
Abstract
BRAF and MEK inhibitors have shown remarkable clinical efficacy in BRAF-mutant melanoma; however, most patients develop resistance, which limits the clinical benefit of these agents. In this study, we found that the human melanoma cell clones, A375-DR and A375-TR, with acquired resistance to BRAF inhibitor dabrafenib and MEK inhibitor trametinib, were cross resistant to other MAPK pathway inhibitors. In these resistant cells, phosphorylation of ribosomal protein S6 (rpS6) but not phosphorylation of ERK or p90 ribosomal S6 kinase (RSK) were unable to be inhibited by MAPK pathway inhibitors. Notably, knockdown of rpS6 in these cells effectively downregulated G1 phase-related proteins, including RB, cyclin D1, and CDK6, induced cell cycle arrest, and inhibited proliferation, suggesting that aberrant modulation of rpS6 phosphorylation contributed to the acquired resistance. Interestingly, RSK inhibitor had little effect on rpS6 phosphorylation and cell proliferation in resistant cells, whereas P70S6K inhibitor showed stronger inhibitory effects on rpS6 phosphorylation and cell proliferation in resistant cells than in parental cells. Thus regulation of rpS6 phosphorylation, which is predominantly mediated by BRAF/MEK/ERK/RSK signaling in parental cells, was switched to mTOR/P70S6K signaling in resistant cells. Furthermore, mTOR inhibitors alone overcame acquired resistance and rescued the sensitivity of the resistant cells when combined with BRAF/MEK inhibitors. Taken together, our findings indicate that RSK-independent phosphorylation of rpS6 confers resistance to MAPK pathway inhibitors in BRAF-mutant melanoma, and that mTOR inhibitor-based regimens may provide alternative strategies to overcome this acquired resistance.
Similar content being viewed by others


Introduction
Melanoma, the most lethal skin cancer, is among the most aggressive and treatment-resistant human cancers [1]. More than 50% of melanomas harbor mutations in the proto-oncogene BRAF [2], with amino acid substitution of valine to glutamic acid at position 600 (V600E) accounting for approximately 75–90% of BRAF mutations [3]. BRAFV600E, acting as a driver mutation, leads to constitutive phosphorylation and activation of mitogen-activated extracellular signal-regulated kinase 1/2 (MEK1/2), which in turn activates extracellular signal-regulated kinase 1/2 (ERK1/2) to phosphorylate downstream effectors [4]. The RAF/MEK/ERK mitogen-activated protein kinase (MAPK) cascade regulates cellular responses, including proliferation, differentiation, and survival signaling [5]. Thus inhibition of this pathway provides new therapeutic opportunities in malignant melanoma.
In recent years, several relevant inhibitors of this signaling pathway have been introduced for treating melanoma and have significantly improved overall survival in metastatic melanoma patients with BRAFV600E mutations. These agents include the selective BRAFV600E inhibitors vemurafenib and dabrafenib [6, 7], the MEK inhibitors trametinib and cobimetinib [8,9,10], and also the combination of dabrafenib/vemurafenib and trametinib/cobimetinib [11,12,13]. Although these single and combinatorial regimens produce robust positive responses, the clinical benefit is usually transient, owing to the rapid emergence of acquired resistance.
Several mechanisms responsible for the acquired resistance to BRAF- or MEK-targeted agents have been reported, including feedback reactivation of the MAPK pathway [14] and activation of parallel signaling pathways [15,16,17]. The tumor micro-environment also plays an important role in drug resistance. Long-term drug exposure leads to increased expression of melanoma antigens and decreased expression of the immunosuppressive cytokines interleukin-6 (IL-6) and IL-8 [3, 18].
Ribosomal protein S6 (rpS6) is a component of the 40S subunit of eukaryotic ribosomes and is therefore thought to be involved in regulating translation [19, 20]. rpS6 can be phosphorylated by p70 ribosomal S6 kinase (P70S6K), which is the downstream effector of mammalian target of rapamycin (mTOR) [20, 21]. Notably, accumulating evidence suggests that p90 ribosomal S6 kinase (RSK), an effector of the MAPK signaling pathway, is also capable of phosphorylating rpS6 on Ser235/236 through an mTOR-independent mechanism [22, 23]. Thus rpS6 represents a point of convergence of mTOR and MAPK signaling pathways. It has been reported that phosphorylated rpS6 is able to interact with cellular proteins and thus affect cellular physiological processes, including cell proliferation, cell size, and glucose homeostasis [24,25,26,27] Phosphorylation can be induced by a wide range of stimuli, such as growth factors, tumor-promoting agents, and mitogens [22, 26, 28]. Therefore, rpS6 has been proved to be instructive for neoplastic transformation.
Here we report a new mechanism of acquired resistance to dabrafenib and trametinib in BRAF-mutant melanoma. We found that constitutive phosphorylation of rpS6 in dabrafenib- and trametinib-resistant cells after treatment with dabrafenib/trametinib assisted cells in breaking through G0/G1 arrest by positively regulating the expression of cell cycle checkpoint proteins, thereby promoting cell proliferation. Furthermore, we demonstrate that constitutive activation of rpS6 in resistant cells is attributable to a switch in the regulation of rpS6 phosphorylation from the RSK to the P70S6K pathway. Importantly, we further show that mTOR inhibitor-based regimens are capable of overcoming acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma.
