scott, O. C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 26, 638–648 (1953).
Rohwer, N. & Cramer, T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. updat. 14, 191–201 (2011).
Zaidi, N., Royaux, I., Swinnen, J. V. & Smans, K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell-and environment-dependent mechanisms. Mol. Cancer Ther. 11, 1925–1935 (2012).
Usman, H., Ameer, F., Munir, R., Iqbal, A., Zaid, M., Hasnain, S. et al. Leukemia cells display lower levels of intracellular cholesterol irrespective of the exogenous cholesterol availability. Clinica Chimica Acta 457, 12–17 (2016).
Ameer, F., Munir, R., Usman, H., Rashid, R., Shahjahan, M., Hasnain, S. et al. Lipid-load in peripheral blood mononuclear cells: impact of food-consumption, dietary-macronutrients, extracellular lipid availability and demographic factors. Biochimie 135, 104–110 (2017).
Author contributions
All authors participated in writing, generation of figures and literature survey for this review article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable
Data availability
Not applicable
Consent for publication
Not applicable
Funding
Work from the author’s laboratory was supported by Alexander von Humboldt Foundation and Higher Education Commission of Pakistan (Project # 2505/R&D/11–2670).
Note
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution 4.0 International (CC BY 4.0).
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Munir, R., Lisec, J., Swinnen, J.V. et al. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer 120, 1090–1098 (2019). https://doi.org/10.1038/s41416-019-0451-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-019-0451-4
This article is cited by
-
Dyslipidemia: prevalence and association with precancerous and cancerous lesions of the cervix; a pilot study
Lipids in Health and Disease (2024)
-
Single cell lineage tracing reveals clonal dynamics of anti-EGFR therapy resistance in triple negative breast cancer
Genome Medicine (2024)
-
Stearoyl-CoA desaturase 1 inhibition induces ER stress-mediated apoptosis in ovarian cancer cells
Journal of Ovarian Research (2024)
-
METTL16 inhibits papillary thyroid cancer tumorigenicity through m6A/YTHDC2/SCD1-regulated lipid metabolism
Cellular and Molecular Life Sciences (2024)
-
Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics
Journal of Hematology & Oncology (2023)