Material and methods
Materials
Dabrafenib, trametinib, BEZ235, and AZD2014 were obtained from Selleckchem (Shanghai, China). LY2584702, BI-D1870, GDC0994 and SCH772984 were obtained from Melonepharma (Dalian, China). Antibodies against rpS6, p-rpS6 (Ser235/236), P70S6K, p-P70S6K (Thr398), β-tubulin, p-ERK1/2 (Thr202/Tyr204), RSK, p-RSK (Ser380), AKT, p-AKT (Ser473), and p-mTOR (Ser2448) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against ERK were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Cell culture and treatment
A375 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. A375-TR (A375-trametinib resistant) and A375-DR (A375-dabrafenib resistant) cell lines were established from A375 cells by chronic treatment with gradually increasing concentrations of trametinib or dabrafenib, as described previously [29], and were selected for monoclones using the limiting dilution method.
Cell proliferation assay
Cell proliferation was determined by sulforhodamine B assay [30]. Cells were incubated with serial dilutions of drugs alone or as two-drug combinations in complete culture medium for 72 h. At least three independent experiments were performed. The curve-fitting software, GraphPad Prism version 5 (GraphPad Software, SanDiego, CA, USA), was used to calculate half-maximal inhibitory concentration values.
Cell cycle analysis
Cells were fixed with 70% ethanol at −20 °C overnight after treatment with drugs or small interfering RNA (siRNA) and then stained with propidium iodide. The cell cycle distribution was determined by fluorescence-activated cell sorting (FACS) using a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA) and analyzed with the ModFit LT 3.0 software (Verity Software House, Topsham, ME, USA) and the CellQuest Pro software (BD Biosciences, San Jose, CA, USA).
Western blotting
Cells were washed with phosphate-buffered saline and lysed with sodium dodecyl sulfate (SDS) sample buffer. Cell lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidine difluoride membranes. Immunoreactive proteins were analyzed using an enhanced chemiluminescence system (Thermo-Fisher Scientific, Waltham, MA, USA).
Clonogenic assay
Cells were seeded in six-well plates at low density (400 cells/well) and cultured for 15 days, after which colonies were fixed with methanol:acetic acid (3: 1) and stained with 1× crystal violet. The number of clones was counted using Photoshop CS5.
siRNA transfection
For siRNA studies, cells were transfected with siRNAs against rpS6 (sirpS6#1, sirpS6#2) or negative control siRNA (GenePharma, Shanghai, China) using the Lipofectamine 2000 transfection reagent (Thermo-Fisher Scientific). The siRNA sequences were as follows: sirpS6#1, 5’-CUAGCAGAAUCCGCAAACUTT-3’ (sense) and 5’-AGUUUGCGGAUUCUGCUAGTT-3’ (antisense); sirpS6#2, 5’-CUUCGUACUUUCUAUGAGATT-3’ (sense) and 5’-UCUCAUAGAAAGUACGAAGTT-3’ (antisense).
Microarray analysis
After treatment with trametinib for 12 h, A375 and A375-TR cells were lysed, and total RNA was extracted using an RNeasy Mini Kit (Qiagen, Germany). Agilent human genome 4×44K microarrays and Agilent 2100 Bioanalyzer were used to analyze the mRNA expression. Gene expression was compared using significant microarray analysis tools and gene set enrichment analysis. All sequencing and data analyses were performed at Shanghai Biotechnology Corporation (Shanghai, China).
Statistical analysis
The significance of differences between indicated groups was determined by Student’s t-tests using the GraphPad Prism Version 5 software (GraphPad Software, Inc., San Diego, USA).
Results
A375-DR and A375-TR cells are specifically resistant to MAPK pathway inhibitors
A375 cells were grown in medium containing increasing concentrations of the MEK inhibitor trametinib and BRAF inhibitor dabrafenib, ultimately yielding A375-TR and A375-DR cell lines, respectively. A375-TR cells were substantially less sensitive to trametinib than parental cells (resistance factor, 16.4) and were cross-resistant to selumetinib (MEK inhibitor), GDC0994 (ERK inhibitor), and dabrafenib (Table 1). Similarly, A375-DR cells were substantially less sensitive to dabrafenib than parental cells (resistance factor, 133.2) and were also cross-resistant to selumetinib, GDC0994, and trametinib. However, the cytotoxic agents, paclitaxel and doxorubicin, and the phosphoinositide-3 kinase (PI3K) inhibitor, GDC0941, exhibited the same potency in the two resistant cells as in the parental cell line (Table 1). These results demonstrate that A375-DR and A375-TR cell lines are specifically resistant to MAPK pathway inhibitors, suggesting that sustained MAPK signaling might underlie acquired resistance to BRAF and MEK inhibitors.
rpS6 is constitutively phosphorylated in resistant melanoma cells
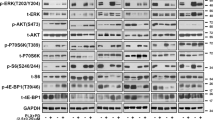
Given that the majority of previously described resistance mechanisms involve reactivation of MAPK or PI3K signaling pathways [14,15,16,17, we next investigated the expression of proteins involved in these two pathways in resistant cells. Phosphorylated ERK (p-ERK), RSK (p-RSK), and rpS6 (p-rpS6) levels were increased in A375-DR cells and p-AKT levels were increased in A375-TR cells, when compared to those of parental cells. However, the observed hyperphosphorylation of AKT was not related to the acquired resistance, as evidenced by the similar potency of GDC0941, in resistant cells and parental cells (Table 1). In addition, the expression levels of p-P70S6K and rpS6 were similar to those of A375 parental cell line (Fig. 1a).

MAPK pathway inhibitors do not inhibit rpS6 phosphorylation in A375-DR and A375-TR cells. a Whole-cell lysates were prepared and analyzed by Western blotting using the indicated antibodies. b A375, A375-DR, and A375-TR cells were treated with the indicated concentrations of trametinib or dabrafenib for 3 h. Cell lysates were analyzed by Western blotting with the indicated antibodies. c A375, A375-DR, and A375-TR cells were treated with the indicated concentrations of GDC0994 for 3 h. Cell lysates were analyzed by Western blotting with the indicated antibodies
Next, we examined the downstream effectors of MAPK signaling following treatment of these two resistant cell lines and parental A375 cells with dabrafenib or trametinib. Surprisingly, both trametinib and dabrafenib blocked ERK activation in resistant cells with a potency similar to that in parental A375 cells but did not block the activation of the downstream effector, rpS6, in either resistant cell line. A similar pattern was observed in resistant cells after treatment with the ERK inhibitor GDC0994, which decreased the levels of p-RSK but not those of p-rpS6 (Fig. 1c). Thus hyperphosphorylation of ERK/RSK/rpS6 in A375-DR cells plays no role in acquired dabrafenib resistance. Taken together, these results suggest that dysregulation of rpS6 phosphorylation might be involved in acquired resistance to MAPK pathway inhibitors.
rpS6 knockdown induces G1 phase arrest and inhibits proliferation of resistant melanoma cells
Previous reports have demonstrated that deletion of rpS6 blocks cellular proliferation [26, 31]. To determine whether rpS6 is essential for promoting proliferation of resistant cells, we next knocked down rpS6 using rpS6-targeted siRNA. Transfection of cells with two different rpS6-trageted siRNAs, sirpS6#1 and sirpS6#2, decreased the expression of rpS6 (Fig. 2a) and inhibited proliferation of both A375-TR and A375-DR cells (Fig. 2b). Resistant cells transfected with control (scrambled) siRNAs exhibited a time-dependent proliferation within 96 h similar to that of untreated parental cells. These results indicate that the melanoma cells resistant to MAPK pathway inhibitors could be targeted by rpS6 inhibition.

rpS6 knockdown suppresses cellular proliferation and induces cell cycle arrest in resistant cells. a After transfecting A375-DR and A375-TR cells with rpS6-targeted siRNA (sirpS6#1, sirpS6#2) or negative control siRNA (siCon), whole-cell lysates were prepared and probed for rpS6 and p-rpS6 by Western blotting. b After transfecting A375-DR and A375-TR cells with rpS6-targeted siRNA (sirpS6#1, sirpS6#2) or negative control siRNA (siCon), cellular proliferation was determined by cell proliferation assay. Results are presented as means ± SD (n = 3). The relative cell counts at 0 h is defined as 1.0. c Twenty-four hours after transfecting A375-DR and A375-TR cells with sirpS6#2 or siCon, cells were collected and analyzed by flow cytometry. Left: Representative experiment. Right: Summary data showing the percentage of G0/G1 phase cells in each cell line transfected with sirpS6#2 or control siRNA, expressed as means ± SD (n = 3; *P < 0.05). d A375, A375-TR, and A375-DR cells were treated with dabrafenib (100 nM) or trametinib (3 nM) for 48 h, then collected and analyzed by flow cytometry. Top: Representative experiment. Bottom: Summary data showing the percentage of G0/G1 phase cells in each cell line, expressed as means ± SD (n = 3; ***P < 0.001; NS, not significant)
Because it has been reported that rpS6 positively regulates the cell cycle [31], we next sought to determine whether the acquired resistance was attributable to rpS6-mediated promotion of cell cycle progression. Consistent with this possibility, siRNA targeting rpS6 (sirpS6#2) strongly induced G0/G1 phase arrest in both A375-DR and A375-TR cells (Fig. 2c). Furthermore, both A375-TR and A375-DR cells could break through the G0/G1 arrest caused by dabrafenib or trametinib; conversely, dabrafenib and trametinib induced a significant G0/G1 arrest in parental A375 cells (Fig. 2d), consistent with previous reports [32, 33]. Taken together, these findings suggest that rpS6 plays an important role in promoting cell cycle progression and proliferation in resistant cells and is involved in acquired resistance to MAPK pathway inhibitors in melanoma cells.
Dysregulation of rpS6-regulated G1 phase-related proteins contributes to the acquired resistance to MAPK pathway inhibitors
To elucidate the mechanism by which constitutive rpS6 phosphorylation causes resistance to trametinib/dabrafenib, we analyzed global changes in gene expression in A375-TR and A375 cells after treatment with trametinib using a human gene expression array. Trametinib treatment led to the alteration of global transcription profile, including upregulation of 1562 genes and downregulation of 1254 genes in A375 cells and upregulation of 771 genes and downregulation of 917 genes in A375-TR cells. Gene Ontology Enrichment analysis showed that the changed genes of A375 cells after trametinib treatment were involved in the regulation of cell proliferation and cell cycle (Fig. 3a), which was in accord with previous studies [34]. Upon further analysis, we found that some cell cycle checkpoint-related genes, including cyclin-dependent kinase 6 (CDK6) and CCND1 (cyclin D1), which are downstream effectors of rpS6 [26, 31], were much less affected by trametinib treatment in A375-TR cells than in A375 cells. Subsequent Western blotting assays confirmed the results of gene chip analyses, showing that trametinib and dabrafenib significantly decreased the expression of the cell cycle-related proteins, CDK6, cyclin D1, and p-RB in A375 cells but caused little change in their expression in both A375-TR and A375-DR cells (Fig. 3b).

Cell cycle checkpoint proteins regulated by constitutively active rpS6 are persistently expressed in resistant cells. a Heatmaps showing changes in genes known to be associated with regulation of cell proliferation in A375 and A375-TR cells. b A375, A375-TR, and A375-DR cells were treated with trametinib (1 nM) or dabrafenib (100 nM) for 48 h, after which cell cycle checkpoint proteins were detected by Western blotting. c Twenty-four hours after transfection of A375-DR and A375-TR cells with rpS6-targeted siRNA (sirpS6#2), cell cycle checkpoint proteins were determined by Western blotting. d A375-TR were incubated with trametinib (Tra; 3 nM) and/or PD0332991 (PD; 100 nM) for 48 h and A375-DR cells with dabrafenib (Dab; 100 nM) and/or PD0332991 (PD; 100 nM) for 48 h. The percentages of cells in different phases were analyzed by flow cytometry. Representative data are presented as means ± SD (n = 3)
To determine whether the lack of change in these cell cycle-related proteins was attributable to the constitutive phosphorylation of rpS6, we assessed the expression of CDK6, cyclin D1, and p-RB after siRNA-mediated knockdown of rpS6. The expression of these proteins was decreased in both A375-TR and A375-DR cells transfected with sirpS6#2 (Fig. 3c). In addition, co-treatment of A375-TR cells with the CDK4/6 inhibitor PD0332991 increased trametinib-induced G0/G1 phase arrest, indicating that targeting CDK4/6 alleviates the resistance of melanoma cells to MAPK pathway inhibitors (Fig. 3d). A similar effect was observed in A375-DR cells co-treated with PD0332991 and dabrafenib (Fig. 3d). Taken together, these observations indicate that acquired resistance to dabrafenib/trametinib is due, in part, to the persistent expression of cell cycle checkpoint proteins mediated by constitutively phosphorylated rpS6.
rpS6 phosphorylation is predominantly dependent on P70S6K in resistant melanoma cells
It has been previously reported that the RAF/MEK/ERK/RSK signaling cascade mediates rpS6 phosphorylation in BRAF-mutant melanoma [22]. Here we found that the RSK inhibitor BI-D1870, at a concentration of 10 μM, significantly decreased the phosphorylated rpS6 levels in parental A375 cells but not in A375-DR or A375-TR cells (Fig. 4a). Furthermore, A375 cells were more sensitive to BI-D1870 than that of A375-DR and A375-TR cells (Fig. 4b). BI-D1870 at a concentration of 1 μM significantly decreased the viability of A375 cells but only slightly decreased the viability of resistant cells (P < 0.05). These results indicate that phosphorylation of rpS6 is less dependent on RSK in these resistant melanoma cells.

Phosphorylation of rpS6 is primarily dependent on P70S6K in resistant cells. a A375, A375-DR, and A375-TR cells were incubated with the indicated concentrations of BI-D1870 for 3 h, after which p-ERK1/2, ERK1/2, p-RSK, RSK, p-rpS6, and rpS6 were determined by Western blotting. b Clonogenic assay of A375, A375-DR, and A375-TR cells after a 15-day treatment with BI-D1870. Top: Representative experiment. Bottom: Summary data showing inhibition rate, expressed as means ± SD (n = 3; *P < 0.05). c A375, A375-DR, and A375-TR cells were incubated with the indicated concentrations of LY2584702 for 3 h, after which p-P70S6K, P70S6K, p-rpS6, and rpS6 were determined by Western blotting. d Clonogenic assay of A375, A375-TR, and A375-DR cells after a 15-day treatment with trametinib (Tra; 3 nM), dabrafenib (Dab; 100 nM), and/or LY2584702 (LY; 3 μM). Top: Representative experiment. Bottom: Summary data showing inhibition rate, expressed as means ± SD (n = 3; *P < 0.05; **P < 0.01). e A375-TR cells were treated with LY2584702 (3 μM) and/or trametinib (10 nM) for 3 h, and A375-DR cells were treated with LY2584702 (3 μM) and/or dabrafenib (100 nM) for 3 h, after which p-rpS6 and rpS6 were determined by Western blotting
Because P70S6K is also an upstream regulator of rpS6 [20], the P70S6K inhibitor LY2584702 was used to investigate the possible modulation of rpS6 in resistant cells. Interestingly, LY2584702 more potently inhibited rpS6 phosphorylation in resistant cells than in parental cells (Fig. 4c). LY2584702 (3 μM) significantly decreased the phosphorylated rpS6 levels in A375-TR and A375-DR cells but not in A375 cells. Consistent with this, clonogenic assays showed that A375-TR and A375-DR cells were more sensitive to LY2584702 than that of A375 cells (Fig. 4d).
Next, we investigated the effects of combined treatment with LY2584702 and dabrafenib or trametinib in resistant cell lines. Combined treatment with dabrafenib/trametinib augmented the inhibitory effects of LY2584702 on rpS6 phosphorylation (Fig. 4e) and cell viability (Fig. 4d) in both A375-DR and A375-TR cells compared with each single agent alone. Although previous studies have demonstrated that rpS6 is predominantly regulated by RSK in cells in which MAPK signaling is hyperactivated [22], our results indicate that rpS6 is predominantly regulated by P70S6K in resistant cells, and chronic inhibition of P70S6K effectively and substantially inhibits the growth of melanoma cells that are resistant to MAPK pathway inhibitors.
Inhibition of mTOR overcomes resistance to MAPK pathway inhibitors
P70S6K is among the best-characterized downstream effectors of mTOR [35, thus, based on results obtained using the P70S6K inhibitor LY2584702, inhibition of mTOR would be expected to overcome the resistance to MAPK pathway inhibitors. Treatment with PI3K/mTOR inhibitor (BEZ235) or mTOR inhibitor (AZD2014) alone significantly reduced colony formation in A375-TR and A375-DR cells. Resistant cells were even more sensitive to BEZ235 and AZD2014 than that of parental A375 cells (Fig. 5a). Consistent with this, the inhibitory effect of rpS6 was more evident in A375-TR and A375-DR cells than in parental A375 cells after treatment with mTOR inhibitors (Fig. 5b). Phosphorylated AKT levels showed the same decreasing trends at the same concentrations of AZD2014 and BEZ235 in all three cell lines, further supporting the conclusion that hyperphosphorylation of AKT is merely an epiphenomenon in A375-TR cells (Fig. 1a). Taken together, these results indicate that mTOR/P70S6K might have roles in resistant cells different from those in parental cells and that rpS6 phosphorylation is more dependent on the mTOR/P70S6K signaling pathway in resistant BRAF-mutant melanoma cells.

Inhibition of mTOR overcomes acquired resistance to MAPK pathway inhibitors. a Clonogenic assay of A375, A375-TR, and A375-DR cells after 15-day treatment with BEZ235 (3 nM) or AZD2014 (100 nM). Top: Representative experiment. Bottom: Summary data showing inhibition rate, expressed as means ± SD (n = 3; **P < 0.01). b A375, A375-TR, and A375-DR cells were treated with the indicated concentrations of AZD2014 or BEZ235 for 3 h, after which whole-cell lysates were analyzed by Western blotting using the indicated antibodies. c A375-DR and A375-TR cells were incubated with the indicated concentrations of each compound or their combinations for 72 h. Inhibition rate, expressed as means ± SD (n = 3), was determined by cell proliferation assay. CI, combination index. d A375-DR and A375-TR cells were treated with trametinib (10 nM), dabrafenib (100 nM), and/or BEZ235 (10 nM) or AZD2014 (100 nM) for 3 h, after which cells were collected and analyzed by Western blotting
Previous studies have shown that combined inhibition of BRAF and mTOR signaling produces a greater response than single-agent treatments in BRAF-mutant cells [36, 37]. In light of this, we evaluated how combining mTOR inhibitors with RAF/MEK inhibitors affected the proliferation of resistant cells. mTOR inhibitor-based regimens were capable of re-sensitizing resistant melanoma cells to trametinib or dabrafenib treatment to a certain extent, as both AZD2014 and BEZ235 exerted synergistic antiproliferative effects on A375-DR and A375-TR cells when combined with trametinib or dabrafenib (Fig. 5c). The combination index (CI) values for BEZ235 and AZ2014 combined with dabrafenib in A375-DR cells were 0.45 and 0.63, and the corresponding CI values for BEZ235 and AZ2014 with trametinib in A375-TR cells were 0.56 and 0.67. To analyze the mechanism underlying these synergistic antiproliferative effects, we performed Western blotting analyses in A375-DR and A375-TR cells. These analyses showed that the combined treatment regimens exerted augmented inhibitory effects on rpS6 phosphorylation in both cell lines compared with each single agent alone (Fig. 5d). Taken together, these findings indicate that constitutive rpS6 phosphorylation plays an important role in therapy-resistant melanoma cells and that mTOR inhibitors alone or combined with MAPK pathway inhibitors are capable of overcoming this resistance.
Discussion
The MAPK signaling pathway is highly activated in the majority of malignant melanomas harboring activating mutations of the BRAF oncogene [4, 38]. Inhibition of key signaling proteins in this signaling pathway therefore represents an attractive strategy for treating melanoma. In the past 10 years, a number of small molecular inhibitors of MAPK signaling have been developed and shown to exert antitumor activity and improved survival of melanoma patients [7, 39, 40]. Unfortunately, the initially impressive response rates are limited by the resistance that rapidly and inevitably emerges, with most patients relapsing after long-term treatment [41]. Therefore, understanding the mechanism underlying drug resistance and developing strategies to overcome it is of paramount clinical importance [42]. Given the heterogeneity of melanomas, additional resistance mechanisms are likely to arise, and novel therapeutic strategies will be needed. In this study, we demonstrate that constitutive activation of rpS6, a downstream effector of RSK, confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma cells and that mTOR inhibitors are capable of overcoming this resistance.
The majority of the previously described resistance mechanisms involve reactivation of MAPK signaling, including amplification and secondary mutations of NRAS, MEK, or BRAF [14, 43], and RSK hyperphosphorylation [44]. In this study, we examined the sequences of NRAS, MEK, and BRAF and found no secondary mutations relevant to resistance in the cell lines with acquired resistance to MAPK pathway inhibitors (data not shown). Different from the previous studies [44], we found that the phosphorylation of ERK and RSK could still be decreased in A375-TR and A375-DR cell lines following treatment with MAPK pathway inhibitors. Activation of parallel signaling pathways, including receptor tyrosine kinase upregulation [17], growth factor secretion [15, 45], and dysregulation of the AKT pathway, have also been reported to be involved in resistance to MAPK pathway inhibitors [16, 46]. Here resistant cells and parental cells were found to be equally sensitive to the PI3K inhibitor GDC0941; moreover, AKT phosphorylation showed the same decreasing tendency in resistant and parental cells after the treatment with BEZ235 or AZD2014. Meanwhile, p-P70S6K, the downstream effector of mTOR, remained unchanged in both A375-TR and A375-DR cells. Collectively, these results indicate that the phosphorylation level of AKT/mTOR/P70S6K signaling pathway does not underlie resistance in these cell lines. Although our study showed that MAPK signaling and AKT/mTOR/P70S6K signaling were not consistently changed in these two resistant cell lines, both cell lines were specifically resistant to MAPK pathway inhibitors, and inhibition of MEK or BRAF decreased phosphorylation of rpS6 protein, a convergence of these two signaling pathways, in parental cells but not in the two resistant cell lines.
rpS6 is involved in mRNA binding and its phosphorylation might have a regulatory role in the translation of some cell cycle-related proteins [26, 31]. There have been some reports that address the contribution of rpS6 to unfavorable survival outcomes in non-small cell lung cancer and gastric cancer patients [31, 47], and constitutive p-rpS6 expression has also been addressed to indicate drug resistance in breast cancer cells [48]. Also, previous study has reported that the primary resistance of some BRAF-mutant melanoma cells to RAF or MEK inhibitor is related to maintenance of rpS6 phosphorylation [49]. Here we delve into the mechanism that could be involved in the regulation of rpS6 phosphorylation in cells with acquired drug resistance and report for the first time that switched modulation of rpS6 phosphorylation from RSK to P70S6K leads to acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma, by inducing persistent expression of cell cycle checkpoint proteins, including CDK6, cyclin D1, and p-RB, thereby attenuating cell cycle arrest. The CDK inhibitor PD0332991 also partly restored sensitivity of resistant cells and overcame acquired resistance to MAPK pathway inhibitors. Thus phosphorylation of rpS6 can be used as a diagnostic marker to evaluate the sensitivity of agents that target MAPK signaling in BRAF-mutant melanoma. This finding falls in line with the role of rpS6 as a predictive biomarker in cancers, proposed by Corcoran RB et al. [49] and Knoll et al. [50].
Cells are in a dynamic state and their signaling properties can be ‘rewired’ at any time. As a nodal cellular protein, p-rpS6 can switch from its reliance on one upstream regulator to another. In fact, rpS6 phosphorylation is predominantly regulated by P70S6K under normal circumstances but is more dependent on RSK under conditions in which MAPK signaling pathway is hyperactivated [28]. Interestingly, as shown in Fig. 6, our study showed that rpS6 phosphorylation was predominantly regulated by mTOR/P70S6K signaling in resistant cells, as evidenced by the stronger inhibition of rpS6 phosphorylation after treatment with mTOR/P70S6K inhibitors, as well as the diminished inhibition of rpS6 phosphorylation by an RSK inhibitor in resistant cells compared with parental cells. Accordingly, resistant cells were more sensitive to both mTOR inhibitors and P70S6K inhibitors and were insensitive to an RSK inhibitor than that of parental cells. Although our study showed that the predominant modulation of rpS6 phosphorylation switched from RSK to P70S6K in resistant cells, it remains important to understand the mechanism underlying this regulation.

Schematic diagram of molecular signaling pathways relevant to the resistance to MAPK pathway inhibitors. Schematic summary of key molecular players in parental and resistant cells. MAPK and mTOR/P70S6K signaling pathways simultaneously promote cellular proliferation. Phosphorylation of rpS6 on Ser235/236 is positively primarily regulated by hyperactivated MAPK signaling in A375 cells. However, in A375-DR and A375-TR cells, phosphorylation of rpS6 at Ser235/236 is more dependent on the mTOR/P70S6K signaling pathway, which mediates the resistance to MAPK pathway inhibitors. Dotted lines indicate weak regulation, and triple arrows denote strongly positive regulation
Several other mechanisms have been reported to account for the acquisition of resistance including dysregulation of metabolic reprogramming [18, 51], negative immune checkpoint regulation [52], and deregulation of enzymes during metastatic transition of melanomas [53]. Our preliminarily microarray analysis results showed obvious dysregulation of pathways associated with chemokine receptor binding and cytokine–cytokine receptor interaction. Accordingly, we speculate that long-term drug exposure leads to metabolic reprogramming, such that some secreted factors disrupt signaling associated with the binding of P70S6K and rpS6. Collectively, our results proved a new paradigm for interactions between mTOR/P70S6K and MAPK signaling cascades in regulating the phosphorylation of rpS6. Further studies will be needed to identify the upstream regulators of rpS6.
Although hyperactive MAPK signaling in melanomas has received the most research attention, accumulating evidence points to an important role for mTOR/P70S6K signaling in melanoma cells [54]. In a recent clinical study, dual inhibition of mTOR and MAPK signaling pathways doubled the progression-free survival benefit relative to either monotherapy [36, 37]. In this study, we found that mTOR inhibitors combined with MAPK pathway inhibitors exerted synergistic antitumor activity and overcame acquired drug resistance in BRAF-mutant melanoma. The combination led to enhanced inhibition of rpS6 phosphorylation, further supporting the role of p-rpS6 in the resistance to BRAF/MEK inhibitors. Thus combined inhibition of mTOR and MAPK signaling may be a promising strategy against BRAF-mutant melanoma, particularly for patients with tumors who are resistant to MAPK pathway inhibitors.
Taken together, our results conclusively prove that RSK-independent constitutive phosphorylation of rpS6 is responsible for the resistance to MAPK pathway inhibitors in BRAF-mutant melanoma and offer new insights into the downstream effectors of the MAPK pathway in cancer cells, especially for those nodal proteins. The predominant P70S6K-dependent phosphorylation of rpS6 in MAPK pathway inhibitor-resistant melanoma may provide a compelling rationale for clinical trials. Moreover, combination strategies targeting these two pathways merit further evaluation as a potential approach for treating melanomas that are refractory to MAPK pathway inhibitors.
References
Lo JenniferA, Fisher DE. The melanoma revolution: from UV carcinogenesis to a new era in therapeutics. Science. 2014;346:945–9.
Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic repro-gramming in melanoma. Trends Mol Med. 2015;21:164–71.
Welsh SJ, Rizos H, Scolyer RA, Long GV. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer. 2016;62:76–85.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Kolch. W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351:289–305.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65.
Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–9.
Rosen LS, LoRusso P, Ma WW, Goldman JW, Weise A, Colevas AD, et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Invest New Drugs. 2016;34:604–13.
Watanabe M, Sowa Y, Yogosawa M, Sakai T. Novel MEK inhibitor trametinib and other retinoblastoma gene (RB)-reactivating agents enhance efficacy of 5-fluorouracil on human colon cancer cells. Cancer Sci. 2013;104:687–93.
Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol. 2014;15:e371–81.
Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9.
Ascierto PA, McArthur GA, Dréno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248–60.
Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, et al. Increased MAPK reactivation in early resistance to dabrafenib/ trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694.
Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4.
Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/ PI3K. Cancer Cell. 2010;18:683–95.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7.
Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225–31.
Wool. IG, Chan. Y-L, Gliick. A. Structure and evolution of mammalian ribosomal proteins. Cell Biol. 1995;73:933–47.
Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342–8.
Khalaileh A, Dreazen A, Khatib A, Apel R, Swisa A, Kidess-Bassir N, et al. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 2013;73:1811–20.
Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–64.
Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1-/-/S6K2-/- mice exhibit perinatal lethality and rapamycin-sensitive 5'-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–24.
Kim TS, Jang CY, Kim HD, Lee JY, Ahn BY, Kim J. Interaction of Hsp90 with ribosomal proteins protects from ubiquitination and proteasome-dependent degradation. Mol Biol Cell. 2006;17:824–33.
Schumacher. AM, Velentza AV, Watterson DM, Dresios J. Death-associated protein kinase phosphorylates mammalian ribosomal protein S6 and reduces protein synthesis. Biochemistry. 2006;45:13614–21.
Volarevic. S, Stewart. MJ, Ledermann. B, Zilberman. F, Terracciano. L, Montini. E, et al. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science. 2000;288:2045–7.
Doerks T, Copley RR, Schultz J, Ponting CP, Bork P. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genome Res. 2002;12:47–56.
Meyuhas O. Chapter 1. Physiological roles of ribosomal protein S6: one of its kind. Int Rev Cel Mol Biol. 2008;268:1–37.
Xie CY, Chen XL, Zheng MY, Liu XH, Wang HB, Lou LG. Pharmacologic characterization of SHR8443, a novel dual inhibitor of phosphatidylinositol 3-kinase and mammalian target of rapamycin. Oncotarget. 2017;8:107977–90.
Zhao ZX, Zhu JM, Quan HT, Wang GM, Li B, Zhu WL, et al. X66, a novel N-terminal heat shock protein 90 inhibitor, exerts antitumor effects without induction of heat shock response. Oncotarget. 2016;7:29648–63.
Chen B, Tan Z, Gao J, Wu W, Liu L, Jin W, et al. Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J Exp Clin Cancer Res. 2015;34:126.
Gibney GT, Zager JS. Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin Drug Metab Toxicol. 2013;9:893–9.
Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000.
Haq R, Yokoyama S, Hawryluk EB, Jonsson GB, Frederick DT, McHenry K, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A. 2013;110:4321–6.
Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21.
Sanchez-Hernandez I, Baquero P, Calleros L, Chiloeches A. Dual inhibition of (V600E)BRAF and the PI3K/AKT/mTOR pathway cooperates to induce apoptosis in melanoma cells through a MEK-independent mechanism. Cancer Lett. 2012;314:244–55.
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/ AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–25.
Satyamoorthy K, Li G, Gerrero MR, Brose MS, Volpe P, Weber BL, et al. Constitutive mitogen-activated protein kinase activation in melanoma is mediated by both BRAF mutations and autocrine growth factor stimulation. Cancer Res. 2003;63:756–9.
Menzies AM, Long GV. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2014;20:2035–43.
Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14.
Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Cancer Res. 2011;29:3085–96.
Obaid NM, Bedard K, Huang WY. Strategies for overcoming resistance in tumours harboring BRAF mutations. Int J Mol Med. 2017;18:E585.
Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93.
Kosnopfel C, Sinnberg T, Sauer B, Niessner H, Schmitt A, Makino E, et al. Human melanoma cells resistant to MAPK inhibitors can be effectively targeted by inhibition of the p90 ribosomal S6 kinase. Oncotarget. 2017;8:35761–75.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9.
Shi H, Hong A, Kong X, Koya RC, Song C, Moriceau G, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4:69–79.
Zheng Z, Zheng Y, Zhang M, Wang J, Yu G, Fang W. Reciprocal expression of p-AMPKa and p-S6 is strongly associated with the prognosis of gastric cancer. Tumour Biol. 2016;37:4803–11.
Yang-Kolodji G, Mumenthaler SM, Mehta A, Ji L, Tripathy D. Phosphory-lated ribosomal S6 (p-rpS6) as a post-treatment indicator of HER2 signalling targeted drug resistance. Biomarkers. 2015;20:313–22.
Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Winokur D, Piris A., et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. https://doi.org/10.1126/scitranslmed.3005753 (2013).
Knoll M, Macher-Goeppinger S, Kopitz J, Duensing S, Pahernik S, Hohenfellner M, et al. The ribosomal protein S6 in renal cell carcinoma: functional relevance and potential as biomarker. Oncotarget. 2016;7:418–32.
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404.
Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang TD, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015;21:1639–51.
Kim H, Frederick DT, Levesque MP, Cooper ZA, Feng Y, Krepler C, et al. Downregulation of the ubiquitin ligase RNF125 underlies resistance of melanoma cells to BRAF inhibitors via JAK1 deregulation. Cell Rep. 2015;11:1458–73.
Karbowniczek M, Spittle CS, Morrison T, Wu H, Henske EP. mTOR is activated in the majority of malignant melanomas. J Invest Dermatol. 2008;128:980–7.
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (No. 81273546) and the Shanghai Science and Technology Committee (No. 14DZ2294100).
Author information
Authors and Affiliations
Contributions
L.-g.L. and C.-y.X. conceived and designed the experiments; M.-z.G. wrote the manuscript, performed experiments, and analyzed the data; H.-b.W., X.-l.C., W.-t.C., L.F., Y.L., and H.-t.Q. performed the experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Gao, Mz., Wang, Hb., Chen, Xl. et al. Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma. Acta Pharmacol Sin 40, 268–278 (2019). https://doi.org/10.1038/s41401-018-0020-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0020-z
Keywords
This article is cited by
-
Overexpressed p-S6 associates with lymph node metastasis and predicts poor prognosis in non-small cell lung cancer
BMC Cancer (2022)
-
Inhibiting insulin and mTOR signaling by afatinib and crizotinib combination fosters broad cytotoxic effects in cutaneous malignant melanoma
Cell Death & Disease (2020)
-
Generation and characterization of MEK and ERK inhibitors- resistant non-small-cells-lung-cancer (NSCLC) cells
BMC Cancer (2018)
